
Is a Key Regulator of Pancreaticobiliary Ductal System Development
The pancreaticobiliary ductal system connects the liver and pancreas to the intestine. It is composed of the hepatopancreatic ductal (HPD) system as well as the intrahepatic biliary ducts and the intrapancreatic ducts. Despite its physiological importance, the development of the pancreaticobiliary ductal system remains poorly understood. The SRY-related transcription factor SOX9 is expressed in the mammalian pancreaticobiliary ductal system, but the perinatal lethality of Sox9 heterozygous mice makes loss-of-function analyses challenging. We turned to the zebrafish to assess the role of SOX9 in pancreaticobiliary ductal system development. We first show that zebrafish sox9b recapitulates the expression pattern of mouse Sox9 in the pancreaticobiliary ductal system and use a nonsense allele of sox9b, sox9bfh313, to dissect its function in the morphogenesis of this structure. Strikingly, sox9bfh313 homozygous mutants survive to adulthood and exhibit cholestasis associated with hepatic and pancreatic duct proliferation, cyst formation, and fibrosis. Analysis of sox9bfh313 mutant embryos and larvae reveals that the HPD cells appear to mis-differentiate towards hepatic and/or pancreatic fates, resulting in a dysmorphic structure. The intrahepatic biliary cells are specified but fail to assemble into a functional network. Similarly, intrapancreatic duct formation is severely impaired in sox9bfh313 mutants, while the embryonic endocrine and acinar compartments appear unaffected. The defects in the intrahepatic and intrapancreatic ducts of sox9bfh313 mutants worsen during larval and juvenile stages, prompting the adult phenotype. We further show that Sox9b interacts with Notch signaling to regulate intrahepatic biliary network formation: sox9b expression is positively regulated by Notch signaling, while Sox9b function is required to maintain Notch signaling in the intrahepatic biliary cells. Together, these data reveal key roles for SOX9 in the morphogenesis of the pancreaticobiliary ductal system, and they cast human Sox9 as a candidate gene for pancreaticobiliary duct malformation-related pathologies.
Published in the journal:
. PLoS Genet 8(6): e32767. doi:10.1371/journal.pgen.1002754
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002754
Summary
The pancreaticobiliary ductal system connects the liver and pancreas to the intestine. It is composed of the hepatopancreatic ductal (HPD) system as well as the intrahepatic biliary ducts and the intrapancreatic ducts. Despite its physiological importance, the development of the pancreaticobiliary ductal system remains poorly understood. The SRY-related transcription factor SOX9 is expressed in the mammalian pancreaticobiliary ductal system, but the perinatal lethality of Sox9 heterozygous mice makes loss-of-function analyses challenging. We turned to the zebrafish to assess the role of SOX9 in pancreaticobiliary ductal system development. We first show that zebrafish sox9b recapitulates the expression pattern of mouse Sox9 in the pancreaticobiliary ductal system and use a nonsense allele of sox9b, sox9bfh313, to dissect its function in the morphogenesis of this structure. Strikingly, sox9bfh313 homozygous mutants survive to adulthood and exhibit cholestasis associated with hepatic and pancreatic duct proliferation, cyst formation, and fibrosis. Analysis of sox9bfh313 mutant embryos and larvae reveals that the HPD cells appear to mis-differentiate towards hepatic and/or pancreatic fates, resulting in a dysmorphic structure. The intrahepatic biliary cells are specified but fail to assemble into a functional network. Similarly, intrapancreatic duct formation is severely impaired in sox9bfh313 mutants, while the embryonic endocrine and acinar compartments appear unaffected. The defects in the intrahepatic and intrapancreatic ducts of sox9bfh313 mutants worsen during larval and juvenile stages, prompting the adult phenotype. We further show that Sox9b interacts with Notch signaling to regulate intrahepatic biliary network formation: sox9b expression is positively regulated by Notch signaling, while Sox9b function is required to maintain Notch signaling in the intrahepatic biliary cells. Together, these data reveal key roles for SOX9 in the morphogenesis of the pancreaticobiliary ductal system, and they cast human Sox9 as a candidate gene for pancreaticobiliary duct malformation-related pathologies.
Introduction
The pancreaticobiliary ductal system refers to the complex network of ducts that compose the hepatopancreatic ductal (HPD) system as well as the intrapancreatic and intrahepatic ductal networks. The HPD system consists of the extrahepatic duct, cystic duct, gallbladder, common bile duct, and extrapancreatic duct. It connects to the intrahepatic biliary ducts to enable bile flow and storage. The intrapancreatic ducts collect the digestive enzymes secreted by the pancreatic acinar cells. Pancreatic juice and bile flow to the hepatopancreatic ampulla to be released into the intestine and allow digestion and absorption of nutrients [1]. Malformations of the pancreaticobiliary ductal system impair the function of digestive organs and are associated with various congenital conditions whose causes are mostly unknown.
In mammals, the transcription factor Sox17 is specifically expressed in a segment of the ventral foregut from which the pancreaticobiliary ductal system derives [2]. This factor has been shown to be a master regulator of pancreaticobiliary ductal system formation by specifying, in conjunction with Hhex and Pdx1, different lineages of the liver, pancreas and HPD system [2]. The liver is specified as a group of cells that expresses Hhex but not Sox17 or Pdx1. The intrahepatic biliary network requires several signaling pathways including TGFβ, Notch and Wnt, to differentiate and mature (for a review, see [3]). In particular, Notch signaling has been shown to promote intrahepatic biliary differentiation and tubulogenesis [4]–[9]. Adjacent to the liver domain, cells expressing both Sox17 and Pdx1 delineate a domain that gives rise to the HPD system and pancreas [2]. After lineage segregation, Sox17+/Pdx1 − cells give rise to the HPD system under the regulation of downstream factors such as HNF6, HNF1β and Hhex [2] which themselves have been shown to play important roles in the development of the HPD system [10]–[12]. As for the intrapancreatic ducts, they arise from a subset of Pdx1+/Sox17 − cells that also express HNF6 and HNF1β [13], [14]. These two transcription factors regulate duct tubulogenesis as well as the differentiation of the epithelial cells lining the ducts [14].
The HPD system in zebrafish is morphologically similar to the one in amniotes. As in mammals, the zebrafish HPD system develops from a specific domain within the foregut endoderm that lies between the emerging liver and ventral pancreas [15]. HPD system patterning and differentiation depend on Fgf10 signaling [15], whereas the specification of the liver and ventral pancreas is regulated by the transcription factors Prox1 [16], [17] and Pdx1 [18], respectively. In the liver, hepatocytes and intrahepatic biliary cells derive from bipotential hepatoblasts [19]. Multiple genes encoding Jagged ligands and Notch receptors are expressed in the zebrafish liver during intrahepatic biliary duct formation [20]. Perturbation of Jagged-mediated Notch signaling impairs differentiation and morphogenesis of the intrahepatic biliary cells, whereas constitutive Notch activation induces ectopic bile duct formation [20], [21]. These studies support an evolutionarily conserved role for Notch signaling in intrahepatic duct development. Regarding the intrapancreatic ducts, live-imaging analyses of the Tg(Nkx2.2a(3.5kb):GFP) line revealed that they derive from cells in the ventral pancreatic bud that migrate towards the pancreatic islet to initiate the formation of a branched network [18], [22]. Molecular mechanisms regulating the development of the intrapancreatic ducts remain poorly understood.
Studies in mouse have shown that the transcription factor SOX9 is expressed in the intrahepatic and intrapancreatic ducts, as well as in the developing HPD system including the common bile duct, gallbladder and hepatopancreatic ampulla [23]–[25]. Sox9 belongs to the SRY-related box (SOX) gene family that encodes transcription factors containing an HMG box DNA-binding domain. In humans, SOX9 is expressed in the fetal brain, liver, testis and skeletal tissue [26]. Haploinsufficiency of SOX9 is associated with campomelic dysplasia (CD, OMIM #114290), which is characterized by severe skeletal malformations and sex reversal [26], [27]. More recently, it has been shown that SOX9 is also expressed in the early human fetal pancreas and analysis of CD individuals have revealed pancreatic defects including islet hypoplasia and reduction of hormone expression [28]. Consistent with the defects observed in CD patients, heterozygous knock-out mice are perinatal lethal due to skeletal abnormalities [29]. Conditional knock-out mice have been generated to study SOX9 function: pancreas-specific inactivation of Sox9 using Pdx1:Cre reveals a critical role in the maintenance of the pancreatic progenitor pool [25], whereas liver-specific inactivation of Sox9 using Albumin/α-fetoprotein (Alfp):Cre shows that it is required for the timely maturation of asymmetrical structures to symmetrical biliary ducts [23]. A potential role for SOX9 in HPD development has not yet been investigated due to the lack of a HPD-specific Cre line.
The zebrafish genome contains two sox9 orthologs, sox9a and sox9b, which exhibit partially overlapping expression patterns in the craniofacial cartilage, otic placodes and pectoral appendages [30]. Null mutants of sox9a exhibit cartilage defects that mimic those observed in human CD [31]. Although a similar phenotype has been reported for the sox9bb971 mutant [30], the chromosomal deletion which underlies the b971 lesion removes eleven other genes, greatly limiting the use of this allele to study the function of Sox9b.
Here, we dissect the requirement for Sox9b in the development of the pancreaticobiliary ductal system in zebrafish. We show that similar to mammalian Sox9, zebrafish sox9b is expressed in the pancreaticobiliary ductal system. Detailed phenotypic analysis of a sox9b TILLING mutant reveals that Sox9b regulates the formation of the HPD system as well as the morphogenesis of the intrapancreatic and intrahepatic ducts. Strikingly, the pancreaticobiliary phenotypes observed in larvae worsen during juvenile stages and lead to cholestasis in the homozygous mutant adult fish. We also observed a positive feedback loop between Sox9b and Notch signaling in the developing intrahepatic biliary cells: Notch signaling regulates sox9b expression, and in turn Sox9b is required to maintain Notch activity in the intrahepatic biliary cells.
Results
sox9b is expressed in the developing pancreaticobiliary ductal system
Intrigued by the recent data revealing Sox9 expression in the ductal trees of the liver and pancreas as well as in the HPD system in mouse [24], we analyzed the expression pattern of sox9b in zebrafish by in situ hybridization. We found that in addition to the head region and pectoral fins, sox9b is specifically expressed in the pancreaticobiliary ductal system (Figure 1A–1D). At 30 hours post fertilization (hpf), sox9b is expressed in a segment of the foregut endoderm (bracket, Figure 1A) that appears to give rise to the liver bud (arrow, Figure 1B) and the HPD primordium (bracket, Figure 1B). At 60 hpf, sox9b expression becomes evident in the intrahepatic ducts (arrow, Figure 1C) and then extends to the extra - and intrapancreatic ducts (white arrow, Figure 1D). In contrast to sox9b, sox9a does not appear to be expressed in the pancreaticobiliary ductal system in zebrafish (Figure S1, left panel). These data show that zebrafish sox9b recapitulates the expression pattern of mammalian Sox9 in the intrapancreatic and intrahepatic ducts as well as in the HPD system [23]–[25].

sox9bfh313 lesion is a nonsense mutation
To investigate the potential role of Sox9b in the formation of the pancreaticobiliary ductal system, we isolated a novel mutation in sox9b, sox9bfh313, in collaboration with the Zebrafish TILLING Consortium. Contrary to sox9bb971 which consists of a deletion of the lower tip of linkage group 3 [30], sox9bfh313 is a point mutation located in the first exon of sox9b (Figure 1E). The A to T transversion at position 302 leads to a premature stop codon at amino acid Lys68. This nonsense mutation likely leads to the synthesis of a truncated protein that lacks the HMG-box DNA binding domain (Figure 1F) and therefore would be non-functional. In situ hybridization analyses revealed a substantial decrease of sox9b expression in sox9bfh313 mutants at 72 hpf (data not shown), possibly via nonsense mediated mRNA decay. In order to analyze a potential redundancy between sox9 gene functions in zebrafish, we examined the expression of sox9a in sox9bfh313 mutants (Figure S1). As in wild-type, sox9a expression appears to be excluded from the digestive organs in sox9bfh313 mutants, suggesting that sox9a expression does not compensate for the reduction of Sox9b function in these mutants. Hence, sox9bfh313 is the first point mutation described for this gene in zebrafish and is likely to represent a severe loss-of-function allele.
sox9b mutants survive to adulthood and exhibit cholestasis associated with hepatic and pancreatic fibrosis and duct dilation
In contrast to a previous report describing the phenotypes of sox9bb971 mutants and sox9b morpholino-injected embryos [30], homozygous sox9bfh313 mutants exhibit a normal external morphology and do not show a curly-down body axis or craniofacial defects (data not shown). sox9b mutants survive to the adult stage but are much thinner than their wild-type or heterozygous siblings (data not shown). Dissection of the digestive system of 5-month old sox9b mutants revealed preserved anatomical relationships, including a three-lobed liver and correctly-looped intestine; however, both the liver and pancreas were strikingly dark green suggesting abnormal bile accumulation (cholestasis) in these organs (Figure 2A–2B). Hematoxylin-and-eosin staining of histological sections of mutant organs showed that both organs exhibited lesions with extensive proliferation and dilation of the ducts, which were surrounded by fibrotic tissue (Figure 2C–2D″). Interestingly, in the liver, ductal defects were restricted to the region that connects to the extrahepatic ductal system (dashed rectangle, Figure 2D) whereas the rest of the organ was much less affected. In contrast, ductal defects in the pancreas were present throughout the organ and worsened towards its distal part (Figure 2D). In the pancreas, the acinar compartment was greatly reduced and secondary islets could not be detected in the sections examined (Figure 2D″).

Sox9b is required for the patterning and differentiation of the HPD system
Due to the robust and highly conserved expression of sox9b in the pancreaticobiliary ductal system and the striking liver and pancreas phenotypes seen in the adult mutant fish, we decided to further investigate the roles of Sox9b in the development of these tissues. In zebrafish, the HPD system exhibits unique gene expression profiles that separate it from the liver and pancreas starting at early developmental stages [15]. At 50 hpf, the primordium of the HPD system can be distinguished by strong labeling with the 2F11 antibody, whose antigen remains to be identified [15], [32] (bracket, Figure 3A, 3A′), and low expression of the transcription factor Prox1 [15] (bracket, Figure 3C). In contrast, the liver and pancreas exhibit moderate labeling of 2F11 (Figure 3A, 3A′), but high expression of Prox1 (Figure 3C). In sox9b mutants, 2F11 labeling was mostly absent from the region where the presumptive HPD primordium resides (bracket, Figure 3B, 3B′). Moreover, we observed elevated expression of Prox1 in the same region (bracket, Figure 3D). 2F11 labeling showed that by 80 hpf, the HPD system in wild-type larvae has developed into different compartments, including the extrahepatic duct, cystic duct, common bile duct, and gallbladder [15] (Figure 3E, 3E′). At the equivalent stage, the differentiation of the HPD system had partially recovered in sox9b mutants as suggested by 2F11 labeling. However, it was severely dysmorphic, with no clear morphological distinction between the cystic duct, extrahepatic duct, and common bile duct (Figure 3F, 3F′). Furthermore, the mutant HPD system often seemed to intrude into the liver (Figure 3F), which was never observed in wild-type larvae. These data indicate that the HPD primordium in sox9b mutants exhibits patterning and differentiation defects.
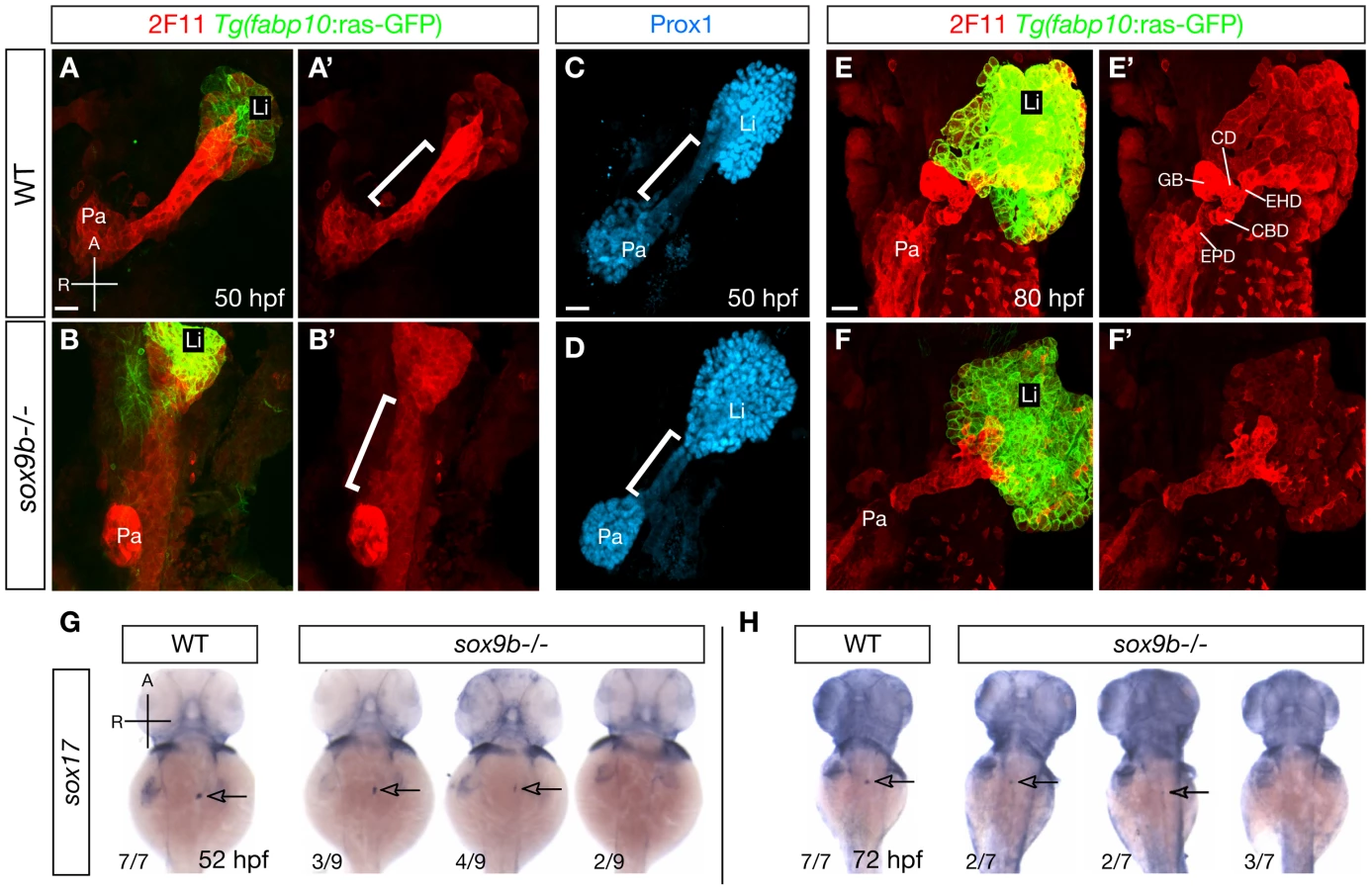
Concordant with the dysmorphic HPD system, the gallbladder in sox9b mutants was often indistinguishable based on morphology (Figure 3F and Figure S3A, S3B). We analyzed the expression of sox17 which marks the gallbladder and its primordium from 36 hpf to 5 days post-fertilization (dpf) [33], and found that it was greatly reduced or absent in sox9b mutants at 52 hpf (Figure 3G) and that it did not recover during later development (Figure 3H). This defect in sox17 expression supports the notion that gallbladder development is severely impaired in sox9b mutants.
sox9b mutants fail to form a complex network of ducts in the pancreas
We then addressed the role of Sox9b in intrapancreatic duct formation by using the double transgenic line Tg(Tp1bglob:GFP);Tg(Tp1bglob:H2B-mCherry) that expresses both GFP and H2B-mCherry under the control of a Notch-responsive element [34], [35]. This line allows the visualization of the shape and nuclei of the intrapancreatic duct cells, as indicated by the overlapping expression of these fluorescent proteins with ductal markers such as E-cadherin and 2F11 [34], [35]. Intrapancreatic ducts derive from cells within the ventral pancreatic bud that migrate towards, and eventually surround, the principal islet at 48 hpf [22]. From 60 hpf, ductal progenitors start to migrate caudally to form a row of cells that give rise to the main intrapancreatic duct [22] (Figure 4A–4A′). The migration of the ductal progenitors did not seem to be impaired in sox9b mutants; however, the number of cells within the intrapancreatic ducts was significantly reduced (Figure 4B–4B′, 4G). In wild-type larvae, at 100 hpf, the pancreatic tail keeps elongating, the number of ductal cells has slightly increased (Figure 4C–4C′, 4G) and secondary branches (arrowheads, Figure 4C–4C′) start to form from the main duct. In contrast, in sox9b mutants, the number of ductal cells did not increase from 80 to 100 hpf, and no secondary branches appeared, resulting in a primitive ductal system (Figure 4D–4D′, 4G). At 120 hpf, the ductal network in wild-type larvae has become more complex with numerous secondary branches (arrowheads, Figure 4E″) spreading over the acinar compartment (Figure 4E–4E′″). In contrast, the intrapancreatic ductal system in sox9b mutants remained poorly developed and clusters of cells could be observed along the main duct (Figure 4F–4F′″), which was still devoid of secondary branches. These data indicate that fewer intrapancreatic duct cells differentiate in the mutants and those that do fail to undergo branching morphogenesis. Furthermore, the number of ductal cells in sox9b mutants did not increase as in wild-types. Such a defect is likely due to a problem with cell differentiation as we did not observe any obvious differences in ductal cell proliferation or survival between wild-types and sox9b mutants (data not shown).

Given that the adult mutant pancreas exhibits a loss of acinar and potentially endocrine tissues, we analyzed the formation of these compartments during larval and juvenile stages. As assessed by Elastase staining, sox9b mutants showed apparently normal proportion of pancreatic acinar tissue at all stages analyzed (Figure 4B, 4D, 4F and Figure S2B″, S2D″, S2F″). As for the endocrine tissue, we investigated the morphology of the primary islet by analyzing TgBAC(neurod:GFP) expression which marks early endocrine cells [36]. At late larval (7 dpf) as well as juvenile (2 and 3 weeks) stages, the area of the primary islet appeared similar in wild-type and sox9b mutant animals (Figure 4H), suggesting that Sox9b is not required for primary islet formation. At 4 weeks of age, the sox9b mutant primary islet was half the size of the wild-type primary islet (Figure 4H). However, it is important to note that at this stage, sox9b mutant pancreata were also much less developed than wild-type pancreata (Figure S2E′″, S2F′″). Indeed, sox9b juvenile mutants often exhibit growth retardation compared to wild-types and thus, the smaller size of the primary islet could be attributed to an overall growth defect.
In addition to the primary islet, we investigated the formation of secondary islets that arise from progenitors in the intrapancreatic ducts [34], [35] and that, during larval and juvenile stages, appear as small clusters of delaminated cells [34]. We decided to also count single TgBAC(neurod:GFP)-positive cells that recently delaminated from the ducts and assumed an endocrine fate. Hence, counting the number of TgBAC(neurod:GFP)-positive cells/clusters along the intrapancreatic ducts (Figure S2A′–S2F′), we observed a difference between wild-type and sox9b mutant animals at two weeks of age. At 3 and 4 weeks of age, this difference became more pronounced with respectively a 50% and 80% decrease in TgBAC(neurod:GFP)-positive cell/cluster number in sox9b mutants (Figure 4I). Given that the mutant intrapancreatic ductal network remained primitive and failed to expand at juvenile stages (Figure S2D′″, S2F′″), the defect in secondary islet formation could be related to the lower number of progenitors within the pancreas. Altogether, these data indicate that Sox9b function is required for the development of the intrapancreatic ductal system as well as - directly or indirectly - for the formation of secondary islets.
Sox9b regulates intrahepatic biliary duct morphogenesis and bile canaliculi formation
To examine the role of Sox9b in intrahepatic biliary development, we used the Tg(Tp1bglob:GFP) line which also marks the intrahepatic biliary cells [21], [35]. During zebrafish liver development, the intrahepatic biliary cells undergo significant morphological changes, whereby these initially contiguous cells separate from one another and interconnect via cytoplasmic processes [21]. By 96 hpf, the wild-type intrahepatic biliary system is composed of a lattice-like network of long ducts joined by short interconnecting ducts [21] (Figure 5A). At the equivalent stage, sox9b mutant livers contained similar numbers of intrahepatic biliary cells and hepatocytes as wild-type (data not shown), suggesting that differentiation of the biliary cells is not affected in the mutants. We did not detect any apoptosis of the biliary cells in wild-type or mutant larvae. Strikingly, we observed that most of the biliary cells in the mutant livers failed to separate from one another (Figure 5B). We quantified the percentage of single intrahepatic biliary cells versus cells in cluster of two, three or four and more cells, and found a significant decrease in the percentage of single intrahepatic biliary cells in sox9b mutants compared to wild-types concomitant with a significant increase in the percentage of cells in clusters of four and more cells (Figure 5C). Moreover, the long bile ducts in the mutants appeared to be wider than those in wild-types (diameters of the mutant ducts: 3.5 µm or wider; wild-type ducts: 2.5 µm or thinner), and were less branched (Figure 5D).

We then used the Tg(fabp10:ras-GFP) line [37] to analyze hepatocyte organization, and co-labeled the animals with an antibody against the bile transporter BSEP to mark the bile canaliculi [38] (Figure 5E, 5G). At 96 hpf, hepatocytes in wild-type livers are arranged as tubules surrounding intrahepatic biliary ducts [20] (Figure 5E). Bile canaliculi are located on the hepatocyte apical membrane which can be marked by the activated leukocyte cell adhesion molecule Alcam [39]. However, in sox9b mutants, hepatocytes often formed spherical rosettes with bile canaliculi and Alcam expression located in the center (Figure 5G). This phenotype coincided with the aberrant clustering of intrahepatic biliary cells. Moreover, we found that the canaliculi in sox9b mutants appeared to be shorter and wider compared to wild-types (arrows, Figure 5F, 5H), which is consistent with recent data showing the highly coordinated development of intrahepatic biliary cells and bile canaliculi [21].
In liver-specific Sox9-inactivated mice, intrahepatic biliary duct morphogenesis is delayed until birth [23], which incited us to track the development of the intrahepatic biliary system in wild-types and sox9b mutants during juvenile stages. We found that intrahepatic biliary duct morphogenesis did not recover in sox9b mutants and that these animals did not generate morphologically normal bile ducts (Figure S2G–S2L). These data show that, despite a conserved requirement for SOX9 in intrahepatic biliary duct development, zebrafish sox9b mutants exhibit a much more severe intrahepatic biliary duct phenotype than the liver-specific knockout mouse model.
Loss of Sox9b function causes severe defects in bile transport
To determine whether the cholestasis-like phenotype observed in adult sox9b mutants occurred during early larval development, we administered fluorescent lipid analogs used to visualize bile transport [40] to 6 dpf-old wild-type and mutant larvae. These fluorescent analogs consist of fatty acids with acyl chains of 5 - and 2-carbons (C5 : 0 and C2 : 0, respectively) tagged with the BODIPY fluorophore. We selected these two analogs because of the different cells and subcellular details each analog reveals following ingestion. BODIPY-FL C5 : 0 reveals a high degree of subcellular detail in hepatocytes and acinar cells, such as lipid droplets and zymogen granules, as well as in the ductal networks in the liver and pancreas. The shorter BODIPY-FL C2 : 0 illuminates the hepatic and pancreatic ducts, as well as the gallbladder providing a functional readout of gallbladder and ductal integrity.
Wild-type larvae fed BODIPY-FL C2 : 0 exhibited a strong fluorescence signal in their gallbladders (Figure S3A), indicating that bile production, drainage and accumulation was normal. Conversely, no gallbladder BODIPY-FL C2 : 0 signal was observed in sox9b mutants, consistent with their defective gallbladder development (Figure S3B). In the pancreas, BODIPY-FL C2 : 0 fluorescence was detected throughout the entire intrapancreatic ductal network in wild-type larvae (Figure S3E, S3E′), while it was restricted to the anterior region of the pancreas in sox9b mutants (Figure S3F, S3F′), suggesting that the distal intrapancreatic ducts were not functional. Moreover, we observed large pools of fluorescent fluid accumulating in the mutants' pancreatic tail (Figure S3F), which is not typically observed in wild-type larvae unless the gallbladder ruptures. This abnormal extracellular accumulation of fluid (likely pancreatic juice or bile) in and around sox9b mutant pancreata is consistent with their malformed pancreaticobiliary ductal system.
Administering BODIPY-FL C5 : 0 to sox9b mutant livers confirmed the dilation and lack of branching morphogenesis of the intrahepatic biliary ducts described above as well as the deformation of bile canaliculi in hepatocytes (Figure S3C, S3C′, S3D, S3D′). Taken together, these data support the notion that the cholestasis-like phenotype observed in the adult sox9b mutants results from defects in early ductal morphogenesis.
Notch regulates sox9b expression during intrahepatic biliary network formation
The defects in intrahepatic biliary ducts and bile canaliculi observed in sox9b mutants are strikingly similar to those reported in the mouse and zebrafish models of Notch deficiency [6], [9], [20], [21], [41]. In particular, it has been shown in zebrafish that Notch signaling directs the segregation of intrahepatic biliary cells between 70 and 96 hpf [21]. Given that the intrahepatic biliary cells in sox9b mutant livers fail to separate from one another, we hypothesized that Sox9b interacts with Notch signaling to regulate the morphogenesis of the intrahepatic biliary ducts. To test whether inhibiting Notch signaling affects sox9b expression, we treated wild-type and sox9b heterozygous animals with a low dose of the γ-secretase inhibitor DAPT from 75 to 99 hpf [42], and assessed sox9b expression by in situ hybridization (Figure 6A–6E). Such DAPT treatment caused a reduction in sox9b expression which was more pronounced in sox9b heterozygotes than in wild-types. We also performed the reverse experiment by using Tg(hsp70l:Gal4);Tg(UAS:myc-Notch1a-intra) hemizygous larvae to induce ubiquitous overexpression of the Notch intracellular domain (NICD) upon heat-shock treatment [43]. These animals were heat-shocked at 80 hpf and sox9b expression was analyzed 26 hours later by in situ hybridization. The heat-shock treatment efficiently induced overactivation of Notch signaling activity in the Tg(Tp1bglob:GFP); Tg(Tp1bglob:H2B-mCherry) larvae (Figure 6H, 6I). We observed an increase in sox9b expression throughout the pancreas, liver and HPD system (bracket) in the double–transgenic larvae compared to their single-transgenic control siblings (Figure 6F, 6G). Expression of sox9b could even be detected in the gallbladder (arrowhead, Figure 6G), which was not seen in control larvae (Figure 6F). This increase in sox9b expression is unlikely due to an NICD-induced proliferation of sox9b-positive cells because we could already detect higher levels of sox9b expression as early as four hours after heat-shock treatment (data not shown). Taken together, these loss - and gain-of-function analyses reveal that Notch signaling regulates sox9b expression during intrahepatic biliary duct morphogenesis. These data are consistent with studies in mouse showing that Notch1 can directly bind to the Sox9 promoter [9].

Sox9b is required to maintain Notch signaling in the intrahepatic biliary duct cells
To further analyze the dynamics of Notch signaling in sox9b mutants, we utilized Tg(Tp1bglob:H2B-mCherry);Tg(Tp1bglob:VenusPest) animals in which the Notch-responsive element drives the expression of both H2B-mCherry and VenusPest fluorescent proteins. Contrary to H2B-mCherry which is very stable, the destabilized fluorescent protein VenusPest has a short half-life (2 hours for GFP-Pest in mammalian cells) [44]. Thus, the Tg(Tp1bglob:H2B-mCherry);Tg(Tp1bglob:VenusPest)-double positive cells are currently Notch responsive, whereas the Tg(Tp1bglob:H2B-mCherry)-positive;Tg(Tp1bglob:VenusPest)-negative cells were positive for Notch signaling in their recent past but have since switched it off [34].
In wild-type and sox9b heterozygous livers, the expression of Tg(Tp1bglob:H2B-mCherry) and Tg(Tp1bglob:VenusPest) largely overlapped at 75 hpf (Figure 7A, 7A′, 7G, and data not shown). Between 99 and 123 hpf, a small proportion of intrahepatic biliary cells switched off Notch signaling and became Tg(Tp1bglog:H2B-mCherry)-single positive (Figure 7B–7C′, G). Up to 99 hpf, sox9b mutant livers exhibited a similar pattern of Notch signaling activity as wild-type with a clear overlap between Tg(Tp1bglob:H2B-mCherry) and Tg(Tp1bglob:VenusPest) expression (Figure 7D–7E′, 7G). However, at 123 hpf, the proportion of Tg(Tp1bglog:H2B-mCherry)-single positive cells was significantly higher in sox9b mutants compared to wild-types or heterozygotes (Figure 7F, 7F′, 7G), suggesting that sox9b mutants fail to maintain Notch signaling in the intrahepatic biliary cells. Interestingly, we did not observe any obvious phenotype when assessing Notch signaling activity in the mutant intrapancreatic ducts.

To follow up this observation, we treated wild-type and mutant larvae with 50 µM DAPT, a dose that only partially inhibits Notch signaling. Upon treatment between 106 and 154 hpf, DAPT-treated wild-type larvae showed a specific loss of Notch signaling activity in the proximal (p) rather than the distal (d) region of the liver (Figure S4C). In DAPT-treated mutant larvae, we observed a drastic loss of Notch signaling throughout the entire liver (Figure S4D, S4E), indicating that sox9b mutant biliary cells are more likely to lose Notch signaling activity than wild-type cells. Altogether, these data indicate that Sox9b is required for the maintenance but not the initiation of Notch signaling in the intrahepatic biliary cells. Considering our previous data showing that Notch signaling regulates sox9b expression, we hypothesize that Notch and Sox9b interact in a positive feedback loop to ensure the development of the intrahepatic biliary network.
To better understand the biological significance of Notch responsiveness during intrahepatic biliary duct morphogenesis, we compared the distribution of Tg(Tp1bglob:H2B-mCherry)-single positive cells and Tg(Tp1bglob:H2B-mCherry);Tg(Tp1bglob:VenusPest)-double positive cells in wild-type livers. At 123 hpf, 89% of the double-positive cells existed as individual cells connecting to one another through cellular extensions (Figure 7C′; 467 cells in 5 larvae were analyzed). On the other hand, 60% of the Tg(Tp1bglob:H2B-mCherry)-single positive cells, which had switched off Notch signaling, were intermingled with each other to form larger groups (Figure 7C′, arrows; 110 cells in 5 larvae were analyzed). Interestingly, these clusters of Tg(Tp1bglob:H2B-mCherry)-single positive cells were mostly present in the multicellular large bile ducts contiguous with the extrahepatic duct, whereas the individual Tg(Tp1bglob:H2B-mCherry);Tg(Tp1bglob:VenusPest)-double positive cells were localized in the distal part of the liver and formed smaller bile ducts (Figure 7C′). These data suggest that Notch responsiveness correlates with the relative position of the intrahepatic biliary cells, with the cells turning off Notch signaling forming the large bile ducts in the proximal region of the liver. In sox9b mutant livers, we observed more clusters of Tg(Tp1bglob:H2B-mCherry)-single positive cells in both the proximal and distal regions (Figure 7F′, arrows; 243 cells in 6 larvae were analyzed).
We then addressed whether Notch responsiveness was related to the proliferation status of the intrahepatic biliary cells, which would correlate with the increase in ductal structures observed in mutant adults. We incubated wild-type and mutant animals with the replication marker 5-ethynyl-2′-deoxyuridine (EdU) during two intervals of larval development, and analyzed EdU incorporation in Tg(Tp1bglob:H2B-mCherry)-single positive and Tg(Tp1bglob:H2B-mCherry);Tg(Tp1bglob:VenusPest)-double positive cells (Figure 7H). In wild-type larvae, approximately 30% of the Tg(Tp1bglob:H2B-mCherry)-single positive cells incorporated EdU after incubation from 96 to 120 hpf. The Tg(Tp1bglob:H2B-mCherry);Tg(Tp1bglob:VenusPest)-double positive cells exhibited a slightly higher percentage of EdU incorporation, although the difference between these two cell populations was not statistically significant (p>0.08). Similar rates of EdU incorporation were observed when we incubated the animals from 120 to 144 hpf. In sox9b mutants, we detected an increase in EdU incorporation in both the single and double positive cells compared to wild-type (Figure 7H), with the increase being more pronounced in the Tg(Tp1bglob:H2B-mCherry)-single positive cells than in the double positive cells. To further support the hypothesis that loss of Notch signaling correlates with increased proliferation of biliary cells, we found that partial inhibition of Notch signaling in wild-type larvae by a low dose DAPT treatment led to an increase in biliary cell proliferation similar to that observed in sox9b mutants (data not shown). Hence, taken together, these data suggest that the reduction in Notch signaling in sox9b mutants promotes the clustering and proliferation of the intrahepatic biliary cells, which is consistent with the biliary duct defects observed in the mutant adults.
Discussion
In this study, we analyzed a novel sox9b mutant in zebrafish, revealing for the first time that global loss-of-function of Sox9b severely impairs the development of the pancreaticobiliary ductal system. In particular, we showed that in the mutant animals, the HPD system is malformed, and the intrahepatic and intrapancreatic ducts fail to form a functional ductal network. We also uncovered the existence of a Notch-Sox9b positive feedback loop that is crucial for intrahepatic biliary duct development. Our study thus brings new insights into our understanding of pancreaticobiliary ductal system formation, an important but understudied process.
We showed that loss of Sox9b function leads to mispatterning of the HPD primordium. Fgf10 signaling also plays a pivotal role in the formation of the HPD system in zebrafish [15]. Zebrafish fgf10 is expressed in the mesenchyme surrounding the HPD system and intestine but not that surrounding the liver or pancreas [17]. fgf10 mutants show a dysmorphic HPD with a reduction or loss of the common bile duct as well as reduced extrapancreatic and extrahepatic ducts. Moreover, fgf10 mutants misexpress hepatic markers such as Prox1 and Hnf4α in their HPD system and pancreas, leading to the ectopic differentiation of some cells in these organs towards a liver fate. Based on the similarities of the fgf10 and sox9b mutant phenotypes, it is possible that Fgf10 and Sox9b interact; for example, Fgf10 signaling could induce or maintain sox9b expression in the HPD primordium to modulate the patterning of this tissue. However, we did not observe any obvious change in sox9b expression in the HPD upon Fgf receptor pharmacological inhibition treatment, nor in fgf10 expression in the surrounding mesenchyme in sox9b mutants. Thus, Fgf10 and Sox9b functions might intersect in other ways.
Our data show that Sox9b is involved in gallbladder development, whose primordium specifically expresses sox17. sox17 is expressed in all endodermal cells during gastrulation [45] and starts to be reexpressed at 36 hpf in a small region of the liver close to the extrahepatic duct [33]. It is then detected in the gallbladder at 60 hpf where it persists until 5 dpf. It will be interesting to determine whether Sox9b directly regulates sox17 expression and also to identify the additional factors involved in inducing sox17 expression in a subset of sox9b positive cells.
The intrapancreatic ductal network is severely disrupted in sox9b mutant larvae, leading to the formation of dilated ducts surrounded by fibrotic tissue in mutant adults. At first glance, this adult phenotype is reminiscent of the one caused by pancreas-specific inactivation of mouse Sox9 [25]; however, given that the pancreatic remnants described in the mouse mutants come from unrecombined Sox9+ progenitor cells, the zebrafish adult phenotype appears much less severe than the one seen in mouse. The discrepancy between the two models could be explained by differences during early pancreas development. Indeed, mouse SOX9 is expressed in pluripotent pancreatic progenitors and is required to stimulate their proliferation and survival [25]. However, in zebrafish at the earliest stages of pancreas development, endocrine cells derive first from the endodermal epithelium [18] and then from the extrapancreatic duct [46]; only during larval stages, do endocrine cells derive from progenitors within the intrapancreatic ducts [34], likely following mechanisms similar to those regulating the secondary transition in mouse. Analysis of zebrafish sox9b expression at early stages suggests that it is present in a subset of ventral pancreatic bud cells, which may correspond to the ductal progenitors. If this interpretation is correct, the lack of Sox9b function in zebrafish would first impair these ductal progenitors, leading to a reduced number of intrapancreatic ductal cells. The first two waves of endocrine cells, as well as the acinar cells, which originate from sox9b negative tissue, would therefore not be affected in a sox9b mutant.
Loss of Sox9b function in zebrafish severely impairs the development of the intrahepatic biliary network including the morphogenesis of the bile canaliculi. These phenotypes are more severe than those seen in liver-specific SOX9-depleted mice [23]. During mouse liver development, SOX9 expression is first detected at E10.5 in the endodermal cells lining the lumen of the liver diverticulum [23]. This expression is lost as the liver cells migrate into the septum transversum, but re-emerges at E11.5 in cells that form the ductal plate. The Alfp:Cre line that was used to recombine the Sox9 locus becomes active at E11.5, thus likely only inhibiting the second phase of SOX9 expression. Therefore, the phenotypic differences between the zebrafish sox9b mutant and the mouse model might be related to the consequences of an earlier depletion in Sox9b in zebrafish than in mouse, illustrating the value of the zebrafish Sox9b global loss-of-function model to uncover functions of this critical transcriptional regulator. However, differences in the expression or function between sox9b (zebrafish) and Sox9 (mouse) may also explain the phenotypic differences between the two models.
Intrahepatic biliary cells in sox9b mutant livers fail to segregate from one another and remain clustered, leading to a primitive ductal network. Such defects are strikingly similar to Notch-deficient zebrafish and mouse models [6], [9], [20], [21], [41] which phenocopy the human Alagille syndrome (OMIM#118450), which itself is associated with JAGGED1 and NOTCH2 mutations. Notably, this developmental disorder is characterized by cholestasis due to a paucity of biliary ducts. Our data indicate that Sox9b interacts with Notch signaling in a positive feedback loop to regulate intrahepatic biliary duct morphogenesis. Studies in mouse have provided possible mechanisms underlying this Sox9-Notch crosstalk: the Sox9 promoter displays ten consensus Rbpj binding sites and is a direct target of Notch signaling [9]. In addition, SOX9 modulates Notch signaling by positively regulating the expression of the Notch downstream target gene Hes1 in the liver [23] as well as in other organs such as the pancreas [25]. These mechanisms are likely to be at play in zebrafish as well, and it will be interesting to delve deeper into the complexities of this positive feedback loop.
Intriguingly, we found that the biliary cells in the proximal region of wild-type livers tend to lose Notch signaling more quickly and are more susceptible to Notch inhibition than the distal cells. Notably, the biliary cells in the proximal region form large ducts whereas the distal cells form small ducts. In sox9b mutants, loss of Sox9b function tempers Notch signaling in all biliary cells. Consequently, the mutant livers exhibit aberrant clusters of Tg(Tp1bglob:H2B-mCherry)-single positive cells in their distal region, suggesting that lack of Sox9b-mediated maintenance of Notch signaling could promote the formation of large ectopic ducts in distal regions of the liver. Interestingly, it has been shown in mouse that SOX9, which is expressed in all biliary cells at E18.5, persists in small ducts but regresses from large ducts after birth [23]. Therefore, it is possible that in wild-type animals, the loss of Notch signaling in the proximal region of the liver induces the local loss of sox9b expression, leading to the formation of large bile ducts. To begin to test this hypothesis, it will be necessary to generate a Sox9b antibody in order to examine Sox9b expression at cellular resolution. The identification of markers that distinguish large and small bile ducts would also greatly facilitate such studies.
Our data suggest that in addition to regulating biliary morphogenesis, the Notch-Sox9b module also influences the proliferation of biliary cells. In sox9b mutants, the biliary cells that turn off Notch signaling exhibit a higher proliferation rate. Recent lineage tracing studies in mouse have shown that Sox9 is expressed in liver progenitor cells that reside within the biliary ducts [47], [48]. It will be interesting to determine whether zebrafish sox9b is also expressed in liver progenitor cells, whether loss of Sox9b function affects their proliferation rate, and investigate into the underlying mechanisms.
In addition to bringing new insights into our understanding of the development of the pancreaticobiliary ductal system, the analysis of zebrafish sox9b mutants should lead one to consider SOX9 as a candidate gene for human diseases associated with HPD, intrapancreatic or intrahepatic duct malformations. In particular, numerous cases of congenital non-syndromic or syndromic extrahepatic biliary atresia have been reported [49] and their causes remain unknown [50]. These conditions likely have multifactorial causes and do not display simple Mendelian inheritance. Given the expression pattern of SOX9 in mammals [24], [26], [28] as well as the phenotypes caused by loss of SOX9 function in zebrafish and mouse, SOX9 is therefore an interesting gene to sequence in those patients. Campomelic dysplasia has been shown to be essentially associated with heterozygous mutations that are predicted to severely disrupt SOX9 protein structure and function [51]; but milder lesions could be associated with pancreaticobiliary duct malformations and contribute to the onset or severity of these malformations without necessarily impairing skeletal development.
Materials and Methods
Zebrafish strains and lines
Embryos and adult fish were raised and maintained under standard laboratory conditions [52]. The sox9bfh313 heterozygote was crossed with Tg(fabp10:ras-GFP)s942 [37], Tg(Tp1bglob:GFP)um14 [35], Tg(Tp1bglob:H2B-mCherry)s939, Tg(Tp1bglob:VenusPest)s940 [34], TgBAC(neurod:GFP)nl1 [53] and genotyped according to the TILLING center protocol with AcuI or SfcI (http://labs.fhcrc.org/moens/Tilling_Mutants/sox9b/allele_1.html). We also used Tg(hsp70l:Gal4)1.5kca4 and Tg(UAS:myc-Notch1a-intra)kca3 [43] hemizygous or double hemizygous fish.
In situ hybridization and immunohistochemistry
Whole-mount in situ hybridizations were performed as described previously [54] using sox9b [30] and sox17 [45] probes. Animals were photographed with a Zeiss Axioplan using an Axiocam digital camera. Immunohistochemistry on whole-mount animals or cryosections was performed as previously described [16], using the following antibodies: chicken polyclonal anti-GFP (1∶1000; Aves Labs, Tigard, OR, USA), rabbit polyclonal anti-Prox1 (1∶1000; Chemicon, Billerica, MA, USA), mouse monoclonal 2F11 (1∶1000; Abcam, Cambridge, UK), rabbit polyclonal anti-dsRed (1∶500; Clontech, Mountain View, CA, USA), rabbit polyclonal anti-ABCB11/BSEP (1∶1000; Kamiya Biomedical), mouse monoclonal anti-Alcam/Zn8 (1∶20; ZIRC), rabbit polyclonal anti-elastase (1∶200; Millipore AB1216) and fluorescently conjugated Alexa antibodies (1∶250; Molecular Probes, Carlsbad, CA, USA). Samples were imaged on a Zeiss Pascal confocal microscope. The width of the bile ducts was measured using the “local thickness” function in Fiji software.
Heat-shock experiment and chemical inhibitor treatment
Heat-shock treatments of Tg(hsp70l:Gal4)1.5kca4 larvae were performed at 38°C as described [16]. To inhibit Notch signaling, larvae were treated with 20 µM or 50 µM DAPT (Sigma) in egg water [42]. Control larvae from the same batch were treated with 0.4% DMSO in egg water. Statistical analyses were performed using the Student's two-tailed t-test.
Histology
Five-months old zebrafish (one wild-type and one mutant) were euthanized and their digestive systems were dissected and fixed overnight with formalin at 4°C. The samples were embedded in paraffin, cut into 5 µm sections, and stained with hematoxylin and eosin.
BODIPY assay
6–7 dpf larvae were fed with BODIPY C2.0 or BODIPY C5.0 as described in [40] for 6–8 hours before being mounted and imaged live. The larvae were subsequently genotyped and 9 wild-type and 9 sox9b mutant animals were analyzed.
EdU cell cycle analysis
To assess the proliferation of the intrahepatic biliary cells, wild-type and sox9b mutant larvae were incubated in 7 µM EdU dissolved in egg water during the stages indicated. Control larvae collected from the same batch were treated with 1.7% DMSO. Animals were fixed after incubation and processed using the Click-iT EdU Imaging Kit (Invitrogen). Quantification of EdU incorporation was conducted using the Cell Counter plug-in in ImageJ.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. RaynaudPCarpentierRAntoniouALemaigreFP 2011 Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol 43 245 256
2. SpenceJRLangeAWLinSCKaestnerKHLowyAM 2009 Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Dev Cell 17 62 74
3. ZongYStangerBZ 2011 Molecular mechanisms of bile duct development. Int J Biochem Cell Biol 43 257 264
4. GeislerFNaglFMazurPKLeeMZimber-StroblU 2008 Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology 48 607 616
5. KodamaYHijikataMKageyamaRShimotohnoKChibaT 2004 The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology 127 1775 1786
6. LozierJMcCrightBGridleyT 2008 Notch signaling regulates bile duct morphogenesis in mice. PLoS ONE 3 e1851 doi:10.1371/journal.pone.0001851
7. McCrightBLozierJGridleyT 2002 A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 129 1075 1082
8. TchorzJSKinterJMullerMTornilloLHeimMH 2009 Notch2 signaling promotes biliary epithelial cell fate specification and tubulogenesis during bile duct development in mice. Hepatology 50 871 879
9. ZongYPanikkarAXuJAntoniouARaynaudP 2009 Notch signaling controls liver development by regulating biliary differentiation. Development 136 1727 1739
10. ClotmanFLannoyVJReberMCereghiniSCassimanD 2002 The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development 129 1819 1828
11. CoffinierCGreshLFietteLTroncheFSchutzG 2002 Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development 129 1829 1838
12. HunterMPWilsonCMJiangXCongRVasavadaH 2007 The homeobox gene Hhex is essential for proper hepatoblast differentiation and bile duct morphogenesis. Dev Biol 308 355 367
13. MaestroMABojSFLucoRFPierreuxCECabedoJ 2003 Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Human Molecular Genetics 12 3307 3314
14. PierreuxCEPollAVKempCRClotmanFMaestroMA 2006 The transcription factor hepatocyte nuclear factor-6 controls the development of pancreatic ducts in the mouse. Gastroenterology 130 532 541
15. DongPDMunsonCANortonWCrosnierCPanX 2007 Fgf10 regulates hepatopancreatic ductal system patterning and differentiation. Nat Genet 39 397 402
16. ChungWSShinCHStainierDY 2008 Bmp2 signaling regulates the hepatic versus pancreatic fate decision. Dev Cell 15 738 748
17. ShinDLeeYPossKDStainierDY 2011 Restriction of hepatic competence by Fgf signaling. Development 138 1339 1348
18. FieldHADongPDBeisDStainierDY 2003 Formation of the digestive system in zebrafish. II. Pancreas morphogenesis. Dev Biol 261 197 208
19. OberEAVerkadeHFieldHAStainierDY 2006 Mesodermal Wnt2b signalling positively regulates liver specification. Nature 442 688 691
20. LorentKYeoSYOdaTChandrasekharappaSChitnisA 2004 Inhibition of Jagged-mediated Notch signaling disrupts zebrafish biliary development and generates multi-organ defects compatible with an Alagille syndrome phenocopy. Development 131 5753 5766
21. LorentKMooreJCSiekmannAFLawsonNPackM 2010 Reiterative use of the notch signal during zebrafish intrahepatic biliary development. Dev Dyn 239 855 864
22. PaulsSZecchinETisoNBortolussiMArgentonF 2007 Function and regulation of zebrafish nkx2.2a during development of pancreatic islet and ducts. Dev Biol 304 875 890
23. AntoniouARaynaudPCordiSZongYTroncheF 2009 Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology 136 2325 2333
24. FuruyamaKKawaguchiYAkiyamaHHoriguchiMKodamaS 2011 Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet 43 34 41
25. SeymourPAFreudeKKTranMNMayesEEJensenJ 2007 SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A 104 1865 1870
26. WagnerTWirthJMeyerJZabelBHeldM 1994 Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 79 1111 1120
27. FosterJWDominguez-SteglichMAGuioliSKwokCWellerPA 1994 Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 372 525 530
28. PiperKBallSGKeelingJWMansoorSWilsonDI 2002 Novel SOX9 expression during human pancreas development correlates to abnormalities in Campomelic dysplasia. Mech Dev 116 223 226
29. BiWHuangWWhitworthDJDengJMZhangZ 2001 Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A 98 6698 6703
30. YanYLWilloughbyJLiuDCrumpJGWilsonC 2005 A pair of Sox: distinct and overlapping functions of zebrafish sox9 co-orthologs in craniofacial and pectoral fin development. Development 132 1069 1083
31. YanYLMillerCTNissenRMSingerALiuD 2002 A zebrafish sox9 gene required for cartilage morphogenesis. Development 129 5065 5079
32. CrosnierCVargessonNGschmeissnerSAriza-McNaughtonLMorrisonA 2005 Delta-Notch signalling controls commitment to a secretory fate in the zebrafish intestine. Development 132 1093 1104
33. ShinDWeidingerGMoonRTStainierDY Intrinsic and extrinsic modifiers of the regulative capacity of the developing liver. Mech Dev 128 525 535
34. NinovNBoriusMStainierDY 2012 Different levels of Notch signaling regulate quiescence, renewal and differentiation in pancreatic endocrine progenitors. Development 139 1557 1567
35. ParsonsMJPisharathHYusuffSMooreJCSiekmannAF 2009 Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech Dev 126 898 912
36. ObholzerNWolfsonSTrapaniJGMoWNechiporukA 2008 Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J Neurosci 28 2110 2118
37. CheungIDBagnatMMaTPDattaAEvasonK 2011 Regulation of intrahepatic biliary duct morphogenesis by Claudin 15-like b. Dev Biol
38. GerloffTStiegerBHagenbuchBMadonJLandmannL 1998 The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem 273 10046 10050
39. SakaguchiTFSadlerKCCrosnierCStainierDY 2008 Endothelial signals modulate hepatocyte apicobasal polarization in zebrafish. Curr Biol 18 1565 1571
40. CartenJDBradfordMKFarberS 2011 Visualizing digestive organ morphology and function using differential fatty acid metabolism in live zebrafish. Dev Biol
41. HofmannJJZoveinACKohHRadtkeFWeinmasterG 2010 Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Development 137 4061 4072
42. GelingASteinerHWillemMBally-CuifLHaassC 2002 A gamma-secretase inhibitor blocks Notch signaling in vivo and causes a severe neurogenic phenotype in zebrafish. EMBO Rep 3 688 694
43. ScheerNGrothAHansSCampos-OrtegaJA 2001 An instructive function for Notch in promoting gliogenesis in the zebrafish retina. Development 128 1099 1107
44. LiXZhaoXFangYJiangXDuongT 1998 Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem 273 34970 34975
45. AlexanderJStainierDY 1999 A molecular pathway leading to endoderm formation in zebrafish. Curr Biol 9 1147 1157
46. HesselsonDAndersonRMBeinatMStainierDY 2009 Distinct populations of quiescent and proliferative pancreatic beta-cells identified by HOTcre mediated labeling. Proc Natl Acad Sci U S A 106 14896 14901
47. CarpentierRSunerREvan HulNKoppJLBeaudryJB Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology 141 1432 1438 1438e 1431–1434
48. FuruyamaKKawaguchiYAkiyamaHHoriguchiMKodamaS 2010 Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet 43 34 41
49. Galan-GomezESanchezEBArias-CastroSCardesa-GarciaJJ 2007 Intrauterine growth retardation, duodenal and extrahepatic biliary atresia, hypoplastic pancreas and other intestinal anomalies: further evidence of the Martinez-Frias syndrome. Eur J Med Genet 50 144 148
50. ChappellLGormanSCampbellFEllardSRiceG 2008 A further example of a distinctive autosomal recessive syndrome comprising neonatal diabetes mellitus, intestinal atresias and gall bladder agenesis. Am J Med Genet A 146A 1713 1717
51. KwokCWellerPAGuioliSFosterJWMansourS 1995 Mutations in SOX9, the gene responsible for Campomelic dysplasia and autosomal sex reversal. Am J Hum Genet 57 1028 1036
52. WesterfieldM 2000 The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio)
53. ObholzerRJNouraeiSAAhmedJKadhimMRSandhuGS 2008 An approach to the management of paroxysmal laryngospasm. J Laryngol Otol 122 57 60
54. AlexanderJStainierDYYelonD 1998 Screening mosaic F1 females for mutations affecting zebrafish heart induction and patterning. Dev Genet 22 288 299
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Rumors of Its Disassembly Have Been Greatly Exaggerated: The Secret Life of the Synaptonemal Complex at the Centromeres
- The NSL Complex Regulates Housekeeping Genes in
- Tipping the Balance in the Powerhouse of the Cell to “Protect” Colorectal Cancer
- Interplay between Synaptonemal Complex, Homologous Recombination, and Centromeres during Mammalian Meiosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy