
Suppresses Senescence Programs and Thereby Accelerates and Maintains Mutant -Induced Lung Tumorigenesis
KRAS mutant lung cancers are generally refractory to chemotherapy as well targeted agents. To date, the identification of drugs to therapeutically inhibit K-RAS have been unsuccessful, suggesting that other approaches are required. We demonstrate in both a novel transgenic mutant Kras lung cancer mouse model and in human lung tumors that the inhibition of Twist1 restores a senescence program inducing the loss of a neoplastic phenotype. The Twist1 gene encodes for a transcription factor that is essential during embryogenesis. Twist1 has been suggested to play an important role during tumor progression. However, there is no in vivo evidence that Twist1 plays a role in autochthonous tumorigenesis. Through two novel transgenic mouse models, we show that Twist1 cooperates with KrasG12D to markedly accelerate lung tumorigenesis by abrogating cellular senescence programs and promoting the progression from benign adenomas to adenocarcinomas. Moreover, the suppression of Twist1 to physiological levels is sufficient to cause Kras mutant lung tumors to undergo senescence and lose their neoplastic features. Finally, we analyzed more than 500 human tumors to demonstrate that TWIST1 is frequently overexpressed in primary human lung tumors. The suppression of TWIST1 in human lung cancer cells also induced cellular senescence. Hence, TWIST1 is a critical regulator of cellular senescence programs, and the suppression of TWIST1 in human tumors may be an effective example of pro-senescence therapy.
Published in the journal:
. PLoS Genet 8(5): e32767. doi:10.1371/journal.pgen.1002650
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002650
Summary
KRAS mutant lung cancers are generally refractory to chemotherapy as well targeted agents. To date, the identification of drugs to therapeutically inhibit K-RAS have been unsuccessful, suggesting that other approaches are required. We demonstrate in both a novel transgenic mutant Kras lung cancer mouse model and in human lung tumors that the inhibition of Twist1 restores a senescence program inducing the loss of a neoplastic phenotype. The Twist1 gene encodes for a transcription factor that is essential during embryogenesis. Twist1 has been suggested to play an important role during tumor progression. However, there is no in vivo evidence that Twist1 plays a role in autochthonous tumorigenesis. Through two novel transgenic mouse models, we show that Twist1 cooperates with KrasG12D to markedly accelerate lung tumorigenesis by abrogating cellular senescence programs and promoting the progression from benign adenomas to adenocarcinomas. Moreover, the suppression of Twist1 to physiological levels is sufficient to cause Kras mutant lung tumors to undergo senescence and lose their neoplastic features. Finally, we analyzed more than 500 human tumors to demonstrate that TWIST1 is frequently overexpressed in primary human lung tumors. The suppression of TWIST1 in human lung cancer cells also induced cellular senescence. Hence, TWIST1 is a critical regulator of cellular senescence programs, and the suppression of TWIST1 in human tumors may be an effective example of pro-senescence therapy.
Introduction
Lung cancer is responsible for more cancer deaths in the US than colorectal, breast, and prostate cancer combined with a dismal overall survival of 15% [1]. The majority of human lung cancers are adenocarcinomas carrying somatic mutations in the genes that encode the EGFR/KRAS/BRAF pathway [2]. Observations in both experimental mouse models and human lung tumors strongly suggest that these pathways are causally responsible for lung tumorigenesis [3], [4], [5], [6], [7].
KRAS mutant lung adenocarcinomas are generally refractory to conventional cytotoxic therapies [8] and currently available small molecule targeted agents [9], [10]. Difficulties in pharmacologically targeting K-RAS have resulted in some labeling the protein “undruggable” [11]. Approaches such as using farnesyl transferase inhibitors to prevent prenylation of Ras for its membrane localization have not shown clinical efficacy [12], [13]. Other potential kinase targets for KRAS mutant tumors have been identified through RNAi screens including: TBK1, STK33 and PLK1 [14], [15], [16]. Rational candidate based approaches that target key pathways required during the process of tumorigenesis for KRAS mutant cancers have not been exhaustive.
One such pathway is oncogene-induced senescence (OIS), a failsafe program that prevents normal cells from progressing towards malignancy following introduction of a mutant form of an oncogene such as KrasG12D [17]. OIS is an irreversible cell cycle arrest that is characterized by cells displaying an enlarged, flattened cytoplasm, increase in senescence associated beta-galactosidase (SA-β-Gal) activity, increased chromatin condensation and changes in gene expression associated with DNA damage checkpoint proteins or cell cycle checkpoint proteins. OIS is thought to be triggered early during tumorigenesis in order to inhibit aberrant cell cycle progression, preventing pre-malignant tumors from progressing to malignancy [17]. OIS seems to be dependent on the p53-p19ARF, p16-Rb and Atf4-p27 pathways to enforce the senescent phenotype, but the requirement of any or all these pathways is highly context dependent [18], [19]. Whether these latent OIS programs can be activated in KRAS mutant cancers to result in a clinical effect has only recently been examined [20], [21].
Recently, Twist1, a basic helix-loop-helix transcription factor that is central to embryogenesis [22], has been shown to suppress OIS associated with KrasG12D and EGFR2 oncogenes in vitro in MEFs [23] and pancreatic epithelial cells [24]. Twist1 protein expression is usually undetectable in most adult tissues, but has been shown to be overexpressed in cancers including prostate, bladder pancreatic, osteosarcomas, melanomas and breast [25], [26], [27], [28], [29], [30], [31]. The high expression of Twist1 in cancers strongly correlates with invasive and metastatic tumor cells. Twist1 is thought to regulate epithelial-mesenchymal transition (EMT) through the down-regulation of key proteins that maintain epithelial cell characteristics and up-regulation of proteins that confer a mesenchymal phenotype [31]. Thus, Twist1 may act both to induce malignancies early in tumorigenesis and also promote tumor progression [32]. To date, there has yet to be reported an autochthonous model to study the role of Twist1 overexpression in the initiation and maintenance of tumorigenesis. Here we report the generation of such a model and through this demonstration we show an important role of Twist1 in suppressing cellular senescence programs.
Results
Generation of an inducible lung epithelium specific Twist1 transgenic mouse model
To produce a useful tool to address Twist1 functions in vitro and in vivo we generated a transgenic founder line, Twist1-tetO7-luc (T), that harbored the mouse Twist1 cDNA under the control of a bidirectional tetracycline operator sequence (tetO7) also regulating the firefly luciferase gene (luc) [33] (Figure 1A). This T founder was crossed to Clara cell secretory protein-reverse tetracycline transactivator protein (CCSP-rtTA or C) mice to generate inducible, double-transgenic (CT) mouse cohorts. CT mice contain both the rtTA activator expressed primarily in lung alveolar Type II pneumocytes [34] and the tetracycline inducible Twist1-tetO7-luc transgene allowing for spatial and temporal expression of Twist1 and luc (Figure 1A).

Inducible regulation in CT mice was verified using serial small animal bioluminescence imaging (BLI) and Western blotting, respectively (Figure 1B–1C). Doxycycline drinking water given to CT mice (CT ON) induced luciferase and Twist1 expression specifically in the lung only (Figure 1C) which reverted to background luciferase and Twist1 expression by 3–7-days after doxycycline withdrawal [34], [35], (Figure 1B–1C).
To address the functional significance of ectopic Twist1 expression in the lung epithelium global gene expression microarray analysis was performed with induced CT mouse lungs versus wildtype mouse lungs. Notably, after performing gene set enrichment analysis (GSEA) [36] with this dataset, we found CT ON lungs had a global gene expression pattern that had a highly significant similarity to two overlapping gene sets for EMT [37] (Figure 1D and Figure S1A) and three EMT related phenotypes (hypoxia, metastasis and wound healing [38], [39], [40]; Figure S1B–S1D). CT ON lungs showed a subset of epithelial cells appeared to lose E-cadherin and gain vimentin staining by immunofluorescence consistent with an EMT (Figure 1E and Figure S1E), strongly supportive of the gene expression data. Thus, our lung specific CT mouse model is capable of enforcing a Twist1-dependent transcriptional program in lung epithelial cells that is consistent with cells that have undergone EMT.
Twist1 accelerates KrasG12D-induced lung tumorigenesis and promotes progression to adenocarincoma
Twist1 has been strongly implicated in tumor progression, but no studies have examined the effect of Twist1 alone for autochthonous tumorigenesis. Twist1 was not a strong oncogene when expressed alone in the lung epithelium. CT ON mice did not develop lung tumors at an increased frequency compared to wildtype FVB/N mice (Figure 2A) [41].

Twist1 cooperated dramatically with KrasG12D expression in the lung. CCSP-rtTA/tetO-KrasG12D (CR) mice developed multiple synchronous lung tumors, mostly adenomas, with a median tumor latency of 32 weeks [3], [35] (Figure 2A and 2E). Triple transgenic mice, CCSP-rtTA/tetO-KrasG12D/Twist1-tetO7-luc (CRT), demonstrated a greatly reduced lung tumor latency compared to CR mice, 15 versus 32 weeks (p<0.0001 by log-rank analysis) (Figure 2A). CRT mice developed numerous lung tumors (Figure 2B) that appeared to be from a type II pneumocyte origin based on CCSP negative and proSpC positive immunohistochemistry (IHC) (Figure 2C). Twist1 cooperation with KrasG12D increased the number and size of lung tumors that developed. At six months of oncogene induction there was a large difference in the total lung tumors per mouse for CRT versus CR, 40 tumors versus 2 tumors (p = 0.03 by t-test) (Figure 2D). Twist1 co-expression with KrasG12D in the lung also appeared to promote transformation of the predominantly benign lung adenoma tumor phenotype of CR mice [3] to a malignant phenotype composed mostly of adenocarcinomas as determined by a veterinarian pathologist [42] (Figure 2E, p<0.0001 Fisher's exact test). A more sensitive marker of this conversion from adenoma to adenocarcinoma was the proliferative rate as we observed much higher proliferative index in CRT versus CR tumors (Figure 2E, p = 0.021 Chi-square and Figure S2A).
Although, we observed a strong genetic interaction between Twist1 and KrasG12D for lung tumorigenesis, we did not see a pronounced effect on distant lung tumor metastases. One CRT mouse did exhibit a macroscopic metastasis to the liver confirmed by pathology (data not shown). However, in general the CRT cohort of mice (n = 33) did not demonstrate increased distant metastasis compared to CR mice (n = 55) when followed for up to 9 months of oncogene induction (data not shown). Taken together, these data suggest that Twist1 does not appear to be a strong oncogene when over-expressed alone in the lung. Twist1 is capable of strong cooperation with KrasG12D for lung tumorigenesis and progression. Despite markedly accelerating tumorigenesis, Twist1 did not promote increased numbers of circulating tumors cells as detected by qPCR specific for the luc transgene (data not shown) and nor did Twist1 promote distant metastasis from primary lung tumors.
Reversible KrasG12D/Twist1-induced lung tumorigenesis
CR lung tumors were fully reversible following 2–3 weeks of KrasG12D oncogene inactivation with the mechanism of tumor regression being a combination of tumor cells undergoing proliferative arrest and apoptosis [3]. We inactivated both Twist1 and KrasG12D from a cohort of CRT lung tumor moribund mice by the removal of doxycycline and monitored them for lung tumor regression at multiple time points using serial non-invasive imaging techniques in addition to final pathologic analysis (n = 4). CRT lung tumors showed dramatic tumor regression by gross examination (compare Figure 3A versus Figure 2B) that could be demonstrated serially with microCT (Figure 3B) and microPET-CT (Figure 3C and Figure S3A) after as little as 1 week of dual oncogene inactivation. By 4 weeks, CRT OFF lungs typically demonstrated no evidence of viable tumor cells on histologic analysis (Figure 3D) even despite CRT mice having considerably more advanced lung tumors than CR at similar time points (Figure 2D). CRT mice with heavy initial tumor burden did have residual fibrotic scars remaining (white spots in Figure 3A and trichome collagen staining in Figure S3B).
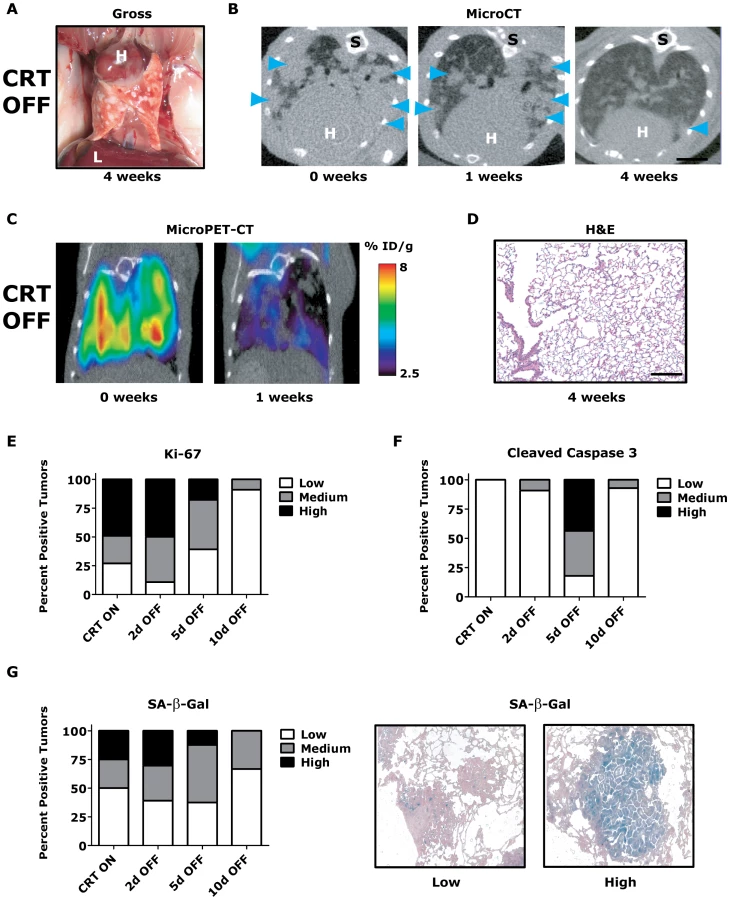
To gain insight into the mechanism of tumor regression, we performed a time course analysis of CRT OFF lung tumors during the first week of oncogene inactivation. CRT OFF lung tumors demonstrated a prominent decrease in proliferation and increase in apoptosis following 5 days of doxycycline withdrawal as measured by Ki-67 and cleaved caspase 3 (CC3) IHC, respectively (Figure 3E–3F, p<0.0001 Chi-square for both Ki-67 and CC3 and Figure S3C–S3D). As mentioned previously, Twist1 has been shown in vitro to suppress KrasG12D oncogene-induced senescence (OIS) [23]. However, we did not see any appreciable increase in senescence associated beta-galactosidase (SA-β-Gal) staining following simultaneous inactivation of Twist1 and KrasG12D in CRT OFF lung tumors (Figure 3G, p = 0.68 Chi-square) or by assessing for markers of cell cycle arrest (data not shown). These data suggest that although CRT lung tumors demonstrate more aggressive histologic appearance than CR tumors, CRT lung tumors are still strictly dependent on initiating oncogenes for tumor maintenance. Furthermore, Twist1 did not alter the mechanism of tumor regression between CR OFF and CRT OFF lung tumors.
Induction of cellular senescence in KrasG12D-induced lung tumors by inactivation of Twist1
The strong dependency or addiction of KrasG12D-initiated lung tumors for KrasG12D [3], [35] may have precluded us from observing any activation of OIS in CRT OFF lung tumors. Furthermore, given the genetic configuration of the CRT mouse model we were not able to examine the effects on lung tumors following inactivation of Twist1 alone.
We addressed in vitro whether activation of rasG12V-induced senescence could be driven by inactivation of Twist1 by using mouse embryonic fibroblasts (MEFs) generated from β-actin-rtTA/Twist1-tetO7-luc (BT) mice. BT MEFs were shown to be inducible with doxycycline in vitro by Western blotting (Figure S4A). As reported previously [23], we found Twist1 was able to fully suppress rasG12V-induced senescence in vitro as shown by proliferation curves and SA-β-Gal staining (Figure S4B–S4D). We removed doxycycline from the media of BT MEFs infected with rasG12V to downregulate expression of Twist1 at Day 12. These de-induced BT MEFs activated OIS in vitro as shown by decreased proliferation and increased SA-β-Gal staining relative to cells maintained in the presence of doxcycline (p = 0.0025 for proliferation and p = 0.0294 for SA-β-Gal; Figure S4E–S4F). These data suggested that at least in vitro inhibition of Twist1 can activate rasG12V-induced senescence.
To examine whether Twist1 inhibition could be a viable therapeutic target in vivo for Kras mutant autochthonous lung cancers, we generated mice in which only Twist1 expression was doxycycline-dependent (Figure 4A). The LSL-KrasG12D (LSL) model allows for conditional activation of an endogenous KrasG12D allele in the lungs following intranasal adenoviral delivery of Cre recombinase (AdCMVCre) [43]. The strain background difference between CT (FVB/N) and LSL (C57BL/6) transgenic models forced us to use first generation progeny from these crosses for all our experiments. We generated tri-transgenic CT-LSL animals (Figure 4A), activated Twist1 expression with doxycycline, then conditionally activated the KrasG12D allele with AdCMVCre and followed these CT-LSL ON mice and similarly treated littermate controls for lung tumor development. Twist1 accelerated conditional KrasG12D-induced lung tumorigenesis in CT-LSL mice (CT-LSL versus LSL, p = 0.0121 by log-rank analysis, Figure 4B–4C, similar to CRT mice, although to a lesser degree. CT-LSL lung tumors were similar to CRT tumors based on histology, expression of type II pneumocyte markers, and increase in the proportion of lung tumors with a higher proliferative index (Figure 4D–4F, p<0.0001 Chi-square). Recently, two groups have demonstrated in a similar LSL-p53 model system that p19ARF is a critical sensor of oncogenic stress from MAPK signaling in adenocarcinomas [44], [45]. We similarly observed overlap of activated p19ARF (nucleolar localization) with areas of intense pErk1/2 staining by IHC in our CT-LSL ON tumor model (Figure 4G).

We next inactivated the expression of Twist1 alone in a cohort of CT-LSL lung tumor moribund mice by withdrawal of doxcycline (CT-LSL OFF, n = 4). Twist1 levels were confirmed in CT-LSL OFF tumors by qPCR (Figure 5A, p = 0.004 by t-test) and by serial BLI (data not shown) to return to levels in wildtype lungs. Interestingly, CT-LSL OFF lung tumors showed tumor stasis by serial microCT over the course of the 4 weeks of Twist1 inactivation in stark contrast to the progressive tumor growth seen for the control LSL OFF tumors (Figure 5B; 18% versus 220% growth, p<0.0001 t-test).

To further characterize in an unbiased manner the mechanism by which Twist1 suppression was inducing tumor stasis we performed microarray analysis. We compared CT-LSL OFF lung tumors with normal lung and microdissected lung tumors from CR, CRT, LSL, CT-LSL ON and CT-LSL OFF mice. The analysis of 2,163 annotated pathways using single sample GSEA (ssGSEA), an algorithm designed for modest samples sizes [as used previously in [16]], found gene sets representing p21 ectopic overexpression to be highly correlated with the CT-LSL OFF lung tumor transcriptional program (Figure S5A–S5B). We used the complimentary Ingenuity Pathway Analysis (IPA) to identify canonical pathways from the differentially expressed genes between CT-LSL ON and CT-LSL OFF tumors. Consistent with ssGSEA we found Twist1 regulated key drivers of cellular senescence (genes encoding p21, p16, p27 and IL-6) and EMT (genes encoding cadherins, vimentin and alpha-catenin) in the context of KrasG12D-driven lung tumors (Figure S6). Directed IHC analysis of CT-LSL OFF tumors confirmed ssGSEA and IPA that molecular changes consistent with activation of OIS, such as marked decreases in proliferation by Ki-67 and pronounced increases in staining for SA-β-Gal, p21 and p16 (Figure 5C–5F and 5H–5K, p<0.0007 Chi-square for 5H–5K).
The single inactivation of Twist1 in our CT-LSL OFF tumors also appeared to decrease the number of adenocarcinomas as shown with decrease in tumors with high proliferative rate to a frequency similar to LSL alone (compare Figure 5C and 5H to LSL from Figure 4F, p = 0.93 Chi-square). In addition MAPK signaling intensity decreased in CT-LSL OFF significantly compaed to CT-LSL ON (Figure 5G and 5L, p<0.015 Chi-square). The decrease of highly proliferative adenoncarcinomas with active MAPK signaling in KrasG12D-induced lung tumors was also seen following p53 restoration [44], [45]. Lastly, apoptosis increased only very slightly in a subset of the CT-LSL OFF tumors as demonstrated by cleaved caspase 3 IHC (data not shown). These data provide the first in vivo evidence that Kras mutant lung adenocarcinomas can be clinically impacted by activating a latent program of cellular senescence via the inhibition of Twist1.
TWIST1 is commonly overexpressed in human lung cancers
The relevance of TWIST1 as a potential therapeutic target in human lung cancers was evaluated by examining public gene expression microarray datasets. We found seven independent human lung cancer gene expression datasets that in total consisted of 394 tumor samples and 159 normal lung samples [46], [47], [48], [49], [50], [51], [52] (Figure 6A). Six out of the 7 datasets, as well as aggregate analysis of all 7 datasets demonstrated TWIST1 overexpression in lung cancers (p = 0.04 for aggregate). The analysis included tumors of adenocarcinoma and squamous cell carcinoma histology which comprise the two most common subtypes encountered in human lung cancer.

This microarray expression data was directly validated using quantitative PCR (qPCR) for TWIST1 on human lung cancer samples. In total we screened by qPCR 164 human lung tumor samples and confirmed that TWIST1 was indeed overexpressed (100/164 or 61% demonstrate at least 3-fold upregulation, 43/164 or 26% at least 10-fold overexpression and in some cases as high as 536 fold overexpression was observed, p<0.0001 by t-test; Figure 6B). TWIST1 was similarly overexpressed in all the histologies examined including adenocarcinoma and squamous cell carcinoma (p<0.0001 by ANOVA) (Figure 6C). The range of relative TWIST1 overexpression observed by qPCR in our 164 primary human lung cancer samples (range 3–536 fold TWIST1 overexpression) was similar to the Twist1 overexpression observed in our mouse KrasG12D-Twist1-induced lung tumors (range 5–960 fold Twist1 overexpression, n = 6). Together these data demonstrate that TWIST1 is commonly overexpressed in human lung cancer and that our KrasG12D-Twist1 mouse models do reflect human lung cancer.
Activation of cellular senescence in human KRAS mutant lung cancer cells by targeting TWIST1
The overexpression of TWIST1 in human lung cancers and our in vivo data from the CT-LSL OFF mouse lung tumors strongly suggested that TWIST1 may be a relevant therapeutic target in human lung cancer. The consequences of knocking down TWIST1 using shRNA technology was tested in human KRAS mutant H460 lung cancer cells. We screened various published shRNAs and found three sequences that were capable of knocking down human TWIST1 as shown by qPCR (Figure 7A, p<0.029 by ANOVA) and at the protein level by Western (Figure 7B). TWIST1 knockdown in H460 cells resulted in marked inhibition of proliferation using all three shRNAs (Figure 7C) and increased staining for the cellular senescence marker SA-β-Gal (Figure 7D–7E, p<0.023 by ANOVA). Other OIS markers p21 and p27 showed upregulation with a subset of the shRNAs examined (Figure 7F). We extended these results in two other human non-small cell lung cancer cell lines, H727 and A549, showing that TWIST1 knockdown resulted in decreased proliferation and increased expression of markers consistent with activation of senescence (Figure S7).

We then confirmed that the TWIST1 shRNA was not having off target effects by performing rescue experiments with mouse Twist1 infected into H460 and A549 cells (Figure S8A and data not shown). Notably, the three shRNAs used in our study were not predicted to knockdown mouse Twist1 cDNA, which was confirmed by qPCR (Figure S8B and data not shown). The anti-proliferative effects of shRNA mediated knockdown of human TWIST1 in H460 and A549 cells was completely rescued by expression of mouse Twist1 (Figure S8C and data not shown). These data provide evidence that inhibition of TWIST1 can activate latent OIS in multiple different human KRAS mutant lung cancer cell lines.
To evaluate if the tumorigenic potential of human NSCLC cells required TWIST1 overexpression, we performed subcutaneous xenografting experiments with A549 cells in immune-compromised NOD-SCID mice. A549 cell infected with sh-Scrambled control shRNA and subcutaneously injected into NOD-SCID mice produced large tumors with high efficiency (5/6 mice developed tumors by ≤4 weeks) necessitating humane euthanasia of the mice. In stark contrast, the identical experiment using sh-TWIST1 produced no tumors in any of the mice injected (Figure 7G, p = 0.015 by Fisher's exact test). These xenografting results confirm that TWIST1 overexpression is required for tumorigenicity in vitro and in vivo in human NSCLC cells.
Discussion
Our results dramatize that suppression of TWIST1 may be an effective pro-senescence therapy for human lung cancer. We provide the first in vivo demonstration that Twist1 plays an important role in both the acceleration and maintenance of KrasG12D-induced autochthonous lung tumorigenesis. Our results illustrate that TWIST1 may be an important target for the treatment of human lung adenocarcinoma. We generated two novel autochthonous transgenic mouse models to demonstrate that Twist1 overexpression cooperates with KrasG12D to markedly accelerate the onset of lung adenocarcinoma. Suppression of Twist1 expression to physiological levels is sufficient to induce lung tumor stasis that was associated with the activation of cellular senescence programs. Importantly, through the transcriptional analysis of over 500 human tumors, human TWIST1 was found to be frequently overexpressed and hence highly relevant to primary human lung cancers. Finally, the knockdown of TWIST1 in human KRAS mutant lung tumor cells was also associated with the loss of their neoplastic properties and the induction of cellular senescence. The generality of our results using different cell types and across species suggest TWIST1 is a potential therapeutic target in KRAS mutant lung cancers.
Oncogene-induced senescence and oncogene-induced apoptosis represent early tumor suppressive barriers that must be overcome for premalignant cells to ultimately emerge as neoplastic. It had been reported previously that Twist1/2 could suppress mutant Kras-induced OIS in vitro [23], [24], but we report for the first time the ability of Twist1 to suppress OIS in vivo using a novel Twist1 lung model in combination with two complementary KrasG12D–induced autochthonous lung tumor models: the inducible transgenic KrasG12D (CR) model and the conditional endogenous KrasG12D (LSL) model. Our results are confirmed by an accompanying paper demonstrating that Twist1 can also accelerate KrasG12D-induced autochthonous breast tumorigenesis (Morel et. al.). Twist1 co-expression accelerated tumorigenesis relative to KrasG12D alone in both lung tumor models. Twist1 acceleration was more pronounced in the CRT model than the CT-LSL model. One explanation for this difference is the greater strength of oncogenic signaling by transgenic KrasG12D versus endogenous KrasG12D [53]. An alterative explanation is that cell type specific chromatin regulation of tumor suppressor loci such as the Ink4a/Arf locus is a key determinant of whether mutant Kras elicits tumor suppressive responses resulting in apoptosis and/or senescence [54]. Another explanation are strain difference effects as we had to use a mixed background for the CT-LSL mouse experiments. These alternatives are not mutually exclusive and further study using additional tissue specific models of KrasG12D and Twist1 expression are needed to define the mechanistic basis for the differences we observed in oncogenic synergy observed between Twist1 and KrasG12D.
The acceleration and progression of KrasG12D -induced lung tumors by Twist1 is reminiscent of that seen with p53 deficiency [3], [44], [45], [55]. Notably, Twist1 may inhibit p53 through several independent mechanisms [23], [56], [57], [58], [59], [60], including direct Twist1-p53 antagonism [61]. One straightforward interpretation of our results is that Twist1 overexpression can phenocopy Trp53 deletion. Twist1 may also accelerate and promote KrasG12D-induced lung tumors by the direct transcriptional regulation of BMI1 [62]. As mentioned above, the control of tumor suppressor loci by chromatin regulatory complexes, such as those containing Bmi1, may be a strong determinant of responses to oncogenic signaling [54]. Interestingly, ectopic expression of Twist1 in lung epithelial cells was associated with the induction of an EMT program. Whether the transdifferentiation program might contribute to accelerated tumor initiation, as proposed by Morel et. al., is also an intriguing possibility. Additional studies are required to define the mechanisms by which Twist1 accelerates KrasG12D-induced lung tumors, as well as explain why different tissues exhibit differing cancer susceptibilities despite harboring the same initiating oncogenic event.
Twist1 has been commonly implicated in metastasis [32]. Thus, our finding that Twist1 expression did not seem to confer increase distant metastases in either the CRT or CT-LSL autochthonous lung tumor models was surprising. We note that Twist1 appears to confer increased prometastatic ability in other models of tumorigenesis as predicted (D. I. Bellovin, P. T. Tran and D. W. Felsher, unpublished data). Hence, Twist1 may have specific effects on metastatic potential.
Our study dramatically illustrates that it is possible to activate a latent senescence program in Kras mutant tumors in vivo by targeting the collaborating oncogene, Twist1. We uncover a newly defined synthetic interaction between mutant Kras and Twist1 resulting not in cell death, but cellular senescence. The activation of this program is evident at the molecular level and most importantly results in marked inhibition of Kras mutant lung tumor growth in vivo. We realize that a possible caveat to this approach is that we first overexpressed Twist1 prior to KrasG12D activation and lung tumor formation and thus may have biased tumors towards dependency for Twist1. However, simply overexpressing an oncogene during tumorigenesis does not per se make tumors dependent or “addicted” to that oncogene as we have shown, in particular for lung tumorigenesis [35], [63]. Finally, we validate that knocking down endogenous TWIST1 in human lung cancer cell lines in vitro and in vivo also results in activation of senescence.
An alternative approach to inducible overexpression using the TET system as we used in our study would be to use genetic ablation of endogenous Twist1 using the Cre-LoxP or a inducible shRNA system following development of KrasG12D–induced lung tumors. As KrasG12D–induced lung tumors are primarily adenomas with low proliferative rates (Figure 2E and Figure 4F), endogenous Twist1 ablation or knockdown would not likely have an effect as has been shown for p53 restoration in adenomas [44], [45]. From a clinical standpoint complete ablation of a gene, such as in mice using the Cre-LoxP system, is therapeutically not possible in humans. In contrast, the TET model system where we can suppress Twist1 overexpression to physiologic levels is more clinically relevant to what is done in the clinic with inhibitors. Others have shown senescence can arise in vivo in established tumors by targeting an initiating oncogene or reconstitution of a tumor suppressor [21], [64], [65], [66]. Our work further highlights the activation of a latent cellular senescence program or pro-senescence therapy as an innovative avenue for cancer therapy [67].
Our results may extend beyond KRAS-mutant lung cancers. Notably, TWIST1 was found to be overexpressed in a majority of human lung cancer samples we tested. This includes not only adenocarcinoma, in which KRAS mutation is commonly observed, but also other major lung cancer histologies including squamous cell carcinoma, in which KRAS mutation is rare. Our preliminary data suggests that TWIST1 knockdown can result in activation of OIS in KRAS wildtype lung cancer cell lines in vitro, but further characterization of these lines for mutations in other components of the EGFR/KRAS/BRAF pathway are needed (T.F. Burns, P. T. Tran and C. M. Rudin, unpublished data). Furthermore, additional preliminary findings suggest that TWIST1 may have a larger role in suppressing OIS following activation of other key driver mutations using other transgenic mouse lines (P. T. Tran and D. W. Felsher, unpublished data). This hypothesis will be further explored in lung cancer through introduction of our inducible Twist1 construct into other relevant transgenic models of lung tumorigenesis. Importantly, regardless of whether there is an exclusive association between KRAS mutation and TWIST1 overexpression in human lung cancer cells, the data presented strongly support that TWIST1 upregulation in KRAS mutant lung cancer represents a novel and particularly promising therapeutic target. These observations have important and immediate translational implications for this particularly refractory subset of lung cancers.
The consequences of systemic transient inhibition of Twist1 in the adult has not been well defined and thus side-effects of such treatment are unknown. Germline deletion of Twist11 in mice is embryonic lethal [22] and loss of function mutations in humans cause a severe developmental disorder. However, postnatal expression of TWIST1 appears to be tightly restricted to a subpopulation of mesoderm derived tissues and limited studies suggest that Twist1 inhibition systemically may be well tolerated [68]. We conclude that TWIST1 may be an effective target for “pro-senescence” therapy for human lung cancers [67]. Our results suggest that it will only be necessary to suppress TWIST1 to a physiological level which may preclude toxicity. Our mouse model will be useful to identify agents that target TWIST1 for the treatment of human cancer.
Materials and Methods
Cell lines
The human non-small cell lung cancer cell lines, H460, H727 and A549; and embryonic kidney cell line HEK 293 T were obtained from ATCC and grown in media as recommended.
MEFs were isolated from E13.5 embryos and propagated as described previously [18]. MEFs were grown for two population doublings and then frozen for future experiments. MEFs were grown in DMEM plus 10% fetal calf serum.
Transgenic mice
The Twist1 cDNA was PCR cloned into the bidirectional tetO7 vector S2f-IMCg [33] at EcoRI and NotI sites, replacing the eGFP ORF. The resultant construct, Twist1-tetO7-luc, was sequence confirmed, digested with KpnI and XmnI to release the bidirectional transgene and then used for injection of FVB/N pronuclei by the Stanford Transgenic Facility. We ultimately obtained three founders from 25 pups after screening by tail genotyping using PCR as described below. These three founders were mated to CCSP-rtTA mice to screen for functional Twist1-tetO7-luc founders. One founder failed to pass the transgene germline and one founder did not report inducible Twist1 or luc expression. The remaining founder was used for all the experiments in this study.
We use the β-actin-rtTA, CCSP-rtTA, tetO-Kras4bG12D and LSL-K rasG12D transgenic lines [3], [34], [69]. Twist1 and/or K-rasG12D expression was activated in the CT, CR, and CRT lung lines by administering doxycycline (Sigma) to the drinking water weekly [2 mg/mL] starting at the age of 3–5 weeks. The conditional LSL-K rasG12D lines were activated by intranasal delivery of adenoviral CMV-Cre [43]. All procedures were performed in accordance with APLAC protocols and animals were housed in a pathogen-free environment.
PCR genotyping
DNA was isolated from mouse tails using the Qiaprep DNeasy kit (Qiagen). The CCSP-rtTA, tetO-K-rasG12D and LSL-K rasG12D transgenic lines were screened as described previously. The Twist1-tetO7-luc line was detected with the following primers: mTwist1-Luc.S2 5′ - CCTTATGCAGTTGCTCTCCAG -3′ and mTwist1-Luc.AS2 5′ - GCTTGCCTATGTTCTTTTGGA -3′. DNA was amplified using PCR and PCR products were resolved on a 2% agarose gel.
SYBR-green quantitative RT–PCR
Total RNA was isolated from tissue using the Qiaprep RNAeasy Kit (Qiagen) according to the manufacturer's directions. Samples were treated with RQ1 RNase-Free DNase (Promega). cDNA was generated from 1 µg of total RNA using the Superscript II kit (Invitrogen Technologies). Control reactions were run without RT enzyme. 50 ng of cDNA equivalents were amplified for the transcript described below in an ABI-prism 7700 for 40 cycles using SYBR green PCR Master mix (Perkin Elmer Applied Biosystems). PCR reactions were performed in duplicate-triplicate in a final volume of 20 µL. Following amplification, the data was processed with the analysis program Sequence Detection Systems v2.2.2 (Perkin Elmer Applied Biosystems). For each sample, the level of RNA for the genes of interest was standardized to a housekeeping gene (ubiquitin or 18S rRNA) within that sample; subsequently, the level of a transcript of interest was normalized to the expression of that transcript from the appropriate comparator sample. Primers for qPCR are listed in the Text S1.
Human normal lung and lung tumor qPCR tissue arrays and TWIST1 qPCR oligos were purchased from OriGene. All relevant clinical information can be found (http://www.origene.com/qPCR/Tissue-qPCR-Arrays.aspx).
Immunoblot analysis
Cells were lysed on ice for 60 min in radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors (Sigma-Aldrich) and clarified by centrifugation. Protein concentrations were determined by Bradford proteinassay (Bio-Rad Laboratories). Equal protein concentrations of each sample were run on NuPAGE bis-Tris gels (Invitrogen) and transferred to membranes. After being blocked with 5% dried milk in TBS containing 0.2% Tween 20, the filters were incubated with primary antibodies. The following primary antibodies were used: goat anti-Actin (C-11, Santa Cruz), mouse anti-Twist1 (TWIST2C1a, Santa Cruz), mouse anti-p21 (Ab-1, Calbiochem)), mouse monoclonal anti-p27 (F-8, Santa Cruz) After washing and incubation with horseradish peroxidase (HRP)-conjugated anti-Goat or anti-mouse IgG (Amersham), the antigen-antibody complexes were visualized by chemiluminescence (ECL detection system; Perkin Elmer).
Histology and immunohistochemistry
Tissues were fixed in 10% buffered formalin for 24 h and then transferred to 70% ethanol until embedded in paraffin. Tissue sections 5 µm thick were cut from paraffin embedded blocks, placed on glass slides and hematoxylin and eosin (H&E) or Masson's trichrome staining was performed using standard procedures. Antibodies used in our study: p21, p27, p16, vimentin (BD Pharmingen) and E-Cadherin (Cell Signaling). We performed IHC, measured K-i67 and CC3-staining as described previously [35]. For immunofluorescence (IF), Alexa488-conjugated anti-mouse and Alexa594-conjugated anti-rabbit (1∶300 dilution, Invitrogen) were used as secondary antibodies and incubated at room temperature for 30 minutes. DAPI was used as a nuclear stain and slides were mounted in aqueous mounting media (Vector Laboratories).
For EMT IF analysis double immunofluorescence was used. Vimetin-expressing cells were labelled with Alexa488 (green) and E-cadherin-expressing cells were labeled with Alexa 594 (red). To quantify cells undergoing EMT, cells that were red(low)green(high) were manually counted. A minimum of seven different fields of view per section from greater than four different animals were analyzed in total.
Lentiviral and retroviral experiments
293T cells were seeded (2.5×106 cells) in T25 flasks. shRNA constructs were obtained from the Broad RNAi Consortium. pLKO.1-shRNA scramble vector was used. Lentivirus was made using a three-plasmid system and infected using the TRC Library Production and Performance Protocols. Twenty-four hours after infection, cells were treated with 1 mg/ml puromycin and passaged once 80% confluent.
Retroviral production used ecotropic and amphotropic Phoenix packaging lines. Early passage MEFs were transduced with pWZL-Hygro vectors expressing HrasG12V or with empty vector for two successive times over a 36-h period and then followed by selection with hygromycin (100 µg/ml) for 4 days. Retroviral infections on H460 cells used pWZL-Hygro vector and pWZL-Hygro/mTwist1 constructs, for two successive times over a 36-h period and then followed by selection with hygromycin (250 µg/ml) for 4 days.
Colony formation and proliferation assays
On Day 6 after infection with the indicated shRNA lentiviruses, cells were plated in 12-well plates at a density of 5E3, 10E3 and 15E3 cells/well. On Day 12, the cells were stained with crystal violet (0.5% in 95% ethanol).
Similar low passage MEFs were used for all proliferation assays. Retroviral infections were performed as above, selection carried out for 4 days and stably selected cells were plated and then treated with or without 2 µg/ml doxyxycline for proliferation assays (Day 1). Sets of cells were removed for trypsinization and counting every 4 days. Values are normalized with Day1 readings.
SA-β-gal staining
Cells were washed twice with phosphate-buffered saline (PBS) and then fixed with PBS containing 2% formaldehyde and 0.2% glutaraldehyde for 5 min. The cells were then incubated at 37°C for 20 hr with staining solution (40 mM citric acid sodium phosphate, pH 6.0, 1 mg/ml 5-bromo-4-chloro-3-isolyl-β-D-galactoside [X-gal, Fisher], 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 150 mM NaCl, 2 mM MgCl2). After incubation, cells were washed twice with PBS and viewed with bright-field microscopy.
Small animal imaging
Micro-computed tomography (μCT) and PET scans were performed on a custom GEHC (London, Ontario) eXplore RS150 cone-beam scanner and an R4 microPET (Siemens Medical Solutions USA, Inc.), respectively, as described previously [35], [70]. Mice were screened serially every 1–2 weeks following doxycycline activation or intranasal adenoviral CMV-Cre and images were reviewed by a board certified radiation oncologist (PTT). PET images were reconstructed using the ordered-subsets expectation maximization algorithm with a spatial resolution of 1.66 to 1.85 mm. No attenuation correction or partial volume corrections were applied.
Lung tumor quantification
Micro-computed tomography (μCT) images were reviewed by a board certified radiation oncologist (PTT) on multiple index tumors in a blinded fashion (n = 2–5 tumors per mouse). Bi-dimensional measurements were made on tumors using serial examinations and tumor volumes calculated using the following equation vol = pi/6×1.65(length×width)×3/2. Volumes were normalized to the starting volume, t = 0 before doxycycline treatment, and percent tumor volume growth was then calculated by (normalized tumor vol.×100%)−100%.
Mouse xenograft model
Female NOD-SCID mice 4–5 weeks old were purchased from Harlan Laboratories. Mice were maintained under pathogen-free conditions and given food and water ad libitum in accordance with guidelines from the Johns Hopkins Animal Care and Use Committee. A549 infected with sh-Scrambled control or sh-TWIST1 shRNA, selected for 4 days as described above and then 5×105 million cells in 100 µL of Hank's solution and Matrigel (BD Biosciences) mixed 1∶1 were injected subcutaneously in the right flank. Tumor measurements were taken every 2–3 days.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. JemalASiegelRXuJWardE 2010 Cancer statistics, 2010. CA Cancer J Clin 60 277 300
2. DingLGetzGWheelerDAMardisERMcLellanMD 2008 Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455 1069 1075
3. FisherGHWellenSLKlimstraDLenczowskiJMTichelaarJW 2001 Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev 15 3249 3262
4. JiHLiDChenLShimamuraTKobayashiS 2006 The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell 9 485 495
5. PolitiKZakowskiMFFanPDSchonfeldEAPaoW 2006 Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev 20 1496 1510
6. LiDShimamuraTJiHChenLHaringsmaHJ 2007 Bronchial and Peripheral Murine Lung Carcinomas Induced by T790M-L858R Mutant EGFR Respond to HKI-272 and Rapamycin Combination Therapy. Cancer Cell 12 81 93
7. RegalesLBalakMNGongYPolitiKSawaiA 2007 Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS ONE 2 e810 doi:10.1371/journal.pone.0000810
8. JankuFStewartDJKurzrockR 2010 Targeted therapy in non-small-cell lung cancer–is it becoming a reality? Nat Rev Clin Oncol 7 401 414
9. PaoWWangTYRielyGJMillerVAPanQ 2005 KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2 e17 doi:10.1371/journal.pmed.0020017
10. EberhardDAJohnsonBEAmlerLCGoddardADHeldensSL 2005 Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 23 5900 5909
11. VerdineGLWalenskyLD 2007 The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin Cancer Res 13 7264 7270
12. MesaRA 2006 Tipifarnib: farnesyl transferase inhibition at a crossroads. Expert Rev Anticancer Ther 6 313 319
13. JohnsonBEHeymachJV 2004 Farnesyl transferase inhibitors for patients with lung cancer. Clin Cancer Res 10 4254s 4257s
14. LuoJEmanueleMJLiDCreightonCJSchlabachMR 2009 A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137 835 848
15. SchollCFrohlingSDunnIFSchinzelACBarbieDA 2009 Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 137 821 834
16. BarbieDATamayoPBoehmJSKimSYMoodySE 2009 Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462 108 112
17. ColladoMSerranoM 2010 Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10 51 57
18. SerranoMLinAWMcCurrachMEBeachDLoweSW 1997 Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88 593 602
19. LinHKChenZWangGNardellaCLeeSW 2010 Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 464 374 379
20. PuyolMMartinADubusPMuleroFPizcuetaP 2010 A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 18 63 73
21. SoucekLWhitfieldJMartinsCPFinchAJMurphyDJ 2008 Modelling Myc inhibition as a cancer therapy. Nature 455 679 683
22. ChenZFBehringerRR 1995 twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev 9 686 699
23. AnsieauSBastidJDoreauAMorelAPBouchetBP 2008 Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 14 79 89
24. LeeKEBar-SagiD 2010 Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell 18 448 458
25. Entz-WerleNStoetzelCBerard-MarecPKalifaCBrugiereL 2005 Frequent genomic abnormalities at TWIST in human pediatric osteosarcomas. Int J Cancer 117 349 355
26. HoekKRimmDLWilliamsKRZhaoHAriyanS 2004 Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res 64 5270 5282
27. KwokWKLingMTLeeTWLauTCZhouC 2005 Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res 65 5153 5162
28. MironchikYWinnardPTJrVesunaFKatoYWildesF 2005 Twist overexpression induces in vivo angiogenesis and correlates with chromosomal instability in breast cancer. Cancer Res 65 10801 10809
29. OhuchidaKMizumotoKOhhashiSYamaguchiHKonomiH 2007 Twist, a novel oncogene, is upregulated in pancreatic cancer: clinical implication of Twist expression in pancreatic juice. Int J Cancer 120 1634 1640
30. ZhangZXieDLiXWongYCXinD 2007 Significance of TWIST expression and its association with E-cadherin in bladder cancer. Hum Pathol 38 598 606
31. YangJManiSADonaherJLRamaswamySItzyksonRA 2004 Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117 927 939
32. SmitMAPeeperDS 2008 Deregulating EMT and senescence: double impact by a single twist. Cancer Cell 14 5 7
33. LoewRVignaELindemannDNaldiniLBujardH 2006 Retroviral vectors containing Tet-controlled bidirectional transcription units for simultaneous regulation of two gene activities. Journal of Molecular and Genetic Medicine 2 107 118
34. PerlAKTichelaarJWWhitsettJA 2002 Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res 11 21 29
35. TranPTFanACBendapudiPKKohSKomatsubaraK 2008 Combined Inactivation of MYC and K-Ras Oncogenes Reverses Tumorigenesis in Lung Adenocarcinomas and Lymphomas. PLoS ONE 3 e2125 doi:10.1371/journal.pone.0002125
36. SubramanianATamayoPMoothaVKMukherjeeSEbertBL 2005 Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102 15545 15550
37. JechlingerMGrunertSTamirIHJandaELudemannS 2003 Expression profiling of epithelial plasticity in tumor progression. Oncogene 22 7155 7169
38. Theilgaard-MonchKKnudsenSFollinPBorregaardN 2004 The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J Immunol 172 7684 7693
39. JiangYZhangWKondoKKlcoJMSt MartinTB 2003 Gene expression profiling in a renal cell carcinoma cell line: dissecting VHL and hypoxia-dependent pathways. Mol Cancer Res 1 453 462
40. CromerACarlesAMillonRGanguliGChalmelF 2004 Identification of genes associated with tumorigenesis and metastatic potential of hypopharyngeal cancer by microarray analysis. Oncogene 23 2484 2498
41. MahlerJFStokesWMannPCTakaokaMMaronpotRR 1996 Spontaneous lesions in aging FVB/N mice. Toxicol Pathol 24 710 716
42. NikitinAYAlcarazAAnverMRBronsonRTCardiffRD 2004 Classification of Proliferative Pulmonary Lesions of the Mouse: Recommendations of the Mouse Models of Human Cancers Consortium. Cancer Res 64 2307 2316
43. TuvesonDAShawATWillisNASilverDPJacksonEL 2004 Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5 375 387
44. FeldserDMKostovaKKWinslowMMTaylorSECashmanC 2010 Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 468 572 575
45. JunttilaMRKarnezisANGarciaDMadrilesFKortleverRM 2010 Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 468 567 571
46. BeerDGKardiaSLHuangCCGiordanoTJLevinAM 2002 Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 8 816 824
47. BhattacharjeeARichardsWGStauntonJLiCMontiS 2001 Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 98 13790 13795
48. LandiMTDrachevaTRotunnoMFigueroaJDLiuH 2008 Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE 3 e1651 doi:10.1371/journal.pone.0001651
49. StearmanRSDwyer-NieldLZerbeLBlaineSAChanZ 2005 Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am J Pathol 167 1763 1775
50. SuLJChangCWWuYCChenKCLinCJ 2007 Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 8 140
51. TalbotSGEstiloCMaghamiESarkariaISPhamDK 2005 Gene expression profiling allows distinction between primary and metastatic squamous cell carcinomas in the lung. Cancer Res 65 3063 3071
52. WachiSYonedaKWuR 2005 Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics 21 4205 4208
53. SarkisianCJKeisterBAStairsDBBoxerRBMoodySE 2007 Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol 9 493 505
54. YoungNPJacksT Tissue-specific p19Arf regulation dictates the response to oncogenic K-ras. Proc Natl Acad Sci U S A 107 10184 10189
55. JacksonELOliveKPTuvesonDABronsonRCrowleyD 2005 The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 65 10280 10288
56. KwokWKLingMTYuenHFWongYCWangX 2007 Role of p14ARF in TWIST-mediated senescence in prostate epithelial cells. Carcinogenesis 28 2467 2475
57. StasinopoulosIAMironchikYRamanAWildesFWinnardPJr 2005 HOXA5-twist interaction alters p53 homeostasis in breast cancer cells. J Biol Chem 280 2294 2299
58. MaestroRDei TosAPHamamoriYKrasnokutskySSartorelliV 1999 Twist is a potential oncogene that inhibits apoptosis. Genes Dev 13 2207 2217
59. Valsesia-WittmannSMagdeleineMDupasquierSGarinEJallasAC 2004 Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 6 625 630
60. VichalkovskiAGreskoEHessDRestucciaDFHemmingsBA 2010 PKB/AKT phosphorylation of the transcription factor Twist-1 at Ser42 inhibits p53 activity in response to DNA damage. Oncogene 29 3554 3565
61. ShiotaMIzumiHOnitsukaTMiyamotoNKashiwagiE 2008 Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene
62. YangMHHsuDSWangHWWangHJLanHY Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol 12 982 992
63. JangJWBoxerRBChodoshLA 2006 Isoform-specific Ras Activation and Oncogene Dependence in MYC and Wnt-induced Mammary Tumorigenesis. Mol Cell Biol
64. WuCHvan RiggelenJYetilAFanACBachireddyP 2007 Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A
65. VenturaAKirschDGMcLaughlinMETuvesonDAGrimmJ 2007 Restoration of p53 function leads to tumour regression in vivo. Nature
66. XueWZenderLMiethingCDickinsRAHernandoE 2007 Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature
67. NardellaCClohessyJGAlimontiAPandolfiPP 2011 Pro-senescence therapy for cancer treatment. Nat Rev Cancer 11 503 511
68. PanDFujimotoMLopesAWangYX 2009 Twist-1 is a PPARdelta-inducible, negative-feedback regulator of PGC-1alpha in brown fat metabolism. Cell 137 73 86
69. SarinKYCheungPGilisonDLeeETennenRI 2005 Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature 436 1048 1052
70. NielsenCHKimuraRHWithofsNTranPTMiaoZ 2010 PET Imaging of Tumor Neovascularization in a Transgenic Mouse Model with a Novel 64Cu-DOTA-Knottin Peptide. Cancer Res 70 9022 9030
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Inactivation of a Novel FGF23 Regulator, FAM20C, Leads to Hypophosphatemic Rickets in Mice
- Genome-Wide Association of Pericardial Fat Identifies a Unique Locus for Ectopic Fat
- Slowing Replication in Preparation for Reduction
- An Essential Role for Katanin p80 and Microtubule Severing in Male Gamete Production
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy