
EMT Inducers Catalyze Malignant Transformation of Mammary Epithelial Cells and Drive Tumorigenesis towards Claudin-Low Tumors in Transgenic Mice
The epithelial-mesenchymal transition (EMT) is an embryonic transdifferentiation process consisting of conversion of polarized epithelial cells to motile mesenchymal ones. EMT–inducing transcription factors are aberrantly expressed in multiple tumor types and are known to favor the metastatic dissemination process. Supporting oncogenic activity within primary lesions, the TWIST and ZEB proteins can prevent cells from undergoing oncogene-induced senescence and apoptosis by abolishing both p53 - and RB-dependent pathways. Here we show that they also downregulate PP2A phosphatase activity and efficiently cooperate with an oncogenic version of H-RAS in malignant transformation of human mammary epithelial cells. Thus, by down-regulating crucial tumor suppressor functions, EMT inducers make cells particularly prone to malignant conversion. Importantly, by analyzing transformed cells generated in vitro and by characterizing novel transgenic mouse models, we further demonstrate that cooperation between an EMT inducer and an active form of RAS is sufficient to trigger transformation of mammary epithelial cells into malignant cells exhibiting all the characteristic features of claudin-low tumors, including low expression of tight and adherens junction genes, EMT traits, and stem cell–like characteristics. Claudin-low tumors are believed to be the most primitive breast malignancies, having arisen through transformation of an early epithelial precursor with inherent stemness properties and metaplastic features. Challenging this prevailing view, we propose that these aggressive tumors arise from cells committed to luminal differentiation, through a process driven by EMT inducers and combining malignant transformation and transdifferentiation.
Published in the journal:
. PLoS Genet 8(5): e32767. doi:10.1371/journal.pgen.1002723
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002723
Summary
The epithelial-mesenchymal transition (EMT) is an embryonic transdifferentiation process consisting of conversion of polarized epithelial cells to motile mesenchymal ones. EMT–inducing transcription factors are aberrantly expressed in multiple tumor types and are known to favor the metastatic dissemination process. Supporting oncogenic activity within primary lesions, the TWIST and ZEB proteins can prevent cells from undergoing oncogene-induced senescence and apoptosis by abolishing both p53 - and RB-dependent pathways. Here we show that they also downregulate PP2A phosphatase activity and efficiently cooperate with an oncogenic version of H-RAS in malignant transformation of human mammary epithelial cells. Thus, by down-regulating crucial tumor suppressor functions, EMT inducers make cells particularly prone to malignant conversion. Importantly, by analyzing transformed cells generated in vitro and by characterizing novel transgenic mouse models, we further demonstrate that cooperation between an EMT inducer and an active form of RAS is sufficient to trigger transformation of mammary epithelial cells into malignant cells exhibiting all the characteristic features of claudin-low tumors, including low expression of tight and adherens junction genes, EMT traits, and stem cell–like characteristics. Claudin-low tumors are believed to be the most primitive breast malignancies, having arisen through transformation of an early epithelial precursor with inherent stemness properties and metaplastic features. Challenging this prevailing view, we propose that these aggressive tumors arise from cells committed to luminal differentiation, through a process driven by EMT inducers and combining malignant transformation and transdifferentiation.
Introduction
While the disruption of embryonic processes has been acknowledged as a cause of the outgrowth of paediatric neoplasms, more recent observations suggest that the aberrant reactivation of developmental regulatory programs might also contribute to progression in the advanced stages of cancers in adults [1]. At the crux of this concept is the subversion of the epithelial-mesenchymal transition (EMT) during tumor progression. This developmental program converts epithelial cells into mesenchymal ones through profound disruption of cell-cell junctions, loss of apical-basolateral polarity and extensive reorganization of the actin cytoskeleton [2]. During embryogenesis, EMT plays critical roles in the formation of the body plan and in the differentiation of most of the tissues and organs derived from the mesoderm and the endoderm [3]. This process is tightly regulated through a delicate interplay between environmental signals from WNT, TGFβ, FGF family members, and a complex network of signaling pathways that converge on the activation of transcription factors that induce EMT through repression of CDH1 (encoding for the E-cadherin) and activation of mesenchymal genes. EMT-inducing transcription factors include several zinc finger proteins (e.g., SNAIL1, SNAIL2), basic helix-loop-helix transcription factors (e.g., TWIST1, TWIST2 and E2A) and zinc-finger and homeodomain proteins (ZEB1, ZEB2/SIP1) [4], [5]. Importantly, while EMT inducers are maintained in a silent state in adult differentiated epithelial cells, their reactivation is commonly observed in a variety of human cancers with a frequent correlation with poor clinical outcome [6]. In the course of tumor progression, the gain of cell motility and the secretion of matrix metalloproteases associated with EMT promote cancer cell migration across the basal membrane and invasion of the surrounding microenvironment, favoring metastatic dissemination. Furthermore, EMT may also facilitate second site colonization by endowing cells with stem-like features including self-renewing properties [7]–[9]. While the involvement of EMT inducers in the invasion-metastasis cascade of epithelial tumors is well delineated, their contribution to tumorigenesis remains unclear. Supporting an oncogenic activity within primary lesions, we recently demonstrated that the TWIST proteins were able to prevent cells from undergoing oncogene-induced senescence and apoptosis by abrogating both p53 - and RB-dependent pathways [10], [11]. As a consequence, TWIST1 and TWIST2 can cooperate with an activated version of RAS to transform mouse embryonic fibroblasts [11]. Furthermore, the ZEB transcription factors were recently shown to overcome EGFR-induced senescence in oesophageal epithelial cells, suggesting that several EMT-inducers might share the property of inhibiting oncogene-induced failsafe programs [12]. On the basis of these findings, we sought to formally assess the oncogenic activity of these EMT-promoting factors in the model of breast tumorigenesis by generating Twist1 transgenic mouse models and by performing cooperation assays in human mammary epithelial cells (HMECs). The focus of this study was underpinned by the common reactivation of ZEB1, ZEB2 and TWIST1 in aggressive and undifferentiated human breast cancers, especially in the newly identified claudin-low intrinsic subtype [13]. Here we demonstrate that commitment to an EMT program favors breast tumor initiation by inhibiting crucial tumor suppressor functions, including PP2A (protein phosphatase 2A) activity, and thus minimizes the number of events required for neoplastic transformation. Importantly, upon aberrant activation of an EMT inducer, a single mitogenic activation is sufficient to transform mammary epithelial cells into malignant cells exhibiting all the characteristic features of claudin-low tumors. These findings extend our understanding of the role of EMT-inducing transcription factors during tumor development and highlight the claudin-low tumor subtype of breast cancers as the first example of human adult malignancies driven by aberrant reactivation of an embryonic transdifferentiation program.
Results
TWIST1 expression promotes primary tumor development in vivo
To gain insight into the role of EMT commitment in tumor initiation and primary tumor growth, we used a Twist1 transgenic mouse model exhibiting a lox-STOP-lox (LSL) version of the active version of the murine TWIST1 (TWIST1-E12 tethered dimer) under a ubiquitous promoter [14]. These mice were crossed with a line expressing a lox-STOP-lox regulated knock-in of the activated K-Ras oncogene (LSL-K-rasG12D) [15], [16] for in vivo oncogenic cooperation experiments. Transgene expression was first induced using a Mouse Mammary Tumor Virus promoter driven Cre recombinase (MMTV-Cre) and thereby restricted to secretory tissues, in particular the mammary gland and skin epithelia, as well as to the hematopoietic system [17], [18]. Neither wild-type nor MMTV-Cre;Twist1 mice exhibited tumor formation by one year of age (n = 47). Expression of knock-in K-rasG12D was associated with low-grade splenic lymphomas as well as anal and oral papillomas (Figure 1). Importantly, papillomas never progressed to a malignant stage but could grow to the point to physical obstruction leading to cachexia and requiring euthanasia (n = 85, median survival 85 days). In contrast, MMTV-Cre;K-rasG12D;Twist1 mice invariably developed aggressive multifocal squamous cell carcinomas (SCC) at very young ages necessitating euthanasia at the significantly earlier median age of 35 days (n = 12, p<0.0001, Figure 1). These observations demonstrated for the first time the oncogenic properties of TWIST1 in vivo and underscored the cooperative effect between K-RAS and TWIST1 in promoting malignant conversion.
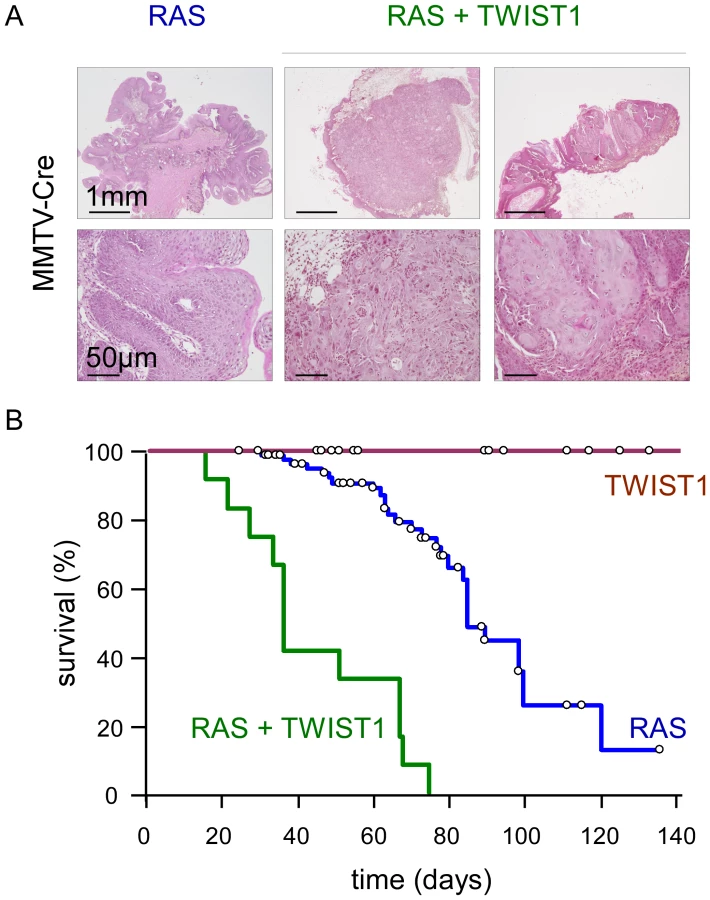
TWIST1 expression in murine differentiated mammary epithelial cells promotes the development of claudin-low breast cancers
Due to the speed with which SCC developed in the MMTV-Cre;K-rasG12D;Twist1 animals, the role of TWIST1 in promoting mammary tumor formation could not be assessed. Consequently, transgene expression was next restricted to differentiated mammary epithelial cells by using mice expressing the Cre recombinase under the control of the Whey Acidic Protein promoter (WAP-Cre) [17]. WAP is a milk protein expressed late in the differentiation pathway of mammary epithelial cells [19]. The 2.6-kb fragment of the mouse WAP gene promoter used in the present study is active in the mammary alveolar epithelium during the second half of pregnancy upon the initiation of differentiation [17], [20]. In virgin animals, the promoter is only transiently activated in a few mammary alveolar and ductal cells, during estrus (average age at first estrus = 35 days), allowing mosaic transgene activation so as to better mimic the emergence of spontaneous oncogenic activations [19]. Wild-type, WAP-Cre;K-rasG12D, and WAP-Cre;Twist1 transgenic females exhibited normal mammary gland development (Figure 2B) and remained healthy for at least 240 days (n = 19). However, all virgin WAP-Cre;K-rasG12D;Twist1 females developed multifocal breast carcinomas by 140 days of age, approximately 105 days after first transgene expression (p<0.001, median survival = 125 days, n = 9) (Figure 2A and 2D). These tumors exhibited metaplastic features with a mixed morphologic aspect that included epithelial-type and spindle shaped cells (Figure 2D). In support of the relevance of EMT in vivo, the presence of the tagged-TWIST1 transgene in both the epithelial and fusiform cancer cell contingents demonstrated that mesenchymal cells arose through transdifferentiation of their epithelial counterparts (Figure 2D). Molecular characterization of human and murine breast tumors led to identifying five intrinsic subtypes (luminal A, luminal B, HER2-enriched, basal-like and claudin-low) [13], [21], [22]. Global gene expression profile analysis (Accession number GSE32905) classified tumors developed by WAP-Cre;K-rasG12D;Twist1 transgenic mice as claudin-low (Figure S1). Immunostaining of epithelial and mesenchymal markers (Figure 2D) and quantitative RT-PCR analysis (lack of E-cadherin and claudin expression, high expression of vimentin; Figure S2) were highly consistent with this classification. Of note, endogenous expression of the Zeb1, Zeb2 and Twist2 EMT inducers was also induced (Figure S2), further supporting the association of the EMT interactome with the claudin-low breast cancer subtype [23].

TWIST1 and ZEB1/2 transcription factors foster malignant transformation of human mammary epithelial cells and provide cells with a basal-like or a claudin-low signature, according to the extent of EMT
Both basal-like and claudin-low subgroups are aggressive, chemoresistant, triple-negative carcinomas (estrogen-receptor-, progesterone-receptor-, and HER2-negative). Yet claudin-low tumors exhibit several characteristic features, including low expression of adherens and tight junction proteins, a low level of luminal/epithelial differentiation, stem cell-like features, and a high frequency of metaplastic differentiation [24]. These tumors are believed to originate from an early epithelial precursor with inherent stemness properties and metaplastic features [24]–[26]. Nevertheless, the observation of claudin-low tumors in WAP-Cre;K-RasG12D;Twist1 transgenic mice suggested that the development of these neoplasms could rely upon an EMT-driven process affecting epithelial cells formerly engaged in differentiation. We sought to test this hypothesis by mimicking in non-stem cells events occurring commonly in this breast cancer subtype. Claudin-low tumors and cell lines frequently exhibit increased levels of the EMT-inducing transcription factors TWIST1, ZEB1, and ZEB2 [13] and show activation of RAS/MAPK pathway components [24], [27]. To functionally reproduce these two common features of claudin-low tumors, we performed oncogenic cooperation assays by transducing genes encoding a single EMT inducer (TWIST1, ZEB1 or ZEB2) and/or an active form of H-RAS (H-RASG12V) into immortalized human mammary epithelial cells (hTERT-HMECs, thereafter named HME cells). As in all experiments the infection efficiency exceeded 80%, the hypothesis of selection of a rare subpopulation of parental cells can be ruled out. Forced expression of an EMT inducer triggered acquisition of EMT features, including significant upregulation of mesenchymal markers (i.e., vimentin, fibronectin) and decreased expression of genes involved in epithelial cell-cell adhesion (i.e., E-cadherin, occludin) (Figure 3). However, the degree of EMT commitment observed after infection was highly dependent on the EMT-inducing transcription factor, both in the absence or in the presence of the active form of RAS. In the absence of the mitogenic oncoprotein, ZEB1 expression was sufficient to promote a complete transdifferentiation process, giving rise to typical spindle-like cell morphology, and a total loss of E-cadherin expression (Figure 3; HME-ZEB1 cells). In contrast, ZEB2-expressing cells (HME-ZEB2 cells) and TWIST1-expressing cells (HME-TWIST1 cells) exhibited intermediate phenotypes, maintaining significant levels of E-cadherin expression and retaining a cobblestone morphology despite increased levels of mesenchymal markers such as vimentin and fibronectin (Figure 3). Transduction of an active form of RAS further promoted EMT induction, as assessed by cell morphology (Figure 3A) and protein expression analysis (Figure 3F and 3G). Nevertheless, HME-ZEB2-RAS and HME-TWIST1-RAS cells still exhibited an intermediate phenotype. Importantly, combining the mitogenic oncoprotein with either TWIST1, ZEB1 or ZEB2 was sufficient to provide cells with transformation potential, as assessed by their ability to form colonies in an assay on semi-solid medium and by acquisition of a characteristic stellate phenotype in 3D cell culture (Figure 3C and 3D). This observation suggested that EMT inducers could exert a potent oncogenic activity in the absence of a complete EMT. Global gene expression array analysis was next performed on freshly established cell lines (Accession number GSE32905). Strikingly, HME-ZEB1 cells and HME-ZEB1-RAS cells were defined as basal-B (P = 2.7×10−20 and 2.4×10−22 respectively), reminiscent of claudin-low tumors [13], [21], HME-TWIST1, HME-TWIST1-RAS and HME-ZEB2 as basal-A (P = 3.6×10−8, P = 1.1×10−2, P = 3.8×10−9 respectively), reminiscent of basal-like tumors [13], [21], while HME-ZEB2-RAS positively correlated with basal-A/basal-like (P = 5.0×10−2) and basal-B/claudin-low (P = 2.3×10−2) (Figure 4 and Figure S3), suggesting a direct link between the extent of EMT and the intrinsic subtype. Confirming this hypothesis, exposure of HME-TWIST1-RAS and HME-ZEB2-RAS to the EMT-promoting cytokine TGFβ triggered a complete EMT with a shift to basal-B/claudin-low (P = 2.3×10−18 for TGFβ-treated HME-TWIST1-RAS cells; P = 6.8×10−22 for TGFβ-treated HME-ZEB2-RAS cells) (Figure 4 and Figure S3).


The extent of the EMT and the basal-B/claudin-low profiling were strongly associated with acquisition of stem cell-like features, as judged by the ability to form mammospheres under non-adherent culture conditions (Figure 3E) and by the fraction of cells exhibiting the CD44+/CD24−/low stem-like antigenic phenotype (respectively 84.5%, 83.2%, 20.6% and 15.3% of HME-ZEB1; HME-ZEB1-RAS, HME-ZEB2-RAS and HME-TWIST1-RAS; Figure 5). The gain of a mammary stem cell signature was also revealed by the use of the recently described Genomic Differentiation Predictor [13], following global gene expression array analysis on freshly established cell lines (Figure 6). Cells exhibiting the more pronounced mesenchymal phenotype (HME-ZEB1, HME-ZEB1-RAS; HME-ZEB2-RAS treated with TGFβ and HME-TWIST1-RAS treated with TGFβ) exhibited a mammary stem like signature, whereas cells with an epithelial or an intermediate phenotype (HME, HME-TWIST1, HME-TWIST1-RAS; HME-ZEB2; HME-ZEB2-RAS) showed a luminal progenitor signature.


As expected from earlier studies [28]–[30], immortalized HMEC cells into which only H-RASG12V had been transduced (HME-RAS) exhibited a low transformation potential (Figure 3D). Characterization of the few colonies growing on soft agar revealed a constant endogenous activation of EMT inducers, including TWIST1, ZEB1 and ZEB2 (Figure S4), and a mesenchymal phenotype, further highlighting the deleterious interplay between the mitogenic oncoprotein and EMT-promoting factors. To confirm the association between the RAS-induced transformation and the endogenous expression of EMT inducers, immortalized HMECs were transduced with an H-RASG12V and sorted by flow cytometry using the EpCAM epithelial antigen. EpCAM-positive epithelial cells were next cultured in the presence of TGFβ. As shown in Figure S5, TGFβ exposure triggered morphological and phenotypical features of EMT, associated with increased expression of TWIST1, TWIST2, ZEB1 and ZEB2 EMT inducers. Reactivation of these transcription factors was associated with the acquisition of a transformed phenotype.
Taken together, these observations showed that activation of EMT inducers, through either forced expression or endogenous induction, fosters malignant transformation of mammary epithelial cells and confers to them basal-like or claudin-low signatures, according to the extent of transdifferentiation.
EMT commitment triggers malignant transformation of human mammary epithelial cells deficient for p53 - and RB-dependent pathways
Our data demonstrated that EMT inducers can promote transformation of mammary epithelial cells. We and others have previously shown that the TWIST and ZEB proteins can functionally inhibit p53 - and RB-dependent pathways, preventing cells from undergoing oncogene-induced senescence and apoptosis [10]–[12]. To test whether the oncogenic properties of EMT-inducing transcription factors act only to lift these two oncosuppressive barriers or whether they might be involved in additional processes, we have generated human mammary epithelial cells deficient in both pathways. The INK4A tumor suppressor, a crucial regulator of the RB-dependent pathway, is known to be silenced by progressive promoter methylation in HMECs escaping from stasis [31]. We depleted these cells of p53 by means of RNA interference (using a shRNA TP53 thereafter named shp53 or, as a control, a scrambled shRNA). Knockdown of p53 was checked by western blotting and by demonstrating that, in response to DNA damage, p53 induction and the resulting G1 growth arrest were abolished (Figure S6). Cells were next infected with H-RASG12V and immortalized by transfection with hTERT to generate shp53/H-RASG12V/hTERT HMECs (hereafter named HME-shp53-RAS cells). Characterization of the colonies generated after growth of these cells in soft agar demonstrated that a vast majority of them expressed mesenchymal markers (Figure S7E). This observation led us to hypothesize that a subset of HME-shp53-RAS cells committed spontaneously to an EMT program and that initiation of the transdifferentiation process promoted cell transformation. In support of the first hypothesis, cells exhibiting a cobblestone phenotype and expressing epithelial markers (E-cadherin+ and EpCAM+) and cells displaying a fibroblastic morphology and exhibiting mesenchymal markers (vimentin+) were found to coexist in HME-shp53-RAS cells (Figure S7C). Epithelial and mesenchymal cell subpopulations were next sorted on the basis of their differential antigenic phenotypes (EpCAM+ and EpCAM− respectively) (Figure S8). The phenotypes of the epithelial and mesenchymal cell populations were confirmed by assessing the expression of additional epithelial markers (β-catenin, E-cadherin, ZO-1, and occludin) and mesenchymal markers (fibronectin and vimentin) by immunofluorescence staining and western blotting (Figure S9). The sorted mesenchymal-cell subpopulations specifically displayed EMT-associated features such as motility, invasiveness, and a stellate phenotype when cultured in 3D (Figure 7). Gene profile analysis classified these mesenchymal cells as claudin-low/basal B, while epithelial HME-shp53-RAS cells were classified as basal-like/basal A (Figure 4 and Figure S3). Although epithelial and mesenchymal HMEC derivatives exhibited similar H-RASG12V expression levels (Figure S10), only mesenchymal cells grew in soft agar and gave rise to tumor formation, within three months, when homotopically xenografted in nude mice (6 of 7 mice, Figure 7).

These observations demonstrated that EMT commitment fosters malignant transformation of human mammary epithelial cells deprived of functional p53 - and RB-dependent pathways. To further confirm this hypothesis sorted epithelial HME-shp53-RAS cells were treated with TGFβ. Exposure to this EMT-inducing cytokine triggered a pronounced shift from epithelial to mesenchymal markers, associated with induction of ZEB1 (200-fold) and ZEB2 (10-fold) (data not shown) and with a dramatic gain in anchorage-independent growth properties (Figure S11). Importantly, forced expression of ZEB1 in sorted epithelial HME-shp53-RAS cells was sufficient to mimic TGFβ exposure, promoting both EMT and cell transformation (Figure S12).
TWIST1 and ZEB1/2 transcription factors downregulate the oncosuppressive PP2A activity
Our observations strongly suggested that, beyond the inhibition of the p53 - and RB-dependent pathways, EMT inducers display additional, as yet unidentified oncogenic activities. It has been previously shown that, in vitro, transformation of normal human epithelial cells, including mammary epithelial cells, requires disruption of the telomere maintenance system and dysregulation of at least four key signaling pathways: activation of the RAS-dependent pathway and inhibition of the p53-, RB-, and protein phosphatase 2A-dependent pathways [28]–[30]. We thus endeavored to analyze the effects of EMT commitment on protein phosphatase 2A (PP2A) activity. PP2A is a ubiquitously expressed serine/threonine phosphatase accounting, with protein phosphatase 1 (PP1), for 90% of all the serine/threonine phosphatase activity in the cell [32]. By using a peptide substrate (synthetic phosphothreonine peptide RRA(pT)VA) compatible with the phosphatase activity of PP2A but not with that of PP1 and by employing experimental conditions ensuring the specificity of PP2A activity (see Materials and Methods), we found sorted mesenchymal HME-shp53-RAS cells to exhibit lower phosphatase activity than sorted epithelial HME-shp53-RAS cells (Figure 8). More importantly, expression of either TWIST1, ZEB1, or ZEB2 in HME cells was sufficient to trigger significant (2-fold) downregulation of serine/threonine phosphatase activity, revealing a novel oncogenic feature of these proteins. This downregulation was similar to that observed in immortalized HMECs transformed with H-RASV12 and the SV40 large T and small t antigens (HMLER cells; Figure 8), the small t antigen being known to inhibit PP2A activity [33]. Notably, claudin-low HME-ZEB1-RAS cells exhibited 4-fold lower phosphatase activity than immortalized HMECs (Figure 8).

Discussion
Whereas TWIST1 has been convincingly implicated in the metastatic dissemination of breast cancer cells, these data underscore the importance of EMT-inducing transcription factors in driving mammary carcinogenesis, with a dual role in cell transformation and dedifferentiation. Tumor development has been portrayed as a multistep processes with a progressive acquisition of genetic and epigenetic abnormalities providing cells with biological capabilities such as sustained proliferation, replicative immortality, survival advantages, angiogenesis and, in some cases, invasive growth and metastasis [34]. According to the Darwinian model of cancer development, each of these acquired traits confers a distinct selective advantage, originating successive waves of clonal expansion that drive tumor progression. It is well known that this complex and time consuming process requires abrogation of several oncosuppressive barriers. In epithelial cells, including mammary epithelial cells, these barriers comprise the p53-, RB - and PP2A-dependent pathways [28]–[30]. We and others have previously demonstrated that TWIST and ZEB transcriptions factors were capable to inhibit p53 - and RB - dependent pathways [10]–[12]. Remarkably, our observations reveal that activation of these factors in HMECs also affects PP2A phosphatase activity. Considerable evidence highlights the tumor-suppressor functions of this serine/threonine phosphatase. For example, it has been shown in vitro that the transforming ability of the SV40 small t antigen requires interactions with PP2A and downregulation of its activity [34]–[36]. In vivo, mutations affecting different components of the PP2A holoenzyme complex have been identified in a variety of human malignancies and, in mouse models, mutation of PP2A favors tumorigenesis [37]. Loss of PP2A during cell transformation triggers multiple events, such as upregulation of kinases involved in mitogenic and survival signaling (e.g. AKT and MAPK), stabilization of protooncogenes (e.g. MYC), destabilization of tumor suppressors (e.g. p53 and RB), and loss of proapoptotic signaling pathways (e.g. BAD) [32]. Modulation of downstream components of the RAS signaling pathway by PP2A might be of particular significance in our model, as the ability of PP2A to antagonize the oncogenic properties of RAS by dephosphorylating crucial downstream effectors such as c-MYC and AKT makes its downregulation a prerequisite to RAS-induced malignant transformation. The modulation of PP2A activity by EMT inducers might thus be an important mechanism underlying the deleterious cooperation of these factors with oncogenic RAS in cell transformation. The inhibition of PP2A activity by EMT inducers might also be relevant during embryogenesis, as PP2A appears as a negative regulator of the WNT signaling cascade [38] which is required for several crucial steps in early development. Further studies are needed, however, to better characterize the mechanisms involved in this regulatory process, as PP2A represents a complex family of holoenzyme complexes known to display different activities and to exhibit diverse substrate specificities [39].
Given the importance of p53-, RB - and PP2A-dependent protective barriers against tumorigenesis and their role in regulating cell differentiation and self-renewal [40], aberrant reactivation of EMT inducers might profoundly affect the multistep nature of tumorigenesis by increasing cell plasticity and leapfrogging the mutation bottleneck toward tumor progression. This view is supported by our in vitro transformation assays demonstrating that, upon a single mitogenic activation, forced expression of either TWIST1 or ZEB1/2 is sufficient to trigger malignant conversion of immortalized human mammary epithelial cells (hTERT-HMECs). It is also consistent with the observed rapid and repeated appearance of multifocal breast carcinomas in WAP-Cre;K-rasG12D;Twist1 mice. It is further supported by the work by Phuoc T. Tran and colleagues, who demonstrated in an elegant inducible transgenic mouse model that TWIST1 overexpression accelerates K-RAS-induced lung tumorigenesis [41]. Several phenomena associated with tumor initiation, such as inflammation [42], physical constrains (including hydrostatic pressure, shear stress and tension forces) [43], abnormal activation of signaling pathways such as those controlled by WNT, NOTCH, or TGFβ [4], [44], and hyperactivation or RAS-ERK1/2 signaling [45] are known to trigger expression of EMT-promoting factors and could thus induce reactivation of these embryonic transcription factors in early stages of tumor development, as previously observed in animal models [46]. Moreover, beyond the deleterious consequences of aberrant reactivation of EMT inducers in differentiated or committed epithelial cells, the ability of EMT inducers to inhibit key oncosuppressive pathways also implies that embryonic or adult stem cells that normally express these factors are particularly vulnerable to cell transformation.
Cooperation assays demonstrate that activation of EMT-inducing transcription factors such as TWIST1 or ZEB2 is sufficient to make cells highly prone to transformation, even in the absence of a complete mesenchymal morphological shift. In line with this view, immunohistochemical analysis of TWIST1 in human non-invasive breast cancers (ductal carcinomas in situ, DCIS) has revealed frequent overexpression of this EMT inducer within the bulk of the primary lesion, while the cancer cells maintain an epithelial phenotype (Figure S13). EMT is known to be a highly dynamic process giving rise to a series of important changes in cell phenotype, including loss of cell polarity, loss of cell-cell adhesion structures, remodeling of the cytoskeleton, and promotion of cell motility. As recently highlighted by Klymkowsky and Savagner, although the term EMT is generally applied as if it were a single conserved process, EMT-related processes can in fact vary in degree from a transient loss of cell polarity to total reprogramming of the cell [47]. The existence of malignant cells with combined epithelial and mesenchymal characteristics has previously been demonstrated in vivo, in both mouse models of EMT and human tumors [48], [49]. Especially, epithelial cells coexpressing cytokeratins 5/19 and vimentin have been identified by dual immunofluorescence labeling in claudin-low and basal-like breast cancers, two breast cancer subtypes frequently exhibiting overexpression of EMT-inducing transcription factors [13]. Overall, these observations strongly suggest that EMT-promoting factors can exert oncogenic functions in cells retaining an epithelial phenotype, in the total absence of morphological features of EMT, and probably long before initiation of the invasion-metastasis cascade.
Previous in vitro studies using human mammary epithelial cells have revealed a link between EMT, malignant transformation, and acquisition of stem cell properties. For example, the transformation of HMECs by means of a combination of hTERT, SV40 large T and small t antigens, and H-RASG12V (HMLER cells) is associated with both mesenchymal and stem-like features [7], [8], [50]. In the absence of oncogenic RAS, introduction of SV40 T and small t antigens and hTERT into mammosphere-derived HMECs also generates malignant cells exhibiting EMT and stem-like properties [51]. Recent reports further demonstrate in human cancer cell lines that spontaneous EMT or TGFβ/TNFα-mediated EMT generates cells with a claudin-low phenotype [52], [53]. Yet the intrinsic role of EMT inducers was not addressed in these studies. We highlight herein a dual role of these factors in cell transformation and dedifferentiation. Remarkably, in the context of a very few genetic events, the aberrant activation of an EMT inducer can initiate mammary epithelial cell transformation in vitro and in vivo and can drive the growth of undifferentiated tumors exhibiting all the characteristic features of claudin-low tumors, including a malignant phenotype, low expression of tight and adherens junction genes, EMT traits, and stem-cell-like characteristics.
The origin of the different intrinsic subtypes of human breast cancer is a topic of contentious debate and remains ill defined. Recent in vitro observations support the view that both luminal and basal-like breast cancers derive from a common luminal progenitor cell, whereas claudin-low tumors, viewed as the most primitive malignancies, originate from a stem/progenitor cell with inherent stemness properties and metaplastic features [24]–[26]. Others suggest that basal-like and claudin-low tumors arise from transformation of a similar stem cell, but that the claudin-low tumors stay arrested in an undifferentiated state, while basal-like cancer cells divide asymmetrically and give off differentiated progeny arresting at the luminal progenitor state [54]. Our observations pave the way for an alternative model highlighting a dynamic process orchestrated by the activity of EMT-inducing transcription factors. According to this model, aberrant activation of EMT inducers in committed cells (e.g. luminal progenitors) might foster initiation of triple-negative breast tumors and confer basal-like or claudin-low signatures, according to the extent of transdifferentiation. Our model also implies that basal-like tumors might progressively evolve towards a claudin-low phenotype through completion of the EMT process. This view is supported by the histopathology of human metaplastic breast tumors: phenotypically, the acquisition of mesenchymal features can occur at various stages of the disease [55], highlighting the dynamic role of transdifferentiation during tumor development and pointing to the interaction between cancer cells and the microenvironment as a key determinant of tumor phenotype and behavior. It is noteworthy that a model of murine claudin-low tumors has recently been described, involving transplantation of p53-null mammary tissues into the cleared fat pads of wild-type recipients [56]. This observation is consistent with the role of p53 loss in EMT induction [57]–[59] and with the spontaneous generation of mesenchymal cells exhibiting a claudin-low phenotype in HME-shp53-RAS cells. Yet in this model described by Herschkowitz and colleagues, p53-null mouse mammary tumors fell into a variety of molecular groups, also including luminal and basal-like subtypes [56]. WAP-Cre;K-rasG12D;Twist1 mice thus appear as the first mouse model consistently generating claudin-low tumors. These transgenic mice might thus serve as valuable preclinical models for testing both potential therapeutic agents targeting these aggressive neoplasms and potential preventive agents.
Materials and Methods
Constructs and mouse strains
The TWIST1-E12 tethered heterodimer was generated by PCR by fusing the human TWIST1 and E12 proteins using a G3-S2-G2-S-G3-S-G3-S2-G2-S-G3-S-G polylinker as described in [60]. The full-length murine HA-tagged ZEB1 and ZEB2 cDNAs were cloned into the pBabe retroviral construct. The TP53 shRNA (shp53) pRETRO SUPER expression construct has been described in [61].
Animal maintenance and experiments were performed in a specific pathogen free animal facility “AniCan” at the CRCL, Lyon, France in accordance with the animal care guidelines of the European Union and French laws and were validated by the local Animal Ethic Evaluation Committee. The heterozygous knock-in LSL-K-rasG12D mouse strain [16] was crossed with CAG-LSL-(Myc)-Twist1 mice (FVB background) [14]. Both TWIST1 monomer and T1-E12 dimers were used producing similar results [60]. K-rasG12D;Twist1 offspring were subsequently crossed with mice carrying the Cre recombinase under the control of the Mouse Mammary Tumor Virus or Whey Acidic Protein promoters (MMTV-Cre (B6129F1 background) or WAP-Cre (c57BL/6 background) [17]; purchased from the NCI-MMHCC. Cre (wild type), Cre;K-rasG12D, Cre;Twist1, and Cre;K-rasG12D;Twist1 virgin animals were maintained and monitored at least weekly for tumor incidence. End points were determined based on tumor diameter (>17 mm) or the sick appearance of an animal. Tissues were harvested and either snap frozen in N2(l) or immersed in formalin until pathological analysis. Genotyping of genomic DNA from tails purified using the NucleoSpin Tissue kit (Macherey-Nagel) was performed with primers described in references [14], [16], [17] using REDTaq 2× ReadyMix (Sigma).
Cell culture
Primary human mammary epithelial cells (HMECs) were provided by Lonza. HMEC-derivatives were cultured in 1∶1 Dulbecco's Modified Eagle's Medium (DMEM)/HAMF12 medium (Invitrogen) complemented with 10% FBS (Cambrex), 100 U/ml penicillin-streptomycin (Invitrogen), 2 mM L glutamine (Invitrogen), 10 ng/ml human epidermal growth factor (EGF) (PromoCell), 0.5 µg/ml hydrocortisone (Sigma) and 10 µg/ml insulin (Actrapid).
Three-dimensional cultures consisted in culturing 5×103 cells/well in 2% growth factor reduced Matrigel (BD Biosciences) on top of a 100% matrigel layer. 20 days after seeding, cells were fixed in 3% paraformaldehyde (Sigma), permeabilized in 0.5% Triton 100X (Sigma) in PBS buffer for 10 min. After several washes in PBS, cells were labeled with 1 µg/ml of TRITC-conjugated Phalloïdin P1951 (Sigma) for 45 min. Following washes in PBS, nuclei were stained with Hoechst 5 µg/ml for 10 min and mounted with Fluoromount-G (SouthernBiotech).
For mammosphere formation, after filtration through a 30 µm pore filter, single-cells were plated at a density of 105 cells/ml in Corning 3261 ultra-low attachment culture dishes. Primary cell spheres were enzymatically dissociated with 0.05% trypsin for 15 min at 37°C to obtain single-cell suspension. The ability to generate mammospheres was defined after three consecutive passages.
Treatment with TGFβ was performed with 2.5 ng/ml of the recombinant cytokine (Peprotech) for a three week period.
Cell distribution was performed using the FITC-EpCAM VU-1D9 (Stem Cell), the FITC-CD44 G44-26 (BD Pharmingen) and the PE-CD24 ML5 (BD Pharmingen) monoclonal antibodies, the FACScan Calibur (Becton Dickinson) and analyzed using the FlowJo software.
Matrigel invasion assays
Matrigel (BD Biosciences) was added to the wells of an eight-well Labtek chamber in a volume of 300 µl/well. A Matrigel plug of about 1 mm diameter was removed. The hole was successively filled with 105 cells and 100 µl of Matrigel. Appropriate growth medium was added on top. Cultures were analyzed after 24 h (Figure 7) or 72 h (Figure S11). Areas of migration were visualized using an Olympus IX50 (NA 0.075). Samples were performed in duplicate.
Transwell migration assay
5×104 cells were placed in the upper chamber of an 8 µM Transwells (BD Biosciences). 24 h later, chambers were washed twice with PBS. The filter side of the upper chamber was cleaned with a cotton swab. The membrane was next cut out of the insert. Cells were fixed in methanol and stained with 5% Giemsa 30 min at room temperature.
Retroviral infection
2×106 Phoenix cells were transfected by calcium-phosphate precipitation with 10 µg of retroviral expression vectors. 48 h post-transfection, the supernatant was collected, filtered, supplemented with 5 µg/ml of polybrene (Sigma) and combined with 106 targeted cells for 6 h. Cells were infected twice and selected 48 h post-second infection with puromycin (0.5 µg/ml), neomycin (100 µg/ml) or hygromycin (25 µg/ml).
Soft-agar colony formation assay
To measure anchorage-independent growth, cells were detached with trypsin and resuspended in growth medium. Plates were prepared with a coating of 0.75% low-melting agarose (Lonza) in growth medium and then overlaid with a suspension of cells in 0.45% low-melting agarose (5×104 cells/well). Plates were incubated for 3 weeks at 37°C and colonies were counted under microscope. Experiments were performed in triplicate.
Mouse injection
Eight-week old female athymic Swiss nude mice (C. River laboratories) were X-irradiated (4 Gy) prior to injection. Single cell suspensions, (5×106 HMEC derivatives resuspended in a PBS-Matrigel (1/1) mixture) were injected into the fat pad of a mammary gland. Tumor incidence was monitored up to 90 days post-injection. Animals were allowed to form tumors up to 1.5 cm in diameter, at which point animals were euthanized. Each tumor was dissected, fixed in paraformaldehyde and processed for histopathology examination.
Immunoblot analysis
Cells were washed twice with phosphate buffered saline (PBS) containing CaCl2 and then lysed in a 100 mM NaCl, 1% NP40, 0.1% SDS, 50 mM Tris pH 8 RIPA buffer supplemented with a complete protease inhibitor cocktail (Roche). Protein expression was examined by western blot using anti-E-cadherin clone 36 (Becton Dickinson), anti-β-catenin clone 14 (Becton Dickinson), anti-fibronectin clone 10 (Becton Dickinson), anti-vimentin clone V9 (Dako), anti-N-cadherin clone 32 (Becton Dickinson), anti-occludin clone OC-3F10 (Zymed Laboratories), anti-β-actin clone AC-15 (Sigma), anti-HA clone 11 (BabCO), anti-TWIST Twist2C1a (Abcam) monoclonal antibodies and a rabbit polyclonal anti-H-RAS clone C20 (Santa Cruz) for primary detection. Horseradish peroxidase-conjugated rabbit anti-mouse and goat anti-rabbit polyclonal antibodies (Dako) were used as secondary antibodies. Western blots were revealed using an ECL detection kit (Amersham) or a western-blotting Luminol reagent (Santa Cruz).
Immunofluorescence
104 cells were seeded on 8-well Lab-TekII chamber slide, fixed in 3% paraformaldehyde (Sigma) and permeabilized in 0.1% Triton 100X (Sigma) in PBS buffer at room temperature for 10 min. The cells were then washed 3 times with PBS and incubated with a blocking solution (10% horse serum in PBS). The cells were then incubated with the anti-E-cadherin clone 36 (Becton Dickinson) or the anti-vimentin clone V9 (Dako) primary antibodies overnight at 4°C. Phalloidin labeling was performed by incubating cells with 1 µg/ml of TRITC-conjugated Phalloïdin P1951 (Sigma) for 30 min. Following extensive washes in PBS, nuclei were stained with Hoechst 5 µg/ml for 10 min and mounted with Fluoromount-G (SouthernBiotech). All matched samples were photographed (control and test) using an immunofluorescence microscope (Leica) and identical exposure times.
Immunohistochemistry on murine tumors
The immunohistochemical study was performed on three microns deparaffinized sections, using the avidin-biotin-peroxidase complex technique (LSAB universal, Dako), after 15 min heat-induced antigen retrieval in 10 mM citrate buffer, pH 6. The primary anti-E-cadherin clone 36 diluted at 1/500 (Becton Dickinson), anti-vimentin clone V9 diluted at 1/200 (Dako) and anti-c-MYC A14 at 1/100 (Santa-Cruz Biotechnology) antibodies were applied 60 min at room temperature.
Immunohistochemistry on xenograft tumors
Paraffin embedded tumors were serially sectioned at a thickness of 4 µm. After deparaffinisation and rehydration, endogenous peroxidases were blocked by incubating the slides in 5% hydrogen peroxide in sterile water. For heat induced antigen retrieval, tissue sections were boiled at 97°C for 40 min either in a 10 mM citrate buffer pH 6 (when anti-cytokeratin and anti-vimentin antibodies were used) or in buffer pH 7 (Dako) (for the anti-E-cadherin antibody) clone 36 (BD Biosciences). Slides were then incubated with the monoclonal pancytokeratin clone AE1/AE3, (Dako), the polyclonal anti-vimentin SC7557 (Santa Cruz) or the monoclonal anti-E-cadherin clone 36 (BD Biosciences) primary antibodies or a non-immune serum used as a negative control, for 1 h at room temperature. Slides were rinsed in phosphate buffered saline, and then incubated with a biotinylated secondary antibody bound to a streptavidin peroxidase conjugate (LSAB+ kit, Dako).
Microarray analysis
Microarray processing and data analysis as well as procedures to classify human cell lines and mammary tumors are described in detail in Text S1.
Protein phosphatase activity
Cells were lysed in a 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1 mM EGTA, 0.3% CHAPS lysis buffer supplemented with a protease inhibitor cocktail (Roche) and cleared by centrifugation. The PP2A activity was assessed using the “Serine/Threonine Phosphatase Assay System” (Promega) according to the manufacture instruction. Briefly, the cleared cell lysate was filtered through a Sephadex G25 column to remove free phosphate. Protein concentration was determined using the Bradford method. 5 µg of cell protein was incubated in presence of the RRA(pT)VA substrate in a 250 mM imidazole pH 7.2, 1 mM EGTA, 2 mM EDTA, 0.1% β-mercaptoethanol, 0.5 mg/ml BSA PP2A-specific reaction buffer at 25°C for 30 min. After incubation with 50 µl of molybdate dye/additive at 25°C for 30 min, optical density was measured at 620 nm. All determinations were performed in triplicate and the absorbance of the reactions was corrected by determining the absorbance of control reactions without phosphoprotein substrate. The PP2A activity was performed in presence of absence of 5 nM of okadoic acid to confirm the specificity of these reaction conditions. The amount of phosphate released (pmol) was calculated from a standard curve (0–2000 pmol) and was normalized with respect to HMEC-hTERT cells.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. YangJManiSADonaherJLRamaswamySItzyksonRA 2004 Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117 927 939
2. ThieryJPAcloqueHHuangRYNietoMA 2009 Epithelial-mesenchymal transitions in development and disease. Cell 139 871 890
3. AcloqueHAdamsMSFishwickKBronner-FraserMNietoMA 2009 Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 119 1438 1449
4. PeinadoHOlmedaDCanoA 2007 Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7 415 428
5. YangJWeinbergRA 2008 Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14 818 829
6. AnsieauSMorelAPHinkalGBastidJPuisieuxA 2010 TWISTing an embryonic transcription factor into an oncoprotein. Oncogene 29 3173 3184
7. MorelAPLievreMThomasCHinkalGAnsieauS 2008 Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3 e2888 doi:10.1371/journal.pone.0002888
8. ManiSAGuoWLiaoMJEatonENAyyananA 2008 The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133 704 715
9. VesunaFLisokAKimbleBRamanV 2009 Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 11 1318 1328
10. Valsesia-WittmannSMagdeleineMDupasquierSGarinEJallasAC 2004 Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 6 625 630
11. AnsieauSBastidJDoreauAMorelAPBouchetBP 2008 Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 14 79 89
12. OhashiSNatsuizakaMWongGSMichayliraCZGruganKD 2010 Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res 70 4174 4184
13. PratAParkerJSKarginovaOFanCLivasyC 2010 Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 12 R68
14. ConnerneyJAndreevaVLeshemYMercadoMADowellK 2008 Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev Biol 318 323 334
15. ClarkGJDerCJ 1995 Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res Treat 35 133 144
16. JacksonELWillisNMercerKBronsonRTCrowleyD 2001 Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15 3243 3248
17. WagnerKUWallRJSt-OngeLGrussPWynshaw-BorisA 1997 Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res 25 4323 4330
18. WagnerKUMcAllisterKWardTDavisBWisemanR 2001 Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res 10 545 553
19. RobinsonGWMcKnightRASmithGHHennighausenL 1995 Mammary epithelial cells undergo secretory differentiation in cycling virgins but require pregnancy for the establishment of terminal differentiation. Development 121 2079 2090
20. MiyoshiKShillingfordJMLe ProvostFGounariFBronsonR 2002 Activation of beta -catenin signaling in differentiated mammary secretory cells induces transdifferentiation into epidermis and squamous metaplasias. Proc Natl Acad Sci U S A 99 219 224
21. NeveRMChinKFridlyandJYehJBaehnerFL 2006 A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10 515 527
22. HerschkowitzJISiminKWeigmanVJMikaelianIUsaryJ 2007 Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 8 R76
23. TaubeJHHerschkowitzJIKomurovKZhouAYGuptaS 2010 Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 107 15449 15454
24. HennessyBTGonzalez-AnguloAMStemke-HaleKGilcreaseMZKrishnamurthyS 2009 Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res 69 4116 4124
25. LimEVaillantFWuDForrestNCPalB 2009 Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 15 907 913
26. KellerPJArendtLMSkibinskiALogvinenkoTKlebbaI 2011 Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci U S A 109 2772 2777
27. HoeflichKPO'BrienCBoydZCavetGGuerreroS 2009 In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res 15 4649 4664
28. HahnWCCounterCMLundbergASBeijersbergenRLBrooksMW 1999 Creation of human tumour cells with defined genetic elements. Nature 400 464 468
29. ElenbaasBSpirioLKoernerFFlemingMDZimonjicDB 2001 Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 15 50 65
30. HahnWCDessainSKBrooksMWKingJEElenbaasB 2002 Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol 22 2111 2123
31. WongDJFosterSAGallowayDAReidBJ 1999 Progressive region-specific de novo methylation of the p16 CpG island in primary human mammary epithelial cell strains during escape from M(0) growth arrest. Mol Cell Biol 19 5642 5651
32. EichhornPJCreyghtonMPBernardsR 2009 Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta 1795 1 15
33. ChenWPossematoRCampbellKTPlattnerCAPallasDC 2004 Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 5 127 136
34. HanahanDWeinbergRA 2011 Hallmarks of cancer: the next generation. Cell 144 646 674
35. PallasDCShahrikLKMartinBLJaspersSMillerTB 1990 Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60 167 176
36. CampbellKSAugerKRHemmingsBARobertsTMPallasDC 1995 Identification of regions in polyomavirus middle T and small t antigens important for association with protein phosphatase 2A. J Virol 69 3721 3728
37. WalterGRuedigerR 2012 Mouse model for probing tumor suppressor activity of protein phosphatase 2A in diverse signaling pathways. Cell Cycle 11 451 459
38. SeelingJMMillerJRGilRMoonRTWhiteR 1999 Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science 283 2089 2091
39. JanssensVGorisJ 2001 Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353 417 439
40. SpikeBTWahlGM 2011 p53, Stem Cells, and Reprogramming: Tumor Suppression beyond Guarding the Genome. Genes Cancer 2 404 419
41. TranPTShroffEHBurnsTFThiyagarajanSDasST 2012 Twist1 Suppresses Senescence Programs and Thereby Accelerates and Maintains Mutant Kras-Induced Lung Tumorigenesis. PLoS Genet 8 e1002650 doi:10.1371/journal.pgen.1002650
42. Lopez-NovoaJMNietoMA 2009 Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med 1 303 314
43. DespratNSupattoWPouillePABeaurepaireEFargeE 2008 Tissue deformation modulates twist expression to determine anterior midgut differentiation in Drosophila embryos. Dev Cell 15 470 477
44. HoriguchiKShirakiharaTNakanoAImamuraTMiyazonoK 2009 Role of Ras signaling in the induction of snail by transforming growth factor-beta. J Biol Chem 284 245 253
45. ShinSDimitriCAYoonSODowdleWBlenisJ 2010 ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 38 114 127
46. HusemannYGeiglJBSchubertFMusianiPMeyerM 2008 Systemic spread is an early step in breast cancer. Cancer Cell 13 58 68
47. KlymkowskyMWSavagnerP 2009 Epithelial-mesenchymal transition: a cancer researcher's conceptual friend and foe. Am J Pathol 174 1588 1593
48. DamontePGreggJPBorowskyADKeisterBACardiffRD 2007 EMT tumorigenesis in the mouse mammary gland. Lab Invest 87 1218 1226
49. SantistebanMReimanJMAsieduMKBehrensMDNassarA 2009 Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res 69 2887 2895
50. ChafferCLBrueckmannIScheelCKaestliAJWigginsPA 2011 Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 108 7950 7955
51. ParanjapeANMandalTMukherjeeGKumarMVSenguptaK 2011 Introduction of SV40ER and hTERT into mammospheres generates breast cancer cells with stem cell properties. Oncogene 30 1896 1909
52. AsieduMKIngleJNBehrensMDRadiskyDCKnutsonKL 2011 TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res 71 4707 4719
53. SarrioDFranklinCKMackayAReis-FilhoJSIsackeCM 2012 Epithelial and mesenchymal subpopulations within normal basal breast cell lines exhibit distinct stem cell/progenitor properties. Stem Cells 30 292 303
54. PratAPerouCM 2011 Deconstructing the molecular portraits of breast cancer. Mol Oncol 5 5 23
55. van DeurzenCHLeeAHGillMSMenke-PluijmersMBJagerA 2011 Metaplastic breast carcinoma: tumour histogenesis or dedifferentiation? J Pathol 224 434 437
56. HerschkowitzJIZhaoWZhangMUsaryJMurrowG 2012 Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci U S A 109 2778 2783
57. ChangCJChaoCHXiaWYangJYXiongY 2011 p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol 13 317 323
58. KimTVeroneseAPichiorriFLeeTJJeonYJ 2011 p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med 208 875 883
59. SiemensHJackstadtRHuntenSKallerMMenssenA 2011 miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 10 4256 4271
60. ConnerneyJAndreevaVLeshemYMuentenerCMercadoMA 2006 Twist1 dimer selection regulates cranial suture patterning and fusion. Dev Dyn 235 1345 1357
61. BrummelkampTRBernardsRAgamiR 2002 A system for stable expression of short interfering RNAs in mammalian cells. Science 296 550 553
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Inactivation of a Novel FGF23 Regulator, FAM20C, Leads to Hypophosphatemic Rickets in Mice
- Genome-Wide Association of Pericardial Fat Identifies a Unique Locus for Ectopic Fat
- Slowing Replication in Preparation for Reduction
- An Essential Role for Katanin p80 and Microtubule Severing in Male Gamete Production
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy