
A Mouse Model of Acrodermatitis Enteropathica: Loss of Intestine Zinc Transporter ZIP4 (Slc39a4) Disrupts the Stem Cell Niche and Intestine Integrity
: Mutations in the human Zip4 gene cause acrodermatitis enteropathica, a rare, pseudo-dominant, lethal genetic disorder. We created a tamoxifen-inducible, enterocyte-specific knockout of this gene in mice which mimics this human disorder. We found that the enterocyte Zip4 gene in mice is essential throughout life, and loss-of-function of this gene rapidly leads to wasting and death unless mice are nursed or provided excess dietary zinc. An initial effect of the knockout was the reprogramming of Paneth cells, which contribute to the intestinal stem cell niche in the crypts. Labile zinc in Paneth cells was lost, followed by diminished Sox9 (sex determining region Y-box 9) and lysozyme expression, and accumulation of mucin, which is normally found in goblet cells. This was accompanied by dysplasia of the intestinal crypts and significantly diminished small intestine cell division, and attenuated mTOR1 activity in villus enterocytes, indicative of increased catabolic metabolism, and diminished protein synthesis. This was followed by disorganization of the absorptive epithelium. Elemental analyses of small intestine, liver, and pancreas from Zip4-intestine knockout mice revealed that total zinc was dramatically and rapidly decreased in these organs whereas iron, manganese, and copper slowly accumulated to high levels in the liver as the disease progressed. These studies strongly suggest that wasting and lethality in acrodermatitis enteropathica patients reflects the loss-of-function of the intestine zinc transporter ZIP4, which leads to abnormal Paneth cell gene expression, disruption of the intestinal stem cell niche, and diminished function of the intestinal mucosa. These changes, in turn, cause a switch from anabolic to catabolic metabolism and altered homeostasis of several essential metals, which, if untreated by excess dietary zinc, leads to dramatic weight loss and death.
Published in the journal:
. PLoS Genet 8(6): e32767. doi:10.1371/journal.pgen.1002766
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002766
Summary
: Mutations in the human Zip4 gene cause acrodermatitis enteropathica, a rare, pseudo-dominant, lethal genetic disorder. We created a tamoxifen-inducible, enterocyte-specific knockout of this gene in mice which mimics this human disorder. We found that the enterocyte Zip4 gene in mice is essential throughout life, and loss-of-function of this gene rapidly leads to wasting and death unless mice are nursed or provided excess dietary zinc. An initial effect of the knockout was the reprogramming of Paneth cells, which contribute to the intestinal stem cell niche in the crypts. Labile zinc in Paneth cells was lost, followed by diminished Sox9 (sex determining region Y-box 9) and lysozyme expression, and accumulation of mucin, which is normally found in goblet cells. This was accompanied by dysplasia of the intestinal crypts and significantly diminished small intestine cell division, and attenuated mTOR1 activity in villus enterocytes, indicative of increased catabolic metabolism, and diminished protein synthesis. This was followed by disorganization of the absorptive epithelium. Elemental analyses of small intestine, liver, and pancreas from Zip4-intestine knockout mice revealed that total zinc was dramatically and rapidly decreased in these organs whereas iron, manganese, and copper slowly accumulated to high levels in the liver as the disease progressed. These studies strongly suggest that wasting and lethality in acrodermatitis enteropathica patients reflects the loss-of-function of the intestine zinc transporter ZIP4, which leads to abnormal Paneth cell gene expression, disruption of the intestinal stem cell niche, and diminished function of the intestinal mucosa. These changes, in turn, cause a switch from anabolic to catabolic metabolism and altered homeostasis of several essential metals, which, if untreated by excess dietary zinc, leads to dramatic weight loss and death.
Introduction
The rare pseudo-dominant genetic disease acrodermatitis enteropathica (AE) is thought to be caused by the inefficient absorption of dietary zinc [1]. AE occurs at a frequency 1 in 500,000 and symptoms in humans usually develop soon after birth in bottle fed infants or after weaning in breast fed infants. The triad of alopecia, eczematous dermatitis and diarrhea are classic symptoms of AE in humans and patients experience growth retardation and a myriad of symptoms of severe zinc deficiency which eventually lead to death unless treated by exogenous zinc [2].
In 2002, the AE gene was mapped to human chromosomal region 8q24.3 and the Zip4 (Slc39a4) locus [3], [4]. Currently, over 32 mutations or variants of ZIP4 have been reported [5]. Missense and nonsense mutations as well as deletions or rearrangements of the gene have all been reported, and hypomorphic as well as complete loss-of-function alleles have been identified.
Recent studies have shed light on the mechanisms of Zip4 regulation and function [6]. Mouse Zip4 is most actively expressed in tissues involved in the absorption of dietary or maternal zinc, but also shows high level expression in other cell-types (e.g. pancreatic islet cells, brain capillaries), and low level expression in other tissues (e.g. liver, kidney) and some cultured cells. Zip4 expression is regulated by cell-specific transcription as well as by multiple posttranscriptional mechanisms in response to zinc availability. ZIP4 protein is at the apical surface of enterocytes and endoderm cells when zinc is deficient, due to increased mRNA and protein stability. During zinc deficiency ZIP4 undergoes processing by removal of the extracellular amino-terminus. In contrast, in the presence of normal levels of zinc Zip4 mRNA is unstable and the protein is internalized and rapidly degraded.
ZIP4 function is critical during periods of rapid growth when zinc requirements are high but this zinc transporter also has important functions when zinc is replete. Zip4 is aberrantly expressed in many cancers [7], [8]. Knockdown of ZIP4 can slow cell cycle and cell migration in mouse Hepa cells and ZIP4 functions to reduce apoptosis and enhance cell cycle in hepatomas and to enhance pancreatic tumor growth in nude mice [7], [8]. Many recent studies have demonstrated that zinc can modulate signal transduction cascades [9].
The essential function of ZIP4 in zinc homeostasis is confirmed in Zip4-intestine knockout mice. Homozygous Zip4-intestine knockout mouse embryos die soon after implantation because this gene is also actively expressed in the visceral endoderm which surrounds the developing mouse embryo and which serves a nutrient uptake function before development of the placenta. The visceral yolk sac is a more vestigial organ in humans and apparently contributes little to providing nutrients for the embryo, explaining why loss of ZIP4 in humans is apparently not embryonic-lethal. Interestingly, heterozygous Zip4-intestine knockout mouse embryos are significantly underrepresented in the population at parturition. These heterozygous embryos display an array of developmental defects including exencephalia, anophthalmia, and severe growth retardation. About 22% of the Zip4-heterozygous mice that survive to weaning age are severely growth retarded and display anopia, anopthalmia, hydrocephalus and heart defects, among other abnormalities [10]. Mice heterozygous for Zip4 knockout are hypersensitive to zinc deficiency. Thus, haploinsufficiency of Zip4 may contribute to growth retardation in humans, an effect that is probably exacerbated by zinc deficiency and/or by modifier genes. Mutations in genes essential for posttranscriptional regulation of ZIP4 may also cause AE although this has not been demonstrated. The finding of Zip4 haploinsufficiency defines AE as a pseudo-autosomal dominant trait.
Given that a global knockout of Zip4 is embryonic lethal in mice, unlike it is in humans, we sought to develop a better mouse model of AE. To that end we created mice with floxed Zip4 genes and bred them with mice that express an ErtCre fusion protein driven by the villin promoter [11] specifically in intestine enterocytes. Using this tamoxifen-inducible Zip4-enterocyte knockout model we provide evidence that intestine expression of Zip4 is essential for the growth and viability unless mice are supplied with excess zinc and that an absence of ZIP4 in the intestine appears to closely mimic the AE phenotype in humans. Moreover we provide evidence that an absence of ZIP4 only in the intestine leads to a rapid switch from anabolic to catabolic metabolism in the animal, to tissue-specific dysregulation of other essential metals and alterations in gene expression. These phenotypes appear to reflect compromised Paneth cell functions which lead to disruption of the intestinal stem cell niche ultimately resulting in loss of intestinal integrity and diminished nutrient uptake.
Results
Knocking out the Zip4 gene in the intestinal epithelium
To enable tissue-specific deletion of the mouse Zip4 gene a targeting construct was created which contained a LoxP site flanked by an XhoI restriction site in intron 5, and a LoxP site just downstream of the last exon (exon 12), followed by an mc1-Neomycin cassette (Figure 1). This construct was targeted in E14 embryonic stem (ES) cells and cells with proper integration of the floxed Zip4 gene were then identified by long-range PCR using primers outside of the engineered targeting construct coupled with overlapping internal primers (Figure 1A). The 5′ integration screen (Figure 1C) amplified a 7.35 kb product from the wild-type and the floxed alleles and cleavage of the floxed allele with XhoI yielded the predicted 5.2 and 2.1 kb restriction fragments indicative of proper insertion of the floxed allele into one of the endogenous Zip4 alleles. This was confirmed by the 3′ integration screen which yielded a 3.4 kb PCR product from the floxed allele and a 2.1 kb PCR product from the wild-type allele. Targeted E14 ES cells were used to create chimeric mice by blastocyst injection and agouti offspring from the chimeric mice were genotyped using primers which flank the LoxP-XhoI insertion site in intron 5 (Figure 1A). Genotyping PCR yielded a 227 bp product from the floxed allele, which could be cleaved by XhoI, and a 187 bp product from the wild-type allele (Figure 1D).

To enable tissue-specific deletion of the floxed Zip4 gene specifically in the intestinal epithelium, Zip4FX/FX mice were crossed with mice that express an estrogen receptor-Cre recombinase fusion protein under control of the villin promoter [11]. Vil-Cre-ERT2 is expressed only in the intestinal epithelium and visceral endoderm. The Cre-ERT2 fusion protein resides in the cytoplasm and is driven to the nucleus by tamoxifen which allows for temporal control of the Zip4 gene deletion [11]. Mice heterozygous for vil-Cre-ERT2 and homozygous Zip4FX/FX were crossed with Zip4FX/FX mice to yield 50% offspring with Zip4FX/FX: vil-Cre-ERT2 alleles and 50% with Zip4FX/FX alleles. The latter provided age and genetically matched controls for all of our experiments and are labeled as control (Con) mice in the figures. Tamoxifen injections resulted in recombination of approximately 50% of the floxed Zip4 genes in the small intestine of recently weaned Zip4FX/FX: vil-Cre-ERT2 mice (Figure 1D: intest KO). This is as expected since the epithelial cells represent about half of the cells in the small intestine. The extent of loss of Zip4 mRNA however provides a suitable assay for the extent of recombination of the floxed Zip4 gene in the epithelium since the expression of this gene in the intestine is restricted to the epithelium. Three injections of tamoxifen resulted in a dramatic, if not complete loss of intestine Zip4 mRNA (Figure 1E: intest KO) which suggests that the vast majority, if not all, of the floxed enterocyte Zip4 genes had undergone recombination. ZIP4 protein in the intestine of mice fed a zinc-adequate diet is present at only low levels but this mRNA and protein are stabilized when zinc is deficient. ZIP4 protein and mRNA are rapidly turned-over when dietary zinc is restored. The loss of Zip4 mRNA is expected to be followed by the turn-over of ZIP4 protein within a few hours [12].
The intestine Zip4 gene controls growth and viability but knockout mice can be rescued by nursing or excess dietary zinc
Symptoms of AE in humans often appear soon after breast feeding is stopped. Human breast milk provides a rich source of zinc that is easily absorbed by the gut [13]. The effect of knocking out intestine Zip4 in neonatal mice (5 days post-partum) was therefore examined. Newborn mice were injected with tamoxifen and allowed to suckle until weaning on day 21 post-partum (Figure 2A). At weaning there was no difference in the average body weight of the control (7.93 g: n = 4) and knockout mice (7.96 g: n = 5) and the intestine knockout mice appeared normal. However, the Zip4-intestine knockout mice rapidly lost weight after weaning (Figure 2A and Figure S1A) and perished within a week unless provided with excess zinc in the drinking water (Figure 2B). Removal of the excess dietary zinc led to precipitous weight loss and lethality. Northern blot hybridization confirmed that the Zip4 gene was effectively knocked out in these neonatal mice (Figure 2B, inset). Not all of the recently weaned Zip4-intestine knockout mice thrived after providing excess zinc at weaning. Those mice could be rescued from lethality for three weeks post-weaning but failed to gain much body weight during that period (Figure 2B).

The function of intestine Zip4 in rapidly growing, recently weaned mice was then examined. Recently weaned mice (5 days post-weaning) were injected three consecutive days with tamoxifen and their body weights were measured beginning 1 day after the first injection (Figure 2C and Figure S1B). These mice continue to grow for two to three days after the first injection and then begin to lose weight precipitously. Within a week they lose over 20% of their body weight although they continue to eat a normal amount of food and by d8 they can weigh only 60% as much as their littermate controls (Figure 2D). On day 6 the tibia and quadriceps muscle from the Zip4-intestine knockout mice weigh 43 to 50% less than controls while body weight has decreased 23 to 30% relative to littermate controls (n = 4–5; p<0.007). Much of the weight loss involves loss of muscle and bone mass as has been reported to occur during dietary zinc deficiency [14]. Thus, knocking out the intestine Zip4 gene causes a switch from anabolic to catabolic metabolism in these mice. During the course of these studies we examined many mice and all of the Zip4-intestine knockout mice died within two to three weeks. The penetrance of this phenotype is 100% in both male and female mice.
Northern blot hybridization to small intestine RNA demonstrated a loss of Zip4 mRNA and a dramatic reduction of Zip5 mRNA (Figure 2C and 2E, insets). ZIP5 is a basolateral zinc transporter expressed predominately in intestinal crypt cells [15]. This mRNA remains unchanged in abundance during severe zinc deficiency in mice [15] but its translation is repressed [12] by a mechanism involving a stem-loop structure in its mRNA [16]. Attenuation of Zip5 mRNA, whose abundance is not responsive to Zn itself, could suggest injury to the small intestine crypts, as is shown below.
Excess dietary zinc can ameliorate many of the symptoms of AE in humans. Therefore, control and Zip4-intestine knockout mice were provided drinking water containing 250 ppm zinc sulfate beginning on the day of the first tamoxifen injection (Figure 2E). A representative animal is shown but 12 mice were analyzed with similar results. Excess zinc prevented the rapid weight loss in the Zip4-intestine knockout mice and allowed them to thrive for 4 weeks in this experiment, but withdrawal of the excess zinc led to a rapid loss of weight and lethality within two weeks. The Zip4 gene remained knocked out for 4 weeks in these mice (Figure 2E, inset) which indicates that it was also knocked out in the intestinal stem cell population, as reported previously using the vil-CreERT2 system [11]. In other experiments it was noted that providing excess zinc could not effectively ameliorate the lethal effects of the Zip4 knockout if provided later than day 6 (data not shown).
The efficacy of one, two or three consecutive injections of tamoxifen on Cre-ERT2 deletion of the Zip4 gene was examined using recently weaned mice. As described above, three consecutive injections resulted in effective deletion of the Zip4 gene even in the intestinal stem cells. In contrast, a single injection of tamoxifen resulted in partial deletion of the Zip4 gene (Figure 2F, also see Figure S1C). This did not result in lethality; instead those mice grew normally for about a week and then began to slowly lose weight. Thereafter, about half of the mice examined recovered vigorous growth and grew to adult weight (two are shown in Figure 2F) while the other half of these mice continued to slowly lose weight during that time (Figure S1C). The resumption of vigorous growth coincided with active expression of Zip4 mRNA which had returned to normal levels by three weeks (Figure 2F, inset). These data show that partial loss-of-function of ZIP4 can exert detrimental effects of growth consistent with the findings of potentially hypomorphic alleles of Zip4 in AE [17] and that heterozygosity of Zip4 renders mice hypersensitive to zinc deficiency [10].
Many patients with AE require lifelong zinc supplementation. Thus, ZIP4 may function not only during periods of rapid growth but also in full grown adults. Adult mice (7 weeks old) were injected with tamoxifen and then monitored for changes in body weight (Figure 2G). The knockout mice began to lose weight within days of the first injection, lost weight precipitously after 1 week and then succumbed soon thereafter. Thus, the loss of intestine ZIP4 led to a switch from anabolic to catabolic metabolism in adult mice.
Loss-of-function of the intestine Zip4 gene has profound tissue-specific effects on the homeostasis of zinc, iron, manganese, and copper
ZIP4 is a zinc-specific transporter [18]. Although a recent study suggests that it may also be able to transport small amounts of free copper the physiological relevance of that activity remains to be determined [19]. Herein the effect of knocking out intestine Zip4 in recently weaned mice, on the homeostasis of several essential metals was examined using ICP-MS. Within 4 days of initiating the knockout, zinc levels in the small intestine, pancreas and liver were significantly reduced (Figure 3A) consistent with a primary function of ZIP4 in the acquisition of dietary zinc. This loss of zinc correlated temporally with the beginning of precipitous body weight loss after the knockout. Although, by day 4 a small but statistically significant loss of copper (Figure 3B) also occurred in these organs, the largest copper loss was in the small intestine which also lost ∼50% of its iron (Figure 3C). Thus, the loss of ZIP4 in the intestine led to large reductions in small intestine zinc, iron and copper content suggesting possible injury to the intestine, as was found to be the case. An increase in manganese was noted in the liver (Figure 3D), but no other significant changes in other elements in these tissues were found on day 4. Remarkably, by day 8 after initiation of the knockout, iron and manganese were twice as high in the liver of Zip4-intestine knockout mice versus control and hepatic copper had also increased significantly (Figure 3E). In contrast, zinc levels were normalized in the liver which suggests that zinc had been redistributed as body weight declined. The accumulation of iron in the liver during zinc deficiency has been reported previously [20], [21]. Taken together, these results show that intestine ZIP4 is essential for the homeostasis of several essential metals.

Loss-of-function of the intestine Zip4 gene rapidly alters gene expression in the small intestine
Being able to temporally control the intestine knockout of Zip4 enables us for the first time to monitor the rapid effects of loss-of-function of a zinc transporter in an animal. This model of AE compresses the time frame from months in humans to a few days in mice. This allows for examining primary versus secondary effects of losing Zip4 function. To that end Northern blot hybridization and quantitative PCR were used to monitor changes in Zip4, Zip5 and metallothionein - I (MT-I) mRNAs, zinc homeostatic genes, in the small intestine (Figure 4). In addition changes in IGF-1 mRNA, a potent mitogen in the intestine [22], were monitored.

A single injection of tamoxifen resulted in a large loss of native Zip4 mRNA in the small intestine 24 hr later (Figure 4). By one day after the second injection (day 2), little or no native Zip4 mRNA was detectable. Zip5 mRNA abundance dropped precipitously by day 3 while MT-I mRNA was reduced in abundance within a day of the first tamoxifen injection but increased again by d4 and remained high (Figure 4). Thus, deletion of the intestine Zip4 gene occurs rapidly in this model and leads to significant changes in the abundance of several mRNAs involved in zinc homeostasis. The loss of Zip5 expression suggests that injury to the intestine may occur rapidly after deletion of the Zip4 gene, as Zip5 mRNA abundance is not zinc-responsive.
Deletion of the intestine Zip4 gene led to a large increase in the abundance of IGF-I mRNA. Several IGF-I transcripts are present in the intestine. Quantitative PCR of the region encoding IGF-I in all of the reported transcripts confirmed this Northern blot finding and established that this mRNA is induced up to 7-fold within a few days (Figure 4). There were no changes in the abundance of IGF-I transcripts in control littermates (Figure 4, Con). The Con blot was over-exposed relative to that of the knockout mice.
Loss-of-function of the intestine Zip4 gene leads to a rapid loss of Paneth cell zinc, repression of Sox9, and induction of mucin in Paneth cells, which is accompanied by disorganization of crypt and villus morphology
Histological examination of the small intestine after Zip4 knockout revealed progressive and profound disorganization of villus (Figure 5) and crypt architecture (Figure 6) and significantly diminished cell division within days of the knockout (Figure 7). Villus histology was apparently normal on day 2 after initiation of the knockout but became progressively disorganized over the next 4 days (Figure 5A). By day 6 the epithelial cells had lost much of their columnar morphology with basal nuclei and had become cuboidal with centric nuclei (Figure 5B). The lamina propria appeared disorganized, occupied substantially more space in the villi and often contained red blood cells and cells with pyknotic nuclei.
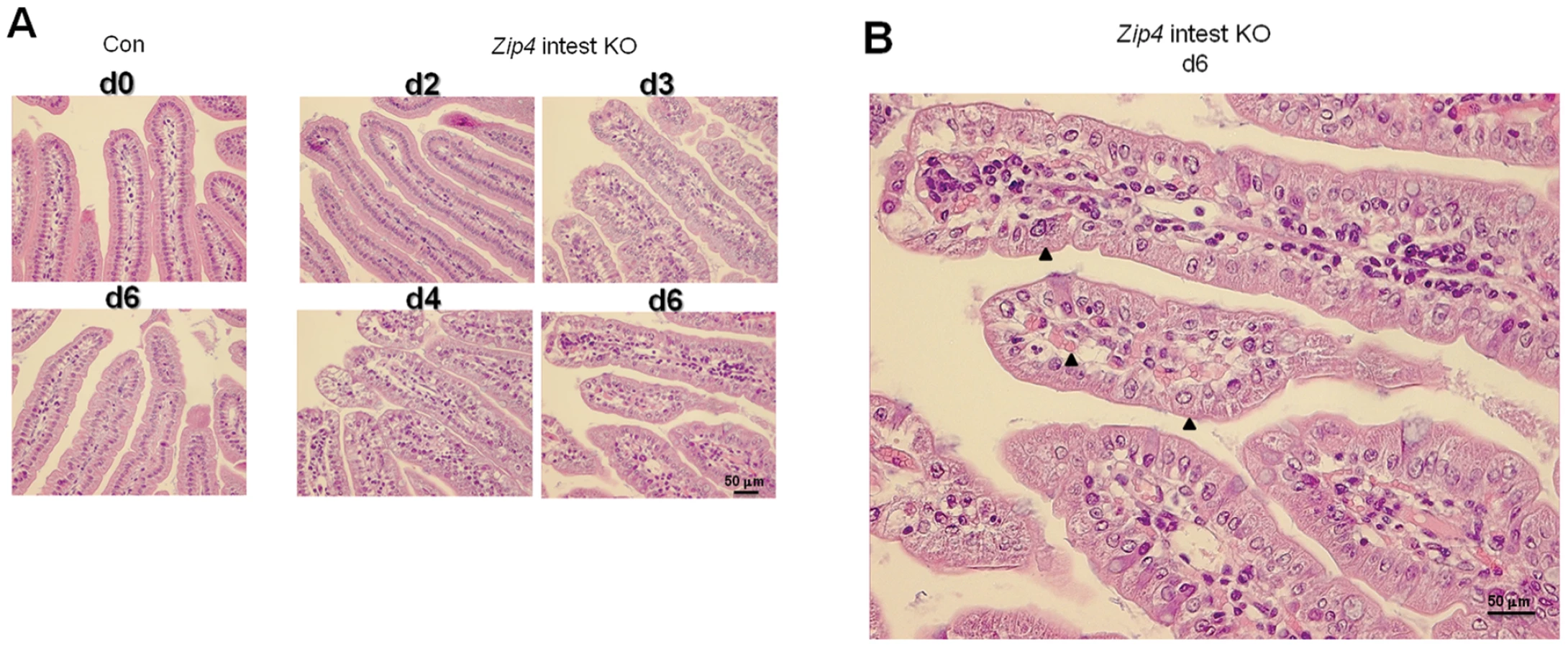

Crypts were examined by localizing the transcription factor Sox9 in sections of small intestine. Sox9 (sex determining region Y - box9) marks the stem/progenitor cell population in the crypts [23] as well as the villus enteroendocrine cells [24] and it regulates cell proliferation and Paneth cell differentiation [25]. In control mice and Zip4-intestine knockout mice before tamoxifen injections and on day 2 after initiation of the knockout, Sox9 was localized to the crypts (Figure 6A) and to dispersed enteroendocrine cells in the villi (not shown), as reported previously [24]. However, after day 2 the architecture of the crypts progressively became disorganized and elongated, and Sox9 positive cells were increasingly detected extending up into the villus epithelium with Sox9 negative cells interspersed among the positive cells (Figure 6A). By days 4 and 6, Sox9 negative cells were found at the base of many crypts (Figure 6A and 6C). The location of these cells at the base of the crypts suggested that these were Paneth cells; however, these cells contained large cytoplasmic vacuoles not found in normal Paneth cells.
Paneth cells contain large amounts of labile zinc which can be detected by staining with the zinc-specific fluorophore ZP1 [26]. ZP1 staining was readily detectable in the intestinal crypts before initiation of the Zip4 knockout (Figure 6B). When viewed under high power ZP1 staining was restricted to one or two cells at the base of the crypts (Figure 6B; 400X panel) whereas under low power the fluorescence signal was dispersed throughout the crypt. ZP1 staining was lost by day 2 after initiation of the knockout and remained undetectable in the Paneth cells on day 4. Thus, zinc is rapidly depleted in Paneth cells when ZIP4 function is lost.
The antibacterial enzyme lysozyme is abundantly produced by Paneth cells [27]. IHC localization of lysozyme in intestine sections from control and Zip4-intestine knockout mice confirmed that Paneth cells accumulate large vacuoles, and have reduced Sox9 and zinc when Zip4 is knocked out (Figure 6C). Paneth cells, enteroendocrine and goblet cells are thought to arise from a common precursor [28]. Paneth cells constitute the niche for Lgr5-positive columnar stem cells in the crypt base [29], [30]. The Lgr5-positive columnar cells can give rise to all epithelial lineages [29]. PAS staining of mucins revealed normal goblet cells interspersed in the villus epithelium of uninjected and knockout mice (Figure S2). PAS staining of the small intestine from Zip4-intestine knockout mice demonstrated positive staining of the large vacuoles in Paneth cells by day 4 (Figure 6C). This staining was resistant to α-amylase and was therefore not due to glycogen (data not shown). Enteroendocrine (Sox9+) cells and goblet (PAS+) cells are long-lived in the villus epithelium and appeared to be normal in numbers after knocking out intestine Zip4, at least within the time period examined (Figure S2). No change in Paneth cells were seen in control littermates (Con) that had been similarly injected with tamoxifen and processed in parallel (data not shown). These results indicate that loss-of-function of intestine ZIP4 leads to a rapid loss of Paneth cell zinc which is followed by changes in Paneth cell morphology and phenotypic reprogramming.
Loss-of-function of the intestine Zip4 gene leads to repressed cell division
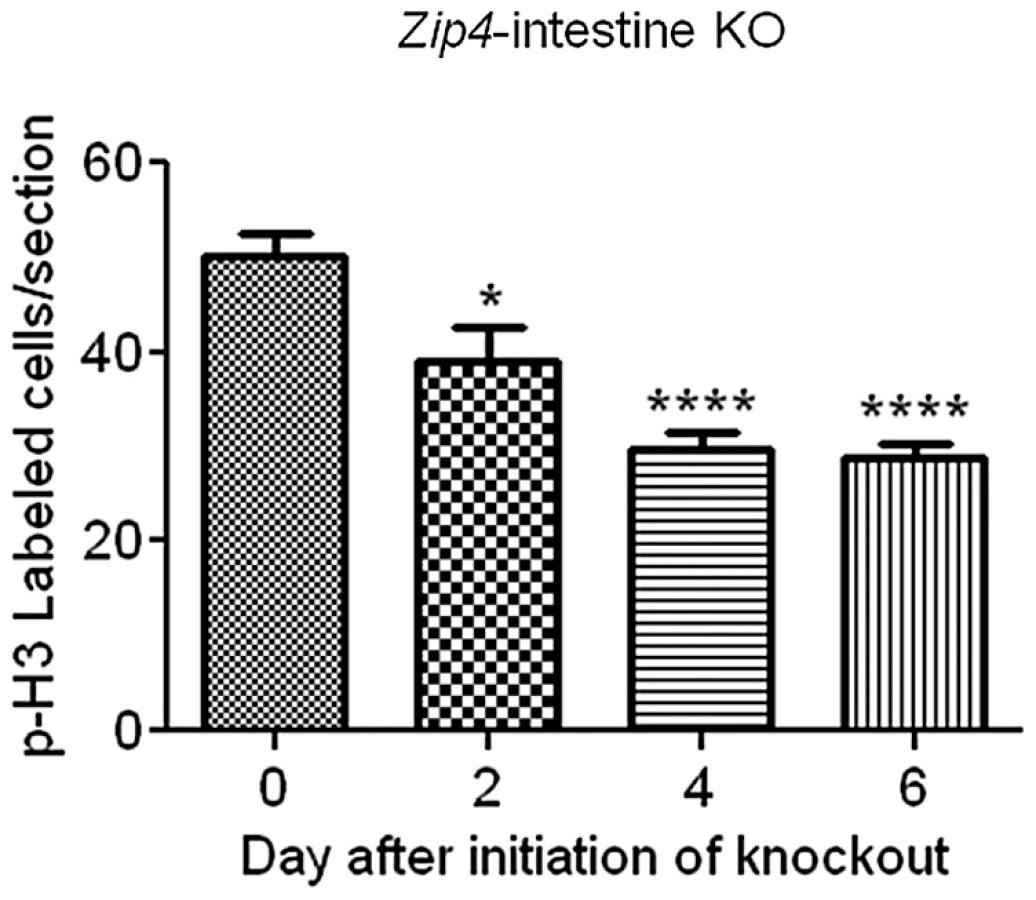
The relative amount of cell division in the small intestine was determined since Sox9 plays a role in intestinal stem cell proliferation and the crypts are critical for renewal of the epithelium. Cell division was monitored by IHC for phosphorylated (Ser 10) histone H3 and quantified by counting positively stained cells in multiple fields of view (5 or more) in sections from several (4 or 5 mice) control and Zip4-intestine knockout mice (Figure 7). On day 2 after initiation of the knockout, cell division was measurably repressed (22%) in the small intestine and by day 4, cell division was reduced by 47% and remained repressed on day 6. No reduction in cell division after tamoxifen injections was noted in control littermates (data not shown). These results reveal that intestine ZIP4 functions to maintain the structural and functional integrity of the small intestine.
Loss-of-function of the intestine Zip4 gene causes a rapid loss of phosphorylated S6 ribosomal protein in the villus epithelium followed by increased phosphorylated S6 ribosomal protein in the crypts
Given the repressive effects of loss-of-function of ZIP4 on small intestine cell division and structural integrity leading to a switch from anabolic to catabolic metabolism in mice, we examined the effects of this knockout on the activity of mammalian target of rapamycin (mTOR1) in the intestine. mTOR1 plays an important role in growth regulation and intestinal cell migration by integrating signals from nutrients, energy status, growth factors and stresses thereby controlling protein translation [31]–[33]. Ribosomal protein S6 kinase is activated by mTOR1 leading to phosphorylation of ribosomal protein S6 and enhanced translation [31]. Therefore, the status of phosphorylated ribosomal protein S6 was monitored by IHC in the small intestine of control and Zip4-intestine knockout mice (Figure 8). Phosphorylated ribosomal protein S6 was readily detectable in the enterocytes of the villus tips in control mice. Little or no staining was detected in crypts (Figure 8A). However, by d2 after initiating the knockout staining in the crypts became apparent while that in the villus tips was diminished (Figure 8B), and by d4 crypt staining was strong and villus staining was weak or absent. On day 6 crypt staining remained strong and cells interspersed in the villus epithelium stained positive for phosphorylated ribosomal S6 protein. Closer examination of the stained crypts revealed an absence of staining in the Paneth cells (Figure 8B). These results indicate that loss of ZIP4 function in enterocytes leads to a rapid and prolonged diminution in mTOR1 activity and impaired protein synthesis in those cells. Remarkably this is accompanied by enhanced mTOR1 activity in the stem cell niche, but not in Paneth cells.

Loss-of-function of the intestine Zip4 gene causes rapid and transient changes in energy metabolism in the liver
Unlike the intestine, the liver in Zip4 intestine knockout mice apparently does not undergo obvious morphological changes and the liver to body weight ratio remains relatively constant during the first week or so after initiating the knockout (data not shown). In contrast there is a rapid loss of bone and muscle mass and body weight in these mice. The liver plays a critical role in metabolism; therefore, the status of AMP-activated protein kinase (AMPK) and mTOR1 activity in the liver were examined by Western blotting (Figure 9). The activity of AMPK is allosterically increased by AMP binding which leads to phosphorylation and activation of AMPK. When ATP generation is attenuated, AMPK becomes activated and phosphorylates many substrates leading to increased activation of catabolic pathways and reduced activation of anabolic pathways [32]. As mentioned above, mTOR1 activity enhances anabolic metabolism by supporting translation and transcription.

Western blot analysis of liver proteins revealed that phosphorylated (T172)-AMPK increased soon after the knockout of intestine Zip4 reaching a peak on day 3 before rapidly declining (Figure 9). This decline was accompanied by increased and prolonged phosphorylation (S2448) of mTOR1. In contrast, little change was noted in the phosphorylation of AKT (S473) or active β-catenin until day 8. Taken together, these results show that the loss-of-function of intestine ZIP4 rapidly causes a switch from anabolic to catabolic metabolism in the liver reflecting an increased AMP/ATP ratio. However, this effect was transient and mTOR1 activity was restored when the mice began wasting muscle and bone which would provide amino acids and other energy sources for liver metabolism. Thus, unlike the small intestine (as well as bone and muscle) the liver is only transiently affected by the loss-of-function of intestine ZIP4.
Discussion
Herein we describe the creation of a mouse model of the rare pseudo-dominant human genetic disorder of zinc metabolism AE. Being able to control the timing and extent of the knockout of enterocyte Zip4 in mice, and the rapid progression of the disease thereafter, allowed us for the first time to effectively address underlying mechanisms of AE. A central remaining question in our understanding of AE has been to define the role of intestinal Zip4 in the etiology of this disease. Mouse Zip4 expression can be detected in several cell-types [18], [34]. Human Zip4 expression is also detected in several organs (NCBI Unigene EST profile) and fibroblasts from AE patients show diminished zinc uptake and zinc content leading to the suggestion that loss-of-function of ZIP4 in organs other than the intestine could contribute to the pathology of AE [35]. Our data demonstrate that the most profound phenotypes of AE, severe growth retardation and morbidity, reflect a loss-of-function of ZIP4 only in intestinal enterocytes, that was predicted from studies of patients with AE nearly 30 years ago [2]. A significant difference between this mouse model and human AE is the lack of severe dermatitis and alopecia in mice. This likely reflects the fact that Zip4 is actively expressed in human skin (NCBI Unigene EST profile) whereas it is not actively expressed in mouse skin [18].
In mice, growth retardation and/or weight loss occur rapidly after enterocyte Zip4 is knocked out and the disease progresses to morbidity within a few days accompanied by a switch from anabolic to catabolic metabolism, unless mice were nursing or given access to high zinc concentrations in their water. Similarly, humans with AE respond favorably to increased dietary intake of zinc. How zinc is absorbed from the gut in the absence of ZIP4 is not known. However, uptake of zinc by L-type calcium channels has been reported [36] and several other Zip and ZnT proteins are expressed in the intestine and could contribute to zinc uptake when zinc levels are very high [37]–[39].
Our studies revealed that one of the most rapid effects of loss-of-function of intestine ZIP4 is the loss of labile zinc from Paneth cells. This may therefore be an initial event in the progression of AE. Studies of patients with AE, also reported nearly 35 years ago, revealed changes in Paneth cells including the accumulation of abnormal inclusion bodies and giant granules, which were reversed when patients were given oral zinc [40]–[42]. Zinc deficiency or prolonged fasting has been shown to cause the loss of Paneth cell zinc, as measured by reactivity with the zinc chelator dithizone [43] and dithizone injection causes selective killing of Paneth cells [44]. However, zinc deficiency alone in rats does not lead to abnormal Paneth cell morphology [45] which suggests that factors in addition to zinc loss may contribute to the pathology of Paneth cells in AE patients.
The loss-of-function of intestine ZIP4 rapidly alters the phenotype of Paneth cells which are long-lived, suggesting that preexisting Paneth cells undergo reprogramming. The rapid loss of zinc in these cells is followed by reduction of Sox9 and the accumulation of mucin positive Paneth cells. Sox9 regulates cell proliferation in the small intestine epithelium as well as Paneth cell differentiation and repression of mucin (Muc2) expression [23], [46]. Sox9 is regulated by the Wnt pathway [23] which suggests that ZIP4 may play a role in regulating signal transduction in Paneth cells via the Wnt-APC pathway. A role of zinc in controlling glycogen-synthase kinase-3β (GSK-3β) activity and, by extension the Wnt-APC pathway, has been reported [47]–[49]. Increased zinc leads to increased phosphorylation of GSK-3β, the accumulation of β-catenin and increased signal transduction. Therefore, our data are consistent with the concept that the loss of zinc in Paneth cells leads to reduced phosphorylation of GSK-3β, increased turn-over of β-catenin, reduced Sox9 expression, and reprogramming of crypt Paneth cells.
Loss-of-function of ZIP4 also resulted in a reduction in cell division in the crypts which may be a consequence of the altered Paneth cell phenotype and crypt dysplasia. A recent report revealed that Paneth cells contribute to the intestinal stem cell niches by expressing several growth factors, most importantly Wnt, which support those stem cells [30]. Perturbed function of Paneth cells would negatively affect Lgr5 columnar stem cells which are interspersed between Paneth cells in the crypts [29], [30]. These stem cells are able to give rise to all the epithelial lineages in vivo, can generate crypt like structures in vitro, and there is a strong correlation between the number of Paneth cells and the number of Lgr5 stem cells in the intestine in vivo [29], [30]. Therefore, impaired Paneth cell function contributes to reduced cell division in the small intestine through its effects on the stem cell niche.
It is interesting to note that Zip4 expression in the mouse intestine is restricted to villus enterocytes and this protein is not detected by IHC in the crypts [15], [18]. Although this cannot exclude the possibility that weak expression of Zip4 in the crypts may contribute to the changes noted in Paneth cells after this gene is knocked out, a more plausible explanation is that loss-of-function of villus enterocyte ZIP4 leads to loss of Paneth cell zinc and it is this loss of zinc which initiates the reprogramming of Paneth cells rather than a loss of a direct signal transduction function of ZIP4 in Paneth cells. The blood circulation provides an intimate connection between the villus and crypt such that enterocyte absorbed nutrients, including Zn, are available at high concentrations to cells in the crypt.
There is evidence for zinc transporters affecting cell signaling. Studies of knockout mice suggest that Slc39a14 (ZIP 14) represses phosphodiesterase activity leading to enhanced systemic growth, but the loss-of-function of ZIP14 is not lethal and has only modest effects on growth [50], unlike the loss of ZIP4 function. Similarly Slc39a13 (ZIP 13) has been shown to play a role in connective tissue development where it affects BMP/TGF-β signaling [51]. The preponderance of data suggests that zinc mediates the effects of these zinc transporters [9]. However, ZnT1 has been shown to directly interact with EVER proteins [52] leading to altered intracellular distribution of zinc, and ZnT1 can interact with Raf-1 kinase through its c-terminal domain leading to activation of ERK signaling [53]. No binding partners of ZIP4 have been reported.
In contrast to the pattern of Zip4 expression in villi, Zip5 expression is most active in crypt cells where it localizes to the basolateral membrane when dietary zinc is adequate [15]. This protein is normally degraded during periods of zinc deficiency but the mRNA stays associated with polysomes [12] and its translation is regulated by a conserved stem-loop structure in the 5′-untranslated region and two microRNAs in response to zinc [16]. The reduction of Zip5 mRNA abundance after knocking out the enterocyte Zip4 gene occurs concomitantly with crypt dysplasia. However, intestine-specific knockout of mouse Zip5 is asymptomatic (Geiser and Andrews, unpublished results) which suggests that loss of Zip5 expression in the crypts of the Zip4-intestine knockout mice reflects a loss of the differentiated state of intestinal crypt cells.
Another rapid effect of loss-of-function of intestine ZIP4 is the attenuation of mTOR1 activity, as measured by phosphorylation of S6 ribosomal protein, in the villus epithelium. This indicates that protein synthesis in villus enterocytes is rapidly diminished after ZIP4 is lost [31]. The intestine has a high rate of protein synthesis to supply digestive enzymes and transporters necessary for enterocyte nutritional functions, and total intestine utilization of amino acids can account for half of whole-body utilization [54]. Attenuation of protein synthesis in the intestine impairs growth via malnutrition. For example, the loss-of-function of intestine Slc6a19, the neutral amino acid transporter, impairs growth and body weight control in mice and reduces mTOR1 activity in the intestine [55] but is not lethal. Whether loss-of-function of ZIP4 attenuates amino acid uptake by the small intestine remains to be determined but amino acids and growth factors play essential roles in mTOR1 signaling [31] and amino acid stimulation of mTOR1 signaling and protein synthesis are required for intestinal cell migration [33]. The attenuation of mTOR1 activity in villus enterocytes likely contributes to the disorganization of the epithelium which occurs after Zip4 is knocked out. Dietary zinc enhances mTOR1 signaling and GSK-3β phosphorylation in skeletal muscle and liver leading to increased protein synthesis [49]. Similar to Paneth cells, villous enterocytes show altered morphology when intestine Zip4 is knocked out. Combined with the reduced rate of cell division and reduced mTOR1 activity, the loss of normal columnar morphology of enterocytes is consistent with a loss of differentiated function, which likely is a major contributor to growth failure in these mice.
Our studies revealed that loss-of-function of intestine ZIP4 leads to transient activation of hepatic AMPK, a cellular energy sensor which is activated when AMP levels increase [32] and which antagonizes mTOR1 activity [32]. Loss of zinc likely leads to attenuated mTOR1 signaling in the liver through reduced production of ATP and activation of AMPK. This is followed by repletion of hepatic zinc due to the redistribution of zinc, and other nutrients, from bone and muscle stores as the knockout mice begin to lose body mass. This leads to attenuation of AMPK activity and increased mTOR1 activity. By extension, the attenuation of villous mTOR1 activity in response to the loss-of-function of ZIP4 in villous enterocytes likely reflects a response to diminished zinc uptake rather than a signaling function of the ZIP4 protein in enterocytes.
In addition to the potential reduction in intestinal uptake of amino acids and zinc which can control mTOR1 activity, it is also possible that loss-of-function of ZIP4 attenuates IGF-1 signaling in the intestine. Intestine IGF-1 mRNA is induced after Zip4 is knocked out and this growth factor is a potent tropic mitogen in the intestine [22]. IGF-1 mRNA is induced in intestinal subepithelial myofibroblasts in response to stimulation by glucagon-like-peptide 2 secreted from intestinal L-cells, and IGF-1 enhances crypt cell proliferation by stimulating β-catenin signaling in non-Paneth cells [22]. The finding that IGF-1 is induced but mTOR1 activity is repressed in the villus epithelium suggests that IGF-1 signaling is attenuated when intestine ZIP4 is lost. Intracellular zinc concentrations have been shown to modulate IGF-1 signaling by affecting protein tyrosine phosphatase activity [56]. High zinc is inhibitory leading to enhanced IGF-1 signaling whereas zinc deficiency impairs IGF-1 signaling [57].
Loss of mTOR1 activity in the villus epithelium occurred soon after Zip4 was knocked out in the intestine. In contrast, the activity of mTOR1 was increased in crypts thereafter but this failed to lead to enhanced cell proliferation. The activation of mTOR1 in crypts may reflect a stress response or a compensatory response to crypt dysplasia in the intestine [32]. Increased IGF-1 signaling may lead to activation of mTOR1 in crypt cells causing enhanced translation but fails to enhance cell division due to impaired β-catenin signaling. Clearly a complicated interplay between loss of zinc and altered signal transduction cascades occurs in the intestine leading to diminished integrity of the small intestine in AE.
Loss-of-function of intestine ZIP4 led to altered homeostasis of several essential metals over time. Although zinc was rapidly lost from intestine, pancreas and liver soon after the Zip4 knockout, normal levels of zinc re-accumulated in the liver as body weight, bone and muscle mass were lost. In contrast, iron and manganese accumulated in the liver. Accumulation of iron in the liver during zinc deficiency has been previously reported but manganese accumulation has not. Zinc deficiency can result in alterations in iron transporter, storage, and regulatory proteins which facilitate iron accumulation [20]. In addition ZIP14 (Slc39a14) has been suggested to play a role in the accumulation of non-transferrin bound iron in the liver [58]. It is possible that reduced expression of ATP13A2, which has been shown to reduce cellular manganese concentrations, occurs in response to severe zinc deficiency in our model [59], and a recent study revealed that ZnT10 (Slc30a10) plays a key role in manganese transport in humans [60], [61]. Whether enhanced expression or activity of ZIP14 and/or repression of ZnT10 play roles in the accumulation of iron and manganese, respectively, in the liver in response to loss-of-function of intestine ZIP4 remains to be determined. Accumulation of very high levels of iron in the liver (20 to 100×) can lead to hemochromatosis and can inhibit manganese uptake by mitochondria leading to mitochondrial dysfunction [62]. Therefore, the slow accumulation of hepatic iron and manganese in the Zip4-intestine knockout mice could eventually contribute to liver injury if very high levels are accumulated later during the disease process. Perhaps humans with AE may be at risk for hemochromatosis at advanced stages of the disease.
In summary, our studies report the first conditional knockout of a member of the mammalian ZIP family of zinc transporters. By controlling the timing and extent of the knockout of the intestine Zip4 gene in mice we were able for the first time to monitor temporal and tissue-specific effects of the loss-of-function of ZIP4 and provide insights into the etiology of AE. The results suggest that Paneth cell dysfunction leading to disruption of the stem cell niche and loss of villus enterocyte integrity are primary causes of AE. These changes likely reflect diminished zinc uptake by ZIP4 and are followed by a switch from anabolic to catabolic metabolism in the mouse leading to dramatic weight loss, dyshomeostasis of several essential metals and ultimately lethality in the absence of excess dietary zinc.
Materials and Methods
Animals
Experiments involving mice were performed in accordance with the guidelines from the National Institutes of Health for the care and use of animals and were approved by the Institutional Animal Care and Use Committee. Mice were maintained on normal mouse chow (zinc adequate:ZnA) and where indicated were also provided with drinking water containing 250 ppm ZnSO4 (zinc excess: ZnE).
Recombineering of a floxed Zip4 (Slc39a4) gene
We previously described the structure of the mouse Zip4 (Slc39a4) gene in detail [15], [18]. The final structure of the floxed Zip4 gene targeting vector created herein and of the targeted chromosomal locus is shown in Figure 1. BAC recombineering [63] using galK selection was employed to manipulate the Zip4 gene [64] A 9180 bp region containing the Zip4 gene plus 2.3 kb of 5′-flanking DNA and 3.2 kb of 3′ flanking DNA was captured from BAC ct7-43303 and gap-repaired in a conditionally amplifiable [65] BAC-based vector (P[acman]-M-KO) [66] that allows for negative selection (HSV-TK) in ES cells and positive selection in bacteria (ampicillin). A LoxP site engineered to contain an Xho I restriction site was inserted into a poorly conserved region in intron 5 and a second LoxP site was inserted immediately downstream of exon 12, the last exon in this gene. An mc1-neomycin cassette was inserted about 250 bp downstream of exon 12. The targeting vector's structure was confirmed for the entire gene including the neomycin cassette by DNA sequencing and the ends of the captured regions were sequenced to verify that no rearrangements or mutations had occurred. In addition, the functionality of the LoxP sites was confirmed by transformation of the final targeting vector into bacteria (EL 350) that express Cre recombinase under control of an arabinose inducible promoter [63].
Targeted insertion of the floxed Zip4 (Slc39a4) gene in embryonic stem cells
The Transgenic and Gene-Targeting Institutional Facility at the KU Medical Center generated targeted ES cell clones and performed blastocyst injections. The Zip4 FX targeting vector was linearized with Not1 and electroporated into E14 embryonic stem (ES) cells. Selected colonies were screened by long range PCR using LA-Taq (TaKaRa Bio, Inc.) and primers which flank the Zip4 region captured and manipulated by recombineering paired with primers within the Zip4 gene itself (Figure 1). Primers flanking the LoxP site in intron 5 were used for routine genotyping of the targeted allele in mice (Figure 1). Homologous recombination of the targeting vector into the endogenous locus resulted in the insertion of an XhoI site engineered into the LoxP sequence in intron 5 which aided in identifying the targeted alleles. Southern blot hybridization was used to screen for the Y-chromosome as described in detail previously [67]. The sequences of oligonucleotides for integration screen and genotyping are shown in Table S1.
Generation of mice with targeted floxed Zip4 (Slc39a4) alleles
Chimeric mice were generated by microinjection of two independent Zip4FX/WT ES cell clones into d4 C57BL/6 blastocysts, followed by transfer to pseudopregnant CD-1 foster mothers. Resulting chimeric mice were crossed with C57BL/6 females (Harlan labs). Germline transmission was confirmed by PCR from tail DNA of agouti offspring (Figure 1). Zip4FX/WT mice were crossed and Zip4FX/FX offspring were identified by PCR amplification of the LoxP:XhoI insertion in intron 5.
Generation of mice for inducible deletion of Zip4FX/FX (Slc39a4) alleles in the intestinal epithelium
Zip4FX/FX mice were crossed to create a working colony of mice homozygous for floxed Zip4 genes. These mice were then crossed with transgenic mice bearing a tamoxifen-dependent Cre recombinase (vil-Cre-ERT2: a kind gift from S. Robine, Institut Curie-CNRS, Paris, France) expressed under the control of the villin promoter to allow for inducible deletion of the Zip4 gene specifically in the intestinal epithelium [11] and selected offspring were backcrossed to yield mice heterozygous for vil-Cre-ERT2 and homozygous Zip4FX/FX. These mice were then crossed with Zip4FX/FX mice to yield 50% offspring with Zip4FX/FX: vil-Cre-ERT2 alleles and 50% with Zip4FX/FX alleles. The latter provided age and genetically matched controls for our experiments and were labeled as control (Con) in all the figures.
Tamoxifen induction of recombination
A tamoxifen stock solution was prepared essentially as described previously [68] by the addition of 100 µl of ethanol to 10 mg of tamoxifen (free base: MP biomedicals, LLC) and heating to 37°C briefly to dissolve the tamoxifen. This solution was then diluted to 1 ml with autoclaved canola oil and heated briefly to 37°C. The stock solution (10 mg tamoxifen/ml) was stored at 4°C for up to two weeks or at −80°C for months. Before injection, the tamoxifen stock solution was heated to 37°C. Adult and recently weaned mice (5 to 8 days post-weaning) were injected I.P. with 100 µl (1 mg tamoxifen) of the tamoxifen stock solution daily for 3 consecutive days unless noted otherwise (e.g. Figure 2F). Neonatal mice (d5 post-partum) were injected I.P. for 5 consecutive days with 10 µl (100 µg) of the tamoxifen stock solution.
Northern blot hybridization
Total RNA was isolated using Trizol reagent (Invitrogen). Total RNA (3–6 µg) was size-fractionated by agarose-formaldehyde gel electrophoresis, transferred and UV cross-linked to a Zeta Probe GT nylon membrane (BioRad). Northern blot membranes were prehybridized in 0.5 M sodium phosphate, pH 7.0, 7% SDS at 65°C for 30 min and then hybridized in 40% formamide, 0.5 M sodium phosphate, pH 7.0, 7% SDS at 65°C for 24 hr. Membranes were then washed twice in 3X SSC, 0.1% SDS for 30 min each followed by two washes in 1X SSC, 0.1% SDS and two washes in 0.3X SSC, 0.1% SDS for 30 min each at 65°C. Hybrids were detected by autoradiography at −80°C. Duplicate gels were stained with acridine orange to ensure equivalent loading and integrity of total RNA. Riboprobes for mouse MT-I, Zip4, and Zip5 [15] and for IGF-1 mRNAs were as described [69]. Probes were used at 2×106 cpm/ml of hybridization solution.
Quantitative RT–PCR of IGF-1 mRNA
Small intestine total RNA was reverse transcribed using Accuscript reverse transcriptase (RT) enzyme (Stratagene) and IGF mRNA and GAPDH mRNA, as an internal control, were amplified using the PerfeCTa Sybr Green FastMix qPCR kit (Quanta Biosciences) and a Miniopticon Real-Time PCR Detection System (BioRad) essentially as described previously [8]. Melting-curve data were collected to confirm PCR specificity. Each cDNA sample was run in triplicate, and the corresponding no-RT sample was included as a negative control. GAPDH primers were included in every sample to control for sample variation. IGF-1 mRNA product in each sample was normalized to that of the GAPDH mRNA. The amount of PCR products was measured by threshold cycle (Ct) values and relative mRNA levels were determined as unit values of 2∧[Ct(GAPDH) -Ct(IGF-1)]. Values presented are fold-change relative to control. The following primers were used: mIGF-1 (s) gctcttcagttcgtgtgtggaccg: mIGF-1 (as) cttctgagtcttgggcatgtcagtgtg. These primers amplify the IGF-1 peptide sequence found in all mIGF-1 variants. GAPDH (s): aggttgtctcctgcgacttca: mGAPDH (as) ccaggaaatgagcttgacaaag.
Antisera
The following antibodies were used at the indicated dilution for Western blots: Total β-catenin (Cell Signaling: 1∶1000), active β-catenin (Millipore: 1∶1000), p(T172)-AMPK (Cell Signaling: 1∶1000), total AMPK (Cell Signaling: 1∶1000), p(S2448) mTOR1 (Cell Signaling: 1∶1000), total mTOR1 (Cell Signaling: 1∶1000), p(s473)-AKT (Cell Signaling: 1∶1000), total AKT (Cell Signaling: 1∶1000). The following antibodies were used at the indicated dilution for immunohistochemistry (IHC): Sox9 (Millipore: 1∶750), lysozyme (DakoCytomation: 1∶150), phospho-S6 ribosomal protein (Cell Signaling: 1∶400), phosphor (Ser10)-histone H3, (Cell Signaling: 1∶100)
Histology/cytochemistry/immunohistochemistry (IHC)
The first ∼5 cm of small intestine (3 to 5 mice per group) was collected, flushed with cold PBS, cut into small pieces and fixed in Bouin's fixative or 4% paraformaldehyde in PBS overnight at 4°C. Fixed tissues were embedded in paraffin and sections (1 µm) were prepared by Histo-Scientific Research Laboratories (HSRL). Bouin's fixed sections were deparaffinized, rehydrated and stained with hematoxylin-eosin for examination of gross morphology. Paraformaldehyde fixed tissues were deparaffinized, rehydrated and stained using the Periodic Acid-Schiff (PAS) staining system (Sigma-Aldrich) to detect mucin (mainly Muc2 and Muc3) and identify goblet cells. Serial sections were pretreated with α-amylase (239 µg/ml for 1 hr at 37°C: Worthington Biochemical Corporation) to test for glycogen contribution to the PAS staining. For IHC, paraformaldehyde fixed serial sections were deparaffinized, rehydrated and antigens were retrieved in 10 mM citrate, pH 6.0, using a 2100 Retriever pressure cooker (PickCell Laboratories). Proteinase K digestion (20 µg/ml in 50 mM Tris base, pH 8.0, 0.1 mM EDTA, 0.5% Triton X - 100 at 37°C for 10 min) was used for antigen retrieval for the lysozyme antibody. Samples were processed using the Histostain Plus LAB SA Detection System (Invitrogen) according to the manufacturer's instructions using the antibodies listed above. Stained slides were counterstained briefly in Mayer's hematoxylin (Sigma) and photographed using a Leica DM 4000B microscope (Leica-microsystems) with Adobe Photoshop image capture software (Adobe).
Detection of Paneth cell zinc
Paneth cell labile zinc was detected using the zinc-specific-responsive fluorescent dye Zinpyr-1(ZP1) (Millitech) essentially as described with modifications [26]. The small intestine from 3 to 5 mice per group was isolated, flushed with cold PBS and cut into small pieces that were dropped into cold 0.6 M sucrose in PBS. Once the small intestine pieces sank (1 to 2 min) they were transferred to a solution of 2 parts 0.6 M sucrose plus 1 part TFM embedding compound (Triangle Biomedical Sciences, Inc.) until they sank (2 to 3 min). The intestine pieces were then immersed in TFM, frozen in dry ice-propanol and 6 µm frozen sections were cut. Frozen sections were thawed for ∼30 seconds at room temperature in a solution containing 20 µM ZP1 and 0.5 µM DAPI in 147 mM NaCl, 4 mM KCl, 3 mM CaCl2, 0.9 mM MgCl2, 11 mM HEPES, pH 7.4, and 10 mM glucose, washed briefly in PBS and then photographed using a Leica DM 4000B microscope (Leica-microsystems) with Adobe Photoshop image capture software (Adobe). ZP1 was visualized using the +L5 (Leica-microsystems) GFP filter cube and DAPI stained nuclei were visualized using the +A4 filter cube (Leica-microsystems). Brightness of the photographs was reduced in Photoshop.
Western blot analysis
Liver proteins were isolated by homogenizing ∼100 mg of tissue in 400 µl of RIPA buffer (150 mM NaCl, 50 mM Tris base, pH 8.0, 1% NP40, 0.5% deoxycholate, 0.1% SDS, plus Complete Protease Inhibitor Cocktail and PhosStop phosphatase inhibitor cocktail (Roche Applied Science). Tissues were homogenized at 4°C for 20 strokes using a teflon-glass homogenizer. Samples were then centrifuged at 10,000× g for 10 min at 4°C, the supernatant was collected and protein concentration was determined using BCA reagent (Pierce Biotech). Proteins (30 µg) were resolved on 7.5% or 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Life Science Products, Inc.) using a Transblot semi-dry transfer apparatus (BioRad) according to the manufacturer's instructions. Membranes were blocked overnight in 5% milk in PBS-T or 5% BSA in PBS-T as suggested by the antibody source and then incubated with primary antibody in blocking solution at the appropriate dilution for 2 h at room temperature. After extensive washing, membranes were incubated with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (1∶2000) and the blot was developed using ECL Plus reagent (Amersham Biosciences) according to manufacturer's instructions and detected using Kodak BioMax MS film (Kodak).
Elemental and essential metal determination
Elemental profiling via inductively coupled plasma mass spectrometry (ICP-MS) was performed at the Purdue University Ionomics Facility, Purdue University, West Lafayette, Indiana as described previously [21]. The following elements were measured: Na, Mg, P, K, Ca, Fe, Co, Cu, Zn, Mn, As, Se, and Mo. Mouse tissues (n = 5 to 10: <100 mg wet weight each sample) were dried at 95°C in a vacuum oven. Dried samples of about 5 to 10 mg were weighed into Pyrex tubes and digested in 0.7 to 1.5 ml concentrated HNO3 at 110°C for 4 h. Each sample was diluted as optimal with 18 MOhm water and analyzed on an Elan DRCe ICP-MS (PerkinElmer). Methane was used as a collision cell gas to measure iron. Gallium and indium were used as internal standards, added to the digestion acid bottle to a concentration of 20 µg l−1. National Institute of Standards and Technology traceable single element ICP standards (Ultrasci) were used to make up the calibration standards. Tissue concentrations were determined as µg g−1 dry weight (ppm) of each element.
Statistical analyses
Graphs were generated and statistical analyses performed using GraphPad Prism5 software (GraphPad Software). Statistical significance was determined using the Unpaired T-test (two-tailed) and values were considered different if P<0.05. Data are expressed as the mean ± S.E.M. *indicates P<0.05; *** indicates P<0.001; **** indicates P<0.0001
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. AthertonDJMullerDPAggettPJHarriesJT 1979 A defect in zinc uptake by jejunal biopsies in acrodermatitis enteropathica. Clin Sci (Lond) 56 505 507
2. MoynahanEJ 1974 Letter: Acrodermatitis enteropathica: a lethal inherited human zinc - deficiency disorder. Lancet 2 399 400
3. KurySDrenoBBezieauSGiraudetSKharfiM 2002 Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet 31 239 240
4. WangKZhouBKuoYMZemanskyJGitschierJ 2002 A novel member of a zinc transporter family is defective in acrodermatitis enteropathica. Am J Hum Genet 71 66 73
5. SchmittSKurySGiraudMDrenoBKharfiM 2009 An update on mutations of the SLC39A4 gene in acrodermatitis enteropathica. Hum Mutat 30 926 933
6. AndrewsGK 2008 Regulation and function of Zip4, the acrodermatitis enteropathica gene. Biochem Soc Trans 36 1242 1246
7. ZhangYChenCYaoQLiM 2010 ZIP4 upregulates the expression of neuropilin-1, vascular endothelial growth factor, and matrix metalloproteases in pancreatic cancer cell lines and xenografts. Cancer Biol Ther 9 236 242
8. WeaverBPZhangYHiscoxSGuoGLApteU 2010 Zip4 (Slc39a4) expression is activated in hepatocellular carcinomas and functions to repress apoptosis, enhance cell cycle and increase migration. PLoS ONE 5 e13158 doi:10.1371/journal.pone.0013158
9. FukadaTYamasakiSNishidaKMurakamiMHiranoT 2011 Zinc homeostasis and signaling in health and diseases: Zinc signaling. J Biol Inorg Chem 16 1123 1134
10. Dufner-BeattieJWeaverBPGeiserJBilgenMLarsonM 2007 The mouse acrodermatitis gene Slc39a4 (ZIP4) is essential for development and heterozygosity causes hypersensitivity to zinc deficiency. Hum Mol Genet 16 1391 1399
11. el MarjouFJanssenKPChangBHLiMHindieV 2004 Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39 186 193
12. WeaverBPDufner-BeattieJKambeTAndrewsGK 2007 Novel zinc-responsive post-transcriptional mechanisms reciprocally regulate expression of the mouse Slc39a4 and Slc39a5 zinc transporters (Zip4 and Zip5). Biol Chem 388 1301 1312
13. KelleherSLSeoYALopezV 2009 Mammary gland zinc metabolism: regulation and dysregulation. Genes Nutr 4 83 94
14. SunJYWangJFZiNTJingMYWengXY 2011 Effects of Zinc Supplementation and Deficiency on Bone Metabolism and Related Gene Expression in Rat. Biol Trace Elem Res 143 394 402
15. Dufner-BeattieJKuoYMGitschierJAndrewsGK 2004 The adaptive response to dietary zinc in mice involves the differential cellular localization and zinc-regulation of the zinc transporters ZIP4 and ZIP5. J Biol Chem 279 49082 49090
16. WeaverBPAndrewsGK 2012 Regulation of zinc-responsive Slc39a5 (Zip5) translation is mediated by conserved elements in the 3′-untranslated region. Biometals 25 319 335
17. WangFDKimBEDufner-BeattieJPetrisMJAndrewsG 2004 Acrodermatitis enteropathica mutations affect transport activity, localization and zinc-responsive trafficking of the mouse ZIP4 zinc transporter. Hum Mol Genet 13 563 571
18. Dufner-BeattieJWangFKuoYMGitschierJEideD 2003 The Acrodermatitis Enteropathica Gene ZIP4 Encodes a Tissue-specific, Zinc-regulated Zinc Transporter in Mice. J Biol Chem 278 33474 33481
19. AntalaSDempskiRE 2012 The human ZIP4 transporter has two distinct binding affinities and mediates transport of multiple transition metals. Biochemistry 51 963 973
20. NilesBJCleggMSHannaLAChouSSMommaTY 2008 Zinc Deficiency-induced Iron Accumulation, a Consequence of Alterations in Iron Regulatory Protein-binding Activity, Iron Transporters, and Iron Storage Proteins. J Biol Chem 283 5168 5177
21. PetersJLDufner-BeattieJXuWGeiserJLahnerB 2007 Targeting of the mouse Slc39a2 (Zip2) gene reveals highly cell-specific patterns of expression, and unique functions in zinc, iron and calcium homeostasis. Genesis 45 339 352
22. RowlandKJBrubakerPL 2011 The “cryptic” mechanism of action of glucagon-like peptide-2. Am J Physiol Gastrointest Liver Physiol 301 G1 G8
23. BlachePvan deWMDulucIDomonCBertaP 2004 SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol 166 37 47
24. FormeisterEJSionasALLoranceDKBarkleyCLLeeGH 2009 Distinct SOX9 levels differentially mark stem/progenitor populations and enteroendocrine cells of the small intestine epithelium. Am J Physiol Gastrointest Liver Physiol 296 G1108 G1118
25. BastidePDaridoCPannequinJKistRRobineS 2007 Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol 178 635 648
26. GiblinLJChangCJBentleyAFFredericksonCLippardSJ 2006 Zinc-secreting Paneth cells studied by ZP fluorescence. J Histochem Cytochem 54 311 316
27. ShortMLNickelJSchmitzARenkawitzR 1996 Lysozyme gene expression and regulation. EXS 75 243 257
28. YangQBerminghamNAFinegoldMJZoghbiHY 2001 Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294 2155 2158
29. BarkerNvan EsJHKuipersJKujalaPvan denBM 2007 Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449 1003 1007
30. SatoTvan EsJHSnippertHJStangeDEVriesRG 2011 Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469 415 418
31. KimJGuanKL 2011 Amino acid signaling in TOR activation. Annu Rev Biochem 80 1001 1032
32. InokiKKimJGuanKL 2012 AMPK and mTOR in Cellular Energy Homeostasis and Drug Targets. Annu Rev Pharmacol Toxicol 52 381 400
33. RhoadsJMWuG 2009 Glutamine, arginine, and leucine signaling in the intestine. Amino Acids 37 111 122
34. Belloni-OliviLMarshallCLaalBAndrewsGKBresslerJ 2009 Localization of zip1 and zip4 mRNA in the adult rat brain. J Neurosci Res 87 3221 3230
35. VazquezFGriderA 1995 The effect of the acrodermatitis enteropathica mutation on zinc uptake in human fibroblasts. Biol Trace Elem Res 50 109 117
36. SegalDOhanaEBesserLHershfinkelMMoranA 2004 A role for ZnT-1 in regulating cellular cation influx. Biochem Biophys Res Commun 323 1145 1150
37. Dufner-BeattieJHuangZLGeiserJXuWAndrewsGK 2006 Mouse ZIP1 and ZIP3 genes together are essential for adaptation to dietary zinc deficiency during pregnancy. Genesis 44 239 251
38. FordD 2004 Intestinal and placental zinc transport pathways. Proc Nutr Soc 63 21 29
39. CraggRAPhillipsSRPiperJMVarmaJSCampbellFC 2005 Homeostatic regulation of zinc transporters in the human small intestine by dietary zinc supplementation. Gut 54 469 478
40. BohaneTDCutzEHamiltonJRGallDG 1977 Acrodermatitis enteropathica, zinc, and the Paneth cell. A case report with family studies. Gastroenterology 73 587 592
41. BraunOHHeilmannKPauliWRossnerJABergmannKE 1976 Acrodermatitis enteropathica: recent findings concerning clinical features, pathogenesis, diagnosis and therapy. Eur J Pediatr 121 247 261
42. PolancoINistalMGuerreroJVazquezC 1976 Letter: Acrodermatitis enteropathica, zinc, and ultrastructural lesions in Paneth cells. Lancet 1 430
43. ElmesME 1976 The Paneth cell population of the small intestine of the rat-effects of fasting and zinc deficiency on total count and on dithizone-reactive count. J Pathol 118 183 191
44. SawadaMTakahashiKSawadaSMidorikawaO 1991 Selective killing of Paneth cells by intravenous administration of dithizone in rats. Int J Exp Pathol 72 407 421
45. WilsonIDMcClainCJErlandsenSL 1980 Ileal Paneth cells and IgA system in rats with severe zinc deficiency: an immunohistochemical and morphological study. Histochem J 12 457 471
46. van EsJHJayPGregorieffAvan GijnMEJonkheerS 2005 Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol 7 381 386
47. AnWLBjorkdahlCLiuRCowburnRFWinbladB 2005 Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3beta in SH-SY5Y neuroblastoma cells. J Neurochem 92 1104 1115
48. IlouzRKaidanovichOGurwitzDEldar-FinkelmanH 2002 Inhibition of glycogen synthase kinase-3beta by bivalent zinc ions: insight into the insulin-mimetic action of zinc. Biochem Biophys Res Commun 295 102 106
49. McClungJPTarrTNBarnesBRScrimgeourAGYoungAJ 2007 Effect of supplemental dietary zinc on the mammalian target of rapamycin (mTOR) signaling pathway in skeletal muscle and liver from post-absorptive mice. Biol Trace Elem Res 118 65 76
50. HojyoSFukadaTShimodaSOhashiWBinBH 2011 The zinc transporter SLC39A14/ZIP14 controls G-protein coupled receptor-mediated signaling required for systemic growth. PLoS ONE 6 e18059 doi:10.1371/journal.pone.0018059
51. FukadaTCivicNFuruichiTShimodaSMishimaK 2008 The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-beta signaling pathways. PLoS ONE 3 e3642 doi:10.1371/journal.pone.0003642
52. LazarczykMPonsCMendozaJACassonnetPJacobY 2008 Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J Exp Med 205 35 42
53. BeharierODrorSLevySKahnJMorM 2012 ZnT-1 protects HL-1 cells from simulated ischemia-reperfusion through activation of Ras-ERK signaling. J Mol Med (Berl) 90 127 138
54. vandSSrReedsPJStollBHenryJFRosenbergerJR 2002 The high metabolic cost of a functional gut. Gastroenterology 123 1931 1940
55. BroerAJuelichTVanslambrouckJMTietzeNSolomonPS 2011 Impaired nutrient signaling and body weight control in a Na+ neutral amino acid cotransporter (Slc6a19)-deficient mouse. J Biol Chem 286 26638 26651
56. HaaseHMaretW 2003 Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp Cell Res 291 289 298
57. HaaseHMaretW 2005 Fluctuations of cellular, available zinc modulate insulin signaling via inhibition of protein tyrosine phosphatases. J Trace Elem Med Biol 19 37 42
58. Pinilla-TenasJJSparkmanBKShawkiAIllingACMitchellCJ 2011 Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am J Physiol Cell Physiol 301 C862 C871
59. TanJZhangTJiangLChiJHuD 2011 Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J Biol Chem 286 29654 29662
60. TuschlKClaytonPTGospeSMJrGulabSIbrahimS 2012 Syndrome of Hepatic Cirrhosis, Dystonia, Polycythemia, and Hypermanganesemia Caused by Mutations in SLC30A10, a Manganese Transporter in Man. Am J Hum Genet 90 457 466
61. QuadriMFedericoAZhaoTBreedveldGJBattistiC 2012 Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet 90 467 477
62. JouihanHACobinePACookseyRCHoaglandEABoudinaS 2008 Iron-mediated inhibition of mitochondrial manganese uptake mediates mitochondrial dysfunction in a mouse model of hemochromatosis. Mol Med 14 98 108
63. CopelandNGJenkinsNACourtDL 2001 Recombineering: a powerful new tool for mouse functional genomics. Nat Rev Genet 2 769 779
64. WarmingSCostantinoNCourtDLJenkinsNACopelandNG 2005 Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33 e36
65. WildJHradecnaZSzybalskiW 2002 Conditionally amplifiable BACs: switching from single-copy to high-copy vectors and genomic clones. Genome Res 12 1434 1444
66. VenkenKJHeYHoskinsRABellenHJ 2006 P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314 1747 1751
67. Dufner-BeattieJHuangZLGeiserJXuWAndrewsGK 2005 Generation and Characterization of Mice Lacking the Zinc Uptake Transporter ZIP3. Mol Cell Biol 25 5607 5615
68. MetzgerDChambonP 2001 Site - and time-specific gene targeting in the mouse. Methods 24 71 80
69. DasSKPariaBCAndrewsGKDeySK 1993 Effects of 9-ene-tetrahydrocannabinol on expression of b - type transforming growth factors, insulin-like growth factor - I and c-myc genes in the mouse uterus. J Steroid Biochem Mol Biol 45 459 465
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Rumors of Its Disassembly Have Been Greatly Exaggerated: The Secret Life of the Synaptonemal Complex at the Centromeres
- The NSL Complex Regulates Housekeeping Genes in
- Tipping the Balance in the Powerhouse of the Cell to “Protect” Colorectal Cancer
- Interplay between Synaptonemal Complex, Homologous Recombination, and Centromeres during Mammalian Meiosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy