
Genome-Wide Identification of Ampicillin Resistance Determinants in
Enterococcus faecium has become a nosocomial pathogen of major importance, causing infections that are difficult to treat owing to its multi-drug resistance. In particular, resistance to the β-lactam antibiotic ampicillin has become ubiquitous among clinical isolates. Mutations in the low-affinity penicillin binding protein PBP5 have previously been shown to be important for ampicillin resistance in E. faecium, but the existence of additional resistance determinants has been suggested. Here, we constructed a high-density transposon mutant library in E. faecium and developed a transposon mutant tracking approach termed Microarray-based Transposon Mapping (M-TraM), leading to the identification of a compendium of E. faecium genes that contribute to ampicillin resistance. These genes are part of the core genome of E. faecium, indicating a high potential for E. faecium to evolve towards β-lactam resistance. To validate the M-TraM results, we adapted a Cre-lox recombination system to construct targeted, markerless mutants in E. faecium. We confirmed the role of four genes in ampicillin resistance by the generation of targeted mutants and further characterized these mutants regarding their resistance to lysozyme. The results revealed that ddcP, a gene predicted to encode a low-molecular-weight penicillin binding protein with D-alanyl-D-alanine carboxypeptidase activity, was essential for high-level ampicillin resistance. Furthermore, deletion of ddcP sensitized E. faecium to lysozyme and abolished membrane-associated D,D-carboxypeptidase activity. This study has led to the development of a broadly applicable platform for functional genomic-based studies in E. faecium, and it provides a new perspective on the genetic basis of ampicillin resistance in this organism.
Published in the journal:
. PLoS Genet 8(6): e32767. doi:10.1371/journal.pgen.1002804
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002804
Summary
Enterococcus faecium has become a nosocomial pathogen of major importance, causing infections that are difficult to treat owing to its multi-drug resistance. In particular, resistance to the β-lactam antibiotic ampicillin has become ubiquitous among clinical isolates. Mutations in the low-affinity penicillin binding protein PBP5 have previously been shown to be important for ampicillin resistance in E. faecium, but the existence of additional resistance determinants has been suggested. Here, we constructed a high-density transposon mutant library in E. faecium and developed a transposon mutant tracking approach termed Microarray-based Transposon Mapping (M-TraM), leading to the identification of a compendium of E. faecium genes that contribute to ampicillin resistance. These genes are part of the core genome of E. faecium, indicating a high potential for E. faecium to evolve towards β-lactam resistance. To validate the M-TraM results, we adapted a Cre-lox recombination system to construct targeted, markerless mutants in E. faecium. We confirmed the role of four genes in ampicillin resistance by the generation of targeted mutants and further characterized these mutants regarding their resistance to lysozyme. The results revealed that ddcP, a gene predicted to encode a low-molecular-weight penicillin binding protein with D-alanyl-D-alanine carboxypeptidase activity, was essential for high-level ampicillin resistance. Furthermore, deletion of ddcP sensitized E. faecium to lysozyme and abolished membrane-associated D,D-carboxypeptidase activity. This study has led to the development of a broadly applicable platform for functional genomic-based studies in E. faecium, and it provides a new perspective on the genetic basis of ampicillin resistance in this organism.
Introduction
Enterococci rank third overall as causative agents of healthcare-associated infections [1], [2]. Up to the late 1980s, Enterococcus faecalis was responsible for practically all enterococcal infections, but starting from the 1990s nosocomial infections with E. faecium became more frequent. Currently E. faecium causes approximately 40% of all enterococcal infections that are acquired during hospital stay [2]–[4]. Clinical isolates of E. faecium have rapidly accumulated antibiotic resistance genes, including those for clinically important antibiotics such as ampicillin and vancomycin, which leads to treatment failure and increased mortality rates [2], [5]–[7]. In the USA, nosocomial infections caused by ampicillin-resistant E. faecium (ARE) were first detected in the 1980s and the resistance rates were steadily increasing up to 80% of E. faecium isolates in the 1990s [8], [9]. Vancomycin-resistant E. faecium (VRE) also emerged in the late 1980s and increased rapidly during the 1990s [9], [10]. Currently, VRE is widespread among clinical E. faecium strains in North America, but less common in hospital-acquired infections in Europe [11]. Ampicillin resistance has spread much further and it is currently being reported in over 80% of clinical E. faecium isolates from all over the world [1], [2] (European Antimicrobial Resistance Surveillance Network: http://www.ecdc.europa.eu/en/activities/surveillance/EARS-Net/Pages/index.aspx). In addition to ARE and VRE, the emergence of E. faecium strains that are resistant to new classes of antibiotics is challenging the few remaining therapeutic options [12]–[14]. Thus, the development of new anti-enterococcal agents may become critical for the successful treatment of infections caused by this multi-drug resistant organism in the future.
The intrinsic resistance to β-lactam antibiotics of enterococci was reported 60 years ago, soon after the introduction of penicillin in the early 1940s, when enterococci were found to be considerably less susceptible to β-lactams than streptococci [15]. Mutations in the high-molecular weight class B penicillin-binding protein 5 (PBP5) have been considered the main cause for the resistance to β-lactams in E. faecium. Upregulated expression of pbp5 and/or mutations in the 3′ end of the gene lead to a further reduced susceptibility to ampicillin [16]–[18]. However, several studies have suggested that the high minimum inhibitory concentration (MIC) of ampicillin against E. faecium is not exclusively due to the presence of low-affinity PBP5 but also to other genes or mechanisms that remain to be identified [19], [20]. Recently, Mainardi et al. [21], [22] showed in a spontaneous mutant that was obtained in the laboratory by selection on agar media containing ampicillin, that the D,D-transpeptidase activity of the PBPs could be bypassed by a β-lactam resistant L,D-transpeptidase (Ldtfm) that catalyses the formation of 3→3 cross-links between peptidoglycan side chains instead of the classical 4→3 cross-links. A D,D-carboxypeptidase, termed DdcY, is an important component in the L,D-transpeptidase mediated pathway of peptidoglycan cross-linking. However, the ddcY gene is only present in a small proportion of E. faecium isolates [23], again suggesting that additional ampicillin resistance determinants in E. faecium remained to be identified and characterized.
Genome-wide studies of clinical E. faecium isolates have long been hampered by a lack of appropriate genetic tools. In this study, we describe the construction of a high density mariner transposon mutant library and the development of a powerful tool for functional genomics, termed Microarray-based Transposon Mapping (M-TraM), in E. faecium. By comparing the mutant library following growth in the presence or absence of ampicillin, we identified a compendium of genes affecting the sensitivity to ampicillin. Targeted mutants of the identified genes with predicted roles in cell wall synthesis were generated for further characterization, which resulted in the identification of several intrinsic ampicillin resistance determinants in E. faecium. These ampicillin resistance determinants may serve as targets for the development of novel antimicrobial therapeutics.
Results
Construction of a high-density transposon mutant library in E. faecium
To attempt genome-wide transposon mutagenesis of the E. faecium genome, we constructed the transposon delivery plasmid pZXL5. As shown in Figure S1, this plasmid was composed of a Gram-positive thermo-sensitive replicon, a gentamicin resistant mariner transposon with two outward-facing T7 promoters, a nisin-inducible mariner transposase, a ColE1 replicon and a cat gene. The sequence of pZXL5 was determined by Sanger-sequencing of both DNA strands (Baseclear; Leiden, The Netherlands) and was deposited in GenBank (GenBank Accession Number: JQ088279).
Using pZXL5, we have produced a transposon mutant library in E. faecium strain E1162, an ampicillin-resistant clinical isolate from a bloodstream infection, for which a draft genome sequence has previously been determined [24]. The randomness of the transposon insertions and the absence of multiple transposon insertion events were determined by randomly selecting 17 mutants from the library and carrying out Southern blot hybridizations, using a fragment of the transposon as a probe (Figure S2A), as well as inverse PCR and sequence analysis to determine the location of the transposon insertion point (Figure S2B). The results showed that each mutant carries a single transposon inserted in the genome, and that the transposon was distributed in different loci in the 17 mutants. PCR footprinting was performed to estimate the genome-wide coverage of transposon insertions in the mutant library (Figure 1). An outward-facing primer was designed based on the mariner transposon sequence. The other PCR primer was designed for three target genes, ddl (which encodes a D-alanine∶D-alanine ligase that is essential for bacterial cell wall biosynthesis [25]), esp (which is non-essential and encodes a large surface protein involved in biofilm formation and infection [26]–[28]) and nox (which is a non-essential gene encoding a predicted NADH oxidase). Genomic DNA isolated from the pooled mutant library was used as a template. A range of products can be amplified by these primers, each corresponding to a transposon insertion mutant in the library. If a gene is essential for survival, its transposon insertion mutants should not be present in the library after overnight growth, and consequently no PCR products should be amplified in the corresponding size range. As expected, no PCR product was detected within the ddl gene (Figure 1) while many PCR bands were found in esp and nox at intervals of less than 100 bp, indicating the transposon insertions covered the nonessential genes of the genome at high density. Furthermore, mapping the transposon insertion sites to the complete genome sequence of E. faecium Aus0004 [29] revealed that transposon insertions were randomly distributed over the genome of this strain and not confined to a specific chromosomal region (data not shown). To establish whether pZXL5 has broad applicability in E. faecium we attempted to transform four other clinical E. faecium isolates from different geographic origins (Table S1) with pZXL5 using our optimized electroporation protocol. All four strains were efficiently transformed with transformation efficiencies ranging between 110 and 105 transformants per µg DNA (Figure S2C). We then continued to generate transposon mutant libraries in two of these strains (Figure S2D). These observations show that the transposon mutagenesis approach that we initially developed for strain E1162, can also be used for functional genomics in other clinical E. faecium isolates.

M-TraM is highly reproducible
As described in the Materials and Methods and in Figure 2, we developed a technique to track the presence of all mutants in the library by simultaneously mapping the transposon insertion sites using microarray hybridization. We termed this technique M-TraM for Microarray-based Transposon Mapping. To validate the reproducibility of M-TraM, we independently grew two aliquots containing approximately 107 cells from the library in 20 ml of BHI broth. After 20 hours of culturing at 37°C, genomic DNA was isolated from the two replicate cultures and used for the generation of cDNA. The cDNA samples were labeled with Cy3 and Cy5 respectively and hybridized to a microarray that was designed using the E. faecium E1162 genome sequence. The result showed that M-TraM is highly reproducible, with a correlation coefficient of 0.94 between the two independent experiments (Figure 2C).

Identification of E. faecium genes involved in ampicillin resistance by M-TraM
To identify genes required for ampicillin resistance, we grew the pool of mutants in the presence or absence of a subinhibitory concentration (20 µg ml−1) of ampicillin, and used M-TraM to determine which mutants were selectively lost during culturing in the presence of ampicillin. Eleven genes belonging to a variety of functional categories were identified to be involved in ampicillin resistance (Table 1). Four genes involved in cell wall biogenesis were identified and we decided to further focus on these genes. The EfmE1162_0447 (ddcP) and EfmE1162_1886 (ldtfm) genes were predicted to encode a D-alanyl-D-alanine carboxypeptidase (D,D-carboxypeptidase, DdcP) and beta-lactam-insensitive peptidoglycan transpeptidase (L,D-transpeptidase, Ldtfm), respectively. Previous studies have shown that a different D,D-carboxypeptidase (DdcY) and Ldtfm were able to bypass the D,D-transpeptidase activity of the PBPs by forming 3→3 cross-links instead of the classical 4→3 cross-links thereby conferring resistance to β-lactams [21], [23]. DdcP and DdcY only share 13.7% amino acid identity and have a completely different protein domain architecture as DdcY is a β-lactam-insensitive VanY-type carboxypeptidase [23], [30], while DdcP belongs to the family of low-molecular-weight (LMW) PBPs [30] (Figure S3). The ddcY gene is absent from 24 (including E1162) of the 29 E. faecium genomes available (on 9 January 2012) at NCBI Genomes. The ddcP gene is conserved in all 29 E. faecium genomes. EfmE1162_0975 (pgt) was predicted to encode a glycosyl transferase group 2 family protein which is 63% identical to GltB, a protein that was proposed to be involved in glycosylation of cell wall teichoic acid in serotype 4b Listeria monocytogenes [31]. EfmE1162_2487 (lytG) was predicted to encode an exo-glucosaminidase that could be acting as a peptidoglycan hydrolase involved in cell wall lysis, remodeling and cell division [32]. An overview of the protein domain architecture and predicted cellular localization of DdcP, Ldtfm, Pgt and LytG is provided in Figure S3. Like the ddcP gene, the ldtfm, pgt and lytG genes are present in all the 29 E. faecium genomes as well, suggesting these genes are part of the E. faecium core genome. Notably the E. faecium ampicillin resistance determinants ddcP, ldtfm and pgt do not have homologs (defined here by proteins with >30% amino acid identity) in E. faecalis. It should be noted that the L,D-transpeptidase from E. faecalis that was biochemically characterized by Magnet et al., [33] has not been experimentally linked to β-lactam resistance in E. faecalis and is only remotely related (26% amino acid identity) to ldtfm.
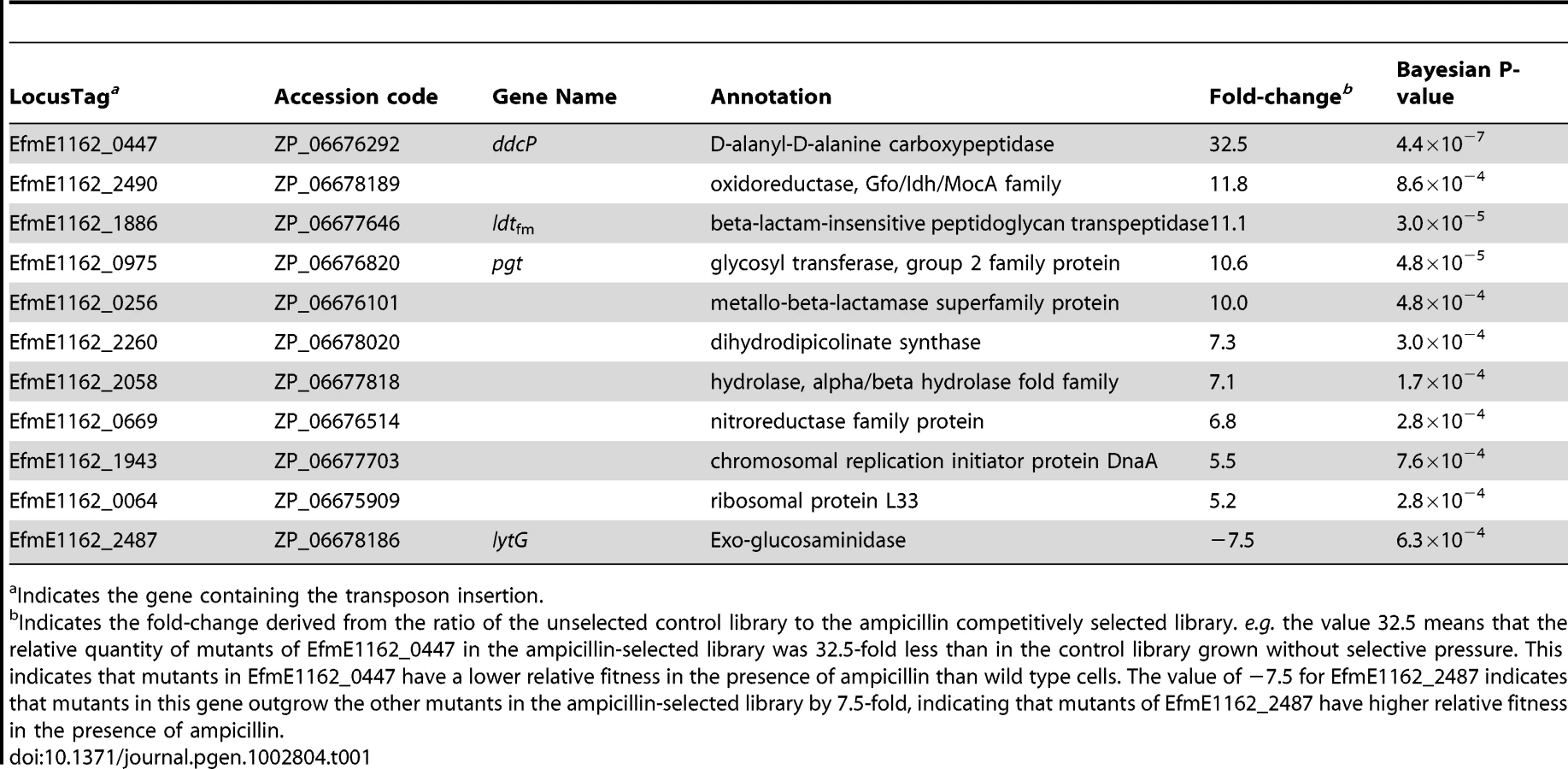
Comparison of ampicillin sensitivities of targeted mutants and wild-type E1162
To validate the results of the M-TraM screen and to further characterize the role of the identified genes in ampicillin resistance, we constructed targeted mutants in the ddcP, ldtfm and pgt genes, which were identified with the most significant P-values (Table 1) and have predicted functions in cell wall biogenesis. We also generated a targeted mutant in the lytG gene of which the inactivation could confer hyper-resistance to ampicillin, as suggested by the M-TraM data. Targeted deletion mutants of ddcP, pgt, and lytG were generated using a novel Cre-lox-based system for the generation of markerless mutants in E. faecium that we developed as part of this study (Figure S4). For ldtfm no double cross-over mutant could be constructed and instead a single cross-over mutant was constructed using the pWS3 vector [34]. Mutants of ddcP, ldtfm, and lytG were also complemented in trans and the ampicillin resistance of E1162 (wild-type), the mutants and the complemented strains was determined. The pgt mutant could not be complemented as constructs containing the pgt gene could not be transformed to either Escherichia coli or E. faecium, presumably due to toxicity of the gene product. In the absence of ampicillin we did not detect significant differences in growth speed or cell density upon entry into stationary phase between wild-type and the mutant strains (Figure S5). When these strains were grown in BHI with 20 µg ml−1 ampicillin, the ΔddcP mutant was dramatically affected in its growth (Figure 3A). Growth of the ldtfm::pWS3 and Δpgt mutants was also poorer than wild-type (Figure 3B and 3C). The in trans complemented strains of the ΔddcP and ldtfm::pWS3 strains could fully or partially restore the ampicillin resistance to wild-type levels (Figure 3A–3C). The ΔlytG mutant had a growth rate that was similar to the parental strain's (0.998±0.007 h−1 for ΔlytG vs. 0.989±0.018 h−1 for E1162) but could grow to slightly higher optical densities (Figure 3D). The in trans complemented lytG mutant exhibited a significantly lower growth rate (0.859±0.017 h−1) in exponential phase (Figure 3D). The empty vector had no effect on the growth of the mutants in BHI supplemented with 20 µg ml−1 ampicillin (data not shown). MICs of ampicillin against the wild-type E1162 and ΔddcP, ldtfm::pWS3, Δpgt and ΔlytG strains were determined by microdilution in cation-adjusted Muller-Hinton broth as 43, 8, 16, 27 and 43 µg ml−1, respectively, which is in accordance with the growth performance of the mutants in BHI with 20 µg ml−1 ampicillin (Figure 3). The seemingly contradictory observation that the ldtfm::pWS3 mutant can grow in BHI medium supplemented with ampicillin at a concentration above the MIC can be explained by the differences in growth media used and the approximately 4-fold larger inoculum size used in the growth experiments in BHI compared to the inoculum size used in the MIC determinations. The MIC of vancomycin was determined to be 0.5 µg ml−1 for all strains.

Comparative analysis of the transcriptome of E. faecium E1162 during exponential growth in the absence and presence of ampicillin
We used microarray-based transcriptome analysis on exponentially growing (OD660 = 0.3) E. faecium E1162 cultures in BHI medium with or without 20 µg ml−1 ampicillin to identify genes that are regulated by the exposure to sub-MIC levels of ampicillin. Compared to the untreated control, only sixteen genes were identified to be differentially regulated between the two conditions and none of these genes were upregulated more than 2.1-fold in the presence of ampicillin (Table S2), indicating that E. faecium does not require major transcriptional rearrangements to cope with the presence of sub-inhibitory levels of ampicillin. None of the genes that were identified by M-TraM were identified to be differentially regulated by the presence of ampicillin, which indicates that the identified ampicillin resistance determinants are constitutively expressed, even in the absence of ampicillin.
Mutations in ddcP and ldtfm increase sensitivity to lysozyme
After the identification of ddcP, ldtfm and pgt as ampicillin resistance determinants, we studied the susceptibility of the wild-type strain E1162 and its mutants to another compound targeting the cell wall, i.e. lysozyme, which is one of the most important antimicrobial enzymes of the host innate immune system. Lysozyme kills Gram-positive bacteria by enzymatic lysis of the bacterial cell wall [35]. The results demonstrated that deletion of pgt had no significant effect on lysozyme resistance, while ΔddcP and ldtfm::pWS3 mutants were significantly more sensitive to lysozyme challenge than the wild-type strain (Figure 4). The in trans complemented strains of ΔddcP and ldtfm::pWS3 could restore the resistance to lysozyme. Hence, ddcP and ldtfm contribute not only to β-lactam resistance but also to the resistance against the peptidoglycan-hydrolyzing enzyme lysozyme.

Disruption of ddcP abolishes membrane-associated D,D-carboxypeptidase activity of E. faecium E1162
Of all novel ampicillin resistance determinants identified in this study, the ddcP gene contributes most to ampicillin resistance in E. faecium E1162. The DdcP protein was annotated as a D-alanyl-D-alanine carboxypeptidase but a functional study confirming this activity has not been performed. To confirm its predicted function, we determined the D-alanyl-D-alanine carboxypeptidase activity in cellular extracts of E1162, the ΔddcP mutant and the in trans complemented strain ΔddcP+ddcP. As shown in Figure 5, the D-alanyl-D-alanine-carboxypeptidase activity of ΔddcP membrane extracts was completely abolished. In the complemented ΔddcP+ddcP strain enzymatic activity was restored in the membrane fraction, revealing that DdcP is responsible for D-alanyl-D-alanine carboxypeptidase activity in E. faecium E1162. The D,D-carboxypeptidase activity was approximately 5 fold lower in the cytoplasmic fractions than in the membrane fractions (data not shown), strongly suggesting that the DdcP protein is associated with the membrane. When E. faecium E1162 was grown in the presence of 20 µg ml−1 ampicillin, all D-alanyl-D-alanine carboxypeptidase activity was undetectable, which is in accordance with the designation of DdcP as a LMW-PBP.

Confirmation of pbp5 as an important ampicillin resistance determinant in E. faecium E1162
Notably, pbp5 was not identified in this M-TraM screen even though this gene has been implicated in high-level ampicillin resistance in E. faecium [16]–[18]. Our failure to identify pbp5 by M-TraM may partially be explained by the presence of an AluI restriction site on one of the two microarray probes for pbp5. The other microarray probe maps to an internal region of the pbp5 gene and is located on a small AluI restriction fragment with a window of 158 nucleotides for the transposon insertion. Previous genome-wide studies using transposon mutagenesis have shown that transposon insertions might fail to fully inactivate the target genes. The failure is commonly found with insertions situated near either end of a gene, but has also been observed with internal insertions [36], [37], after which genes might be capable of intracistronic complementation. We constructed targeted insertional mutants of pbp5, ddcP and ldtfm by the insertion of the plasmid pWS3 5′ to the gene probes and close to the central region of the gene. Consistent with the M-TraM results and the phenotypes of the markerless mutants, the insertional mutants of ddcP (data not shown) and ldtfm (Figure 3B) were sensitized to ampicillin. Surprisingly the insertional mutation in pbp5 did not significantly affect the sensitivity to ampicillin (Figure 6), implying that insertional mutation (by either a plasmid or a transposon) could not fully disrupt pbp5. Therefore a markerless deletion mutant for pbp5 was generated to completely abolish the function of this gene using the Cre-lox system described above. To our knowledge, a targeted mutant of the pbp5 gene has not been generated previously in E. faecium. The pbp5 mutant was unable to grow in cultures containing 20 µg ml−1 ampicillin and the ampicillin resistant phenotype could be partially be restored by in trans complementation of Δpbp5 with the pbp5 gene (Figure 6). The MIC of ampicillin for Δpbp5 was determined by broth microdilution to be only 0.2 µg/ml. Our results revealed that the relative contribution of pbp5, ddcP, ldtfm and pgt to ampicillin resistance in E. faecium E1162 can be summarized as pbp5≫ddcp>ldtfm>pgt. The pbp5 mutant was also assayed for its survival in the presence of lysozyme, but no significant difference was found with the parental strain E1162 (data not shown).

Discussion
Ampicillin resistance in E. faecium has emerged in the late 1970s and has spread rapidly since [38]. Practically all clinical isolates are currently resistant to ampicillin. The resistance to β-lactams of E. faecium complicates the treatment of infections with this organism, particularly when resistance to other antibiotics has also been acquired. The goal of the research described here was to identify genes involved in ampicillin resistance in E. faecium in a high-throughput fashion. We developed a system for the generation of a large random transposon mutant library in E. faecium, coupled to a microarray-based screening approach (termed M-TraM) to simultaneously monitor the relative fitness of individual mutants undergoing selection by growth in the presence of ampicillin.
The lack of appropriate genetic tools has long been a bottleneck for the studies of E. faecium. In this study, we constructed a random high-density transposon mutant library in E. faecium, developed a powerful screening technique to track transposon mutants and adapted the Cre-lox [39] recombination system to construct targeted, markerless mutants in E. faecium, which enabled us to perform high-throughput genome-wide analysis and specific targeted investigations in a clinical E. faecium isolate.
When E. faecium is exposed to ampicillin the D,D-transpeptidase activity of PBPs is inhibited, with the exception of the low-affinity PBP5, which can catalyze the last cross-linking step of the D,D-transpeptidation pathway of cell wall assembly [17]. This implies that any D,D-transpeptidase activity conferred by genes other than pbp5, is not essential for ampicillin resistance. Consequently, no genes encoding D,D-transpeptidases were identified in this study. However, in our transposon mutant screen, we identified a novel D-alanyl-D-alanine carboxypeptidase (DdcP) that plays an important role in resistance to ampicillin. DdcP is the only gene that is responsible for D-alanyl-D-alanine carboxypeptidase activity during exponential growth of strain E1162. The observation that the enzymatic activity of DdcP is abolished in the presence of ampicillin is in accordance with the prediction that DdcP is a LMW-PBP. The in vivo functional roles of LMW-PBPs are relatively poorly understood but they are generally not essential for survival and are thought to contribute to peptidoglycan-remodelling in both Gram-positive and Gram-negative bacteria [40]. Deletion of the gene encoding a LMW-PBP can lead to an increased sensitivity towards β-lactam antibiotics [41]–[42]. Although a mechanistic understanding for this sensitive phenotype is currently lacking, it has been proposed that these LMW-PBPs may enzymatically inactivate β-lactams or, alternatively, save other PBPs from inactivation by sequestration of the β-lactams to the LMW-PBPs [41]. Further functional characterization of DdcP in E. faecium, also involving strains that have higher and lower resistance towards ampicillin than strain E1162, is needed to identify the exact mechanism by which DdcP contributes to ampicillin resistance in E. faecium. Previous biochemical studies have indicated that Ldtfm is a crucial component of the β-lactam-insensitive L,D-transpeptidation pathway, which catalyzes the cross-links of tetrapeptides [21]–[23]. However, genetic evidence for the role of Ldtfm in ampicillin resistance was so far lacking. Here, we have constructed a mutant, confirming the role of this pathway in ampicillin resistance in E. faecium. The identification of pgt as an ampicillin resistance determinant in E. faecium suggests that wall teichoic acid is involved in β-lactam resistance which is in line with a similar observation in methicillin-resistant Staphylococcus aureus (MRSA) strains [43]. None of the mutations had an effect on vancomycin resistance of E. faecium, presumably because tetrapeptide precursors for peptidoglycan crosslinks are present at levels that are too low to confer resistance to this antibiotic in wild-type and the mutant strains.
We did not generate mutants or performed functional analyses to confirm the function of the other genes that were identified in our M-TraM screening (Table 1). However, it seems likely that at least some of these genes also contribute to ampicillin resistance in E. faecium, in particular EfmE1162_2490 (predicted to function as an NAD - or NADP-dependent oxidoreductase) and EfmE1162_0256 (possibly acting as a zinc-dependent β-lactamase), because the transposon mutants in these genes appear to have a similar loss in fitness in the presence of ampicillin as the transposon mutants in ldtfm and pgt (Table 1). The lytG gene was the only gene that was identified in our screen which, upon inactivation by a transposon, appeared to result in a hyper-resistant phenotype. However, this phenotype could not be confirmed in a markerless deletion mutant of lytG, which had the same MIC for ampicillin as the wild-type strain. The observed slightly higher optical density reached by ΔlytG as compared to wild-type and the slower growth rate of the in trans complemented lytG mutant may suggest that there is a subtle role for lytG in ampicillin resistance in E. faecium but further experiments would be required to exactly determine its role.
The recent emergence of E. faecium as a major nosocomial pathogen can be explained by the acquisition of genes that contribute to colonization or infection [26], [44] and by the acquisition of resistance to antibiotics, particularly to ampicillin and vancomycin. Interestingly, resistance against ampicillin and vancomycin emerged predominantly in E. faecium while both resistances are virtually absent in E. faecalis [8]. This study provides insights into the genetic basis of intrinsic β-lactam resistance in E. faecium and identified the ampicillin resistance determinants DdcP, Ldtfm and Pgt in this organism. All three genes are conserved in E. faecium but absent from E. faecalis, indicating that E. faecium possesses more innate β-lactam resistance determinants than E. faecalis. This observation supports the concept that E. faecium has a higher potential to develop high-level β-lactam resistance than E. faecalis, thereby explaining the faster emergence of ampicillin resistance in E. faecium than in E. faecalis [1], [2], [45].
We have identified several novel mechanisms, besides the low-affinity penicillin-binding protein PBP5, that contribute to ampicillin resistance in E. faecium. These proteins could serve as targets for the development of novel therapeutics against this multi-resistant organism. Our study showed that DdcP and Ldtfm also contribute to resistance to lysozyme of E. faecium. An inhibitor of these proteins may thus provide the dual benefit of compromising resistance to the innate immune system as well as enhancing antibiotic susceptibility. In the course of this study we have also developed a number of novel genetic tools for E. faecium allowing for genome-wide analysis of this bacterium. Further functional genomic-based studies to understand the mechanisms involved in colonization, infection and antibiotic resistance of this important nosocomial pathogen are now a realistic opportunity for future research.
Materials and Methods
Bacterial strains, plasmids, and growth conditions
E. faecium and E. coli strains used in this study are listed in Table S1. The ampicillin-resistant E. faecium strain E1162 was used throughout this study. This strain was isolated from a bloodstream infection in France in 1996 and its genome has recently been sequenced [24]. Unless otherwise mentioned, E. faecium was grown in brain heart infusion broth (BHI; Oxoid) at 37°C. The E. coli strains DH5α (Invitrogen) and EC1000 [46] were grown in Luria-Bertani (LB) medium. Where necessary, antibiotics were used at the following concentrations: chloramphenicol 4 µg ml−1 for E. faecium and 10 µg ml−1 for E. coli, gentamicin 300 µg ml−1 for E. faecium and 25 µg ml−1 for E. coli, spectinomycin 300 µg ml−1 for E. faecium and 100 µg ml−1 for E. coli, and erythromycin 50 µg ml−1 for E. faecium (with added lincomycin at 50 µg ml−1) and 150 µg ml−1 for E. coli. All antibiotics were obtained from Sigma-Aldrich (Saint Louis, MO). Growth was determined by measuring the optical density at 660 nm (OD660).
Construction of an E. faecium in vivo transposon mutagenesis system
The transposon delivery plasmid, pZXL5, was constructed in several steps. The Gram-positive lacZ gene of pCJK72 [47] was PCR (Accuprime DNA polymerase, Invitrogen) amplified using the primers pCJK72_PstI_lacZ_F and pCJK72_KpnI_lacZ_R (primer sequences are listed in Table S3) and cloned into pTEX5500ts [48] between PstI and KpnI sites to create pZXL1. A fragment containing the chloramphenicol resistance (Chlr) cassette, the lacZ gene and a gram-positive thermosensitive origin of replication (repAts, functional at 30°C, but not at 37°C) from pZXL1 was PCR amplified using the pZXL1_EcoRI _cm_ori_F and pZXL1_EcoRI _cm_ori_R. Meanwhile, another fragment containing a nisin inducible mariner transposase C9 and the nisRK genes (encoding a two-component system required for the transcriptional activation of the transposase gene in the presence of nisin), and the ColE1 origin of replication was PCR amplified from pCJK55 [47] using the primers pCJK55_EcoRI_tps_F and pCJK55_EcoRI_tps_R. These two fragments were digested with EcoRI and then ligated together to generate pZXL2. To construct a mariner transposon [49], the 5′ and 3′ ITRs of Himar1-mariner were amplified from pMMOrf [50] using the primer pMMOrf_SacII_ITR that resulted in two SacII recognition sites at both ends of the amplified DNA. The amplified fragment was cloned into pGEM-T Easy (Promega) forming pGEM-ITR. A gentamicin-resistance cassette was PCR amplified from pAT392 [51] using primers pAT392_EheI_T7_genta_F and pAT392_EheI_T7_genta_R, resulting in a gentamicin-resistance cassette with outward-facing T7 promoters on both ends of the cassette, which allows for the generation of RNA products corresponding to the regions flanking the site of transposon integration in genomic DNA. This fragment was digested with EheI and cloned into a SmaI site present between the 5′ and 3′ ITRs in pGEM-ITR, thereby forming the transposon cassette. This transposon cassette was then cut out with SacII and subcloned into the SacII site of pZXL2 producing pZXL3. This vector was electroporated to E. faecium E1162 but pZXL3 was found to be able to replicate at 37°C in E. faecium E1162 and blue/white screening using lacZ proved to be ineffective (data not shown). We therefore replaced the repAts and lacZ gene of pZXL3 by the repAts from pAW068 [52]. To this aim, a fragment of pZXL3 containing the nisin inducible mariner transposase and the transposon cassette was PCR amplified by primers pZXL3_BfrI_tn_F and pZXL3_BfrI_tn_R. Another fragment carrying the repAts and Chlr cassette was amplified from pAW068 by PCR using primers pAW068_BfrI_cm_ori_F and pAW068_BfrI_cm_ori_R. After digestion with BfrI these two PCR products was ligated together, resulting in the generation of pZXL5. All restriction enzymes were obtained from New England Biolabs.
Transposon mutant library construction and evaluation
Electrotransformation of the different E. faecium strains (Table S1) with the plasmid pZXL5 was performed according to previously described methodologies [26], [34] with optimizations in preparing electrocompetent cells and the cell-plasmid mixture. To obtain the electrocompetent cells, overnight cell culture from BHI was diluted 1000 fold in 25 ml BHI supplemented with 1% of glycine and 200 mM sucrose and again grown overnight at 37°C. Cells were then resuspended in same volume of pre-warmed BHI supplemented with 1% glycine and 200 mM sucrose and incubated at 37°C for 1 hour. Cells were washed three times with ice-cold wash buffer (500 mM of sucrose and 10% glycerol), and resuspended with 1.25 ml ice-cold wash buffer. A 100 µl aliquot of the cell suspension was mixed with 0.1–1 µg of plasmid and transferred into an ice-cooled electroporation cuvette (2-mm gap) and kept on ice for 20 minutes before electroporation. Gentamicin-resistant transformants were grown overnight in BHI broth supplemented with 300 µg ml−1 gentamicin and 10 µg ml−1 chloramphenicol at the permissive temperature of 28°C, after which 100 µl (approximately 108 viable cells) of this overnight culture were inoculated in 200 ml of pre-warmed BHI broth supplemented with gentamicin and 25 ng ml−1 nisin and grown overnight at the non-permissive temperature of 37°C with shaking at 150 rpm. Subsequently, 100 µl of this culture was transferred to 200 ml of fresh pre-warmed BHI broth and similarly grown overnight without nisin. Cultures were then stored at −80°C in BHI broth containing 50% (v/v) glycerol in 1 ml aliquots as mutant library stocks.
To evaluate the randomness and coverage of transposition, we performed Southern blot analysis, identified the sites of transposon insertion and used PCR footprinting. Southern blot analysis was performed as described previously [34]. Genomic DNA of 17 arbitrarily picked gentamicin-resistant colonies from the library was isolated using the Wizard Genomic DNA Purification kit (Promega), digested with HaeIII and BamHI. The probe consisting of a 414 bp fragment within the gentamicin-resistance gene was amplified from pZXL5 by PCR, using the primer pair genta_probe_F/R. To map the sites of transposon insertion, genomic DNA of 17 mutants from the library was digested with HaeIII and then self-ligated, forming circular DNA. Loci in which the transposon had inserted were amplified using the transposon-specific primer pair IPCR_HaeIII_R/F with AccuPrime DNA polymerases (Invitrogen) with the following conditions: 94°C for 1 min; 32 cycles of 94°C for 18 sec, 53°C for 30 sec, 68°C for 10 min; and 68°C for 7 min. Sequencing of the PCR product was performed using the primer IPCR_HaeIII_R and/or IPCR_HaeIII_F. PCR footprinting was conducted on genomic DNA of mutant library as described elsewhere [53] with a transposon-specific primer, ftp_tn, and gene-specific primers, ftp_ddl, ftp_nox or ftp_esp, respectively.
Simultaneously mapping of transposon insertion sites by M-TraM
Transposon insertion mapping was based on the previously published method of Genomic Array Footprinting (GAF) [54]. Because we observed that T7 polymerase will transcribe E. faecium genomic DNA aspecifically (data not shown), it was required to modify GAF to specifically enrich for the junction sites of the transposon and the flanking E. faecium DNA. Genomic DNA from mutant libraries was isolated using the Wizard Genomic DNA Purification kit (Promega), digested with AluI (New England Biolabs) and then purified on a Qiagen QIAquick PCR Purification column (Qiagen). 200 ng of the digested DNA was self-ligated by the Quick Ligation Kit (New England Biolabs) in a reaction volume of 20 µl. This ligation reaction was directly used as template for PCR amplification of the transposon–chromosome junctions with primer pair IPCR_AluI_F and IPCR_AluI_R in a reaction volume of 200 µl, using AccuPrime Taq DNA polymerases High Fidelity (Invitrogen) with the following conditions: 94°C for 1 min; 26 cycles of 94°C for 18 sec, 56.5°C for 30 sec, 68°C for 50 sec; and 68°C for 7 min. PCR products were purified using Qiagen QIAquick PCR Purification Kit. After purification, 200–500 ng DNA was redigested by AluI and used for in vitro transcription (IVT) in a volume of 20 µl using the T7 MEGAshortscript kit (Ambion) at 37°C for 6 hours. The RNA was first treated with DNase (Ambion) and then purified with the MEGAclear Kit (Ambion). 5–10 µg of the purified RNA was used for generating labeled cDNA using the FairPlay III Microarray Labeling Kit (Agilent Technologies) as described in the manufacturer's protocol. Samples of both conditions (grown in BHI and BHI with 20 µg ml−1 ampicillin) were labeled with Cy3 or Cy5. Dyes were swapped between samples to minimize the effect of dye bias. Microarray hybridizations were carried out using the Gene Expression Hybridization Kit (Agilent) following the manufacturer's instructions, using 60 ng of labeled cDNA. The experiment was performed with four biologically independent replicates.
The microarrays used in this study were custom-made E. faecium E1162 microarrays using Agilent's 8×15 K platform. Probes were designed by Agilent's eArray server. As probes 60-mer oligonucleotides were designed on coding sequences (CDS) only. A total of 2650 CDS are covered by 2 probes (98.4% of the total number of CDS in the E. faecium E1162 genome sequence; NCBI accession number NZ_ABQJ00000000), which were spotted in duplicate. A total of 11 CDS are covered by a single probe (0.4% of the total number of CDS) and these probes were spotted in quadruplicate. For 33 CDS no probes could be designed.
Microarray data were extracted and normalized using Agilent Feature Extraction Software Version 10.7.1.1 (FE 10.7.1). Statistical differences in hybridization signals between the conditions were analyzed using Cyber-T [55] (http://cybert.microarray.ics.uci.edu/). Probes exhibiting Bayesian P-value<0.001 were deemed statistically significant. A gene with two identical probes or all four probes meeting this criterion were classified as significantly selected during exposure to ampicillin.
Screening for genes involved in ampicillin resistance
To carry out identification of genes required for ampicillin resistance, aliquots containing approximately 107 CFU from the mutant pool stored at −80°C were diluted 1 to 1000 in 20 ml of BHI broth or BHI broth with 20 µg ml−1 ampicillin. Cells were grown at 37°C for 8 hours, after which 1 ml of the bacteria cultures were spun down and used for the extraction of genomic DNA, which was then further processed as described above. The same protocol (except that the cells were grown for 20 hours instead of 8 hours) was used to determine the reproducibility of the M-TraM screening in which two independent mutant libraries cultures were mapped using the approach described above.
Construction of targeted, markerless deletion mutants and in trans complementation
For this study, we developed a new method to construct markerless mutants in E. faecium based on the Cre-lox recombination system [39]. The 5′ and 3′ flanking regions (approximately 500 bp each) of the target genes were PCR amplified with the primers in Table S3. The two flanking regions were then fused together by fusion PCR (generating an EcoRI site between both fragments) and cloned into pWS3. Then a gentamicin-resistant cassette was PCR amplified from pAT392 using primers pAT392_EcoRI_lox66_genta_F and pAT392_EcoRI_lox71_genta_R, resulting in a gentamicin-resistant cassette flanked by lox66 and lox71, which allows for the deletion of the gentamicin-resistant cassette in the presence of Cre recombinase. This fragment was digested with EcoRI and cloned into the EcoRI site that was generated between the 5′ and 3′ flanking regions in the pWS3 construct and then electrotransformed into E. faecium as previously described [26], [48]. A transformant containing the plasmid was grown overnight in BHI broth at 30°C supplemented with gentamicin. The cell culture was then diluted 10,000-fold in prewarmed BHI broth and grown at 37°C overnight without antibiotics. The cells were then plated on BHI agar plates with gentamicin and incubated at 37°C. Colonies were then restreaked on BHI agar plates with spectinomycin and BHI agar plates with gentamicin, respectively. The gentamicin-resistant but spectinomycin-susceptible colonies were supposed to be marked deletion mutants and checked by PCR (Table S3). To remove the marker and obtain the markerless mutants a Cre cassette was cut from pRAB1 [56] by digestion with PstI and SacI, blunted by Quick Ligation Kit (New England Biolabs) and cloned into the EcoRV site of pWS3 producing pWS3-Cre, which was subsequently electrotransformed into the marked mutants. Spectinomycin-resistant transformants containing pWS3-Cre were then grown overnight in BHI broth at 30°C supplemented with spectinomycin and then diluted 10000 fold in pre-warmed BHI broth and grown at 37°C overnight without antibiotics. These cultures were plated on BHI agar plates and incubated at 37°C for 18–24 h. Single colonies were then restreaked on BHI agar with spectinomycin, BHI agar with gentamicin and BHI agar without antibiotics. The colonies that were susceptible to both gentamicin and spectinomycin resulted from a recombination event catalyzed by Cre and subsequent loss of the thermosensitive plasmid, resulting in a markerless deletion mutant of the gene of interest. This was verified by PCR and sequencing.
Insertional mutagenesis was performed as previously described [34]. Internal DNA fragments of target genes were PCR amplified using primers listed in Table S3, cloned to a Gram-positive thermosensitive plasmid and electrotransformed into E. faecium as previously described [26], [48]. After electrotransformation, the cells were recovered for 2 hours at 30°C, after which the cells were plated on BHI plates supplemented with 300 µg ml−1 spectinomycin at 30°C to select for transformants. Spectinomycin-resistant colonies were picked and grown overnight in 200 ml of BHI broth at an elevated temperature (37°C) to cure the plasmid. The cells were then plated on BHI agar plates with spectinomycin at 37°C. Single-cross-over integrations into the target genes were verified by PCR with a pWS3-specific primer, check_pWS3, and a gene-specific primer (Table S3).
Plasmids for the in trans complementation of the ddcP, ldtfm, lytG and pbp5 mutants were produced by PCR amplification of the genes using the primers listed in Table S3. PCR products were ligated into the downstream region of PnisA promoter of pMSP3535 [57]. The resulting plasmids were introduced into the appropriate host strains by electroporation as described above.
Determination of growth curves and MIC
A BioScreen C instrument (Oy Growth Curves AB, Helsinki, Finland) was used to monitor effects of ampicillin on bacterial growth. Wild-type E. faecium, mutants and in trans complemented strains were grown overnight in BHI and BHI containing appropriate antibiotics. Cells were inoculated at an initial OD660 of 0.0025 into 300 µl BHI and BHI with ampicillin 20 µg ml−1 and 1 µg ml−1.The cultures were incubated in the Bioscreen C system at 37°C with continuous shaking, and absorbance of 600 nm (A600) was recorded every 15 min for 9 hours. Each experiment was performed in triplicate.
MIC of ampicillin of the wild-type and mutants were determined in triplicate by broth microdilution in cation-adjusted Muller-Hinton broth as previously described [58].
Transcriptome profiling
E. faecium E1162 was incubated in BHI broth and BHI broth supplemented with 20 µg ml−1 ampicillin for 18 hours. Cultures were then diluted to OD660 0.025 in 20 ml of prewarmed BHI broth and BHI broth containing 20 µg ml−1 ampicillin respectively, and grown until OD660 0.3. Cells were centrifuged for 12 seconds at 169000 g at room temperature, and pellets were flash frozen in liquid N2 prior to RNA extraction. RNA was isolated using TRI Reagent (Ambion) according to the manufacturer's protocol. RNA quantity and quality was determined by spectrophotometry (Nanodrop 1000, Thermo Scientific, Wilmington DE, USA) and by Bioanalyzer 2100 analysis (Agilent). Labeling of 5 µg of total RNA, hybridization and data analysis were performed as described above. Genes for which all four probes exhibited a Bayesian P<0.001 in Cyber-T [55] were deemed differentially expressed.
Assay for lysozyme sensitivity
To compare the lysozyme sensitivity of the parental strain E1162, the mutant strains and in trans complemented strains, overnight cell cultures were diluted 100 fold in fresh BHI and grown to OD660 0.5. Two ml of the cell cultures were harvested by centrifugation. The pellets were resuspended in 1 ml phosphate buffered saline (PBS; NaCl 137 mM; 2.7 mM KCl; 10 mM Na2HPO4; 2 mM KH2PO4; pH 7.4) as negative control and in 1 ml PBS containing 0.5 mg ml−1 lysozyme. After a 30-minute incubation at 37°C, cells were washed with PBS and resuspended in 1 ml of PBS. Survival of the strains was determined following serial dilution and plating on BHI agar plates. The experiment was performed in triplicate and statistical analysis of the data was performed using a two-tailed Student's t-test.
Determination of D,D-carboxypeptidase activity in enterococcal extracts
The enzymatic activities in the enterococcal extracts of wild-type, ΔddcP and ΔddcP+ddcP were assayed as described previously with slight modifications [59], [60]. In short, strains were grown until an OD600 of 0.7. Bacteria were then harvested by centrifugation and lysed by treatment with lysozyme at 37°C for 1 hour followed by sonication. The membrane fraction was then pelleted by ultracentrifugation (100,000 g, 45 min). The supernatant (cytoplasmic fractions) was collected and the pellet (membrane fractions) was resuspended in 0.1 M phosphate buffer (pH 7.0) and both fractions were assayed for D,D-carboxypeptidase activity [59], [60]. The amounts of D-Ala released from the pentapeptide (Ala-D-γ-Glu-Lys-D-Ala-D-Ala, Sigma-Aldrich) by D,D-carboxypeptidases were determined by using D-amino acid oxidase and horseradish peroxidase in a colorimetric assay.
Microarray data accession numbers
The microarray data generated in this study have been deposited in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress) under accession numbers E-MEXP-3501 for the M-TraM screening for ampicillin resistance determinants, E-MEXP-3502 for the assay of the reproducibility of the M-TraM procedure and E-MEXP-3564 for the transcriptome analysis data.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. HidronAIEdwardsJRPatelJHoranTCSievertDM 2008 NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol 29 996 1011
2. WillemsRJvan SchaikW 2009 Transition of Enterococcus faecium from commensal organism to nosocomial pathogen. Future Microbiol 4 1125 1135
3. AmmerlaanHSTroelstraAKruitwagenCLKluytmansJABontenMJ 2009 Quantifying changes in incidences of nosocomial bacteraemia caused by antibiotic-susceptible and antibiotic-resistant pathogens. J Antimicrob Chemother 63 1064 1070
4. TopJWillemsRBlokHde RegtMJalinkK 2007 Ecological replacement of Enterococcus faecalis by multiresistant clonal complex 17 Enterococcus faecium. Clin Microbiol Infect 13 316 319
5. LeavisHLBontenMJWillemsRJ 2006 Identification of high-risk enterococcal clonal complexes: global dispersion and antibiotic resistance. Curr Opin Microbiol 9 454 460
6. van SchaikWWillemsRJ 2010 Genome-based insights into the evolution of enterococci. Clin Microbiol Infect 16 527 532
7. AriasCAContrerasGAMurrayBE 2010 Management of multidrug-resistant enterococcal infections. Clin Microbiol Infect 16 555 562
8. MurdochDRMirrettSHarrellLJMonahanJSRellerLB 2002 Sequential emergence of antibiotic resistance in enterococcal bloodstream isolates over 25 years. Antimicrob Agents Chemother 46 3676 3678
9. GraysonMLEliopoulosGMWennerstenCBRuoffKLDe GirolamiPC 1991 Increasing resistance to beta-lactam antibiotics among clinical isolates of Enterococcus faecium: a 22-year review at one institution. Antimicrob Agents Chemother 35 2180 2184
10. MurrayBE 2000 Vancomycin-resistant enterococcal infections. N Engl J Med 342 710 721
11. WernerGCoqueTMHammerumAMHopeRHryniewiczW 2008 Emergence and spread of vancomycin resistance among enterococci in Europe. Euro Surveill 13: pii 19046
12. MonteroCIStockFMurrayPR 2008 Mechanisms of resistance to daptomycin in Enterococcus faecium. Antimicrob Agents Chemother 52 1167 1170
13. ScheetzMHKnechtelSAMalczynskiMPostelnickMJQiC 2008 Increasing incidence of linezolid-intermediate or -resistant, vancomycin-resistant Enterococcus faecium strains parallels increasing linezolid consumption. Antimicrob Agents Chemother 52 2256 2259
14. AriasCAPanessoDMcGrathDMQinXMojicaMF 2011 Genetic basis for in vivo daptomycin resistance in enterococci. N Engl J Med 365 892 900
15. MurrayBE 1990 The life and times of the Enterococcus. Clin Microbiol Rev 3 46 65
16. FontanaRGrossatoARossiLChengYRSattaG 1985 Transition from resistance to hypersusceptibility to beta-lactam antibiotics associated with loss of a low-affinity penicillin-binding protein in a Streptococcus faecium mutant highly resistant to penicillin. Antimicrob Agents Chemother 28 678 683
17. WilliamsonRle BouguenecCGutmannLHoraudT 1985 One or two low affinity penicillin-binding proteins may be responsible for the range of susceptibility of Enterococcus faecium to benzylpenicillin. J Gen Microbiol 131 1933 1940
18. FontanaRAldegheriMLigozziMLopezHSucariA 1994 Overproduction of a low-affinity penicillin-binding protein and high-level ampicillin resistance in Enterococcus faecium. Antimicrob Agents Chemother 38 1980 1983
19. Galloway-PenaJRRiceLBMurrayBE 2011 Analysis of PBP5 of Early U.S. Isolates of Enterococcus faecium: Sequence Variation Alone Does Not Explain Increasing Ampicillin Resistance over Time. Antimicrob Agents Chemother 55 3272 3277
20. RiceLBBellaisSCariasLLHutton-ThomasRBonomoRA 2004 Impact of specific pbp5 mutations on expression of beta-lactam resistance in Enterococcus faecium. Antimicrob Agents Chemother 48 3028 3032
21. MainardiJLLegrandRArthurMSchootBvan HeijenoortJ 2000 Novel mechanism of beta-lactam resistance due to bypass of DD-transpeptidation in Enterococcus faecium. J Biol Chem 275 16490 16496
22. MainardiJLMorelVFourgeaudMCremniterJBlanotD 2002 Balance between two transpeptidation mechanisms determines the expression of beta-lactam resistance in Enterococcus faecium. J Biol Chem 277 35801 35807
23. SaccoEHugonnetJEJosseaumeNCremniterJDubostL 2010 Activation of the L,D-transpeptidation peptidoglycan cross-linking pathway by a metallo-D,D-carboxypeptidase in Enterococcus faecium. Mol Microbiol 75 874 885
24. van SchaikWTopJRileyDRBoekhorstJVrijenhoekJE 2010 Pyrosequencing-based comparative genome analysis of the nosocomial pathogen Enterococcus faecium and identification of a large transferable pathogenicity island. BMC Genomics 11 239
25. WalshCT 1989 Enzymes in the D-alanine branch of bacterial cell wall peptidoglycan assembly. J Biol Chem 264 2393 2396
26. HeikensEBontenMJWillemsRJ 2007 Enterococcal surface protein Esp is important for biofilm formation of Enterococcus faecium E1162. J Bacteriol 189 8233 8240
27. HeikensESinghKVJacques-PalazKDvan Luit-AsbroekMOostdijkEA 2011 Contribution of the enterococcal surface protein Esp to pathogenesis of Enterococcus faecium endocarditis. Microbes Infect 13 1185 1190
28. LeendertseMHeikensEWijnandsLMvan Luit-AsbroekMTeskeGJ 2009 Enterococcal surface protein transiently aggravates Enterococcus faecium-induced urinary tract infection in mice. J Infect Dis 200 1162 1165
29. LamMMSeemannTBulachDMGladmanSLChenH 2012 Comparative Analysis of the First Complete Enterococcus faecium Genome. J Bacteriol 194 2334 2341
30. SauvageEKerffFTerrakMAyalaJACharlierP 2008 The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32 234 258
31. LeiXHFiedlerFLanZKathariouS 2001 A novel serotype-specific gene cassette (gltA-gltB) is required for expression of teichoic acid-associated surface antigens in Listeria monocytogenes of serotype 4b. J Bacteriol 183 1133 1139
32. HorsburghGJAtrihAWilliamsonMPFosterSJ 2003 LytG of Bacillus subtilis is a novel peptidoglycan hydrolase: the major active glucosaminidase. Biochemistry 42 257 264
33. MagnetSArbeloaAMainardiJLHugonnetJEFourgeaudM 2007 Specificity of L,D-transpeptidases from gram-positive bacteria producing different peptidoglycan chemotypes. J Biol Chem 282 13151 13159
34. ZhangXVrijenhoekJEBontenMJWillemsRJVan SchaikW 2011 A genetic element present on megaplasmids allows Enterococcus faecium to use raffinose as carbon source. Environ Microbiol 13 518 528
35. CallewaertLMichielsCW 2010 Lysozymes in the animal kingdom. J Biosci 35 127 160
36. KangYDurfeeTGlasnerJDQiuYFrischD 2004 Systematic mutagenesis of the Escherichia coli genome. J Bacteriol 186 4921 4930
37. ChristenBAbeliukECollierJMKalogerakiVSPassarelliB 2011 The essential genome of a bacterium. Mol Syst Biol 7 528
38. Galloway-PenaJRNallapareddySRAriasCAEliopoulosGMMurrayBE 2009 Analysis of clonality and antibiotic resistance among early clinical isolates of Enterococcus faecium in the United States. J Infect Dis 200 1566 1573
39. SauerB 1987 Functional expression of the cre-lox site-specific recombination system in the yeast Saccharomyces cerevisiae. Mol Cell Biol 7 2087 2096
40. GhoshASChowdhuryCNelsonDE 2008 Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends Microbiol 16 309 317
41. SarkarSKChowdhuryCGhoshAS 2010 Deletion of penicillin-binding protein 5 (PBP5) sensitises Escherichia coli cells to beta-lactam agents. Int J Antimicrob Agents 35 244 249
42. MemmiGFilipeSRPinhoMGFuZCheungA 2008 Staphylococcus aureus PBP4 is essential for beta-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob Agents Chemother 52 3955 3966
43. CampbellJSinghAKSanta MariaJPJrKimYBrownS 2011 Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem Biol 6 106 116
44. LeavisHLWillemsRJvan WamelWJSchurenFHCaspersMP 2007 Insertion sequence-driven diversification creates a globally dispersed emerging multiresistant subspecies of E. faecium. PLoS Pathog 3 e7 doi:10.1371/journal.ppat.0030007
45. BushLMCalmonJCherneyCLWendelerMPitsakisP 1989 High-level penicillin resistance among isolates of enterococci. Implications for treatment of enterococcal infections. Ann Intern Med 110 515 520
46. LeenhoutsKBuistGBolhuisAten BergeAKielJ 1996 A general system for generating unlabelled gene replacements in bacterial chromosomes. Mol Gen Genet 253 217 224
47. KristichCJNguyenVTLeTBarnesAMGrindleS 2008 Development and use of an efficient system for random mariner transposon mutagenesis to identify novel genetic determinants of biofilm formation in the core Enterococcus faecalis genome. Appl Environ Microbiol 74 3377 3386
48. NallapareddySRSinghKVMurrayBE 2006 Construction of improved temperature-sensitive and mobilizable vectors and their use for constructing mutations in the adhesin-encoding acm gene of poorly transformable clinical Enterococcus faecium strains. Appl Environ Microbiol 72 334 345
49. AkerleyBJLampeDJ 2002 Analysis of gene function in bacterial pathogens by GAMBIT. Methods Enzymol 358 100 108
50. LampeDJAkerleyBJRubinEJMekalanosJJRobertsonHM 1999 Hyperactive transposase mutants of the Himar1 mariner transposon. Proc Natl Acad Sci U S A 96 11428 11433
51. ArthurMDepardieuFSnaithHAReynoldsPECourvalinP 1994 Contribution of VanY D,D-carboxypeptidase to glycopeptide resistance in Enterococcus faecalis by hydrolysis of peptidoglycan precursors. Antimicrob Agents Chemother 38 1899 1903
52. WilsonACPeregoMHochJA 2007 New transposon delivery plasmids for insertional mutagenesis in Bacillus anthracis. J Microbiol Methods 71 332 335
53. ChaudhuriRRAllenAGOwenPJShalomGStoneK 2009 Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH). BMC Genomics 10 291
54. BijlsmaJJBurghoutPKloostermanTGBootsmaHJde JongA 2007 Development of genomic array footprinting for identification of conditionally essential genes in Streptococcus pneumoniae. Appl Environ Microbiol 73 1514 1524
55. BaldiPLongAD 2001 A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics 17 509 519
56. LeibigMKrismerBKolbMFriedeAGotzF 2008 Marker removal in staphylococci via Cre recombinase and different lox sites. Appl Environ Microbiol 74 1316 1323
57. BryanEMBaeTKleerebezemMDunnyGM 2000 Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44 183 190
58. AndrewsJM 2001 Determination of minimum inhibitory concentrations. J Antimicrob Chemother 48 Suppl 1 5 16
59. LebretonFDepardieuFBourdonNFines-GuyonMBergerP 2011 D-Ala-D-Ser VanN-Type Transferable Vancomycin Resistance in Enterococcus faecium. Antimicrob Agents Chemother 55 4606 4612
60. ArthurMDepardieuFReynoldsPCourvalinP 1996 Quantitative analysis of the metabolism of soluble cytoplasmic peptidoglycan precursors of glycopeptide-resistant enterococci. Mol Microbiol 21 33 44
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Rumors of Its Disassembly Have Been Greatly Exaggerated: The Secret Life of the Synaptonemal Complex at the Centromeres
- The NSL Complex Regulates Housekeeping Genes in
- Tipping the Balance in the Powerhouse of the Cell to “Protect” Colorectal Cancer
- Interplay between Synaptonemal Complex, Homologous Recombination, and Centromeres during Mammalian Meiosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy