
Inactivation of a Novel FGF23 Regulator, FAM20C, Leads to Hypophosphatemic Rickets in Mice
Family with sequence similarity 20,-member C (FAM20C) is highly expressed in the mineralized tissues of mammals. Genetic studies showed that the loss-of-function mutations in FAM20C were associated with human lethal osteosclerotic bone dysplasia (Raine Syndrome), implying an inhibitory role of this molecule in bone formation. However, in vitro gain - and loss-of-function studies suggested that FAM20C promotes the differentiation and mineralization of mouse mesenchymal cells and odontoblasts. Recently, we generated Fam20c conditional knockout (cKO) mice in which Fam20c was globally inactivated (by crossbreeding with Sox2-Cre mice) or inactivated specifically in the mineralized tissues (by crossbreeding with 3.6 kb Col 1a1-Cre mice). Fam20c transgenic mice were also generated and crossbred with Fam20c cKO mice to introduce the transgene in the knockout background. In vitro gain - and loss-of-function were examined by adding recombinant FAM20C to MC3T3-E1 cells and by lentiviral shRNA–mediated knockdown of FAM20C in human and mouse osteogenic cell lines. Surprisingly, both the global and mineralized tissue-specific cKO mice developed hypophosphatemic rickets (but not osteosclerosis), along with a significant downregulation of osteoblast differentiation markers and a dramatic elevation of fibroblast growth factor 23 (FGF23) in the serum and bone. The mice expressing the Fam20c transgene in the wild-type background showed no abnormalities, while the expression of the Fam20c transgene fully rescued the skeletal defects in the cKO mice. Recombinant FAM20C promoted the differentiation and mineralization of MC3T3-E1 cells. Knockdown of FAM20C led to a remarkable downregulation of DMP1, along with a significant upregulation of FGF23 in both human and mouse osteogenic cell lines. These results indicate that FAM20C is a bone formation “promoter” but not an “inhibitor” in mouse osteogenesis. We conclude that FAM20C may regulate osteogenesis through its direct role in facilitating osteoblast differentiation and its systemic regulation of phosphate homeostasis via the mediation of FGF23.
Published in the journal:
. PLoS Genet 8(5): e32767. doi:10.1371/journal.pgen.1002708
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002708
Summary
Family with sequence similarity 20,-member C (FAM20C) is highly expressed in the mineralized tissues of mammals. Genetic studies showed that the loss-of-function mutations in FAM20C were associated with human lethal osteosclerotic bone dysplasia (Raine Syndrome), implying an inhibitory role of this molecule in bone formation. However, in vitro gain - and loss-of-function studies suggested that FAM20C promotes the differentiation and mineralization of mouse mesenchymal cells and odontoblasts. Recently, we generated Fam20c conditional knockout (cKO) mice in which Fam20c was globally inactivated (by crossbreeding with Sox2-Cre mice) or inactivated specifically in the mineralized tissues (by crossbreeding with 3.6 kb Col 1a1-Cre mice). Fam20c transgenic mice were also generated and crossbred with Fam20c cKO mice to introduce the transgene in the knockout background. In vitro gain - and loss-of-function were examined by adding recombinant FAM20C to MC3T3-E1 cells and by lentiviral shRNA–mediated knockdown of FAM20C in human and mouse osteogenic cell lines. Surprisingly, both the global and mineralized tissue-specific cKO mice developed hypophosphatemic rickets (but not osteosclerosis), along with a significant downregulation of osteoblast differentiation markers and a dramatic elevation of fibroblast growth factor 23 (FGF23) in the serum and bone. The mice expressing the Fam20c transgene in the wild-type background showed no abnormalities, while the expression of the Fam20c transgene fully rescued the skeletal defects in the cKO mice. Recombinant FAM20C promoted the differentiation and mineralization of MC3T3-E1 cells. Knockdown of FAM20C led to a remarkable downregulation of DMP1, along with a significant upregulation of FGF23 in both human and mouse osteogenic cell lines. These results indicate that FAM20C is a bone formation “promoter” but not an “inhibitor” in mouse osteogenesis. We conclude that FAM20C may regulate osteogenesis through its direct role in facilitating osteoblast differentiation and its systemic regulation of phosphate homeostasis via the mediation of FGF23.
Introduction
FAM20C is a member of the “family with sequence similarity 20”. In mammals, this evolutionarily conserved protein family consists of three members: FAM20A, FAM20B and FAM20C. FAM20A was originally observed in the lung and liver and displays obvious differential expression in hematopoietic cells undergoing myeloid differentiation [1]. A viral mRNA transgenic mouse line with an accidental deletion of a 58-kb fragment in chromosome 11E1 encompassing part of the Fam20a gene and its upstream region showed growth disorder [2]. Recently, it was found that FAM20A is also expressed in ameloblasts and its mutations are associated with human amelogenesis imperfecta and gingival hyperplasia syndrome [3]. More recently, FAM20B was shown to be involved in cartilage matrix production and the ultimate regulation on the timing of skeletal development [4]. FAM20C is highly expressed in the mineralized tissues and identified as the causal gene for lethal osteosclerotic bone dysplasia (Raine Syndrome, OMIM 259775) [1], [5]–[7]. Given the high level of conservation in the C-terminal domains among the three FAM20 members and the their roles observed in the hard tissues, it is tempting to speculate that this evolutionarily conserved family might be a new cluster of molecules performing important functions in the development of the mineralized tissues.
Mouse FAM20C, also known as “dentin matrix protein 4” (DMP4) [5], contains 579 amino acid residues, including a putative 26-amino acid signal peptide at the N-terminus. A C-terminal region of approximately 350 amino acids (corresponding to residue218-residue569 in the mouse FAM20C sequence) has been named the “conserved C-terminal domain” (CCD), which is highly conserved among different species [1].
In a previous study, we systematically analyzed the expression and distribution of FAM20C in mouse bone and tooth using in situ hybridization (ISH) and immunohistochemistry (IHC) methods [7], which showed that FAM20C was highly expressed in the mineralized tissues; it was detected in the osteoblasts/osteocytes, odontoblasts, ameloblasts, and cementoblasts, as well as in the matrices of bone, dentin, and enamel. FAM20C was also detected in the epithelium of early-stage tooth germs and in the chondrogenic cells of long bones. The high expression levels of FAM20C in the mineralized tissues strongly suggest that it may play an important role in the formation and/or mineralization of these tissues.
Hao et al. showed that overexpression of mouse FAM20C accelerated the odontoblast differentiation process and silencing this molecule by siRNA inhibited cell differentiation, implying that this protein may be a factor promoting odontoblast differentiation [5]. Subsequently, Simpson et al. reported that the loss-of-function mutations in the FAM20C gene were associated with lethal/non-lethal osteosclerotic bone dysplasia (Raine Syndrome) [6], [8], an autosomal recessive disorder characterized by a generalized increase in the density of all bones; these data indicated that FAM20C might be a down-regulator of biomineralization, which apparently contradicts the mineralization-promoting properties of FAM20C observed by Hao et al.
In this study, we sought to determine the biological functions of FAM20C via generation and characterization of Fam20c conditional knockout (cKO) mice. Our data showed remarkable skeletal defects, along with a significant reduction of serum phosphate and a dramatic elevation of serum fibroblast growth factor 23 (FGF23) in the homozygous Fam20c cKO mice. The phenotypic profiles of the Fam20c-deficient mice resemble those of hereditary hypophosphatemic rickets in humans and rodents resulting from mutations in molecules affecting the regulation of FGF23 [9]–[15].
Results
Validation of FAM20C inactivation in conditional knockout mice
The mouse Fam20c gene consists of 10 exons and spans approximately 55-kb. To generate a conditional knockout allele for Fam20c, we constructed a targeting vector with loxP sites floxing exons 6∼9 which are highly conserved across species (Figure 1A); a number of mutations were identified in this region of the human FAM20C gene in patients with lethal osteosclerotic bone dysplasia [6]. The correct targeting events were confirmed by polymerase chain reaction (PCR) screening, and the presence of 5′ and 3′ loxP sites was determined by PCR product sequencing. Two correctly targeted ES cell clones were identified (Figure 1B, Clones 286 and 297), and both went through germline transmission. F1 Fam20cflox/+ heterozygous mice were crossbred with Sox2 promoter-Cre transgenic mice to generate “Sox2-Cre-Fam20cΔ/Δ” mice, in which exons 6∼9 were removed from both alleles of the Fam20c gene in the epiblasts at post coitum day 6.5 (E6.5). The presence of the floxed alleles and the absence of exons 6∼9 in the null alleles were confirmed by PCR genotyping (Figure 1C).

The lack of Fam20c mRNA in the Sox2-Cre-Fam20cΔ/Δ mice was shown by reverse transcription PCR (RT-PCR) performed with two sets of primers using mRNA extracted from the long bones (Figure 2A), as well as by in situ hybridization (ISH) carried out on the long bones (Figure 2B). The lack of FAM20C protein was determined by immunohistochemistry (IHC) analyses (Figure 2C) performed on the long bones using an affinity-purified anti-FAM20C polyclonal antibody [7].

Both male and female Sox2-Cre-Fam20cΔ/Δ (homozygous cKO) mice are infertile, while the Sox2-Cre-Fam20cΔ/+ (heterozygous cKO) mice have normal fertility. The Fam20cflox/flox mice and the heterozygous cKO mice did not demonstrate any phenotypic changes compared with their wild type (WT) littermates (data not shown), while the homozygous cKO mice displayed remarkable skeletal defects, indicating that the haploinsufficiency of Fam20c has no significant effects on the bone formation. We also bred the Fam20cflox/flox mice with the 3.6 kb Col 1a1-Cre mice to generate Col1a1-Cre-Fam20cΔ/Δ mice, which displayed skeletal defects similar to those observed in the Sox2-Cre-Fam20cΔ/Δ mice. In this report, we described in detail the analyses of phenotypic changes in the Sox2-Cre-Fam20cΔ/Δ mice while the X-ray and histology data of the long bone from the Col1a1-Cre-Fam20cΔ/Δ mice were included in one set of the figures to show the similarity between the global and mineralized tissue-specific cKO mice. The data regarding the Fam20c cKO mice refer to the analyses of the Sox2-Cre-Fam20cΔ/Δ mice unless otherwise stated.
Inactivation of FAM20C leads to skeletal defects
Inactivation of FAM20C leads to growth retardation
At the gross level, the 4-week-old Sox2-Cre-Fam20cΔ/Δ mice demonstrated prominent dwarfism and flat faces (Figure 3A and 3B). The body weight of the Fam20C-deficient mice was significantly lower than that of their WT littermates, indicating retardation in the growth of the mutant mice (Figure 3C). X-ray examination showed that the 5-month-old Sox2-Cre-Fam20cΔ/Δ mice had smaller skeletons, lower levels of mineralization and distorted spines (Figure 3D). The Alizarin Red/Alcian Blue staining of the skeletons from 1-week-old mice revealed that the Sox2-Cre-Fam20cΔ/Δ mice had smaller stature (Figure 4A), delayed ossification in the ribs (Figure 4B), long bones (Figure 4C) and carpus (Figure 4D), along with smaller skulls and delayed cranial suture closure (Figure 4E). These observations indicated that the Fam20c-deficient mice had extensive defects in both endochondral and intramembranous ossifications, resulting in a generalized hypomineralization in the axial skeleton and craniofacial complex.


Inactivation of FAM20C leads to rickets/osteomalacia
Plain X-ray examination did not reveal obvious skeletal abnormalities in the Sox2-Cre-Fam20cΔ/Δ mice at birth (data not shown). However, the bone defects in the Sox2-Cre-Fam20cΔ/Δ mice could be easily identified by X-ray radiography after postnatal 4 days, and the phenotypic changes in the skeleton became more profound as the animals aged. Multiple fractures were often seen in the Sox2-Cre-Fam20cΔ/Δ mice after the age of 3 weeks (data not shown). At the age of 6 weeks, X-ray radiography revealed lower density in the bones of the Sox2-Cre-Fam20cΔ/Δ (global cKO) mice (indicating a lower level of mineralization), along with a delay of the secondary ossification centers in the epiphysis in the long bones (left image in Figure 5A) and vertebrae (left image in Figure 5B). Similar bone defects were observed in the 3.6 kb Col1a1-Cre induced mineralized tissue-specific cKO mice (middle image in Figure 5A). In addition to the delayed ossification, micro-CT (μ-CT) analyses showed increased porosity in the diaphyses and malformed epiphyses of the global cKO mice (Figure 5C and 5D). The appearance of more porous areas in both the outer and inner bone surfaces of the mutant mice seen in the μ-CT radiogram was a result of increased areas of hypomineralization. The μ-CT quantitative analyses showed a significantly lower mineral Apparent Density and Material Density in the tibia midshaft region of the 3-week - and 6-week-old global cKO mice (Table 1).


Histological analyses with H&E staining showed that the long bone of the global cKO mice had a thinner cortical bone along with a reduced proliferative zone and an enlarged hypertrophic zone in the growth plates (left image in Figure 5E–5G). Similar histological defects were observed in the Col1a1-Cre induced mineralized tissue-specific cKO mice (middle image in Figure 5E–5G). Goldner's Masson Trichrome staining showed that the diaphysis region of the tibia in the Sox2-Cre-Fam20cΔ/Δ mice had more osteoid/hypomineralized areas (stained red) in the cortical bones (Figure 6A and 6B). The broad areas with larger amounts of osteoid/hypomineralized tissues in the Sox2-Cre-Fam20cΔ/Δ showed a remarkable increase in the immunostaining for biglycan (Figure 6C and 6D) [16]. The double fluorochrome labeling analyses showed that the Sox2-Cre-Fam20cΔ/Δ mice had a significantly lower mineral deposition rate compared with their WT littermates (Figure 6E and 6F). Taken together, the phenotypic changes in the skeletons of the global and mineralized tissue-specific cKO mice were consistent with a diagnosis of rickets/osteomalacia.

Inactivation of FAM20C leads to defects in the growth plate
X-ray analyses revealed hypomineralized metaphysis and growth plate in the Sox2-Cre-Fam20cΔ/Δ mice (Figure 5A–5D). Histological examination displayed an altered thickness of the proliferative and hypertrophic zones in the long bones of the Sox2-Cre-Fam20cΔ/Δ mice (Figure 5F). To determine the molecular changes associated with the growth plate defects, we examined the differentiation, proliferation and apoptosis of the chondrocytes in the growth plates. In situ hybridization (ISH) analyses showed that type IIα collagen (differentiation marker of proliferating and mature chondrocytes) and type X collagen (differentiation marker of hypertrophic chondrocytes) were downregulated in the growth plates of the Sox2-Cre-Fam20cΔ/Δ mice (Figure 7A–7D), indicating that both the early stage differentiation and the late-stage maturation of chondrocytes were affected. BrdU labeling revealed that the absolute number of labeled proliferating chondrocytes in the Sox2-Cre-Fam20cΔ/Δ mice was lower (Figure 7E and 7F), but there was no statistical difference in the ratio of BrdU labeled cells to the total number of cells in the proliferative zone between the mutant mice and their WT littermates (data not shown). TUNEL assay showed that the percentage of apoptotic chondrocytes in the hypertrophic zone was reduced by ∼70% in the Sox2-Cre-Fam20cΔ/Δ mice (Figure 7G and 7H), which was consistent with the reports that hypophosphatemia impairs caspase-mediated apoptosis in hypertrophic chondrocytes [17], [18]. The reduction of apoptosis might be responsible for the formation of a thicker growth plate in the cKO mice.
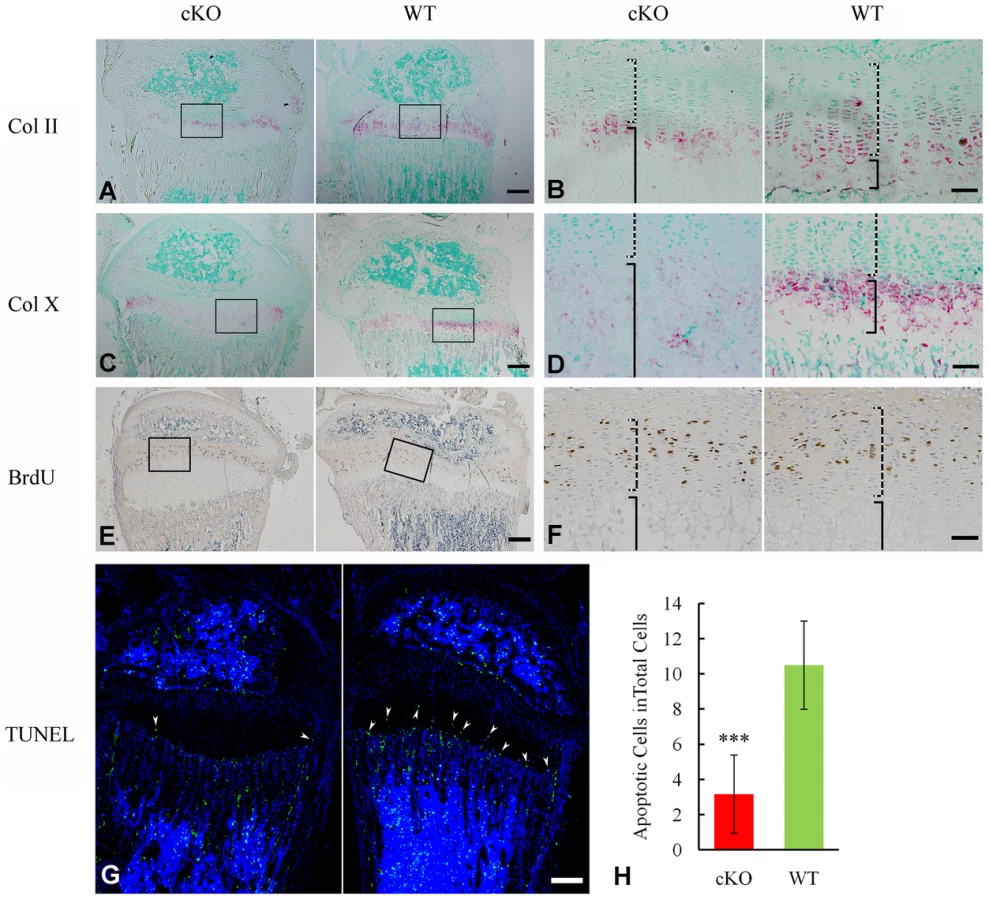
Inactivation of FAM20C leads to cell differentiation defects
The osteocytes in Fam20cΔ/Δ mice lost normal morphology and appeared immature as shown by resin-casted scanning electron microscopy (SEM) analyses (Figure 8A and 8B), indicating a faulty maturation process from osteoblasts to osteocytes. Backscatter SEM analyses revealed periosteocytic lesions (“halo”) surrounding the osteocytes in the Fam20c cKO mouse bone (Figure 8C and 8D). To determine the molecular changes associated with the immaturity of osteoblasts/osteocytes, we examined their terminal differentiation markers: type Ia collagen, dentin matrix protein 1 (DMP1), and osteocalcin (OCN). ISH (Figure 8E–8J), and real-time PCR analyses (Table 2) revealed a significant downregulation of these markers in the Sox2-Cre-Fam20cΔ/Δ mice. Microarray analyses using total RNA extracted from the calvaria of 3-week-old Sox2-Cre-Fam20cΔ/Δ mice and their WT littermates indicated that among the ∼45,000 molecules evaluated, 350 genes were upregulated by over 2.0 folds and 185 were downregulated. Real-time PCR analyses on selected genes confirmed the significant changes in a number of biomineralization regulators and key players in the Wnt and TGF-β signaling pathway associated with cell differentiation (Table 2) [9]–[15], [19]–[27], suggesting an essential role of FAM20C in the differentiation and mineralization of osteogenic cells. Notably, the most striking transcriptional alteration was FGF23 (upregulated by ∼110 folds), a phosphorus regulator mainly produced by osteoblasts/osteocytes [11], [28], [29]. Immunohistochemistry against FGF23 confirmed the dramatic elevation in the bone cells and bone matrix of Fam20c cKO mice (Figure 8K and 8L). The transcript levels of the above genes in the Sox2-Cre-Fam20cΔ/+ (heterozygous cKO) mice showed no difference from the WT mice (data not shown). Given the many similarities among the Fam20c cKO mice, Dmp1 KO mice and Hyp mice, we examined the expression levels of Fam20c in the Dmp1 - and Phex - deficient mice, and the levels of Dmp1 and Phex in the Fam20c cKO mice by real-time PCR analyses. The Fam20c expression was not altered in the Dmp1 KO mice and Hyp mice (data not shown). The expression of Dmp1 was significantly downregulated (Table 2, Figure 8) while that of Phex was not affected (data not shown) in the Fam20c cKO mice. Ectonucleotide pyrophosphatase/phosphodiesterase (Enpp1), another molecule involved in regulating phosphorus homeostasis was slightly downregulated in the bone of the Fam20c cKO mice, but the change (∼1.4 folds) was not statistically significant from the WT (data not shown).


The shRNA knockdown of FAM20C in vitro leads to similar changes in the human and mouse osteogenic cell lines
The bone phenotypes in the Fam20c cKO mice appear opposite to those observed in the patients associated with the human FAM20C mutations [6]. These contradictory results raise the question of whether FAM20C functions differently between the two species. The lentiviral shRNA-mediated “knockdown” of FAM20C in mouse preosteoblasts MC3T3-E1 cells, human Saos-2 cells (osteoblasts isolated from human osteosarcoma) and human mesenchymal stem cells (hMSC) revealed a remarkable downregulation of DMP1 (Figure 9A–9C), along with a significant upregulation of FGF23 in both the human and mouse cell lines (Figure 9D–9F), indicating that FAM20C may function similarly in humans and mice.

Recombinant FAM20C promotes cell differentiation in vitro
To examine the role of FAM20C during osteoblast proliferation and differentiation, recombinant mouse FAM20C protein was generated by insect cells using a Bac-to-Bac baculovirus system. The recombinant FAM20C added to the culture of MC3T3-E1 preosteoblasts promoted the mineral deposition (nodule formation) in a dose-dependent manner (Figure 10A), and significantly enhanced the transcription of DMP1, osteocalcin (OCN), and bone sialoprotein (BSP) (Figure 10B). Adding recombinant FAM20C to MC3TC-E1 cells did not alter the expression of FGF23 and the proliferation rate of the cells at all tested concentrations (data not shown).

Inactivation of FAM20C leads to hypophosphatemia and elevation of serum FGF23
Seeing the significant elevation of FGF23 in the bone cells of Fam20c cKO mice, we performed serum biochemistry analyses in 18-day - and 42-day-old mice (Table 3). The circulating FGF23 level was remarkably elevated in both the 18-day-old (∼200 folds) and 42-day-old (∼60 folds) cKO mice. Accordingly, the serum phosphorus level significantly decreased at both ages (∼2.5 folds in 18-day-old cKO, ∼2 folds in the 42-day-old cKO mice). The circulating PTH level was significantly elevated in both the18-day-old (∼8 folds) and the 42-day-old (∼5 folds) cKO mice. The serum 1,25(OH)2D3 level was significantly reduced (∼2 folds) in the 18-day-old cKO mice, while the serum 1,25(OH)2D3 level in the 42-day-old cKO mice was slightly higher than that of the WT, but the change in the older mice was not statistically significant. The serum calcium level slightly decreased in the 18-day - and 42-day-old cKO mice, but the reduction was not statistically significant from the WT mice. The blood urea nitrogen (BUN) level in cKO mice had no statistic difference from that of the WT mice at both ages, indicating that no renal failure was occurring in the cKO mice. The serum biochemistry results of the Sox2-Cre-Fam20cΔ/+ (heterozygous cKO) mice were not different from their WT literates (data not shown).

The transcriptional levels of renal Klotho and NaPi-2a were slightly lower in the 18-day-old Fam20c cKO mice, and significantly downregulated (∼3 folds) in the 42-day-old cKO mice. The renal 1α-hydroxylase level was significantly reduced (∼2 folds) in the cKO mice at both ages. We also observed remarkable upregulation of renal 24-hydroxylase in the 18-day-old cKO mice (∼30 folds) as well as in the 42-day-old cKO mice (∼7 folds) (Table 4). The transcriptional levels of these genes in the heterozygous Fam20c cKO mice had no difference from their WT littermates (data not shown).

Expression of the Fam20c transgene rescued the defects of Sox2-Cre-Fam20cΔ/Δ mice
Conditional transgenic (cTg) mice expressing the full length FAM20C were generated to test the gain of function in vivo. We obtained 15 lines of cTg mice, and three of them with the transgene expression levels of 4∼8 folds over those of the WT littermates were further analyzed. One line with the highest expression level of the transgene (approximately 8 folds over the WT, Figure 11A) was characterized in detail. No abnormalities were observed in the bone of any of the cTg mice by postnatal 6 weeks (Figure 11B). In addition, by breeding the cTg mice with the Fam20c cKO mice, we obtained mice expressing the transgene in the Fam20c knockout background (designated “Sox2-Cre-Fam20cΔ/Δ-cTg mice”). The Sox2-Cre-Fam20cΔ/Δ-cTg mice had no abnormalities in the skeleton by postnatal six weeks (Figure 11B and 11C), indicating that expressing the transgene fully rescued the defects of the Fam20c-deficient mice. Additionally, IHC staining using the anti-FGF23 antibodies showed that the expression of the Fam20c transgene rescued the altered expression of FGF23 in the Fam20c-deficient bone (Figure 11D). The fact that overexpressing FAM20C in the WT background did not cause defects in the bone, along with the observation that overexpressing the transgene rescued the Fam20c-deficient abnormalities, has provided further evidence that the defects in the hard tissues of the Sox2-Cre-Fam20cΔ/Δ mice resulted from the loss-of-function, and were not due to the gain-of-function.

In summary, the multipronged approaches in this study demonstrated that inactivation of FAM20C in mice led to rickets/osteomalacia, along with altered levels of serum phosphate and FGF23. The manifestations of the Fam20c-deficient mice are consistent with a diagnosis of hypophosphatemic rickets. The Fam20c-deficient cells in the mineralized tissues appeared immature and incapable of forming healthy tissues. It is likely that a combination of cell differentiation failure and hypophosphatemia resulting from the FGF23 excess led to the skeletal defects in the Fam20c-deficient mice.
Discussion
Little is known about FAM20C, a new molecule. In vitro studies have shown that it promotes the differentiation and mineralization processes of undifferentiated mesenchymal cells and odontoblasts [5], whereas human genetic studies suggested that FAM20C might be a down-regulator (inhibitor) of bone formation and/or mineralization [6]. To answer critical questions regarding the biological roles of FAM20C, we generated Fam20c conditional knockout mice, in which exons 6–9 (majority of the conserved CCD region) were ablated. It is worth noting that most of the mutations identified in the patients with lethal osteosclerotic bone dysplasia were in exons 6–9 [6]. The Fam20c conditional knockout mice developed hypophosphatemic rickets but not osteosclerosis. We believe that the abnormalities in the Fam20c cKO mice resulted from the loss-of-function and were not due to the gain-of-function for this protein. This belief is based on the following observations: 1) deleting exons 6–9 (majority of the CCD) in the Fam20c cKO mice was most likely to inactivate this molecule; 2) the inheritance of the phenotypic changes in Fam20c cKO mice occurred in an autosomal recessive trait, while the gain-of-function is usually inherited in an autosomal dominant manner; 3) transgenic mice overexpressing the Fam20c transgene were normal; 4) the overexpression of the Fam20c transgene fully rescued the phenotypic changes in the Fam20c cKO mice; and 5) recombinant FAM20C promoted the differentiation and mineralization of MC3T3-E1 cells in a dose-dependent manner. These data combined with a significant downregulation of osteoblast differentiation markers in cKO mice suggest that FAM20C is essential to the differentiation of mineralizing cells and promotes the formation and mineralization of hard tissues, and thus, inactivation of this molecule leads to differentiation failure of the cells forming these tissues. Additionally, the Fam20c cKO mice developed hypophosphatemia with a remarkable elevation of the serum FGF23 level. We believe that a combination of cell differentiation failure and hypophosphatemia caused by the increase of serum FGF23 led to the skeletal defects in the Fam20c conditional knockout mice.
In 2007, Simpson et al. reported that the mutations of human FAM20C are associated with an osteosclerotic phenotype in some patients [6]. In a later study by the same group [8], Simpson et al. identified FAM20C mutations in two patients whom they believed were suffering from a “different type of Raine Syndrome”; these two patients did not show a generalized increase in bone density, with one case showed “manifestations consistent with a diagnosis of hypophosphatemic rickets”, as the authors stated. The osteosclerotic phenotype in some patients with FAM20C mutations appears opposite to that observed in the Fam20c-deficient mice. These contradictory results raise questions of whether different domains/fragments of FAM20C protein have different functions or if their functions are different between humans and mice.
In previous reports, the human FAM20C mutations in the osteosclerotic patients include point missense mutations and “splicing” mutations [6], [8]. The point mutations were located in different regions of the gene, including the region encoding the N-terminal portion of the protein and that corresponding to the C-terminal part. The 1309G→A mutation (D437N, in exon 7) observed in the “hypophosphatemic rickets”-like patient (Case 1 in [8]) is located between the two missense mutations 1121T→G (L374R, in exon 6) and 1603C→T (R535W, in exon 10) and was very close to a splicing mutation C1322-2A→G (in intron 7). The latter three mutations initially reported by Simpson et al. were associated with a generalized hypermineralization in the patients [6], while the former one was associated with “hypophosphatemic rickets” [8]. The secreted form of mouse FAM20C contains 553 amino acid residues (excluding a putative 26-amino acid signal peptide), and its calculated molecular mass is approximately 63 kDa. In our previous study [7], Western immunoblotting analyses of the culture medium from HEK-293 cells transfected with a pMES construct containing full-length mouse FAM20C cDNA demonstrated a single protein band at approximately 65 kDa, consistent with the expected mass of full-length mouse FAM20C. We did not observe any lower molecular weight protein bands that could be recognized by the anti-FAM20C antibodies. Similar results were documented in the analyses of the mouse C3H10T1/2 cells and MC3T3-E1 cells transfected with the FLAG-tagged FAM20C [5]. These observations indicate that mouse FAM20C may not be proteolytically processed into fragments. Taken together, these human and mouse data do not support the contention that different domains or fragments of FAM20C may perform different functions.
In this study, the lentiviral shRNA-mediated knockdown of FAM20C in the human mesenchymal stem cells and human osteoblasts led to a remarkable downregulation of DMP1, along with a significant upregulation of FGF23 (Figure 9B, 9C, 9E, and 9F). The findings in the human cells are consistent with the results in the shRNA-knockdown of FAM20C in mouse MC3T3-E1 cells (Figure 9A and 9D) and with the observations in the Fam20c conditional knockout bone (Figure 8, Table 2); these results indicate that FAM20C is likely to function similarly in humans and mice. Clearly, more studies are warranted to further clarify the discrepancy between the human and mouse data.
As a growth factor, FGF23 principally functions as a phosphaturic hormone via binding to the Klotho/FGF receptor (FGFR) complexes in the kidney [30], [31]. The binding of FGF23 to FGFR accelerates phosphate excretion into the urine, thereby inducing a negative phosphate balance, which helps maintain the serum phosphate levels in the normal range under physiological conditions (Figure 12). Elevation of the FGF23 plasma level is known to lead to renal phosphate-wasting and hypophosphatemia [11]–[15], [28], [32]–[35]. The main sources of FGF23 are the osteoblasts and osteocytes in the skeleton [11], [28], [29], and a number of studies have shown that inactivating mutations in certain molecules expressed by these bone cells increase the plasma level of FGF23, which leads to hereditary hypophosphatemic rickets [10], [11], [13], [14]. Inactivating mutations in the phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX) cause X-linked hypophosphatemic rickets (OMIM 307800) [10], and loss of DMP1 activity results in autosomal-recessive hypophosphatemic rickets (OMIM 241520) [11], [13]. The phenotypic changes in the Fam20c-conditional knockout mice share many similarities (hypomineralization, elevation of FGF23, hypophosphatemia) with those observed in the PHEX - and DMP1-deficient subjects. As in the cases of PHEX - and DMP1-deficiency, FGF23 was overexpressed in the bones of the Fam20c cKO mice (Figure 8K and 8L, Table 2). The overproduction of FGF23 by the bone cells is likely to be responsible for the elevation of this protein in the serum.
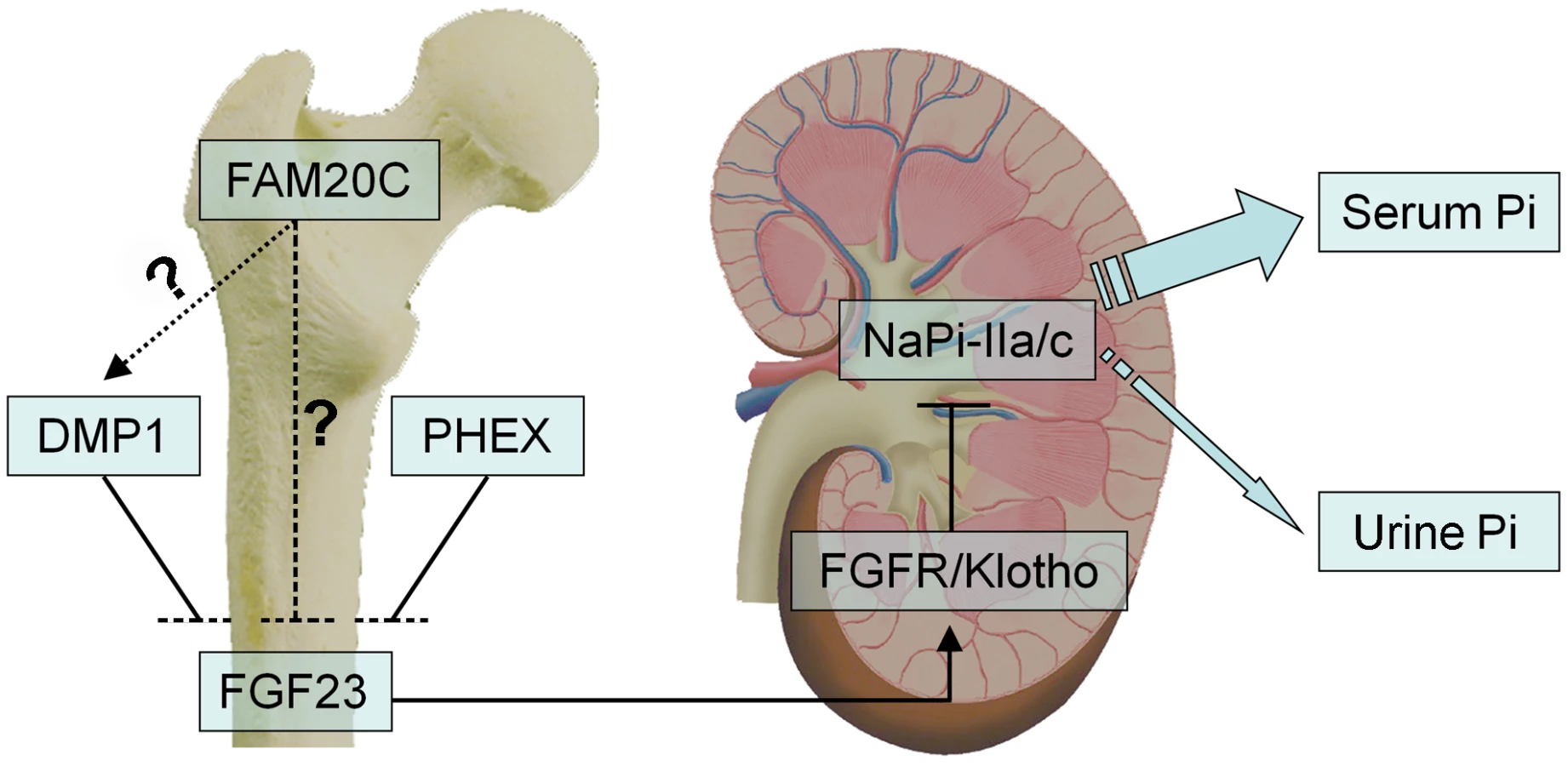
Mutations in four genes, FGF23 itself, PHEX, DMP1 and ENPP1, have been reported to remarkably increase the plasma levels of FGF23, leading to hereditary hypophosphatemic rickets [9]–[11], [13]–[15]. Dmp1-, Phex - and Fam20c-deficient mice shared similarities in osteomalacia, hypophosphatemia and the remarkable elevation of FGF23 in the circulation and skeleton. Interestingly, an alteration of Fam20c expression was not observed in the Dmp1 KO mice or Hyp mice, while remarkable downregulation of Dmp1 (but not Phex) was observed in the Fam20c cKO mice. An up-regulation of Dmp1 was observed in MC3T3-E1 cells treated with recombinant FAM20C. On the other hand, a remarkable down-regulation of Dmp1 was seen in human and mouse osteogenic cell lines treated with FAM20C-shRNA. These findings, along with the similarities of skeletal and serum changes between the Fam20c-deficient and Dmp1-deficient mice, raise the question of whether FAM20C regulates DMP1 (Figure 12). Clearly, further studies are warranted to answer this question. Additionally, there are still a pool of patients with hereditary hypophosphatemic rickets whose etiology is unknown [35], [36], and our discovery that the inactivation of Fam20c in mice results in hypophosphatemic rickets necessitates a consideration of screening FAM20C in such patients.
High level FGF23 reduces the expression of renal vitamin D 1α-hydroxylase and increases the expression of the catabolic 25-hydroxyvitamin D 24-hydroxylase, thus leading to decreased levels of 1,25(OH)2D3 in the serum [15], [37], [38]. In the 18-day-old Fam20c cKO mice, the 1,25(OH)2D3 level was significantly lower, while in the 42-day-old cKO mice the 1,25(OH)2D3 level managed to return to the normal range (or a not significantly higher level). Similar shifts in the serum 1,25(OH)2D3 level with aging have been observed in other hypophosphatemic models such as the Fgf23 transgenic mice, Hyp mice and Dmp1-KO mice that have high levels of circulating FGF23 [11], [12], [39].
While elevation of serum FGF23 reduces the expression of the renal 1α-hydroxylase, hypophosphatemia is normally a stimulator for renal 1α-hydroxylase expression to increase circulating 1,25(OH)2D3 [40]; the stimulating effect of hypophosphatemia on the 1α-hydroxylase expression has been well illustrated in the NaPi2a knockout mice (with lower phosphate and lower FGF23 levels in the serum), in which the serum 1,25(OH)2D3 level is elevated due to the increased 1α-hydroxylase expression stimulated by hypophosphatemia [41]. The Fam20c-cKO mice displayed a decreased level of renal 1α-hydroxylase in the presence of hypophosphatemia, indicating that in these mutant mice, the negative modulation of FGF23 on the expression of the 1α-hydroxylase may outweigh the stimulating effect of hypophosphatemia on the 1α-hydroxylase expression.
In comparison with the Fgf23 transgenic mice, Dmp1-KO mice and Hyp mice, a more remarkable upregulation of 24-hydroxylase was observed in the kidney of the Fam20c-cKO mice, which may be due to the fact that Fam20c-cKO mice have a higher serum FGF23 level than the former three [11], [12], [33]. A higher level of 24-hydroxylase in the 18-day-old cKO mice than that in the 42-day-old Fam20c cKO mice (∼30-fold versus ∼7-fold elevation) may be responsible for the significantly lower serum 1,25(OH)2D3 level in the younger animals. In the older Fam20c cKO mice, a significant reduction of FGF23 co-receptor Klotho, along with a relatively lower serum FGF23 level than that in the younger cKO mice, may attenuate the FGF23-elevation effects on circulating 1,25(OH)2D3 and thus may help maintain a relatively normal serum 1,25(OH)2D3 level in the older cKO mice. More likely, the lower serum level of 1,25(OH)2D3 in the younger Fam20c KO mice may have triggered the overproduction of PTH, as in the cases of Fgf23 transgenic mice and Hyp mice. The secondary hyperparathyroidism may play a critical role for reversing the 1,25(OH)2D3 level to the normal range in the older Fam20c cKO mice [42], [43]. In addition, the elevated PTH may synergize with the high level of serum FGF23 to increase renal phosphate excretion by reducing the expression of NaPi2a in the proximal tubules [38], [44]; a significant reduction of NaPi2a was observed in the kidney of the Fam20c cKO mice. In the end, 1,25(OH)2D3 may maintain a relatively normal level in the older Fam20c cKO mice at the expense of a significant phosphorus wasting.
The skeletal defects of the PHEX - and DMP1-deficient subjects are believed to be due to the combined effects of two factors: 1) the intrinsic defects of the PHEX - and DMP1-deficient cells that prevent them from forming and mineralizing ECM properly and 2) hypophosphatemia [11], [33], [45], [46]. The Fam20c-deficient cells responsible for forming the mineralized tissues appeared immature and showed altered expression levels for molecules associated with cell differentiation. While hypophosphatemia in the Fam20c conditional knockout mice can be attributed to the overproduction of FGF23 in the abnormal skeleton, the direct cause of cell differentiation failure may be complicated. As stated above, the defects in the mineralized tissues of the Fam20c conditional knockout mice could be the combined results of cell differentiation failure and hypophosphatemia. Although the way FAM20C regulates cell differentiation has not yet been defined in this study, our data suggest that FAM20C may regulate the differentiation and function of the mineralizing cells by participating in certain signaling pathways.
Several lines of evidence suggested that FAM20C might be associated with the canonical Wnt signaling pathway. The Wnt canonical pathway inhibitors, secreted frizzled related protein 1 (Sfrp1) and Sfrp3, were upregulated in the Fam20c-deficient mice. Accordingly, the downstream target genes of Wnt pathway, leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5) and lymphoid enhancer-binding factor 1 (Lef1) were significantly downregulated in the Fam20c-deficient bone. Lgr5 is a stem cell marker and a Wnt pathway regulator which has been identified in multiple tissues including bone marrow cells [23], [24]. Lef1 is a Wnt-responsive transcription factor that associates with β-catenin and has been documented to increase osteoblast activity and trabecular bone mass [25]. However, not all of the findings support the postulation that FAM20C is a participant in the Wnt signaling pathway. For example, the level of axis inhibition protein 2 (Axin2), a putative Wnt downstream target gene, was unchanged in the Fam20c-deficient bone. In addition to the molecules in the Wnt signaling pathway, Follistatin (Fst), a potent inhibitor of Activin and the TGF-β pathway, was significantly upregulated in the bones of 3-week and 6-week-old Fam20c-deficient mice. Fst has been reported to inhibit ameloblast and osteoblast differentiation [19], [20], suggesting a possible association between FAM20C and the TGF-β pathway.
Type II and Type X collagen were downregulated in the growth plates of the Fam20c conditional knockout mice. The downregulation of these genes may arise from the intrinsic defects of chondrocytes or occur as a systematic consequence of hypophosphatemia. It has been well documented that hypophosphatemia significantly decreases programmed cell death in growth plates by impairing caspase-mediated apoptosis of hypertrophic chondrocytes [17], [18].
In conclusion, the results in this investigation have demonstrated the crucial role of FAM20C in osteogenesis. Our findings indicate that FAM20C is essential to the differentiation of osteoblasts/osteocytes and is involved in the regulation of phosphate homeostasis via the mediation of FGF23.
Methods
Ethics statement
All animal procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Texas A&M Health Science Center, Baylor College of Dentistry (Dallas, TX, USA).
Generation of Fam20c conditional knockout mice lacking exons 6∼9
To generate the Fam20c conditional knockout mice, a 2.2 kb targeting fragment spanning exons 6∼9 of mouse Fam20c was produced by PCR using the genomic DNA of WW6 ES cells as a template (forward primer: 5′-CTCTCGGGTGAGGCTGTAAG-3′; reverse primer: 5′-AGATCTCTTAGGGAAGAGGGGTCAGG-3′). The fragment was subcloned into a floxed BamHI site upstream of an Frt flanked mcl-Neo cassette in the conditional targeting vector pFlox-Frt-Neo [47]. A 2.5 kb 5′ homologous arm was generated by PCR (forward primer: 5′-CTCGAGTGGGTGTGTCAGGAATCGTA-3′; reverse primer: 5′-CTCGAGACCCGAGAGCAACCACATAC-3′), and subcloned into the XhoI site of the targeting vector. A 4.2 kb 3′ homologous arm with EagI sites at both ends was generated by PCR (forward: 5′-TCGGCCGTTGGACATAGGCTCCCAAAG-3′; reverse: 5′-TGTGCAGGATTGAGAACCAG-3′), and subcloned into the NotI site of the targeting vector. Finally, a negative selection PGK-DTA (diphtheria toxin A) cassette was subcloned into the NotI site downstream of the 3′ homologous arm in the targeting vector (Figure 1A). The final targeting construct was linearized with SacII and electroporated into W4 ES cells (Transgenic Mouse Core Facilities, University at Albany, Rensselaer, NY, USA). Clones were picked after G418 selection. Genomic DNA was extracted from the ES cells in duplicate plates, and PCR analyses were performed to screen the targeted clones (5′ screen primers: 5′S-F: 5′-TTTCTGTCCTAGGTAAGGGTGAAG-3′, 5′S-R: 5′-ACTGCTCGATGAAGTTCCTATTCT-3′; 3′ screen primers: 3′S-F: 5′-TGTTCGGATCGAAGTTCCTATACT-3′, 3′S-R: 5′-ACAGCTTCTTGAATTGGGATAAAG-3′) (Figure 1A and 1B). The 5′ and 3′ screening PCR products were sequenced to confirm the correct targeting and the presence of 5′ and 3′ loxP sites.
Two correctly targeted ES cell clones (Clone 286 and 297, Figure 1B) were identified. The neo cassette was removed from the targeted ES clones by transient transfection of pCAGGS-flpE-puro vector (Addgene plasmid 20733) [48]. The removal of the neo cassette was confirmed by PCR (forward: 5′-GCATCTGCAGACCGAGCCCA-3′, reverse: 5′-CCCCCTGTCCTGAGGGCTGA-3′). Random integration of pCAGGS-flpE-puro DNA into ES cell genome was excluded by PCR analyses (forward: 5′-GCATGGCCGAGTTGAGCGGT-3′, reverse: 5′-GGTGACGGTGAAGCCGAGCC-3′). The targeted ES clones recovered from the master plates were injected into the blastocysts of C57BL/6 mice in the Transgenic Core Facility of the University of Texas Southwestern Medical Center at Dallas. Male chimeras were crossbred with C57BL/6 females to produce F1 agouti offspring. The floxed alleles of F1 agouti mice were genotyped by PCR analyses (see below). To generate Fam20c conditional knockout mice, the F1 heterozygous mice (Fam20cflox/+) were first crossbred with Sox2 promoter-driven Cre transgenic mice (Jackson Laboratory) that express the Cre recombinase transgene in the epiblasts at E6.5; this breeding gave rise to Sox2-Cre-Fam20cΔ/+ mice in which exons 6∼9 were removed from one allele of the Fam20c gene. The Sox2-Cre-Fam20cΔ/+ mice were inbred to produce Fam20c conditional knockout (cKO) mice designated as “Sox2-Cre-Fam20cΔ/Δ mice”, in which exons 6∼9 were removed from both alleles of the Fam20c gene.
Genotypes were determined by PCR analyses using genomic DNA extracted from the mouse tails. The floxed allele was distinguished from the wild type (WT) allele by PCR analyses using a mixture of three primers (a: 5′-TCCAGCTTGCTAGGGCTCTGACC-3′, b: 5′-CTATGTCCAACGGCCGCAGCTT-3′, and c: 5′-GTCCTGAGGGCTGACCCAAGACTA-3′) (Figure 1A and 1C). The null allele (i.e., that with exons 6∼9 removed) arising from Cre-loxP recombination events was detected by PCR using primers Rec-F: 5′-GTGGTCTCTGCCGCTGATGTACC-3′ and Rec-R: 5′-TTTGGGAGCCTATGTCCAACGGCC-3′ (Figure 1A and 1C). Genotyping for the Cre transgene was determined by PCR using primers Cre-F: 5′-CCCGCAGAACCTGAAGATG-3′ and Cre-R: 5′-GACCCGGCAAAACAGGTAG-3′.
We also used the 3.6 kb Col 1a1 promoter-Cre transgenic mice (The Jackson Laboratory) to generate “Col1a1-Cre-Fam20cΔ/Δ mice”, in which FAM20C was inactivated in tissues expressing type I collagen. We first crossbred the Fam20cflox/+ mice with 3.6 kb Col 1a1 -Cre mice, and then inbred the offspring of the Col1a1-Cre-Fam20cΔ/+ mice to get Col1a1-Cre-Fam20cΔ/Δ mice.
Generation of Fam20c conditional transgenic mice and mice expressing the transgene in the Fam20c conditional knockout background
To generate Fam20c conditional transgenic mice, the coding sequence of mouse Fam20c cDNA was subcloned into a bicistronic pMES vector [49] downstream to a chicken β-actin promoter and upstream to an IRES-EGFP cassette as previously described [7]. A floxed STOP cassette was inserted between the β-actin promoter and the Fam20c sequence to block the transcription of the transgene. The Fam20c transgene can be activated only after the floxed STOP cassette is removed by Cre recombinase [7]. This Fam20c conditional transgenic construct was linearized and injected into the pronuclei of fertilized eggs from the C57BL/6 mice in the Transgenic Core Facility of the University of Texas Southwestern Medical Center at Dallas. Fifteen lines of transgenic mice with the conditional Fam20c transgene were identified by PCR genotyping using primers located in the exogenous EGFP sequences (GFP-forward: 5′-ACGTAAACGGCCACAAGTTC-3′ and GFP reverse: 5′-TGCTCAGGTAGTGGTTGTCG -3′). The mice carrying the conditional transgene were crossbred with the Sox2-Cre transgenic mice to remove the floxed STOP cassette between the chicken β-actin promoter and the Fam20c cDNA, thereby allowing the Fam20c transgene to be transcribed. These conditional transgenic mice (cTg mice) were genotyped using the aforementioned GFP primers and Cre primers. The expression level of the Fam20c transgene in the long bones of each line was evaluated by quantitative real-time PCR. Three lines with the transgene expression levels of 4∼8 folds over that of the WT mice were further analyzed.
To generate mice expressing the transgene in the Fam20c conditional knockout background, we crossbred Sox2-Cre-Fam20cΔ/+ mice with cTg mice expressing the highest level of the trangene to obtain Sox2-Cre–Fam20cΔ/+-cTg mice, which were then inbred to produce mice expressing the transgene in the Fam20c conditional knockout background (designated “Sox2-Cre-Fam20cΔ/Δ-cTg mice”). PCR analyses with primers used in the identification of the Fam20c conditional knockout mice and cTg mice were employed in the genotyping of the Sox2-Cre-Fam20cΔ/Δ-cTg mice.
All mice in this study were fed with Teklad 6% fat mouse/rat diet (Harlan, IN) and some of the chow contents are as follows: Calcium 2.4%, phosphorus 1.5%, vitamin D 3.0 IU/g.
RT–PCR
Femurs from the Fam20c conditional knockout mice, Sox2-Cre-Fam20cΔ/Δ-cTg mice and the WT littermates were dissected, and total RNA was extracted using an Rneasy Mini Kit (Qiagen) according to the manufacturer's instructions. The total RNAs were converted into cDNAs using a Reverse Transcription Kit (Qiagen). RT-PCR was performed to examine the lack of Fam20c mRNA in the cKO mice using two sets of primers: Set 1-F: 5′-TGCGGAGATCGCTGCCTTCC-3′, Set 1-R: 5′-GCCACTGTCGTAGGGTGGCG-3′; Set 2-F: 5′-GAGAGCAGGAGACGCCGCCT-3′, and Set 2-R: 5′-CCACCACACTGCTCAGCCCG -3′ (Figure 2A).
Alizarin Red/Alcian Blue staining of the skeleton
One-week-old Sox2-Cre-Fam20cΔ/Δ mice and WT littermates were skinned, eviscerated and fixed in 95% ethanol. Alizarin Red/Alcian Blue staining of the skeletons was performed to visualize the skeleton and the overall mineralization levels, as described previously [50].
X-ray radiography
The narcotized mice or the dissected jaws and long bones from hind legs were analyzed using X-ray radiography (Faxitron MX-20DC12). Micro-computed tomography (Micro-CT) analyses were performed using a Scanco micro-CT35 imaging system (Scanco Medical) with a medium-resolution scan (7.0 µm slice increment) on the dissected tissues, as previously reported [51]. The images were reconstructed with the EVS Beam software using a global threshold at 240 Hounsfield units.
Preparation of decalcified sections and H&E staining
Tibia and jaw tissues dissected from the mice were fixed with 4% paraformaldehyde in 0.1% diethyl pyrocarbonate (DEPC)-treated PBS solution at 4°C overnight and then were decalcified in 0.1% DEPC-treated 15% EDTA (pH 7.4) at 4°C for 8 days. The tissues were processed for paraffin embedding, and serial 5 µm sections were prepared for histological analyses. H&E staining was performed as previously described [17].
Cell proliferation and TUNEL assays
BrdU was administrated to 3-week-old Sox2-Cre-Fam20cΔ/Δ mice and WT littermates at a dosage of 1 ml per 100 g body weight by intraperitoneal (i.p.) injection according to the manufacturer's instructions (Invitrogen). Two hr after the injection, the mice were sacrificed. Tibias were dissected and processed for paraffin embedding, and 5 µm sections were prepared for BrdU detection using a Zymed BrdU staining kit (Invitrogen) following the manufacturer's instructions. Apoptosis in growth plates was examined by TUNEL assay using the ApopTag Plus Fluorescein In Situ Apoptosis Detection Kit (Millipore) according to the manufacturer's instructions. Six serial sections from each of six individual samples of Sox2-Cre-Fam20cΔ/Δ mice and WT littermates were counted, and the data were analyzed statistically.
Immunohistochemistry (IHC) staining
The IHC experiments were carried out using an ABC kit and a DAB kit (Vector Laboratories) according to the manufacturer's instructions. A polyclonal C-terminal anti-FAM20C antibody was used at a concentration of 1 µg IgG/ml for the IHC experiments, as previously described [7]. A monoclonal FGF23 antibody (Cell Essentials) was used at a dilution of 1∶400 following the manufacturer's instruction. A polyclonal biglycan antibody (LF-159) was kindly provided by Dr. Larry Fisher (NIDCR, National Institutes of Health) [52]. Methyl green was used for counterstaining.
In situ hybridization (ISH)
A 380 bp fragment from the region of exons 6–9 of Fam20c cDNA was obtained by PCR using forward primer 5′ - CCGAGCATGCCCTGTGTGGG -3′ and reverse primer 5′ - TGCAGCACTGATGAAGAGGAGCG -3′. The PCR product was subcloned into the pCRII-TOPO vector (Invitrogen) and then linearized with EcoRV to synthesize the antisense RNA probes using the Sp6 RNA polymerase or with HindIII to synthesize the sense RNA probes using the T7 RNA polymerase. The constructs used to generate RNA probes for DMP1, osteocalcin, collagen type I, collagen type II, and collagen type X were provided by the laboratory of Dr. Jian Q. Feng. The constructs were linearized and labeled with digoxigenin (DIG) using a RNA Labeling Kit (Roche, Indianapolis, IN) as previously described [7]. DIG-labeled RNA probes were detected by an enzyme-linked immunoassay with a specific anti-DIG-AP antibody conjugate (Roche) and an improved substrate (Vector Laboratories), which produces a red color for positive signals, according to the manufacturer's instructions. Methyl green was used for counterstaining.
Preparation of undecalcified histology sections
Tibias dissected from 6-week-old mice were fixed in 4% paraformaldehyde overnight. The specimens were dehydrated through a graded series of ethanol (70–100%) and embedded in methylmethacrylate (MMA) without prior decalcification, as previously described [53]. Ten µm sections were prepared for Goldner staining and double-labeling fluorescent analysis.
Double fluorochrome labeling
Double fluorescence labeling was performed as previously described [54]. Briefly, calcein (5 mg/kg i.p.; Sigma-Aldrich) was administered to the 5-week-old mice, followed by injection of an Alizarin Red label (20 mg/kg i.p.; Sigma-Aldrich) 7 days later. The mice were sacrificed 48 hr after the injection of the second label and the tibias were embedded in MMA; 10 µm sections were then prepared. The unstained sections were viewed under epifluorescent illumination using a Nikon E800 microscope, interfaced with Osteomeasure histomorphometry software (version 4.1, Atlanta, GA). The mean distance between the two fluorescent labels was determined and divided by the number of days between labels to calculate the mineral deposition rate (µm/day).
Goldner's Masson Trichrome staining
Ten µm undecalcified sagittal sections from the tibias were stained using Goldner-Masson trichrome assay, as previously described [55]. The cortical bone areas in the midshaft were photographed using a Nikon microscope at 10× with Bioquant OSTEO v.7.20.10 (R&M Biometrics) software. Unmineralized osteoid stains red, and mineralized bone stains green/blue.
Resin-casted scanning electron microscopy (SEM) and backscattered SEM
For resin-casted osteocyte lacunocanalicular SEM, the surface of the MMA embedded tibia was polished, acid-etched with 37% phosphoric acid for 2–10 s, washed with 5% sodium hypochlorite for 5 min and then coated with gold and palladium and examined by FEI/Philips XL30 Field emission environmental SEM. Backscattered SEM was performed as we previously described [56]
Quantitative real-time PCR and microarray analyses
Total RNAs were isolated from the calvaria bones and decapsulated kidneys of 3-week-old mice and cultured cells. The kits for RNA extraction and reverse transcription were the same as in the RT-PCR experiments. Quantitative real-time PCR was performed on a Bio-Rad CFX96 system (Bio-Rad) using SYBR Green Master Mix (Stratagene). The Ct values were normalized to the reference gene 18s rRNA (SABiosciences), and then expressed as fold changes compared with the experimental controls. The primers for human 18s rRNA, mouse 18s rRNA, mouse Fam20c (NM_030565) and human FGF23 (NM_020638) were bought from SABiosciences. All other primers were synthesized by Integrated DNA Technologies (Table S1).
Microarray analyses were performed in the Microarray Core Facility of University of Texas Southwestern Medical Center at Dallas, using total RNA extracted from the calvaria of 3-week-old mice. GeneChip Mouse Genome 430 2.0 Array (Affymetrix) was employed in the microarray analyses, following the manufacturer's instructions. Data analyses were performed using GeneSpring software (Agilent Technologies). The microarray results were uploaded to MAGE-TAB ArrayExpress database (accession number E-MTAB-772).
Serum biochemistry
Serum phosphorus was measured using the phosphomolybdate-ascorbic acid method, as previously described [57]. Serum calcium was measured using a colorimetric calcium kit (Stanbio Laboratory). The serum FGF23 and PTH levels were measured using a full-length FGF23 ELISA kit (Kainos Laboratories) and a mouse intact PTH ELISA kit (Immutopics). Serum 1,25(OH)2D3 was measured using a 1,25 Dihydroxy Vitamin D EIA Kit (Immunodiagnostic Systems). Blood urea nitrogen (BUN) was measured using a BUN Reagent Kit (BQ Kits).
Generation of recombinant mouse FAM20C
The recombinant mouse FAM20C was expressed by insect cells using a Bac-to-Bac baculovirus expression system (Invitrogen). Briefly, the N-terminal of mouse FAM20C (signal peptide removed) was fused with a baculovirus signal sequence gp67 (envelope glycoprotein), 6xHis (tag), SUMOstar (Small Ubiquitin-like Modifier), and a TEV (Tobacco Etch Virus) cleavage site. The fusion gene was inserted into the pFastDual vector (Invitrogen) downstream of a polyhedrin promoter, and a GFP cDNA was inserted downstream of a P10 promoter serving as a baculovirus indicator. The construct was transformed into DH10Bac E. Coli cells (Invitrogen), in which the fusion gene was introduced into BacMid via homologous recombination. The BacMid was extracted from DH10Bac E. Coli cells and transfected into Sf21 insect cells (Invitrogen) to produce the baculovirus. The insect cells infected with the baculovirus secreted the recombinant FAM20C into the SFX-insect cell culture medium (Hyclone). After two rounds of scale-up, the cell culture medium was collected and subjected to a one-step Ni-NTA purification. Turbo-TEV (Eton Bioscience) was used to release mouse FAM20C from the fusion protein, and reverse Ni-NTA purification was performed to remove Turbo-TEV, His tag, and SUMOstar.
Cell culture and in vitro gain - and loss-of-function analyses
Mouse MC3T3-E1 cells were grown in α-MEM medium (Gibco) supplemented with 10% fetal bovine serum (Hyclone) and antibiotics (Gibco); human Saos-2 cells (ATCC, osteoblasts from human osteosarcoma) were grown in McCoy's 5a medium (Gibco) supplemented with 15% fetal bovine serum; human mesenchymal stem cells (hMSC) (Lonza) were maintained in MSCGM BulletKit medium (Lonza). Cells were treated with recombinant FAM20C or the viruses when they reach 80% confluence. For the gain-of-function analyses, MC3T3-E1 cells were treated with mouse recombinant FAM20C at the concentrations of 200 ng/ml, 400 ng/ml and 800 ng/ml. For the loss-of-function studies, MC3T3-E1, Saos-2 and hMSC cells were infected with the mouse or human shRNA-lentiviruses (all from Santa Cruz) containing a mixture of three target-specific shRNA sequences against the mouse or human FAM20C. A lentivirus expressing the scrambled shRNA (with no specific target in the genome) served as the control virus (Santa Cruz). The infection rate was monitored by infecting these cells with another control virus (Santa Cruz) expressing the GFP indicator. After 1-week selection with 5–8 µg/ml concentrations of puromycin (Santa Cruz), the control virus with GFP showed a nearly 100% infection rate. To induce osteogenic differentiation for the MC3T3 cells and Saos-2 cells, the culture medium was supplemented with 100 µg/ml ascorbic acid, 10 mM β-glycerophosphate and 30 nM dexamethasone; the osteogenic differentiation of human MSC was induced using the Osteogenic BulletKit (Lonza) following the manufacturer's instruction. Total RNA was extracted from the cells at different time points. For the gain-of-function analyses, RNA was extracted after 3 weeks of induction. For the “shRNA knockdown” analyses, RNA was extracted at 3 days after the lentiviral infection and before the osteogenic medium (inducing the osteogenic differentiation) addition, and at 1, 2 and 3 weeks after the start of osteogenic induction. Real-time PCR was performed to evaluate the mRNA levels of the selected genes. The mineral deposition rate was determined by nodule formation and Alizarin red concentration in each well were measured using the Osteogenesis Quantitation kit (Millipore) for MC3T3-E1 cells treated with recombinant FAM20C (the gain-of-function experiments). MC3T3-E1 cells and hMSC cells infected with the FAM20C-shRNA viruses became unhealthy (showing lot of cell death) after 4-week culture. The Saos-2 cells could not survive in the osteogenic medium for longer than 2 weeks. The data collected from the culture of cells at the “unhealthy stages” were discarded.
Statistics
The data are expressed as the mean ± SD of at least 6 individual determinations in all experiments unless otherwise indicated. We statistically evaluated the data employing ANOVA to test for any differences among the sample groups. When a difference was determined, 2-sample t tests were employed to evaluate all possible pairs of samples.
Supporting Information
Zdroje
1. NalbantDYounHNalbantSISharmaSCobosE 2005 FAM20: an evolutionarily conserved family of secreted proteins expressed in hematopoietic cells. BMC Genomics 6 11
2. AnCIdeYNagano-FujiiMKitazawaSShojiI 2010 A transgenic mouse line with a 58-kb fragment deletion in chromosome 11E1 that encompasses part of the Fam20a gene and its upstream region shows growth disorder. Kobe J Med Sci 55 4 E82 92
3. O'SullivanJBituCCDalySBUrquhartJEBarronMJ 2011 Whole-Exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am J Hum Genet 88 5 616 620
4. EamesBFYanYSwartzMLevicDSKnapikEW 2011 Mutations in fam20b and xylt1 reveal that cartilage matrix controls timing of endochondral ossification by inhibiting chondrocyte maturation. PLoS Genet 7 e1002246 doi:10.1371/journal.pgen.1002246
5. HaoJNarayananKMuniTRamachandranAGeorgeA 2007 Dentin matrix protein 4, a novel secretory calcium-binding protein that modulates odontoblast differentiation. J Biol Chem 282 15357 15365
6. SimpsonMAHsuRKeirLSHaoJSivapalanG 2007 Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am J Hum Genet 81 906 912
7. WangXHaoJXieYSunYHernandezB 2010 Expression of FAM20C in the osteogenesis and odontogenesis of mouse. J Histochem Cytochem 58 11 957 967
8. SimpsonMAScheuerleAHurstJPattonMAStewartH 2009 Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin Genet 75 271 276
9. ADHR Consortium. 2000 Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26 3 345 348
10. The HYP Consortium. 1995 A gene (PEX) with homologies to endopeptidases is mutated in patients with X linked hypophosphatemic rickets. Nat Genet 11 130 136
11. FengJQWardLMLiuSLuYXieY 2006 Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38 1310 1315
12. LarssonTMarsellRSchipaniEOhlssonCLjunggrenO 2004 Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 145 3087 3094
13. Lorenz-DepiereuxBBastepeMBenet-PagèsAAmyereMWagenstallerJ 2006 DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet 38 1248 1250
14. Lorenz-DepiereuxBSchnabelDTiosanoDHäuslerGStromTM 2010 Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 86 2 267 272
15. WhiteKECarnGLorenz-DepiereuxBBenet-PagesAStromTM 2001 Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int 60 2079 2086
16. BoskeyAL 1997 Amorphous calcium phosphate: the contention of bone. J Dent Res 76 8 1433 1436
17. YeLMishinaYChenDHuangHDallasSL 2005 Dmp1-deficient mice display severe defects in cartilage formation responsible for a chondrodysplasia-like phenotype. J Biol Chem 280 7 6197 6203
18. SabbaghYCarpenterTODemayMB 2005 Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A 102 27 9637 9642
19. AbeYAbeTAidaYHaraYMaedaK 2004 Follistatin restricts bone morphogenetic protein (BMP)-2 action on the differentiation of osteoblasts in fetal rat mandibular cells. J Bone Miner Res 19 8 1302 1307
20. WangXPSuomalainenMJorgezCJMatzukMMWernerS 2004 Follistatin regulates enamel patterning in mouse incisors by asymmetrically inhibiting BMP signaling and ameloblast differentiation. Dev Cell 7 5 719 730
21. FinchPWHeXKelleyMJUrenASchaudiesRP 1997 Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc Natl Acad Sci USA 94 6770 6775
22. LeynsLBouwmeesterTKimSHPiccoloSDe RobertisEM 1997 Frzb-1 is a secreted antagonist of Wnt signaling expressed in the Spemann organizer. Cell 88 747 756
23. BarkerNvan EsJHKuipersJKujalaPvan den BornM 2007 Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449 7165 1003 1007
24. HsuSYLiangSGHsuehAJ 1998 Characterization of two LGR genes homologous to gonadotropin and thyrotropin receptors with extracellular leucine-rich repeats and a G protein-coupled, seven-transmembrane region. Mol Endocrinol 12 12 1830 1845
25. HoeppnerLHSecretoFJRazidloDFWhitneyTJWestendorfJJ 2011 Lef1DeltaN binds beta-catenin and increases osteoblast activity and trabecular bone mass. J Biol Chem 286 13 10950 10959
26. LeeNKSowaHHinoiEFerronMAhnJD 2007 Endocrine regulation of energy metabolism by the skeleton. Cell 130 3 456 469
27. NakashimaKZhouXKunkelGZhangZDengJM 2002 The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108 17 29
28. LiuSGuoRSimpsonLGXiaoZSBurnhamCE 2003 Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem 278 37419 37426
29. SitaraDRazzaqueMSHesseMYoganathanSTaguchiT 2004 Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phexdeficient mice. Matrix Biol 23 421 432
30. KurosuHOgawaYMiyoshiMYamamotoMNandiA 2006 Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281 6120 6123
31. UrakawaIYamazakiYShimadaTIijimaKHasegawaH 2006 Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444 7120 770 774
32. ShimadaTMizutaniSMutoTYoneyaTHinoRTakedaS 2001 Cloning and characterization of FGF23 as a causative factor of tumor induced osteomalacia. Proc Natl Acad Sci USA 98 6500 6505
33. LiuSZhouJTangWJiangXRoweDW 2006 Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab 291 E38 49
34. LiuSTangWFangJRenJLiH 2009 Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol 23 9 1505 1518
35. BastepeMJüppnerH 2008 Inherited hypophosphatemic disorders in children and the evolving mechanisms of phosphate regulation. Rev Endocr Metab Disord 9 2 171 180
36. Alizadeh NaderiASReillyRFMedscape 2010 Hereditary disorders of renal phosphate wasting. Nat Rev Nephrol 6 11 657 665
37. MasuyamaRStockmansITorrekensSVan LooverenRMaesCCarmelietP 2006 Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest 116 12 3150 3159
38. FarrowEGWhiteKE 2010 Recent advances in renal phosphate handling. Nat Rev Nephrol 6 4 207 17
39. MeyerRAJrMeyerMHGrayRWBrunsME 1987 Evidence that low plasma 1,25-dihydroxyvitamin D causes intestinal malabsorption of calcium and phosphate in juvenile X-linked hypophosphatemic mice. J Bone Miner Res 2 1 67 82
40. ZhangMYWangXWangJTCompagnoneNAMellonSH 2002 Dietary phosphorus transcriptionally regulates 25-hydroxyvitamin D-1α-hydroxylase gene expression in the proximal renal tubule. Endocrinology 143 587 595
41. TenenhouseHSMartelJGauthierCZhangMYPortaleAA 2001 Renal expression of the sodium/phosphate cotransporter gene, Npt2, is not required for regulation of renal 1α-hydroxylase by phosphate. Endocrinology 142 1124 1129
42. MeyerRAJrMeyerMHMorganPLKiebzakGMRoosBA 1996 Effects of altered diet on serum levels of 1,25-dihydroxyvitamin D and parathyroid hormone in X-linked hypophosphatemic (Hyp and Gy). Bone 18 23 28
43. KiebzakGMRoosBAMeyerRAJr 1982 Secondary hyperparathyroidism in X-linked hypophosphatemic mice. Endocrinology 111 650 652
44. GattineniJBatesCTwombleyKDwarakanathVRobinsonML 2009 FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol 297 F282 291
45. QuarlesLD 2008 Endocrine functions of bone in mineral metabolism regulation. J Clin Invest 118 12 3820 3828
46. LuYFengJQ 2011 FGF23 in skeletal modeling and remodeling. Curr Osteoporos Rep 9 2 103 108
47. LiangXZhouQLiXSunYLuM 2005 PINCH1 plays an essential role in early murine embryonic development but is dispensable in ventricular cardiomyocytes. Mol Cell Biol 25 3056 3062
48. BeardCHochedlingerKPlathKWutzAJaenischR 2006 Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44 1 23 28
49. SwartzMEberhartJMastickGSKrullCE 2001 Sparking new frontiers: using in vivo electroporation for genetic manipulations. Dev Biol 233 13 21
50. InouyeM 1976 Differential staining of cartilage and bone in fetal mouse skeleton by alcian blue and alizarin red-S. Congen Anom 16 171 173
51. SunYPrasadMGaoTWangXZhuQ 2010 Failure to process dentin matrix protein 1 (DMP1) into fragments leads to its loss of function in osteogenesis. J Biol Chem 285 41 31713 31722
52. FisherLWStubbsJYoungMF 1995 Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop Scand 66 61 65
53. SunYLuYChenLGaoTD'SouzaR 2011 DMP1 processing is essential to dentin and jaw formation. J Dent Res 90 5 619 624
54. MillerSCOmuraTHSmithLJ 1985 Changes in dentin appositional rates during pregnancy and lactation in rats. J Dent Res 64 1062 1064
55. ZhouXZhangZFengJQDusevichVMSinhaKZhangH 2010 Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc Natl Acad Sci U S A 107 29 12919 12924
56. MaDZhangRSunYRiosHFHaruyamaN 2011 A novel role of periostin in postnatal tooth formation and mineralization. J Biol Chem 286 6 4302 4309
57. YuanBTakaiwaMClemensTLFengJQKumarR 2008 Aberrant Phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest 118 2 722 734
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Inactivation of a Novel FGF23 Regulator, FAM20C, Leads to Hypophosphatemic Rickets in Mice
- Genome-Wide Association of Pericardial Fat Identifies a Unique Locus for Ectopic Fat
- Slowing Replication in Preparation for Reduction
- An Essential Role for Katanin p80 and Microtubule Severing in Male Gamete Production
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy