
Congenital Heart Disease–Causing Gata4 Mutation Displays Functional Deficits
Defects of atrial and ventricular septation are the most frequent form of congenital heart disease, accounting for almost 50% of all cases. We previously reported that a heterozygous G296S missense mutation of GATA4 caused atrial and ventricular septal defects and pulmonary valve stenosis in humans. GATA4 encodes a cardiac transcription factor, and when deleted in mice it results in cardiac bifida and lethality by embryonic day (E)9.5. In vitro, the mutant GATA4 protein has a reduced DNA binding affinity and transcriptional activity and abolishes a physical interaction with TBX5, a transcription factor critical for normal heart formation. To characterize the mutation in vivo, we generated mice harboring the same mutation, Gata4 G295S. Mice homozygous for the Gata4 G295S mutant allele have normal ventral body patterning and heart looping, but have a thin ventricular myocardium, single ventricular chamber, and lethality by E11.5. While heterozygous Gata4 G295S mutant mice are viable, a subset of these mice have semilunar valve stenosis and small defects of the atrial septum. Gene expression studies of homozygous mutant mice suggest the G295S protein can sufficiently activate downstream targets of Gata4 in the endoderm but not in the developing heart. Cardiomyocyte proliferation deficits and decreased cardiac expression of CCND2, a member of the cyclin family and a direct target of Gata4, were found in embryos both homozygous and heterozygous for the Gata4 G295S allele. To further define functions of the Gata4 G295S mutation in vivo, compound mutant mice were generated in which specific cell lineages harbored both the Gata4 G295S mutant and Gata4 null alleles. Examination of these mice demonstrated that the Gata4 G295S protein has functional deficits in early myocardial development. In summary, the Gata4 G295S mutation functions as a hypomorph in vivo and leads to defects in cardiomyocyte proliferation during embryogenesis, which may contribute to the development of congenital heart defects in humans.
Published in the journal:
. PLoS Genet 8(5): e32767. doi:10.1371/journal.pgen.1002690
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002690
Summary
Defects of atrial and ventricular septation are the most frequent form of congenital heart disease, accounting for almost 50% of all cases. We previously reported that a heterozygous G296S missense mutation of GATA4 caused atrial and ventricular septal defects and pulmonary valve stenosis in humans. GATA4 encodes a cardiac transcription factor, and when deleted in mice it results in cardiac bifida and lethality by embryonic day (E)9.5. In vitro, the mutant GATA4 protein has a reduced DNA binding affinity and transcriptional activity and abolishes a physical interaction with TBX5, a transcription factor critical for normal heart formation. To characterize the mutation in vivo, we generated mice harboring the same mutation, Gata4 G295S. Mice homozygous for the Gata4 G295S mutant allele have normal ventral body patterning and heart looping, but have a thin ventricular myocardium, single ventricular chamber, and lethality by E11.5. While heterozygous Gata4 G295S mutant mice are viable, a subset of these mice have semilunar valve stenosis and small defects of the atrial septum. Gene expression studies of homozygous mutant mice suggest the G295S protein can sufficiently activate downstream targets of Gata4 in the endoderm but not in the developing heart. Cardiomyocyte proliferation deficits and decreased cardiac expression of CCND2, a member of the cyclin family and a direct target of Gata4, were found in embryos both homozygous and heterozygous for the Gata4 G295S allele. To further define functions of the Gata4 G295S mutation in vivo, compound mutant mice were generated in which specific cell lineages harbored both the Gata4 G295S mutant and Gata4 null alleles. Examination of these mice demonstrated that the Gata4 G295S protein has functional deficits in early myocardial development. In summary, the Gata4 G295S mutation functions as a hypomorph in vivo and leads to defects in cardiomyocyte proliferation during embryogenesis, which may contribute to the development of congenital heart defects in humans.
Introduction
Congenital heart defects (CHD) are the most prevalent of all human birth defects with an estimated incidence of 6–8 per 1,000 live births [1], . Defects of cardiac septation, which encompass atrial and ventricular septal defects, may occur as an isolated defect or in combination with other cardiac malformations. Defects of atrial and ventricular septation are the most common type of CHD and account for 50% of all cases of CHD. If unrepaired, these defects result in ventricular dilation and heart failure, pulmonary overcirculation leading to pulmonary vascular disease, atrial enlargement predisposing to atrial arrhythmias and ultimately a decreased life expectancy. The etiology for atrial and ventricular septal defects is multifactorial with genetic and environmental factors playing important roles [3], [4].
Monogenic etiologies for atrial and ventricular septal defects have been primarily discovered by studying large families with autosomal dominant forms of septal defects using traditional linkage approaches [5], [6]. The first genetic etiology for atrial septal defects was the disovery that mutations in the transcription factor, TBX5, are a cause of septation defects in the setting of Holt-Oram syndrome, which is characterized by cardiac and upper limb malformations [7]. Tbx5 haploinsufficiency in mice accurately mimics the phenotype found in patients with Holt-Oram syndrome [8]. Mutations in the cardiac transcription factor, NKX2-5, were identified in families who primarily exhibited non-syndromic atrial septal defects and atrioventricular conduction abnormalities [9]. While targeted deletion of Nkx2-5 in mice causes developmental arrest during heart tube looping, haploinsufficiency of Nkx2-5 results in only subtle defects of atrial septation [10], [11]. Similarly, mutations in the cardiac transcription factor, GATA4, have also been linked to atrial and ventricular septal defects [12], [13], [14], [15], [16]. Gata4 is necessary for normal cardiac development as mice with targeted deletion of Gata4 display embryonic lethality and defects in ventral morphogenesis associated with failure to form a single ventral heart tube [17], [18]. Subsequent studies have demonstrated that Tbx5, Nkx2-5, and Gata4 interact to regulate distinct developmental processes during heart development [19], [20], [21]. While many of the human mutations are predicted to result in haploinsufficiency, little is understood about the underlying mechanism by which reduced transcription factor dosage causes defects in cardiac septation.
We reported a large pedigree with autosomal dominant congenital heart disease that was associated with a mutation of a highly conserved glycine residue to a serine at codon 296 (G296S) [12]. The affected family members had a spectrum of cardiac phenotypes, primarily atrial and ventricular septal defects and pulmonary valve stenosis [12]. In vitro experiments demonstrated that the mutant Gata4 protein had a greatly reduced affinity for its binding element with an associated decrease in transcriptional activity and disrupted a novel interaction between Gata4 and Tbx5 [12]. Subsequently, two other families have been reported with a GATA4 G296S mutation, and affected members display a similar phenotype of atrial septal defects and incompletely penetrant pulmonary valve stenosis [22]. While Gata4 has been shown to be important for several critical processes during heart development [23], [24], [25], [26], the pathogenesis of heart malformations in humans with the G296S missense mutation in GATA4 is not as well understood.
To identify the functional deficits of the GATA4 G296S mutation in vivo, we generated and characterized transgenic mice that contain the orthologous G295S mutation in the Gata4 murine locus. Here, we show that homozygous Gata4 G295S knock-in (ki) mice (Gata4 G295Ski/ki) display normal ventral morphogenesis but early embryonic lethality after the linear heart tube stage. Histologic analysis demonstrates a thin ventricular myocardium in the Gata4 G295Ski/ki embryos, which is associated with a cardiomyocyte proliferation defect. Molecular characterization of these mutant embryos demonstrates that expression of Gata4 target genes is decreased in the heart. Echocardiographic examination of heterozygote (Gata4 G295Ski/wt) mice found subtle atrial septal defects and semilunar valve stenosis. Embryonic cardiomyocytes from heterozygote mice display cardiac proliferation deficits. Consistent with functional deficits for the Gata4 G295S mutation, compound heterozygote mice harboring only a single Gata4 G295S mutant allele in the early myocardium recapitulated the phenotype seen in Gata4 G295Ski/ki embryos. These studies demonstrate the generation of a mouse model for human cardiac malformations and suggest that abnormal cardiomyocyte proliferation may contribute to human atrial and ventricular septal defects caused by mutations in GATA4.
Results
Generation and phenotypic characterization of heterozygote Gata4 G295Ski/wt mice
In order to determine the in vivo functional deficits of the human CHD-causing GATA4 G296S mutation, we generated mice harboring the orthologus mutation at codon 295 in the murine Gata4 gene (G295S). Using a previously published targeting construct, we mutated the nucleotide from G to A resulting in a glycine to serine substitution in exon 3 which contains the C-terminal zinc finger of Gata4 [27] (Figure 1A). Successfully targeted ES cell clones were identified by Southern analysis and direct sequencing and injected into host blastocysts to generate chimeras. Germline transmission of the targeted G295S knock-in allele (G295Ski) was detected by Southern blotting in chimeric mice and confirmed by direct sequencing in heterozygous and homozygous mice (Figure 1B–1C and data not shown).

Although the Gata4 G295Ski/wt heterozygote mice appeared grossly normal, we examined these mutant mice for cardiac structural and functional abnormalities using transthoracic echocardiography. In a genotype-blinded fashion, M-Mode, 2-D pulsed and color-flow Doppler studies were performed in 8 and 16 week old Gata4 G295Ski/wt mice and their wildtype littermates. Intermittent shunting of blood was noted between the left and right atria in 10/12 Gata4 G295Ski/wt mice as compared to only 1/9 wildtype littermates (p value<0.05) (Figure 2A–2C). The intermittent atrial communication defect is a patent foramen ovale, which was also found in mice heterozygous for Nkx2-5, a gene also implicated in human atrial septal defects [11]. Quantitative pulsed Doppler recordings across the pulmonary and aortic valves demonstrated mild aortic stenosis in 4/12 Gata4 G295Ski/wt mice and pulmonary stenosis in 2/12 Gata4 G295Ski/wt mouse (Figure 2A and 2D–2M). No evidence of aortic or pulmonary valve stenosis was noted in wildtype littermates (p value<0.05). The left ventricular function, chamber size and wall thickness in Gata4 G295Ski/wt mice was not statistically different from wildtype littermates (Figure S1). Histologic analysis of Gata4 G295Ski/wt hearts demonstrated the patent foramen ovale (Figure S2A–S2D) along with thickened aortic (Figure S2E–S2F) and pulmonary valve leaflets (Figure S2G–S2H).

Phenotypic characterization of homozygous Gata4 G295Ski/ki mice
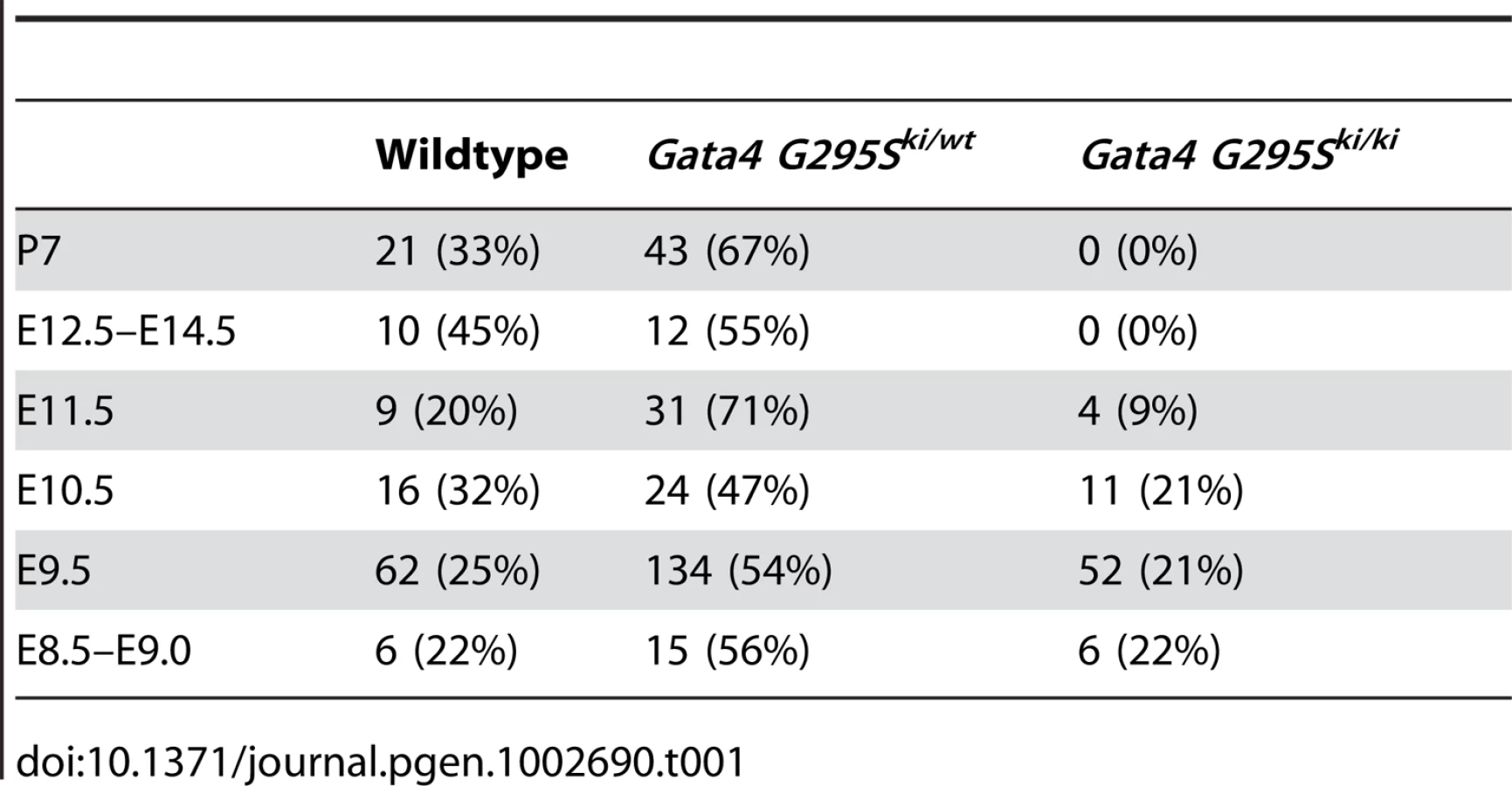
To determine the in vivo functional deficits of the Gata4 G295S mutation, we interbred Gata4 G295Ski/wt to generate Gata4 G295Ski/ki mice. Analysis at postnatal day 7 demonstrated no G295Ski/ki pups indicating that the homozygous knock-in allele is embryonic or early neonatal lethal. To determine the timing of lethality, we performed timed mating and found that Gata4 G295Ski/ki embryos did not survive beyond embryonic day (E)11.5, and normal Mendelian ratios were noted from E8.5–E10.5 (Table 1). To determine the expression levels of Gata4 mutant protein in Gata4 G295Ski/ki embryos, we extracted protein from the hearts of E9.5 Gata4 G295Ski/ki embryos and wildtype littermates. Immunoblotting demonstrated that total Gata4 protein levels in G295Ski/ki embryos were unchanged as compared to wildtype littermates, suggesting that the G295S mutant mRNA was not undergoing decay and resulting in a Gata4-deficient mouse (Figure 1D) [28].
Gross examination of Gata4 G295Ski/ki embryos demonstrated severe growth retardation compared to wildtype and heterozygote littermates at E9.5 and E10.5 (Figure 3 and Figure S3). The mutant embryos did not display defective heart tube fusion or cardiac bifida, as described in mice with targeted deletion of Gata4 [17], [18]. The mutant embryos displayed a linear heart tube and variable amounts of cardiac looping, with some appearing normal while others demonstrate incomplete or delayed looping (Figure 3A–3I and Figure S3).

To further define the morphologic defects in Gata4 G295Ski/ki embryos, histologic examination of wildtype and mutant embryos was performed. The embryonic hearts of knock-in homozygotes did not reveal any obvious defects in cardiac morphogenesis at E9.0 and E9.5 (Figure 3J, 3L and 3N and data not shown). However by E10.5, severe thinning of myocardium and associated decreased wall thickness was noted in the ventricles of Gata4 G295Ski/ki embryos compared with heterozygote littermate controls (Figure 3K, 3M and 3O). The delayed lethality and the presence of a fused heart tube as compared to Gata4-null mice suggested that the Gata4 G295S mutant protein was functioning as a hypomorph in vivo.
Expression of Gata4 target genes
In the early embryo, Gata4 is expressed in developing heart along with the visceral and parietal endoderm [29]. Numerous cardiac genes, including α-myosin heavy chain (α-MHC), cardiac troponin-C (cTNC), atrial natriuretic factor (ANF), have been shown to be direct transcriptional targets of Gata4 [29]. In vitro transactivation studies suggested that the Gata4 G295S mutation had decreased ability to activate downstream target genes [12]. To determine if the expression of direct transcriptional targets of Gata4 were altered in the Gata4 G295Ski/ki mutant hearts, we extracted RNA from E9.5 hearts and analyzed the expression of α-MHC, cTnC, ANF, and myosin light chain 3 (Myl3) by quantitative RT-PCR. The expression levels of all four genes were significantly decreased in Gata4 G295Ski/ki mutant hearts as compared to wildtype controls (Figure 4A). In contrast, the expression of non-Gata4 target genes, Tbx5 and β-MHC, was unchanged (Figure 4B). Of note, the expression of Nkx2.5, and Mef2c, cardiac transcription factors which are proposed to be direct Gata4 targets, was also unchanged (Figure 4B) [30], [31]. We also did not find any change in expression of Gata5 and Gata6 by quantitative RT-PCR (Figure S4). Radioactive in situ hybridization was performed to determine if cardiomyoctyes in Gata4 G295Ski/ki hearts displayed normal markers of differentiation. At E9.5, the expression of Tbx5, Hand1, and Hand2 was similar in G295Ski/ki hearts as compared to control littermates (Figure S5).

The cardiac bifida found in Gata4-null embryos occurs secondary to loss of Gata4 in the embryonic endoderm [17], [18], [32], [33], [34]. Gata4 G295Ski/ki embryos undergo normal heart tube fusion, suggesting that they overcome this endoderm-mediated defect. We examined the expression of Gata4-responsive endoderm genes, alpha-fetoprotein (Afp) and Sox17, along with the endoderm transcription factors, Hex1 and Hnf4, that are not direct transcriptional targets of Gata4 [35], [36]. Afp and Sox17 did not have decreased levels of expression by qRT-PCR in Gata4 G295Ski/ki embryos when compared to wildtype littermates at three different embryonic timepoints (Figure S6). Actually, the expression levels of Afp and Sox17 were somewhat increased in G295Ski/ki embryos similar to Hex1 and Hnf4, but this maybe secondary to the growth retardation found in mutant embryos (Figure S6).
Gata4 G295S mutation and deficits in cardiomyocyte proliferation
In order to determine the etiology of the thin ventricular myocardium found in E10.5 Gata4 G295Ski/ki hearts, we measured the levels of cardiomyocyte proliferation and apoptosis in the mutant hearts. In Gata4 G295Ski/ki embryos, we observed decreased cardiomyocyte proliferation, as assessed by phosphohistone H3 staining at E9.5 in Gata4 G295Ski/ki embryos as compared to littermate controls (Figure 5A–5C), but no differences in apoptosis as assessed by TUNEL staining were found (data not shown). Consistent with this finding, the mRNA levels of cyclin D2, a direct transcriptional target of Gata4 that is critical for cell proliferation, was downregulated in homozygous knock-in embryos by qRT-PCR (Figure 5D) [37]. Additionally, the ability of the Gata4 G295S mutant protein to activate cyclin D2 beta-gal reporter in HeLa cells was significantly reduced as compared to wildtype Gata4 (Figure 5E).

Interestingly, we found that mRNA levels of cyclin D2 were also decreased in the hearts of heterozygote Gata4 G295Ski/wt embryos (Figure 5D). In order to determine if subtle cardiomyocyte proliferation deficits existed in Gata4 G295Ski/wt embryos, we assessed cell proliferation utilizing a fluorescence activated cell sorting (FACS)-based strategy. Hearts were dissected from E11.5 and E13.5 embryos and cardiomyocytes were purified based on cardiac troponin-T expression [38]. Gata4 G295Ski/wt embryonic atrial and ventricular cardiomyocytes displayed decreased cell proliferation as compared to cardiomyocytes from wildtype littermates at both E11.5 and E13.5 (Figure 6A–6K). Accordingly, we found that Gata4 G295Ski/wt embryos had thinner atrial and compact ventricular layers at E12.5 (Figure 6L–6Q).

In vivo analysis of cell lineage deficits of the Gata4 G295S protein
To assess the functional deficit of the Gata4 G295S mutant protein in different cell lineages in vivo, we first generated mice that were compound heterozygotes for the Gata4 G295S mutant allele and a tissue-specific Cre, and lineage-specific deletion of the Gata4 was performed by crossing them with mice with a floxed Gata4 allele [23]. One fourth of the resultant progeny were predicted to harbor only the mutant allele in specific cardiac cell types. Tissue specific deletion was obtained by expressing Cre under the regulation of Tie2, which is expressed in endocardium, endothelium along with a subset of hematopoietic cells (presumed to be circulating endothelial progenitor cells); αMHC, which is specific for late embryonic myocardium, with robust Cre-mediated excision starting at E9.5; and Nkx2-5, which is expressed in early embryonic myocardium starting at E8.0 and also in the pharynx and liver [39], [40], [41]. Immunohistochemistry for Gata4 demonstrated decreased Gata4 expression in E10.5 embryonic hearts with the Nkx2-5 Cre (Figure S7A–S7B) and Tie2-Cre (Figure S7C–S7D) in the myocardium and endocardium, respectively. With the αMHC-Cre, expression of Gata4 was only mildly decreased in the myocardium likely related to the later onset of Cre expression (Figure S7E–S7F). Analysis of these compound heterozygotes allowed for the identification of functional deficits of the Gata4 G295S protein that were present specific to the early myocardium (emGata4G295S), late myocardium (lmGata4G295S) and endocardium (enGata4G295S). We found that only the emGata4G295S was lethal by post-natal day 10 while the lmGata4G295S and enGata4G295S demonstrated partial lethality (Table S1). Genotyping of E10.5 embryos from the crosses using the Tie2-Cre (enGata4G295S) and αMHC-Cre (lmGata4G295S) showed the expected genotypes were present in normal Mendelian ratios with no evidence of growth retardation (Figure 7A–7I). This contrasts with the severe growth retardation found in Gata4G295Ski/ki embryos (Figure 3 and Figure S3). However, embryos obtained from crosses with Gata4flox/flox and Gata4 G295Ski/wt;Nkx2-5-Cre+ (emGata4G295S) demonstrated partial lethality by E10.5 and evidence of severe growth retardation (Figure 7A and 7J–7M). Histologic examination of these three genotypes demonstrated a normal myocardial thickness in enGata4G295S and lmGata4G295S, but myocardial thinning in emGata4G295S embryos (Figure 7E, 7I, and 7M), similar to Gata4 G295Ski/ki embryos (Figure 3K and 3M). These studies support that the Gata4 G295S mutant protein has functional deficits in the early myocardium that contributes to a thin myocardium.

Discussion
The GATA4 G296S mutation has been associated with atrial septal defects and pulmonary valve stenosis in multiple human families [12], [22]. In vitro studies suggested that GATA4 G296S mutant protein resulted in specific functional deficits including diminished DNA binding affinity, reduced transcriptional activity and loss of a protein-protein interaction with TBX5. Here, we have generated a knock-in mouse harboring the corresponding G295S mutation in Gata4. Phenotypic characterization of these mice demonstrate that the Gata4 G295S mutation functions as a hypomorph in vivo as evidenced by the Gata4 G295Ski/ki embryos displaying prolonged survival as compared to Gata4-null embryos and decreased expression of Gata4 transcriptional target genes. Consistent with the cardiac phenotype seen in humans with GATA4 mutations, Gata4 G295Ski/wt mice also display cardiac abnormalities. In addition, we found that the G295S mutation of Gata4 results in defects of embryonic cardiomyocyte proliferation both in vitro and in vivo. These findings suggest a potential role for abnormal cardiomyocyte proliferation in the development of atrial and ventricular septal defects caused by mutations in GATA4.
The G295S mutation in Gata4 may result in multiple functional deficits in the embryonic heart. Mice homozygous for the Gata4 G295S mutation suffer embryonic lethality before E10.5, which limited our analysis to early myocardial development and precluded analysis of potential functional deficits at later stages of heart development. Conditional deletion of Gata4 in the endocardium results in defective endocardial-mesenchymal transition (EMT) and hypoplastic endocardial cushions [26]. In addition, we have previously shown the G295S mutation in Gata4 inhibits an interaction with Tbx5 and Gata4-Tbx5 compound heterozygotes display atrioventricular septal defects [12], [21]. It remains unclear if the GATA4 G296S mutation has a role in formation of the endocardial cushions but one family member with the G296S mutation did have an atrioventricular septal defect [12] and other non-related individuals with GATA4 mutations have atrioventricular septal defects [13]. In our analysis of potential cell lineage deficits for the Gata4 G295S protein, we did find that enGata4G295S mice, where the endocardium/endothelium expresses only the mutant protein, have somewhat hypocellular endocardial cushions and display partial lethality by post-natal day 10. This finding suggests that the Gata4 G295S mutation may have functional deficits in the endocardium, potentially by disrupting EMT but additional study is required (Figure 7). A role for Gata4 and Tbx5 in the endocardium for formation of the atrial septum by regulation of endothelial nitric oxide synthase (Nos3) has been proposed, but the early lethality of the Gata4 G295Ski/ki embryos did not allow for analysis of this pathway [42]. While this early lethality precluded examination of the development of the cardiac outflow tract and semilunar valves in the Gata4 G295Ski/ki embryos, the Gata4 G295Ski/wt heterozygotes did develop stenosis of the semilunar valves. This phenotype along with recent publications demonstrating bicuspid aortic valve and aortic valve stenosis in Gata5-null and Gata4;Gata5 compound heterozygote mice suggest that endocardial specific deficits of the G296S protein may contribute to the aortic and pulmonary valve stenosis [43], [44].
The phenotypic analysis of Gata4 G295Ski/ki mice demonstrates that the Gata4 G295 mutant allele is not a loss of function allele. Initial human genetic studies demonstrated that haploinsufficiency of GATA4 resulted in cardiac malformations and there was a possibility that the Gata4 G295 was a null allele [45]. Our data demonstrate that the G295S mutation in Gata4 results in a selective loss of some Gata4 functions. Specifically, the Gata4 G295S protein is able to activate downstream target genes in the developing endoderm but not in the cardiac mesoderm. Potential mechanisms for this difference may lie in the inability of Gata4 G295S protein to interact with Tbx5 in the mesoderm to activate downstream targets or that Gata4 may function as a transcriptional co-activator in the endoderm and DNA binding is not necessary. The findings from the conditional deletion crosses, as discussed above, also support that the G295S mutant protein has defined functional deficits in different cell lineages. Additional investigation is needed to identify all the potential functions of Gata4 and then determine which are loss with the Gata4 G295S mutation.
Cardiomyocyte proliferation is critical for normal cardiac development, and our findings provide evidence that the Gata4 G295S mutation results in myocardial hypoplasia due to diminished cardiac proliferation. This phenotype is at least partially mediated by reduced expression of cyclin D2, a member of the D-cyclin family of cell cycle regulators. Mice lacking any single D-cyclin are viable and do not display obvious cardiac defects [46]. However, compound mutation of all three D-cyclin genes results in embryonic lethality due to cellular proliferation defects, including reduced cardiomyocyte cell division [47]. Our data demonstrate that both homozygote and heterozygote Gata4 G295Ski mice display cardiomyocyte proliferation deficits and suggest that this is a possible mechanism for the atrial septal defects seen in these mice.
The generation of the Gata4 G295Ski mice provides a mouse model to study human congenital heart defects. This mouse model will be of significant value to study genetic and environment modifiers for cardiac malformations along with allowing for a more mechanistic understanding of the embryologic basis of septation and valvular defects. While the dosage sensitivity of cardiac transcription factors for normal cardiac morphogenesis is generally well accepted, this mouse model demonstrates that specific mutations may have limited functional deficits. The Gata4 G295Ski mice which encodes a partially functional mutant protein, offers us a tool to define these abnormalities in vivo.
Materials and Methods
Ethics statement
Research was approved by the Institutional Animal Care and Use Committee at University of Texas Southwestern Medical Center (Protocol No. 2008-0094) and Research Institute at Nationwide Children's Hospital (Protocol No. AR09-00040) and conforms to the Guide for the Care and Use of Laboratory Animals.
Gene-targeted mutagenesis
A strategy similar to Crispino et al., 2001 was used to generate knock-in mice harboring the Gata4 G295S mutation. The targeting vector contains an 8.2 kb mouse genomic fragement, which has the 2nd–6th exons of murine Gata4. The construct contains a Neomycin resistance cassette flanked by loxP sites, as a positive selection marker, and HSV-tk, as a negative selection marker. Genomic DNA containing the N-terminal of the zinc finger domain of Gata4 was subcloned into pBluescript II KS (+/−) phagemid (Stratagene). By site-directed mutagenesis, glycine at codon 295 was changed to serine (the codon GGC was changed to AGC). The targeting construct was linearized with PvuI and electroporated into 129SvES cells. Targeted clones were identified by Southern blotting. Six successful targeted ES clones identified by Southern blotting and direct sequencing for the G295S point mutation. Three clones were injected into C57BL/6 blastocysts to generate chimeric mice.
Mouse strains and genotyping
Germline transmission was achieved by mating to C57BL/6 mice. Mice used in this study were on a mixed 129SvEv/C57BL6 genetic background. For genotyping, allelic discrimination assay was used to detect this single nucleotide change in Gata4 locus by using fluorescent probes. This method combines PCR and mutation detection in a single step. Two TaqMan (Applied Biosystems, CA) probes were used, one for each allele. This method is implemented using the Applied Biosystems 7500Fast and TaqMan reagents to detect this point mutation in Gata4. The probe and primer sequences are shown in Table S2.
Breeding and collection of mouse embryos
Mice were maintained on a 0600 to 1800h light–dark cycle, with noon of the day of observation of a vaginal plug defined as E0.5. Mice heterozygous for Gata4 G295S mutation were generated and genotyped as described above. Mice heterozygote for Gata4 G295S were mated to generate G295S homozygote embryos. Pregnant mothers were sacrificed at various embryonic timepoints. Littermates were used as controls for histologic sections, gene expression studies and FACS analysis.
Echocardiographic imaging
Two dimensional and Doppler in vivo ultrasound images were obtained in 8 and 16 week old mice using a VisualSonics Vevo2100 imaging system (Ontario, Canada) with a mechanical transducer (MS400). Mice were anesthetized with isoflourane and echocardiograms were performed in a genotype-blinded fashion. Statistical analysis was performed using Fisher's exact test.
Histologic section and radioactive in situ hybridization
For histological analysis, embryos and adult hearts were fixed in 4% paraformaldehyde and paraffin embedded. Hematoxylin and eosin (H and E) staining was carried out on heart sections using standard methodology. In situ hybridization was performed as described previously [48] using 35S-labeled antisense probes synthesized with T3, T7, or SP6 RNA polymerase (Maxiscript; Ambion Inc., Austin, TX) from mouse Hand1, Hand2 and Tbx5 cDNA.
Proliferation and apoptosis assays
For the immunostaining studies, histologic sections were deparafinnized in xylene and rehydrated in phosphate buffered saline (PBS). Proliferation assays were performed using the phosphohistone H3 (PH3) antibody (Upstate Cell Signaling Solutions, Temecula, CA). The sections were permeabilized in 0.3% Triton X-100 in PBS. Sections were then blocked by 3.5% donkey serum in PBS followed by incubation with 1% rabbit anti-phosphohistone H3 antibody overnight at 4°C. Sections were then washed in PBS and Cy3 (1%) secondary antibodies (Vector Laboratories, Burlingame, CA) for 30 min. For cell proliferation studies, contiguous sections were stained for monoclonal mouse anti-human desmin using Cy3-conjugated antibody (Dako, Carpinteria, CA) to label the cardiomyocytes. The percentage of PH3-stained ventricular cardiomyocytes/total number of ventricular cardiomyocytes was calculated by analyzing a minimum of three embryos for each genotype. A minimum of four sections per embryo were analyzed, and the means and standard deviations are shown. Apoptosis (TUNEL) assays were performed using the In Situ Cell Death Detection Kit, Fluorescein (Roche) according to manufacturer instructions. Labeled ventricular cardiomyocytes were counted on a minimum of six sections of control and mutant embryonic hearts. Statistical analysis was performed using Student's t-test.
Gene expression analysis
RNA was purified from embryonic hearts (E8.5–E10.5) from mutant embryos and their respective wildtype littermates using Trizol (Invitrogen). Real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed using the Taqman Universal PCR Master Mix kit (Applied Biosystems, Foster City, CA). 100 ng of total RNA was used for reverse transcription and amplification in each real-time PCR reaction using Applied Biosystems 7500 real-time PCR machine. Commercially available SYBR Green (Applied Biosystems) PCR mix was utilized for the following genes: ANF, α-MHC, cTnC, Myl3, CyclinD2, Nkx2-5, Tbx5, Mef2C, β-MHC, HNF4, Hex1, alpha-fetoprotein and Sox17. The sequences for primers are in Table S2. Mean relative gene expression was calculated from wildtype and mutant hearts after normalization to 18S ribosomal RNA, minimum of n = 3 per group. Statistical analysis was performed using Student's t-test, and a p value of less than 0.05 was considered significant.
Transactivation assays
HeLa cells were transfected using Fugene 6 (Roche) according to manufacturer's instructions with 200 ng of Gata4 wildtype and Gata4 G295S myc-tagged expression vectors [12], 200 ng of Cyclin D2 pAUG-β-gal reporter vector [37]. Immunoblots were used to verify appropriate expression. Cells were cultured for 48 h after transfection, harvested and cellular extracts were prepared by sonication and normalized as described previously [49]. Chemiluminescence β-galactosidase (β-Gal) assays were performed using the luminescent β-Gal detection system (Clontech) according to the manufacturer's recommendations, and relative light units were detected using a Tropix TR717 microplate luminometer (PE Applied Biosystems).
Flow cytometry
Embryonic heart samples for fluorescence-activated cell sorting (FACS) were prepared in the following manner: 15–20 embryonic hearts of each genotype were dissected, dissociated to a single cell solution, digested with collagenase type II (Worthington) solution, washed, spun down and resuspended in cardiomyocyte staining buffer. Cells were fixed with BD Cytofix/CytopermTM solution, permeabilized and incubated with monoclonal mouse anti-troponin T (Abcam, Cambridge, MA) and Ki67 (Abcam, Cambridge, MA ). Experiments were performed in triplicate and cells were analysed on a LSRII with DiVa software (BD Biosciences, San Jose, CA, USA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HoffmanJIKaplanS 2002 The incidence of congenital heart disease. J Am Coll Cardiol 39 1890 1900
2. BottoLDCorreaAEricksonJD 2001 Racial and temporal variations in the prevalence of heart defects. Pediatrics 107 E32
3. PierpontMEBassonCTBensonDWJrGelbBDGigliaTM 2007 Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115 3015 3038
4. JenkinsKJCorreaAFeinsteinJABottoLBrittAE 2007 Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115 2995 3014
5. SrivastavaD 2006 Making or breaking the heart: from lineage determination to morphogenesis. Cell 126 1037 1048
6. GargV 2006 Insights into the genetic basis of congenital heart disease. Cell Mol Life Sci 63 1141 1148
7. BassonCTBachinskyDRLinRCLeviTElkinsJA 1997 Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15 30 35
8. BruneauBGNemerGSchmittJPCharronFRobitailleL 2001 A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106 709 721
9. SchottJJBensonDWBassonCTPeaseWSilberbachGM 1998 Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281 108 111
10. LyonsIParsonsLMHartleyLLiRAndrewsJE 1995 Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev 9 1654 1666
11. BibenCWeberRKestevenSStanleyEMcDonaldL 2000 Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circulation research 87 888 895
12. GargVKathiriyaISBarnesRSchlutermanMKKingIN 2003 GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424 443 447
13. RajagopalSKMaQOblerDShenJManichaikulA 2007 Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol 43 677 685
14. Tomita-MitchellAMaslenCLMorrisCDGargVGoldmuntzE 2007 GATA4 sequence variants in patients with congenital heart disease. J Med Genet 44 779 783
15. ZhangWLiXShenAJiaoWGuanX 2008 GATA4 mutations in 486 Chinese patients with congenital heart disease. Eur J Med Genet 51 527 535
16. ButlerTLEspositoGBlueGMColeADCostaMW 2010 GATA4 mutations in 357 unrelated patients with congenital heart malformation. Genet Test Mol Biomarkers 14 797 802
17. MolkentinJDLinQDuncanSAOlsonEN 1997 Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev 11 1061 1072
18. KuoCTMorriseyEEAnandappaRSigristKLuMM 1997 GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev 11 1048 1060
19. HiroiYKudohSMonzenKIkedaYYazakiY 2001 Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet 28 276 280
20. MoskowitzIPKimJBMooreMLWolfCMPetersonMA 2007 A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell 129 1365 1376
21. MaitraMSchlutermanMKNicholsHARichardsonJALoCW 2009 Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol 326 368 377
22. SarkozyAContiENeriCD'AgostinoRDigilioMC 2005 Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet 42 e16
23. PuWTIshiwataTJuraszekALMaQIzumoS 2004 GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Dev Biol 275 235 244
24. WattAJBattleMALiJDuncanSA 2004 GATA4 is essential for formation of the proepicardium and regulates cardiogenesis. Proc Natl Acad Sci U S A 101 12573 12578
25. ZeisbergEMMaQJuraszekALMosesKSchwartzRJ 2005 Morphogenesis of the right ventricle requires myocardial expression of Gata4. J Clin Invest 115 1522 1531
26. Rivera-FelicianoJLeeKHKongSWRajagopalSMaQ 2006 Development of heart valves requires Gata4 expression in endothelial-derived cells. Development 133 3607 3618
27. CrispinoJDLodishMBThurbergBLLitovskySHCollinsT 2001 Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev 15 839 844
28. ChangYFImamJSWilkinsonMF 2007 The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76 51 74
29. MolkentinJD 2000 The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem 275 38949 38952
30. BrownCO3rdChiXGarcia-GrasEShiraiMFengXHSchwartzRJ 2004 The cardiac determination factor, Nkx2-5, is activated by mutual co-factors GATA-4 and Smad1/4 via a novel upstream enhancer. J Biol Chem 279 10659 10669
31. DodouEVerziMPAndersonJPXuSMBlackBL 2004 Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development 131 3931 3942
32. RojasASchachterleWXuSMMartinFBlackBL 2010 Direct transcriptional regulation of Gata4 during early endoderm specification is controlled by FoxA2 binding to an intronic enhancer. Dev Biol 346 346 355
33. HoltzingerARosenfeldGEEvansT 2010 Gata4 directs development of cardiac-inducing endoderm from ES cells. Dev Biol 337 63 73
34. DuncanSANagyAChanW 1997 Murine gastrulation requires HNF-4 regulated gene expression in the visceral endoderm: tetraploid rescue of Hnf-4(−/−) embryos. Development 124 279 287
35. ArtusJPiliszekAHadjantonakisAK 2011 The primitive endoderm lineage of the mouse blastocyst: sequential transcription factor activation and regulation of differentiation by Sox17. Dev Biol 350 393 404
36. SoudaisCBielinskaMHeikinheimoMMacArthurCANaritaN 1995 Targeted mutagenesis of the transcription factor GATA-4 gene in mouse embryonic stem cells disrupts visceral endoderm differentiation in vitro. Development 121 3877 3888
37. RojasAKongSWAgarwalPGillissBPuWT 2008 GATA4 is a direct transcriptional activator of cyclin D2 and Cdk4 and is required for cardiomyocyte proliferation in anterior heart field-derived myocardium. Mol Cell Biol 28 5420 5431
38. WalshSPontenAFleischmannBKJovingeS 2010 Cardiomyocyte cell cycle control and growth estimation in vivo–an analysis based on cardiomyocyte nuclei. Cardiovasc Res 86 365 373
39. KisanukiYYHammerREMiyazakiJWilliamsSCRichardsonJA 2001 Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 230 230 242
40. GaussinVVan de PutteTMishinaYHanksMCZwijsenA 2002 Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc Natl Acad Sci U S A 99 2878 2883
41. McFaddenDGBarbosaACRichardsonJASchneiderMDSrivastavaD 2005 The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development 132 189 201
42. NadeauMGeorgesROLaforestBYamakALefebvreC 2010 An endocardial pathway involving Tbx5, Gata4, and Nos3 required for atrial septum formation. Proc Natl Acad Sci U S A 107 19356 19361
43. LaForestBAndelfingerGNemerM 2011 Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest 131 2876 2887
44. LaForestBNemerM 2011 GATA5 interacts with GATA4 and GATA6 in outflow tract development. Dev Biol 358 368 378
45. PehlivanTPoberBRBruecknerMGarrettSSlaughR 1999 GATA4 haploinsufficiency in patients with interstitial deletion of chromosome region 8p23.1 and congenital heart disease. Am J Med Genet 83 201 206
46. SherrCJRobertsJM 2004 Living with or without cyclins and cyclin-dependent kinases. Genes Dev 18 2699 2711
47. KozarKCiemerychMARebelVIShigematsuHZagozdzonA 2004 Mouse development and cell proliferation in the absence of D-cyclins. Cell 118 477 491
48. GargVYamagishiCHuTKathiriyaISYamagishiH 2001 Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev Biol 235 62 73
49. SchlutermanMKKrysiakAEKathiriyaISAbateNChandaliaM 2007 Screening and biochemical analysis of GATA4 sequence variations identified in patients with congenital heart disease. Am J Med Genet Part A 143A 817 823
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Inactivation of a Novel FGF23 Regulator, FAM20C, Leads to Hypophosphatemic Rickets in Mice
- Genome-Wide Association of Pericardial Fat Identifies a Unique Locus for Ectopic Fat
- Slowing Replication in Preparation for Reduction
- An Essential Role for Katanin p80 and Microtubule Severing in Male Gamete Production
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy