
Protective Coupling of Mitochondrial Function and Protein Synthesis via the eIF2α Kinase GCN-2
Cells respond to defects in mitochondrial function by activating signaling pathways that restore homeostasis. The mitochondrial peptide exporter HAF-1 and the bZip transcription factor ATFS-1 represent one stress response pathway that regulates the transcription of mitochondrial chaperone genes during mitochondrial dysfunction. Here, we report that GCN-2, an eIF2α kinase that modulates cytosolic protein synthesis, functions in a complementary pathway to that of HAF-1 and ATFS-1. During mitochondrial dysfunction, GCN-2–dependent eIF2α phosphorylation is required for development as well as the lifespan extension observed in Caenorhabditis elegans. Reactive oxygen species (ROS) generated from dysfunctional mitochondria are required for GCN-2–dependent eIF2α phosphorylation but not ATFS-1 activation. Simultaneous deletion of ATFS-1 and GCN-2 compounds the developmental defects associated with mitochondrial stress, while stressed animals lacking GCN-2 display a greater dependence on ATFS-1 and stronger induction of mitochondrial chaperone genes. These findings are consistent with translational control and stress-dependent chaperone induction acting in complementary arms of the UPRmt.
Published in the journal:
. PLoS Genet 8(6): e32767. doi:10.1371/journal.pgen.1002760
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002760
Summary
Cells respond to defects in mitochondrial function by activating signaling pathways that restore homeostasis. The mitochondrial peptide exporter HAF-1 and the bZip transcription factor ATFS-1 represent one stress response pathway that regulates the transcription of mitochondrial chaperone genes during mitochondrial dysfunction. Here, we report that GCN-2, an eIF2α kinase that modulates cytosolic protein synthesis, functions in a complementary pathway to that of HAF-1 and ATFS-1. During mitochondrial dysfunction, GCN-2–dependent eIF2α phosphorylation is required for development as well as the lifespan extension observed in Caenorhabditis elegans. Reactive oxygen species (ROS) generated from dysfunctional mitochondria are required for GCN-2–dependent eIF2α phosphorylation but not ATFS-1 activation. Simultaneous deletion of ATFS-1 and GCN-2 compounds the developmental defects associated with mitochondrial stress, while stressed animals lacking GCN-2 display a greater dependence on ATFS-1 and stronger induction of mitochondrial chaperone genes. These findings are consistent with translational control and stress-dependent chaperone induction acting in complementary arms of the UPRmt.
Introduction
Mitochondrial dysfunction and altered protein homeostasis are associated with numerous developmental and age-related diseases as well as the general process of aging [1]. The mitochondrial protein-folding environment is maintained by nuclear-encoded mitochondrial chaperones, which promote efficient protein folding, and proteases that degrade those proteins that fail to fold or oligomerize correctly [1], [2], [3]. Protein folding is compartmentalized in eukaryotic cells and facilitated by compartment-specific folding machinery in the cytosol, endoplasmic reticulum (ER) and mitochondria. As threats to protein homeostasis affect the folding compartments differently, each compartment has dedicated stress responses or unfolded protein response (UPR) signaling pathways to transcriptionally regulate organelle-specific molecular chaperones and reduce the protein-folding load on the resident protein folding machinery.
Dysfunction and accumulation of misfolded proteins in the ER triggers a multi-pronged unfolded protein response (UPRER) that combines the upregulation of molecular chaperones to accommodate the folding requirements in the organelle with a reduction of cytosolic translation and ER protein import [4]. Activation of the transmembrane kinase PERK phosphorylates the cytosolic translation initiation factor eIF2α, thus attenuating general mRNA translation and reducing the load of incoming unfolded polypeptides [5]. In a complementary branch of the UPRER, the transcription factor XBP-1 is activated and mediates the induction of ER-resident chaperones [6]. Thus, by coordinating signaling through parallel pathways, stress is relieved and organelle function restored. In contrast to these ER-protective mechanisms, signaling pathways that protect the mitochondrial protein-folding environment are only beginning to emerge.
Maintenance of mitochondrial metabolic function depends on the efficient assembly of the mitochondrial proteome, which is comprised of nuclear-encoded as well as mitochondrial-encoded polypeptides [7]. Those proteins encoded by the nucleus are translated in the cytosol and post-translationally imported into mitochondria in an unfolded or unstructured state where they interact with the network of mitochondria-resident molecular chaperones. Failure of mitochondrial proteins to properly fold or oligomerize can result in electron transport chain (ETC) defects and accumulation of ROS, which further impacts additional mitochondrial activities including metabolic function. In order to respond to mitochondrial-specific stresses caused by the accumulation of unfolded proteins, depletion of mtDNA, defects in respiration or altered ROS metabolism, mitochondria have evolved stress response pathways that upregulate mitochondrial molecular chaperones to restore organelle homeostasis [8], [9], [10]. One of these pathways, termed the mitochondrial unfolded protein response (UPRmt), couples the status of the mitochondrial protein-folding environment to the transcription of mitochondrial chaperone genes [8], [9]. The complement of nuclear-encoded mitochondrial chaperones, such as mtHsp70 and HSP-60, assist in import, folding, and assembly of multi-protein complexes in the matrix and on the matrix side of the inner mitochondrial membrane [2]. Increased levels of mitochondrial dysfunction perturb the balance between chaperones and their client proteins, leading to activation of the UPRmt and upregulation of mitochondrial chaperone genes to re-establish homeostasis [8], [9], [10].
Our previous genetic studies in C. elegans have identified several proteins required for signaling the response including the mitochondrial inner membrane-localized peptide transporter HAF-1 and the bZip transcription factor ZC376.7 [11], which was recently renamed ATFS-1 (Activating Transcription Factor associated with Stress-1). Mitochondrial dysfunction triggers the HAF-1-dependent nuclear accumulation of ATFS-1, resulting in the upregulation of mitochondrial chaperone genes including HSP-60 and mtHsp70. Activation of this pathway occurs in response to elevated levels of mitochondrial stress, which can be the result of accumulation of unfolded proteins beyond the capacity of mitochondrial molecular chaperones [8] as well as increased levels of oxidative stress [9], respiratory chain dysfunction and by mtDNA depletion [10]. Thus, this mitochondrial stress response pathway, although termed a UPR because of conceptual similarities with the XBP-1 branch of the UPRER, responds to diverse insults to mitochondrial function.
In addition to chaperone induction, the UPRER also mediates the attenuation of cytosolic translation to protect the ER during stress. Similarly, inhibition of cytosolic translation has been suggested to promote mitochondrial function in yeast and Drosophila models of mitochondrial stress, although a potential regulatory mechanism(s) remained to be elucidated [12], [13]. Cytosolic translation attenuation via PERK-1-mediated eIF2α phosphorylation promotes ER function during stress by reducing the client load on ER-resident chaperones [5], [14]. Additionally, in C. elegans genetic manipulations that reduce cytosolic translation rates provide resistance to numerous stresses including heat shock and also extend lifespan [15], [16], [17]. Several signaling pathways are known to regulate translation rates in eukaryotic cells including TOR-regulated phosphorylation of S6 kinase and 4E-BP [16], [17], [18], however a mechanism to couple cytosolic translation rates to mitochondrial function has not been demonstrated.
Phosphorylation of eIF2α by four dedicated kinases (GCN2, PERK, HRI and PKR) serves to attenuate cytosolic translation in response to a variety of cellular stresses including starvation, oxidative stress, viral infection and unfolded protein stress in the ER [19], [20], . In yeast and mammals, GCN-2 phosphorylates eIF2α in response to conditions of low free amino acid levels and oxidative stress [22], [23]. Here we describe experiments demonstrating that in C. elegans, translation attenuation via GCN-2-dependent eIF2α phosphorylation acts in a responsive and adaptive protective pathway during mitochondrial stress to promote mitochondrial function. Phosphorylation levels of eIF2α are increased during mitochondrial stress, which requires ROS generated from dysfunctional mitochondria. Our data demonstrate that GCN-2-dependent translational control acts in a mitochondrial protective signaling pathway complementary to the regulation of mitochondrial chaperone gene expression mediated by HAF-1 and ATFS-1.
Results
Mitochondrial Chaperone Induction by ATFS-1 Is Required for Development during Mitochondrial Stress
We have previously described a mitochondrial stress response pathway that upregulates mitochondrial chaperone genes in response to multiple perturbations in mitochondrial function [8], [11], [24]. RNAi experiments indicated a requirement for the bZip transcription factor ATFS-1 in mitochondrial chaperone induction as demonstrated by quantitative PCR experiments as well as using reporter strains where the hsp-60 promoter regulates expression of GFP (hsp-60pr::gfp) [11]. In order to corroborate the requirement for ATFS-1, we obtained the atfs-1(tm4525) deletion strain which lacks 432 base pairs and most of exons 2–4, and crossed it into the reporter strain. Unlike wild-type worms, atfs-1(tm4525) animals were unable to induce hsp-60pr::gfp when raised on spg-7(RNAi), a mitochondrial protease required for ETC quality control and mitochondrial ribosome biogenesis [25]. These results confirm the requirement for ATFS-1 in stress-induced mitochondrial chaperone gene induction (Figure 1A).
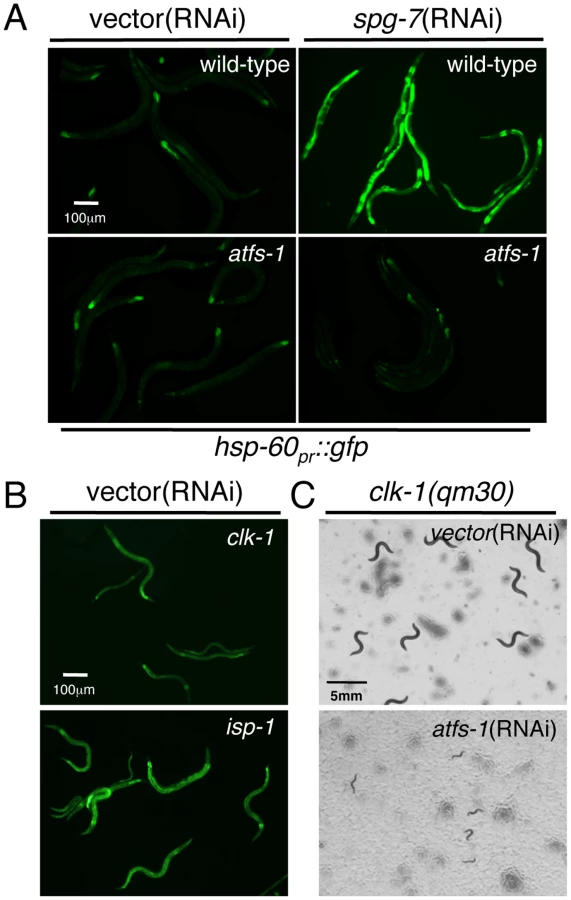
We next investigated ATFS-1-dependent hsp-60pr::gfp activation in strains harboring the well-characterized clk-1(qm30) or isp-1(qm150) mutations [26], [27]. clk-1 encodes a mitochondrial protein required for ubiquinone synthesis [28], which acts as a lipid antioxidant throughout the cell and an electron transporter within the electron transport chain. isp-1 encodes an iron-sulfur component of complex III in the ETC. As both mutations affect respiration and display impaired development [22], [23], we hypothesized that they would cause activation of the UPRmt. Indeed, hsp-60pr::gfp expression was consistently elevated in both strains consistent with the presence of mitochondrial stress. The isp-1(qm150) mutation caused considerably stronger hsp-60pr::gfp induction suggestive of a larger impact on mitochondrial function [29] (Figure 1B). Chaperone induction in both mutants required ATFS-1 as animals raised on atfs-1(RNAi) were unable to induce expression of hsp-60pr::gfp (data not shown).
To determine if the ATFS-1-dependent regulation of mitochondrial chaperone genes has a protective role during mitochondrial stress we examined the effect of atfs-1(RNAi) on the development of wild-type and mitochondrial stressed worms. As previously demonstrated, both clk-1(qm30) and isp-1(qm150) worms developed considerably slower than wild-type animals [22], [28]. Consistent with ATFS-1 being a stress responsive transcription factor, wild-type worms fed atfs-1(RNAi) developed at similar rates to wild-type animals (data not shown). However, feeding clk-1(qm30) and isp-1(qm150) worms atfs-1(RNAi) dramatically impaired their developmental rates (Figure 1C and Figure S1), indicating a requirement for ATFS-1 during development in the presence of mitochondrial stress.
An RNAi Screen Suggests a Role for Translational Regulation in Protecting against Mitochondrial Stress
In addition to ATFS-1-regulated mitochondrial chaperone expression, we sought to identify additional components that promote mitochondrial protein homeostasis by acting in complementary pathways. To identify signaling pathways that act in parallel to ATFS-1 we generated and screened an RNAi sub-library consisting of all C. elegans kinases and phosphatases [30]. We took advantage of hsp-60pr::gfp activation as a sensitive readout for the status of mitochondrial function to identify signaling components that promoted or impaired mitochondrial protein homeostasis. The clk-1(qm30) strain was chosen for the RNAi screen as it displayed mild hsp-60pr::gfp induction, potentially allowing for the identification of candidates whose knockdown by RNAi either decreased or further increased hsp-60pr::gfp expression (Figure 1B). We hypothesized that RNAi knockdown of candidates that act in a complementary protective signaling pathway would show enhanced hsp-60pr::gfp activation in the presence of stress because of an increased substrate load on the mitochondrial protein folding machinery. Alternatively, if components exist whose knockdown somehow enhances the protein-folding capacity, then those RNAi may suppress hsp-60pr::gfp activation in the clk-1(qm30) background.
Interestingly, RNAi of several kinases required for protein synthesis reduced hsp-60pr::gfp expression in the clk-1(qm30) background (Figure 2A), which was also confirmed using the isp-1(qm150) strain (Figure S2A), suggesting that knockdown of these components protected the mitochondrial folding environment. The reduced hsp-60pr::gfp expression was not simply due to a reduction in translation as separate GFP reporters under the myo-3, ges-1 or ER stress-inducible hsp-4 promoters were unaffected by the RNAi candidates (data not shown). These findings are consistent with previous experiments demonstrating that translation attenuation is protective against mitochondrial stress in yeast and Drosophila [12], [13]. Similarly, reduced translation has been associated with longevity and stress resistance in C. elegans. For example, knockdown of the C. elegans target of rapamycin ortholog (TOR), CeTor, which regulates mRNA translation in response to nutrient cues [31], or knockdown of rsks-1, the ribosomal S6 kinase, slows development and extends lifespan in C. elegans [16]. Because the long-lived clk-1(qm30) mitochondrial mutants have increased levels of mitochondrial stress and the stress responsive hsp-60pr::gfp reporter was specifically reduced by CeTor and rsks-1(RNAi), these findings support the hypothesis that reduced translation is beneficial to mitochondrial protein homeostasis. However, because the TOR-signaling pathway impacts many biological processes in addition to translation, other possibilities exist. Because all components identified in our RNAi screen affect protein synthesis, we sought to further characterize the role of translation attenuation in maintaining the mitochondrial protein-folding environment.

GCN-2 Phosphorylates eIF2α in Response to Mitochondrial Stress
In addition to CeTor, rsks-1 and cel-1, we identified components that are known to regulate translation initiation by modulating the phosphorylation status of the translation initiation factor eIF2α. RNAi-knockdown of the eIF2α kinase General Control Non-derepressible-2 (GCN-2) further increased hsp-60pr::gfp expression in clk-1(qm30) animals, suggesting a role for GCN-2 in promoting mitochondrial protein homeostasis or function (Figure 2A and 2B). The effect of gcn-2(RNAi) on hsp-60pr::gfp expression was not due to direct effects on GFP translation as gcn-2(RNAi) did not cause induction of the ER stress reporter hsp-4pr::gfp (Figure 3A) suggesting a specific role for GCN-2 in promoting mitochondrial protein homeostasis. In unstressed animals, gcn-2(RNAi) did not effect hsp-60pr::gfp expression, suggesting its primary role is during stress (Figure 2B). Contrary to gcn-2(RNAi), our RNAi screen identified gsp-1(RNAi), which reduced hsp-60pr::gfp expression in both the clk-1(qm30) and isp-1(qm150) strains (Figure 2A, 2B and Figure S2B). GSP-1 encodes a protein phosphatase (PP1) required for numerous cellular dephosphorylation events [32], [33] and is homologous to the yeast phosphatase required for eIF2α dephosphorylation [34].

To determine if GCN-2 and GSP-1 regulate eIF2α phosphorylation in C. elegans, we examined the phosphorylation status of eIF2α in whole worm lysates. We utilized an antibody that specifically recognizes the highly conserved serine that is phosphorylated by the repertoire of eIF2α kinases (S51 in mammals and S49 in C. elegans (Figure 3B)). Consistent with previous reports, we detected phosphorylated eIF2α in otherwise unstressed worms (Figure 3C and 3D) [35], [36], [37], which was reduced when the lysate was incubated with calf-intestine phosphatase (CIP), confirming the specificity of the antibody for the phosphorylated form of eIF2α (Figure 3C). Furthermore, in a deletion mutant lacking 1482 bases of gcn-2 (gcn-2(ok871)), the level of steady-state phospho-eIF2α was reduced relative to wild-type worms (Figure 3D). In C. elegans, the only other known eIF2α kinase is PEK-1 (homologous to mammalian PERK [38]). Indeed, phospho-eIF2α was further reduced relative to levels of total eIF2α protein and mRNA in a strain lacking both kinases (Figure 3D and Figure S3B), further supporting the specificity of the phospho-eIF2α antibody and demonstrating the contribution of both kinases to steady state levels of eIF2α phosphorylation.
In contrast to inhibition of GCN-2 and PEK-1, GSP-1 knockdown resulted in increased levels of phospho-eIF2α consistent with it acting as a constitutive eIF2α phosphatase (Figure 3E). In either the gcn-2(ok871) or pek-1(zcdf2) deletion strains fed gsp-1(RNAi) there was still an increase in steady state levels of eIF2α phosphorylation likely reflecting the ability of both kinases to constitutively phosphorylate eIF2α in the absence of exogenous stress (Figure 3E). As increased phospho-eIF2α results in reduced cytosolic translation [5], [39], these data suggest that gsp-1(RNAi) reduces hsp-60pr::gfp induction through attenuation of cytosolic translation, thus reducing the load on the mitochondrial protein folding machinery similar to eIF2α phosphorylation and translation attenuation in the UPRER [5], [6].
The data presented above suggest that GCN-2 activity promotes mitochondrial protein folding during mitochondrial stress. Therefore, we hypothesized that eIF2α phosphorylation would increase in a GCN-2-dependent manner in response to mitochondrial dysfunction. Indeed, phospho-eIF2α levels were increased relative to total eIF2α protein levels in the clk-1(qm30)mutant, which was absent in the gcn-2(ok871) mutant strain (Figure 4A). In contrast, gsp-1(RNAi) caused a further increase in phospho-eIF2α levels (Figure 4A). A similar result was observed in the isp-1(qm150) mutant, supporting the role of GCN-2 in eIF2α phosphorylation in response to stress (Figure 4B). As gcn-2(RNAi) perturbs the mitochondrial protein folding environment and GSP-1 knockdown promotes mitochondrial protein homeostasis as indicated by reduced hsp-60pr::gfp expression (Figure 2B and Figure S2B), these data suggest a correlation between an increase in phospho-eIF2α and a more favorable mitochondrial protein-folding environment.

It should be noted that deletion or knockdown of the other C. elegans eIF2α kinase PEK-1 had no obvious effect on hsp-60pr::gfp induction during mitochondrial stress (data not shown). Furthermore, the increase in eIF2α phosphorylation observed in the clk-1(qm30) animals was not dependent on pek-1 indicating GCN-2 is the primary eIF2α kinase involved in maintaining mitochondrial protein homeostasis (Figure 4C). In pek-1 deletion worms, steady state levels of phospho-eIF2α were reduced (Figure 3E), however these animals still induce eIF2α phosphorylation in response to mitochondrial dysfunction supporting the specific role for GCN-2 during mitochondrial stress (Figure 4C). A similar relationship has been described with PEK-1 and the induction of ER chaperones during ER stress. PEK-1 is specifically activated during ER stress and animals lacking PEK-1 display stronger induction of ER chaperone genes including hsp-4 during ER stress [38]. The UPRER reporter hsp-4pr::gfp is induced during mild heat stress, a condition known to activate the UPRER but not the UPRmt [9], [11]. Incubation of hsp-4pr::gfp animals at 30°C for 3 hours mildly induced GFP expression (Figure 3A and Figure S3A). However, worms raised on pek-1(RNAi) displayed a much stronger induction of the UPRER reporter upon heat exposure consistent with PEK-1 activity protecting ER protein homeostasis [19], [38]. Unlike pek-1(RNAi), gcn-2(RNAi) had no impact on hsp-4pr::gfp during heat stress. These results indicate that the effect of gcn-2(RNAi) on hsp-60pr::gfp induction was not due to dysregulation of global translation further supporting a mitochondrial stress-specific role for GCN-2.
Loss of GCN-2 Sensitizes Worms to Conditions That Induce Mitochondrial Stress
As our data indicated that GCN-2 phosphorylates eIF2α in response to mitochondrial stress, we sought to determine the role of GCN-2 in development and mitochondrial maintenance during mitochondrial stress. gcn-2 deletion or RNAi had no observable effect on worm development in the absence of stress (Figure S4). However, in the presence of mitochondrial stress caused by either the isp-1(qm150) or clk-1(qm30) mutations, gcn-2 deletion significantly slowed development (Figure 5A and 5B). Furthermore, exposure to the NADH ubiquinone oxidoreductase (complex I) inhibitor rotenone or spg-7(RNAi) also significantly delayed development of gcn-2(ok871) worms relative to wild-type worms (Figure 5C and data not shown) indicating a protective role for GCN-2 during mitochondrial stress.
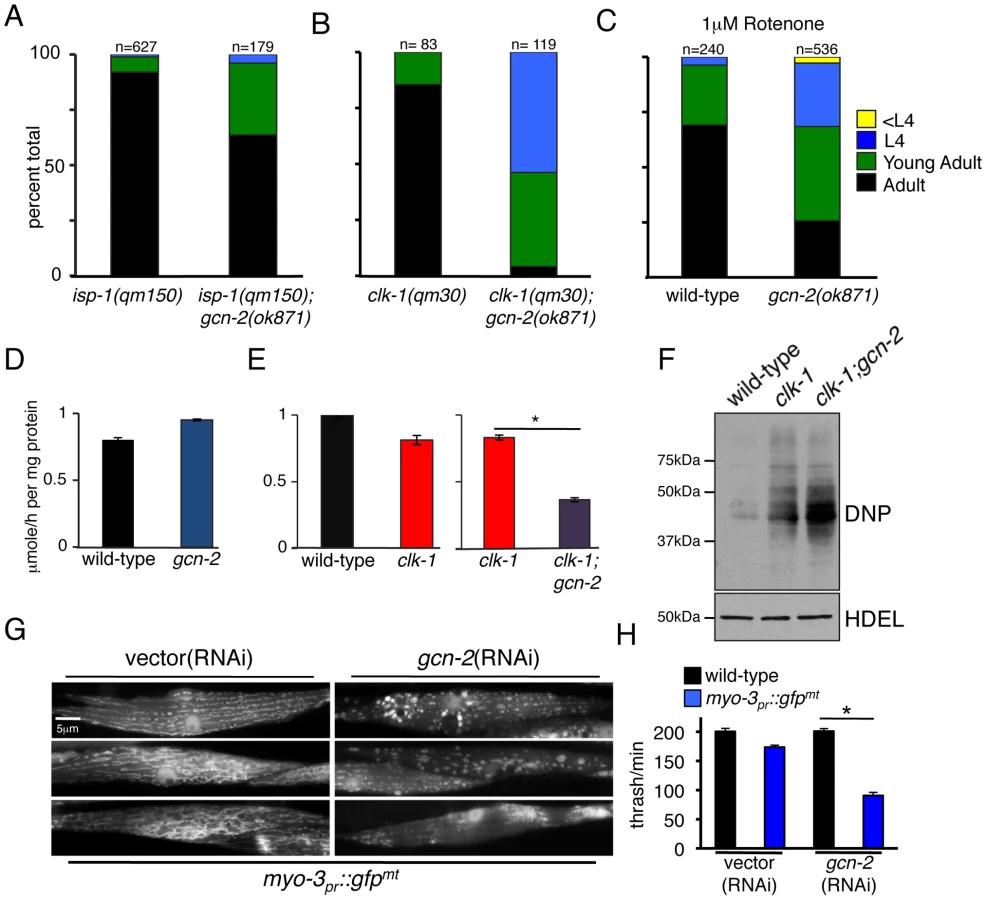
To further assess the role of GCN-2 in maintaining mitochondrial function during mitochondrial stress we examined the effect of gcn-2 deletion on oxygen consumption in wild-type as well as mitochondrial stressed worms. We observed no difference in the rates of oxygen consumption between wild-type worms and those lacking gcn-2 (Figure 5D), consistent with the gcn-2(ok871) deletion having no effect on worm development (Figure S4). clk-1(qm30) worms displayed a slight reduction in oxygen consumption when compared to wild-type worms consistent with mild mitochondrial dysfunction (Figure 5E) [40]. Impressively, clk-1(qm30) worms lacking gcn-2 had a much lower rate of oxygen consumption than worms harboring either the gcn-2-deletion or clk-1(qm30) alone (Figure 5E), supporting a role for GCN-2 in promoting mitochondrial function during mitochondrial stress.
Elevated ROS produced by dysfunctional mitochondria can damage proteins through the formation of irreversible carobonyl modifications on lysine, cysteine, proline and threonine residues [41], [42], [43]. In order to examine levels of oxidative damage in mitochondrial stressed worms, we visualized the accumulation of carbonylated proteins using the Oxyblot system [44]. Consistent with the clk-1(qm30) mutation causing mitochondrial dysfunction, significantly more carbonylated material was detected in lysates from clk-1(qm30) worms than lysates from wild-type worms (Figure 5F). clk-1(qm30);gcn-2(ok871) worms displayed even more oxidative damage than worms harboring clk-1(qm30) alone. Because oxidative damage can perturb protein folding, these data support a role for GCN-2 in protecting the folding environment as well as mitochondrial function.
To further assess the contribution of GCN-2 in maintaining mitochondrial protein homeostasis we targeted GFP to the mitochondrial matrix via the strong muscle-specific myosin promoter (myo-3). High-level expression of mitochondria-targeted GFP challenges the organelle's protein folding environment by increasing the load of unfolded proteins [24], [45]. While wild-type worms were able to accommodate the increased folding load and maintain mitochondrial morphology, myo-3pr::gfpmt worms raised on gcn-2(RNAi) displayed severely perturbed mitochondrial morphology consistent with a loss of protein homeostasis and mitochondrial function [24], [46] (Figure 5G). Furthermore, in the absence of GCN-2, developmental rates (data not shown) and muscle cell function were severely reduced as determined by a motility or thrashing assay (Figure 5H). Together these data indicate that GCN-2 protects mitochondrial function during increased load of mitochondrial unfolded proteins.
GCN-2 Is Required for the Lifespan Extension Associated with Mitochondrial Dysfunction
clk-1(qm30) and isp-1(qm150) animals, which activate ATFS-1-dependent hsp-60pr::gfp expression and GCN-2-dependent eIF2α phosphorylation, are among the numerous C. elegans mitochondrial mutants that exhibit lifespan extension [47], [48], [49]. It was recently reported that ubl-5, a small ubiquitin-like protein required for UPRmt signaling [45], was required for lifespan extension in several mitochondrial mutants highlighting the importance of maintaining mitochondrial protein homeostasis [50]. Consistent with these studies, knockdown of ubl-5 prevented hsp-60pr::gfp induction in the long-lived clk-1(qm30) worms (Figure S5) similar to atfs-1(RNAi).
Additionally, cytosolic translation attenuation also contributes to longevity in several animal models [15], [16], [17]. As GCN-2 slows cytosolic translation [51] in response to mitochondrial dysfunction, we examined the role of GCN-2 in lifespan extension associated with mitochondrial dysfunction. Interestingly, GCN-2 knockdown in clk-1(qm30) animals reduced their lifespan to that of wild-type worms (Figure 6A) consistent with a role for GCN-2 in lifespan extension associated with mitochondrial dysfunction. gcn-2(RNAi) was not generally toxic, as it did not affect lifespan or development in the absence of stress (Figure 6B and Figure S4). gcn-2(RNAi) also shortened the lifespan of isp-1(qm150) animals, but because the animals were very sick with considerable developmental defects, we were unable to determine a role for GCN-2 in longevity of these animals (data not shown). These data are consistent with GCN-2 and increased eIF2α phosphorylation contributing to the lifespan extension observed in mitochondrial mutants and further emphasizes the importance of protein homeostasis in aging.

Because gsp-1(RNAi) caused an increase in eIF2α phosphorylation in the absence or presence of mitochondrial stress (Figure 3E, Figure 4A and 4B), we hypothesized gsp-1(RNAi) would promote lifespan extension. However, the lifespan of wild-type or clk-1(qm30) worms on gsp-1(RNAi) were severely shortened (Figure S6). Interpretation of this result is complicated by the pleitropic, non-specific effects of GSP-1 knockdown. gsp-1(RNAi) also prevents C. elegans germline formation (data not shown) and is required for a variety of cellular processes including mitosis [33]. Therefore, we were unable to determine if increased eIF2α phosphorylation was sufficient to extend lifespan.
ROS Are Required for GCN-2–Dependent eIF2α Phosphorylation during Mitochondrial Stress
We next sought to determine how phospho-eIF2α status is linked to mitochondrial dysfunction. While the most well-studied mechanism of GCN-2 activation is through starvation or amino acid depletion, hydrogen peroxide exposure also stimulates GCN-2 activity through a mechanism that requires the tRNA synthetase domain [52], [53]. Because clk-1(qm30) and isp-1(qm150) worms produce increased levels of ROS (Figure 5F) that are also required for their extended longevity [22], [23], we hypothesized that ROS generated from dysfunctional mitochondria act as an upstream signaling molecule coupling mitochondrial dysfunction to GCN-2 activation. If ROS are required for the observed increase in eIF2α phosphorylation during mitochondrial stress, then treatment with ROS scavengers would phenocopy GCN-2 inhibition with respect to hsp-60pr::gfp activation and the reduced accumulation of phospho-eIF2α in the presence of mitochondrial stress. Impressively, incubation of clk-1(qm30) animals with the ROS scavenger ascorbate resulted in increased hsp-60pr::gfp activation, similar to gcn-2(RNAi) (Figure 7A and Figure 2B). Ascorbate had no effect on the induction of hsp-60pr::gfp in unstressed animals (data not shown) as observed with gcn-2(RNAi) (Figure 2B). We next examined the impact of ascorbate on eIF2α phosphorylation in clk-1(qm30) and isp-1(qm150) animals. Ascorbate treatment, like GCN-2 inhibition, caused a reduction of eIF2α phosphorylation in both mutants supporting a role for ROS in GCN-2 signaling during mitochondrial stress (Figure 7B).

Our data indicate a requirement for ROS in GCN-2-dependent eIF2α phosphorylation observed in response to mitochondrial dysfunction in clk-1(qm30) and isp-1(qm150) mutants. In addition to these two mutants which generate ROS, the herbicide paraquat is known to generate excessive ROS and extend C. elegans lifespan [22], [23]. Interestingly, similar to the clk-1(qm30) and isp-1(qm150) mutants, exposure of wild-type worms to paraquat increased eIF2α phosphorylation in a GCN-2-dependent manner (Figure 7C). In sum, these data support a protective upstream signaling role for mitochondria-generated ROS in GCN-2 activation during mitochondrial stress.
GCN-2 Acts in a Complementary Pathway to That of ATFS-1 and Mitochondrial Chaperone Induction
The above data are consistent with GCN-2-dependent eIF2α phosphorylation and translation attenuation playing a protective role in maintaining mitochondrial function similar to the protection provided by the induced mitochondrial chaperone expression regulated by HAF-1 and ATFS-1 [11]. Therefore, we sought to determine the potential interaction or relationship between GCN-2 and ATFS-1/HAF-1. If they act in complementary pathways, we hypothesized that loss-of-function of both should be more detrimental than loss of either individual pathway. Inhibition of one pathway would cause more stress placing additional burden on the other pathway to maintain the mitochondrial protein-folding environment resulting in further activation of the complementary pathway. As indicated in Figure 2B, in the presence of stress, GCN-2 inhibition results in further activation of hsp-60pr::gfp expression. Similarly, reducing eIF2α phosphorylation by inhibiting ROS accumulation resulted in increased activation of hsp-60pr::gfp activation by ATFS-1 (Figure 7A). To determine if inhibition of mitochondrial chaperone induction during stress caused a further upregulation of the GCN-2 pathway and an increase in eIF2α phosphorylation, we examined phospo-eIF2α levels in clk-1(qm30) animals lacking HAF-1 or ATFS-1. clk-1(qm30) animals displayed an increase in eIF2α phosphorylation which was further increased in combination with the haf-1(ok705) deletion or when fed atfs-1(RNAi) (Figure 8A), consistent with GCN-2 acting in a separate and complementary pathway to that of ATFS-1 and HAF-1.

We next investigated the potential synthetic interaction between GCN-2 and ATFS-1 during worm development. Either individual mutation had no obvious growth or developmental defect in the absence of stress (Figure 8B and Figure S4). However, gcn-2(ok871);atfs-1(tm4525) animals or the gcn-2(ok871) strain fed atfs-1(RNAi) developed somewhat slower in the absence of exogenous stress (Figure 8B and 8C). These data suggest the presence of low levels of mitochondrial stress during development that required the activity of either GCN-2 or ATFS-1.
In the presence of stress caused by spg-7(RNAi) or the clk-1(qm30) mutation, development of worms lacking gcn-2 and atfs-1 was severely compromised. When raised on spg-7(RNAi), most worms arrested at the L1 or L2 larval stage and no animals were able to reach adulthood (Figure 8C and Figure S7A). Furthermore, clk-1(qm30) (Figure 8D and Figure S7B) or isp-1(qm150) (data not shown) animals lacking both GCN-2 and ATFS-1 developed more slowly than worms lacking either individual gene. No synthetic interactions were observed between ATFS-1 and PEK-1 as atfs-1(tm4525) animals raised on pek-1(RNAi) developed at similar rates to atfs-1(tm4525) animals raised on a control RNAi in the absence or presence of stress (data not shown). Despite the developmental defect observed in animals lacking both ATFS-1 and GCN-2 (Figure 8B and 8C), animals lacking both genes had similar lifespans to those of wild-type worms (Figure 8E) suggesting the primary role for each pathway in the absence of exogenous stress is during development, when the majority of mitochondrial biogenesis occurs [54]. Together, these results support a model in which ATFS-1 and GCN-2 act in different yet complementary mitochondrial stress response pathways to regulate mitochondrial chaperone expression and cytosolic translation to protect mitochondrial function (Figure 8F).
Discussion
The experiments described here implicate the eIF2α kinase GCN-2 in the maintenance of mitochondrial function and protein homeostasis. Development of worms lacking GCN-2 was impaired in the presence of mitochondrial stress which caused further induction of ATFS-1-dependent mitochondrial chaperone genes consistent with perturbed mitochondrial protein homeostasis. Furthermore, simultaneous deletion or knockdown of GCN-2 and ATFS-1 has a negative synergistic effect on animal development suggesting that GCN-2-dependent translation attenuation and ATFS-1-dependent mitochondrial chaperone gene induction act in parallel pathways to maintain mitochondrial protein homeostasis. Additionally, GCN-2 was required for development and lifespan extension in the presence of mitochondrial stress suggesting it is responsive to and protective against mitochondrial dysfunction. gcn-2(RNAi) or deletion inhibited eIF2α phosphorylation during mitochondrial stress. These results, along with recent experiments in yeast and flies [12], [13], support our conclusion that attenuation of cytosolic translation is protective during mitochondrial dysfunction.
Our results demonstrate that ROS generated from stressed or dysfunctional mitochondria [22], [23] are required for GCN-2-dependent eIF2α phosphorylation (Figure 7B and 7C). Furthermore, treatment with ROS inhibitors phenocopied gcn-2(RNAi) further exacerbating mitochondrial chaperone induction in the presence of stress (Figure 7A) suggesting ROS are required for GCN-2-dependent eIF2α phosphorylation but not ATFS-1-mediated induction of mitochondrial chaperone gene transcription. While these data support a model in which ROS act as an upstream signaling molecule, the mechanism of GCN-2 activation remains unclear. GCN-2 activation through amino acid depletion is thoroughly characterized, and requires interaction between uncharged tRNA and the tRNA synthetase domain of GCN-2 [55]. GCN-2 activation by peroxide exposure is less well understood, however it also requires the tRNA binding domain [53], [56]. Interestingly, both increased ROS and alterations in amino acid levels are known to occur in clk-1(qm30) and isp-1(1qm150) mutant worms, suggesting that ROS could participate in GCN-2 activation either through a direct interaction with the GCN-2 tRNA synthetase domain or through effects on amino acid metabolism [22], [23], [57], [58], [59]. Regardless, our results suggest a protective role for mitochondrial-generated ROS by influencing eIF2α phosphorylation, consistent with recent data indicating that low levels of ROS participate in beneficial cyto-protective stress-signaling pathways [22], [23], [60].
Attenuation of cytosolic translation slows mitochondrial import, thus reducing the folding load on mitochondrial chaperones. However, continued translation of proteins encoded by the mitochondrial genome could become detrimental when the expression of cytosolic components required for ETC complex formation is reduced. Interestingly, mitochondrial translation is tightly linked to the accumulation of imported ETC subunits and complex assembly. In their absence, mitochondrial translation is also attenuated [61]. We hypothesize that translation attenuation in the cytosol slows mitochondrial protein import leading to translational repression within mitochondria, thus reducing the overall burden on the mitochondrial protein folding and complex assembly machinery.
Protection of mitochondrial protein homeostasis and function appears to be a novel role for GCN-2 in addition to its established role during starvation [62]. The GCN-2 signaling pathway is complementary to the signaling pathway that transcriptionally upregulates mitochondrial chaperone genes during stress, which requires the mitochondrial peptide transporter HAF-1 and transcription factor ATFS-1 (Figure 8F) [11]. This parallel relationship between a reduction in organelle protein folding load and the regulation of organelle-specific protein folding machinery is similar to mechanisms that regulate ER protein homeostasis, in which another eIF2α kinase, PEK-1 (PERK in mammals), responds directly to unfolded protein stress within the ER. PEK-1-mediated translation attenuation complements the IRE-1/XBP-1 branch of the UPRER, which regulates expression of ER chaperones and additional protein handling machinery [38].
In addition to a protective role during development, GCN-2 also contributes to the lifespan extension of clk-1(qm30) animals (Figure 6A). These mutants have disrupted mitochondrial function and elevated levels of mitochondrial chaperones (Figure 1B), consistent with a recent report that indicated a requirement for mitochondrial chaperone induction in the lifespan extension of several mitochondrial mutants [50]. Additionally, clk-1(qm30) animals display elevated levels of ROS that have also been shown to contribute to longevity [22], [23]. Our data support a model in which ROS and GCN-2 activate a pathway that contributes to lifespan extension, in parallel to the requirement for transcriptional induction of mitochondrial chaperone genes. The contribution of GCN-2 is most likely through cytosolic translation attenuation, which promotes stress resistance and extends lifespan in C. elegans [15], [16], [17].
An additional eIF2α-dependent protective activity not addressed here involves the preferential translation of mRNAs with small upstream open reading frames (uORFs). A number of uORF containing transcripts have been identified in S. cerevisiae [63] including the well-characterized transcription factor Gcn4 [64]. Homology searches did not reveal an obvious Gcn4 orthologue in C. elegans, and this avenue was not further pursued. Our RNAi screen identified components that when knocked down slow cytosolic translation as suppressors of hsp-60pr::gfp activation in stressed animals supporting a role for translation attenuation in promoting mitochondrial protein homeostasis.
Our finding that GCN-2-dependent eIF2α phosphorylation protects mitochondrial protein homeostasis raises the possibility that manipulation of phospho-eIF2α status may be a therapeutic entry point for the diverse number of degenerative diseases associated with mitochondrial dysfunction [65]. At least two strategies to accomplish this seem plausible: (1) caloric restriction to reduce cytosolic amino acid levels and activate GCN-2 to increase eIF2α phosphorylation independent of mitochondrial stress or (2) small molecule inhibition of stress-dependent eIF2α dephosphorylation to increase phospho-eIF2α levels through phosphatase inhibition [37]. It will be interesting to determine the viability of these possibilities in future studies.
Materials and Methods
C. elegans Strains and Growth Conditions
Reporter strains hsp-60pr::gfp(zcIs9)V, myo-3pr::gfpmt(zcIs14) and hsp-4pr::gfp(zcIs4)V have been described previously [9], [24], [45]. Where indicated, the hsp-60pr::gfp(zcIs9)V transgene was crossed into individual mutant strains of interest, with the exception of atfs-1(tm4525)V, which was backcrossed with N2 three times prior to crossing into the hsp-60pr::gfp background. The clk-1(qm30), isp-1(qm150) and gcn-2(ok871) strains were obtained from the Caenorhabditis Genetics Center (Minneapolis, MN) and the atfs-1(tm4525) strain was obtained from the National BioResource Project (Tokyo, Japan). RNAi feeding experiments were performed as described [9] with constructs from the Ahringer and Vidal libraries [66], [67].
Development and Lifespan Analysis
Worms were synchronized via bleaching and allowed to develop on the described RNAi plate or condition. For development in the presence of oxidative stress, rotenone was applied to vector(RNAi) plates and allowed to soak in prior to seeding eggs. At the time points indicated, the numbers of L1, L2, L3, L4, young adult (non-gravid) or gravid adult worms were counted on each plate and quantified as a percent of the total number of animals. For each plate, the worms in 6–8 individual fields of view were counted, and the total number combined.
For lifespan analysis, worms were synchronized as eggs and allowed to develop under the described condition for two days. At that point, 100 L4 animals were transferred to new RNAi plates and subsequently transferred to fresh plates every day for the next 5–6 days and every two days thereafter. The numbers of dead and censored worms were counted every second day for the duration of the assay [68]. Survival curves and statistical analysis were generated using Prism 5.0b software (Graphpad). Each experiment was repeated 3 times.
Western Blots
Worms were grown under the described conditions and collected at the L4 stage for analysis as previously described [9], [24]. Phospho-eIF2α antibody (#3597S) was obtained from Cell Signaling Technology (Danvers, MA) and observed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Rockville, IL). GFP and HDEL immunoblots were visualized using Odyssey Infrared Imager (Li-Cor Biosciences, Lincoln, NE). Total eIF2α was assessed as described [36]. Due to limited amounts of pan-eIF2αantibody, western analysis was only performed during select experiments to confirm specificity of the phospho-eIF2α antibody in wild-type lysates and during analysis phospho-eIF2α levels in clk-1(qm30) animals.
For the eIF2α dephosphorylation assay, 150 µg of worm lysate was treated with calf intestinal phosphatase (CIP) for 30 minutes at 30°C prior to SDS-PAGE analysis. For ascorbate treatment, synchronized worms were grown to adulthood in liquid medium, when 25 mM ascorbate was added for 16 hours prior to western analysis.
RNA Isolation, cDNA Synthesis, and Quantitative RT–PCR
Total RNA was isolated using RNA STAT (Tel-Test Inc, Friendswood, TX). RNA samples were prepared from the described worms at the L4 stage. cDNA was then synthesized from total RNA using a iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Following mRNA isolation and cDNA synthesis, qPCR was used to determine the expression level of eif2α using iQ sybr green supermix and MyiQ2 Two-Color Real-Time PCR Detection System (Bio-Rad). Actin was used as a control. Fold changes in gene expression were calculated using the comparative CtΔΔCt method.
Microscopy
Fluorescent photomicrographs were obtained using a Zeiss AxioCam MRm mounted on a Zeiss Imager.Z2 microscope or Zeiss M2 Bio stereo microscope (Carl Zeiss Imaging, Thornwood, NY).
Oxygen Consumption and Examination of Oxidative Protein Modification
Oxygen consumption assays were performed as described [11] using a Clark type electrode [40]. To determine the accumulation of oxidative protein modifications, synchronized wild-type, clk-1(qm30) and clk-1(qm30);gcn-2(ok871) worms were harvested once they reached the L4 stage. Worm lysates were separated by SDS-Page and treated according to the Oxyblot manufacturer (Millipore, Billerica, MA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. TatsutaTLangerT 2008 Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J 27 306 314
2. NeupertWHerrmannJM 2007 Translocation of proteins into mitochondria. Annu Rev Biochem 76 723 749
3. BakerBMHaynesCM 2011 Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci 36 254 261
4. WalterPRonD 2011 The unfolded protein response: from stress pathway to homeostatic regulation. Science 334 1081 1086
5. HardingHPZhangYRonD 1999 Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397 271 274
6. CalfonMZengHUranoFTillJHHubbardSR 2002 IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415 92 96
7. HartlFUNeupertW 1990 Protein sorting to mitochondria: evolutionary conservations of folding and assembly. Science 247 930 938
8. ZhaoQWangJLevichkinIVStasinopoulosSRyanMT 2002 A mitochondrial specific stress response in mammalian cells. EMBO J 21 4411 4419
9. YonedaTBenedettiCUranoFClarkSGHardingHP 2004 Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 117 4055 4066
10. MartinusRDGarthGPWebsterTLCartwrightPNaylorDJ 1996 Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem 240 98 103
11. HaynesCMYangYBlaisSPNeubertTARonD 2010 The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell 37 529 540
12. WangXZuoXKucejovaBChenXJ 2008 Reduced cytosolic protein synthesis suppresses mitochondrial degeneration. Nat Cell Biol 10 1090 1097
13. LiuSLuB 2010 Reduction of protein translation and activation of autophagy protect against PINK1 pathogenesis in Drosophila melanogaster. PLoS Genet 6 e1001237 doi:10.1371/journal.pgen.1001237
14. ShiYVattemKMSoodRAnJLiangJ 1998 Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol 18 7499 7509
15. SyntichakiPTroulinakiKTavernarakisN 2007 eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 445 922 926
16. PanKZPalterJERogersANOlsenAChenD 2007 Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell 6 111 119
17. HansenMTaubertSCrawfordDLibinaNLeeSJ 2007 Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6 95 110
18. ZoncuREfeyanASabatiniDM 2011 mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12 21 35
19. RonDWalterP 2007 Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8 519 529
20. TaylorSSHasteNMGhoshG 2005 PKR and eIF2alpha: integration of kinase dimerization, activation, and substrate docking. Cell 122 823 825
21. DeverTE 1999 Translation initiation: adept at adapting. Trends Biochem Sci 24 398 403
22. YangWHekimiS 2010 A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol 8 e1000556 doi:10.1371/journal.pbio.1000556
23. LeeSJHwangABKenyonC 2010 Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol 20 2131 2136
24. HaynesCMPetrovaKBenedettiCYangYRonD 2007 ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell 13 467 480
25. NoldenMEhsesSKoppenMBernacchiaARugarliEI 2005 The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 123 277 289
26. WongABoutisPHekimiS 1995 Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 139 1247 1259
27. FengJBussiereFHekimiS 2001 Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell 1 633 644
28. EwbankJJBarnesTMLakowskiBLussierMBusseyH 1997 Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science 275 980 983
29. YangWHekimiS 2010 Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 9 433 447
30. ManningG 2005 Genomic overview of protein kinases. WormBook 1 19
31. VellaiTTakacs-VellaiKZhangYKovacsALOroszL 2003 Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426 620
32. RutledgeEDentonJStrangeK 2002 Cell cycle - and swelling-induced activation of a Caenorhabditis elegans ClC channel is mediated by CeGLC-7alpha/beta phosphatases. J Cell Biol 158 435 444
33. HsuJYSunZWLiXReubenMTatchellK 2000 Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 102 279 291
34. WekRCCannonJFDeverTEHinnebuschAG 1992 Truncated protein phosphatase GLC7 restores translational activation of GCN4 expression in yeast mutants defective for the eIF-2 alpha kinase GCN2. Mol Cell Biol 12 5700 5710
35. HardingHPZhangYScheunerDChenJJKaufmanRJ 2009 Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci U S A 106 1832 1837
36. NukazukaAFujisawaHInadaTOdaYTakagiS 2008 Semaphorin controls epidermal morphogenesis by stimulating mRNA translation via eIF2alpha in Caenorhabditis elegans. Genes Dev 22 1025 1036
37. TsaytlerPHardingHPRonDBertolottiA 2011 Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332 91 94
38. ShenXEllisRELeeKLiuCYYangK 2001 Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107 893 903
39. HinnebuschAG 1994 The eIF-2 alpha kinases: regulators of protein synthesis in starvation and stress. Semin Cell Biol 5 417 426
40. BraeckmanBPHouthoofdKDe VreeseAVanfleterenJR 2002 Assaying metabolic activity in ageing Caenorhabditis elegans. Mech Ageing Dev 123 105 119
41. GruneTJungTMerkerKDaviesKJ 2004 Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol 36 2519 2530
42. NystromT 2005 Role of oxidative carbonylation in protein quality control and senescence. EMBO J 24 1311 1317
43. BalabanRSNemotoSFinkelT 2005 Mitochondria, oxidants, and aging. Cell 120 483 495
44. GrunewaldAVogesLRakovicAKastenMVandebonaH 2010 Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS ONE 5 e12962 doi:10.1371/journal.pone.0012962
45. BenedettiCHaynesCMYangYHardingHPRonD 2006 Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174 229 239
46. IchishitaRTanakaKSugiuraYSayanoTMiharaK 2008 An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J Biochem 143 449 454
47. LeeSSLeeRYFraserAGKamathRSAhringerJ 2003 A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33 40 48
48. LakowskiBHekimiS 1998 The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A 95 13091 13096
49. DillinAHsuALArantes-OliveiraNLehrer-GraiwerJHsinH 2002 Rates of behavior and aging specified by mitochondrial function during development. Science 298 2398 2401
50. DurieuxJWolffSDillinA 2011 The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144 79 91
51. SonenbergNHinnebuschAG 2009 Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136 731 745
52. BerlangaJJRiveroDMartinRHerreroSMorenoS 2010 Role of mitogen-activated protein kinase Sty1 in regulation of eukaryotic initiation factor 2alpha kinases in response to environmental stress in Schizosaccharomyces pombe. Eukaryot Cell 9 194 207
53. MascarenhasCEdwards-IngramLCZeefLShentonDAsheMP 2008 Gcn4 is required for the response to peroxide stress in the yeast Saccharomyces cerevisiae. Mol Biol Cell 19 2995 3007
54. TsangWYLemireBD 2002 Mitochondrial genome content is regulated during nematode development. Biochem Biophys Res Commun 291 8 16
55. WekSAZhuSWekRC 1995 The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol 15 4497 4506
56. WekRCRamirezMJacksonBMHinnebuschAG 1990 Identification of positive-acting domains in GCN2 protein kinase required for translational activation of GCN4 expression. Mol Cell Biol 10 2820 2831
57. ReinkeSNHuXSykesBDLemireBD 2010 Caenorhabditis elegans diet significantly affects metabolic profile, mitochondrial DNA levels, lifespan and brood size. Mol Genet Metab 100 274 282
58. LiuDYangJLiLMcAdooDJ 1995 Paraquat–a superoxide generator–kills neurons in the rat spinal cord. Free Radic Biol Med 18 861 867
59. FalkMJZhangZRosenjackJRNissimIDaikhinE 2008 Metabolic pathway profiling of mitochondrial respiratory chain mutants in C. elegans. Mol Genet Metab 93 388 397
60. HekimiSLapointeJWenY 2011 Taking a “good” look at free radicals in the aging process. Trends Cell Biol 21 569 576
61. MickDUFoxTDRehlingP 2011 Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation. Nat Rev Mol Cell Biol 12 14 20
62. DeverTEFengLWekRCCiganAMDonahueTF 1992 Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68 585 596
63. IngoliaNTGhaemmaghamiSNewmanJRWeissmanJS 2009 Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324 218 223
64. HinnebuschAG 2005 Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59 407 450
65. WallaceDC 2005 A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39 359 407
66. RualJFCeronJKorethJHaoTNicotAS 2004 Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14 2162 2168
67. KamathRSFraserAGDongYPoulinGDurbinR 2003 Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421 231 237
68. HsinHKenyonC 1999 Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399 362 366
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Rumors of Its Disassembly Have Been Greatly Exaggerated: The Secret Life of the Synaptonemal Complex at the Centromeres
- The NSL Complex Regulates Housekeeping Genes in
- Tipping the Balance in the Powerhouse of the Cell to “Protect” Colorectal Cancer
- Interplay between Synaptonemal Complex, Homologous Recombination, and Centromeres during Mammalian Meiosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy