
Deletion of PTH Rescues Skeletal Abnormalities and High Osteopontin Levels in Mice
Maintenance of normal mineral ion homeostasis is crucial for many biological activities, including proper mineralization of the skeleton. Parathyroid hormone (PTH), Klotho, and FGF23 have been shown to act as key regulators of serum calcium and phosphate homeostasis through a complex feedback mechanism. The phenotypes of Fgf23−/− and Klotho−/− (Kl−/−) mice are very similar and include hypercalcemia, hyperphosphatemia, hypervitaminosis D, suppressed PTH levels, and severe osteomalacia/osteoidosis. We recently reported that complete ablation of PTH from Fgf23−/− mice ameliorated the phenotype in Fgf23−/−/PTH−/− mice by suppressing serum vitamin D and calcium levels. The severe osteomalacia in Fgf23−/− mice, however, persisted, suggesting that a different mechanism is responsible for this mineralization defect. In the current study, we demonstrate that deletion of PTH from Kl−/− (Kl−/−/PTH−/− or DKO) mice corrects the abnormal skeletal phenotype. Bone turnover markers are restored to wild-type levels; and, more importantly, the skeletal mineralization defect is completely rescued in Kl−/−/PTH−/− mice. Interestingly, the correction of the osteomalacia is accompanied by a reduction in the high levels of osteopontin (Opn) in bone and serum. Such a reduction in Opn levels could not be observed in Fgf23−/−/PTH−/− mice, and these mice showed sustained osteomalacia. This significant in vivo finding is corroborated by in vitro studies using calvarial osteoblast cultures that show normalized Opn expression and rescued mineralization in Kl−/−/PTH−/− mice. Moreover, continuous PTH infusion of Kl−/− mice significantly increased Opn levels and osteoid volume, and decreased trabecular bone volume. In summary, our results demonstrate for the first time that PTH directly impacts the mineralization disorders and skeletal deformities of Kl−/−, but not of Fgf23−/− mice, possibly by regulating Opn expression. These are significant new perceptions into the role of PTH in skeletal and disease processes and suggest FGF23-independent interactions of PTH with Klotho.
Published in the journal:
. PLoS Genet 8(5): e32767. doi:10.1371/journal.pgen.1002726
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002726
Summary
Maintenance of normal mineral ion homeostasis is crucial for many biological activities, including proper mineralization of the skeleton. Parathyroid hormone (PTH), Klotho, and FGF23 have been shown to act as key regulators of serum calcium and phosphate homeostasis through a complex feedback mechanism. The phenotypes of Fgf23−/− and Klotho−/− (Kl−/−) mice are very similar and include hypercalcemia, hyperphosphatemia, hypervitaminosis D, suppressed PTH levels, and severe osteomalacia/osteoidosis. We recently reported that complete ablation of PTH from Fgf23−/− mice ameliorated the phenotype in Fgf23−/−/PTH−/− mice by suppressing serum vitamin D and calcium levels. The severe osteomalacia in Fgf23−/− mice, however, persisted, suggesting that a different mechanism is responsible for this mineralization defect. In the current study, we demonstrate that deletion of PTH from Kl−/− (Kl−/−/PTH−/− or DKO) mice corrects the abnormal skeletal phenotype. Bone turnover markers are restored to wild-type levels; and, more importantly, the skeletal mineralization defect is completely rescued in Kl−/−/PTH−/− mice. Interestingly, the correction of the osteomalacia is accompanied by a reduction in the high levels of osteopontin (Opn) in bone and serum. Such a reduction in Opn levels could not be observed in Fgf23−/−/PTH−/− mice, and these mice showed sustained osteomalacia. This significant in vivo finding is corroborated by in vitro studies using calvarial osteoblast cultures that show normalized Opn expression and rescued mineralization in Kl−/−/PTH−/− mice. Moreover, continuous PTH infusion of Kl−/− mice significantly increased Opn levels and osteoid volume, and decreased trabecular bone volume. In summary, our results demonstrate for the first time that PTH directly impacts the mineralization disorders and skeletal deformities of Kl−/−, but not of Fgf23−/− mice, possibly by regulating Opn expression. These are significant new perceptions into the role of PTH in skeletal and disease processes and suggest FGF23-independent interactions of PTH with Klotho.
Introduction
Maintaining normal mineral ion homeostasis is crucial for essential biological activities that include but are not limited to energy metabolism, signaling activities, and normal skeletal growth, development and function. Blood calcium and phosphate levels are determined by counterbalance between absorption from the intestine, mobilization from bone and excretion from the kidney into urine [1]. This complex process is regulated by several endocrine factors, including parathyroid hormone (PTH), FGF23 and active Vitamin D, which have been widely studied [2]–[5]. More recently, another protein, Klotho, has been suggested to have an important role in regulating calcium and phosphate homeostasis.
Klotho is a type-I membrane protein mainly expressed in kidneys, parathyroid glands, and the choroid plexus [6]. It is also related to β-glucosidases and is found in a soluble form in blood and cerebrospinal fluid [7], [8]. Klotho forms a complex with the FGF receptor 1c (FGFR1c), thereby converting this canonical FGF receptor into a receptor specific for FGF23 [9], a negative regulator of serum phosphate. FGF23 uses the FGFR1c/Klotho complex to directly target the kidney where it induces phosphate wasting by decreasing the expression of the sodium-dependent phosphate co-transporters NaPi2a and NaPi2c [10], [11]. Klotho also regulates serum calcium by affecting both parathyroid gland and kidney independent of FGF23. When serum calcium is low, Klotho hydrolyzes extracellular sugar residues on the renal transepithelial calcium channel TRPV5, entrapping the channel in the plasma membrane [12]. This maintains continuous calcium channel activity and membrane calcium permeability, leading to an increase in tubular reabsorption of calcium in the kidneys and finally increased serum calcium. In the parathyroid gland, Klotho recruits Na+/K+-ATPase to the cell surface, which results in an increase in PTH production, which in turn elevates serum calcium level [13], [14]. In addition to Klotho's independent effect on PTH induction, it can act together with FGF23 to decrease PTH levels [15], [16]. An in vivo study has shown that injection of FGF23 protein into rats can lower PTH secretion and expression [15]. This was confirmed by in vitro experiments using parathyroid gland cultures [16].
The function of Klotho as a cofactor of FGF23 was confirmed in studies by us and others showing that genetic ablation of either Fgf23 or Klotho results in a similar phenotype [17]–[19]. Both Klotho knockout (Kl−/−) and Fgf23 knockout (Fgf23−/−) mice exhibit hypercalcemia, hyperphosphatemia with low to undetectable PTH levels [20]–[22], and severe osteomalacia. We have previously described [23] that deletion of PTH in Fgf23−/− mice ameliorated the abnormal phenotype by normalizing serum Ca2+ and lowering serum vitamin D levels, however, the severe osteomalacia persisted in Fgf23−/−/PTH−/− mice. Because Klotho also has FGF23-independent functions, we thought it would be important to investigate the effects of deleting PTH from Kl−/− mice. We demonstrate that the skeletal mineralization defect in Kl−/−/PTH−/− mice was completely rescued and that this phenomenon was accompanied by a reduction in the high levels of osteopontin in bone and serum, a finding that could not be observed in Fgf23−/−/PTH−/− mice. We also present data showing that continuous infusion of Kl−/− mice with PTH results in an elevation in OPN levels and subsequently increased osteoid volume. Our finding demonstrates for the first time that the skeletal abnormalities and the bone mineralization defect in Kl−/− can be rescued by ablation of PTH actions. Our data suggest regulatory actions on osteopontin by PTH, an important observation with clinical significance. The identical levels in serum calcium, phosphate and vitamin D in Fgf23−/−/PTH−/− and Kl−/−/PTH−/− preclude any effects of these parameters on the regulation of skeletal mineralization and/or Opn levels in these mice. Additional studies are required to identify the mechanisms by which PTH affects osteopontin and mineralization in Kl−/− but not in Fgf23−/−mice.
Results
Deletion of PTH results in healthier Kl−/−/PTH−/− double-knockout animals
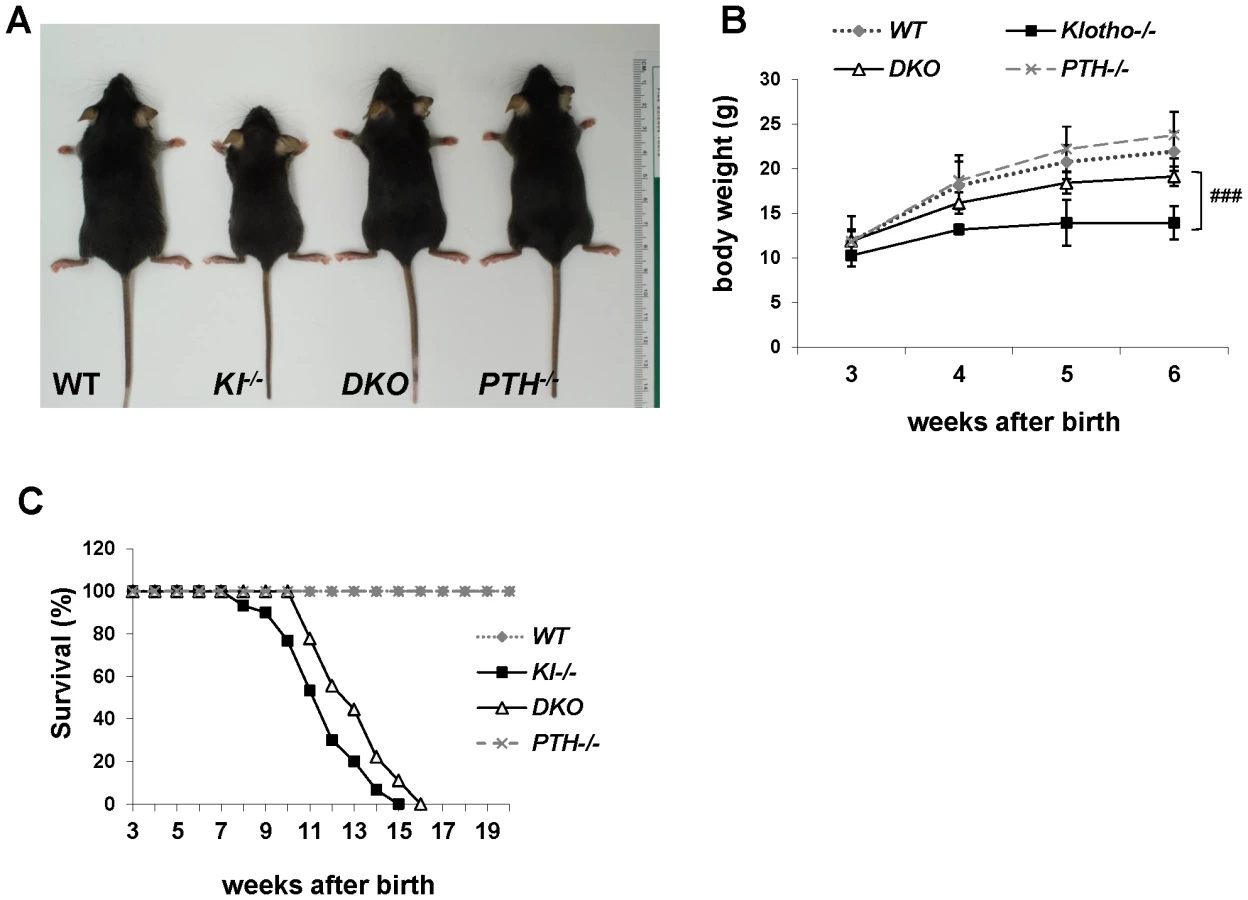
We successfully generated Kl−/−/PTH−/− (DKO) mice by interbreeding heterozygous Kl+/− and PTH+/− mice. DKO mice were more active, healthier and larger in size than Kl−/− mice and more comparable to wild-type and PTH−/− single knock-out littermates (Figure 1A). DKO mice did not show any obvious gross abnormalities with regard to movement and physical activities, whereas Kl−/− littermates were severely weakened, showing restricted movement as well as sluggish physical activities. DKO mouse body weight was significantly higher than that of Kl−/− mice (Figure 1B). Compared to the Kl−/− mice, DKO mice also showed a clear improvement in life span as evidenced by a right shift of the survival curve (Figure 1C). All mice, however, died before 16 weeks of age, probably due to the severe soft tissue calcifications such as found in kidney and lung of both Kl−/− and DKO mice (Figure S1).
Serum biochemistry
Six-week old Kl−/− and PTH−/− mice were severely hypercalcemic (10.86±0.52 mg/dL) and hypocalcemic (6.85±1.17 mg/dL), respectively. However DKO animals were normocalcemic (8.78±0.42 mg/dL) at 6 weeks, comparable to WT control animals (9.52±0.41 mg/dL), (Figure 2A). Serum phosphate levels in both Kl−/− (14.61±0.48 mg/dL) and PTH−/− (14.52±1.87 mg/dL) mice were significantly higher compared to those in WT mice (9.75±1.34 mg/dL), (Figure 2B). Interestingly, DKO exhibited a further increase in serum phosphate to levels (17.65±1.86 mg/dL) far exceeding those in single Kl−/− or PTH−/− mice. We determined the total mineral content by calculating the calcium/phosphate product and found that Kl−/− and DKO mice exhibited similarly high levels (Figure 2C). Measurements of serum 1,25(OH)2D levels showed increased amounts in Kl−/− single knockout mice compared to those in WT and PTH−/− mice. Serum 1,25(OH)2D levels in DKO mice were significantly reduced compared to those of Kl−/− single knockout mice, but were still significantly higher than in wild-type or PTH−/− mice (Figure 2D). We also measured intact serum FGF23 levels and found that DKO mice had a 50% decrease in serum FGF23 compared to Kl−/− mice, however the levels were still significantly higher (1000 fold) than those in wild-type or PTH−/− mice (Figure 2E).

Deletion of PTH rescues skeletal abnormalities of Kl−/− mice
We performed peripheral quantitative computerized tomography (pQCT) to analyze the bone density in the femurs of all genotypes. Kl−/− mice showed decreased total BMD in the distal femur metaphysis (Figure 3A). Ablation of the PTH gene from these mice significantly increased the BMD, which was now comparable to that of WT and PTH−/− mice.

Radiographs showed that the length and radiopacity of the tibiae from DKO mice were increased and comparable to those of WT and PTH−/− mice (Figure 3B). We performed Alizarin red S and Alcian blue staining to determine the mineralization pattern of the bones. As shown in Figure 3C, Kl−/− mice exhibited abnormally widened ribs, but this abnormality could not be observed in the DKO mice, suggesting an improved skeletal architecture.
MicroCT (μCT) analysis of the femurs was performed on all four genotypes. Representative images of distal femoral metaphyses and midshaft cortex are shown in Figure 3D and 3E. Quantification of trabecular bone volume fraction demonstrated that there is no significant difference between each genotype (Figure 3F). As μCT can only detect mineralized bone, Kl−/− mice didn't show increased trabecular volume. However, the midshaft cortical thickness of the Kl−/− mice (0.132±0.013 mm) was significantly reduced compared to the other groups (Figure 3G). It was restored in DKO mice (0.170±0.015 mm) to a volume comparable to that in WT (0.184±0.011 mm) and PTH−/− (0.186±0.016 mm) mice (Figure 3G).
We further analyzed the skeletal properties by generating undecalcified methylmethacrylate sections from the distal ends of femurs to confirm the observed improvement in bone mass compared to Kl−/− mice (Figure 4A and 4B). Most importantly, the severe osteoidosis seen in the secondary spongiosa of Kl−/− mice was completely absent in DKO mice (Figure 4B), indicating the mineralization defect of Kl−/− mice was rescued by PTH ablation. To quantify this observation, we performed histomorphometric analyses (Figure 4C–4N). The increased trabecular bone volume observed in Kl−/− mice (18.1±4.4) was restored in DKO mice (13.5±1.9) to values close to those of PTH−/− mice (11.4±1.6). Interestingly, deletion of PTH also rescued the severe mineralization defect of Kl−/− mice as evidenced by normalized osteoid volume (OV/TV), osteoid surface (OS/BS) and thickness (OTh) in DKO mice (Figure 4D–4F). DKO mice also showed normal trabecular thickness (Tb.Th.), trabecular number (Tb.N.) and separation (Tb.Sp.), as well as osteoclast surface (Oc.S/BS) and osteoclast numbers (N.Oc/B.Pm) (Figure 4G–4K). We also found that the dynamic parameters, including mineral surface (MS/BS), bone formation rate (BFR/BS) and mineral apposition rate (MAR) (Figure 4L–4N), were normalized in DKO mice while the bone labeling in Kl−/− mice was unsuccessful due to the severe mineralization defect.

We next analyzed the concentration of serum markers for bone turnover. Consistent with histomorphometric data, serum levels of the carboxyl-terminal telopeptide of type 1 collagen (CTX), a biomarker of bone resorption activity, were comparable in DKO (31.5±17.2 ng/ml), WT (40.8.5±16.2 ng/ml) and PTH−/− (31.5±3.6 ng/ml) mice (Figure 5A). Similarly, circulating levels of N-terminal propeptide of type I procollagen (PINP), a reliable and sensitive marker of bone formation, were significantly elevated in Kl−/− mice (21.8±6.0 ng/ml) (Figure 5B). PINP levels were restored in DKO mice (14.5±5.0 ng/ml) to levels observed in WT (14.2±4.6 ng/ml) and PTH−/− (17.0±1.6 ng/ml).

Rescued bone mineralization is accompanied by normalized expression of Opn in DKO mice
To explain the rescue in bone mineralization in DKO mice, we compared the expression of osteopontin and other factors associated with skeletal mineralization in all genotypes. As shown by in situ hybridization and immunohistochemical staining on decalcified paraffin sections (Figure 6A and 6B), the expression of Opn, an inhibitor of osteogenic mineralization and member of the SIBLING protein family, is abnormally high in the bone. Interestingly, its expression was normalized in DKO mice to levels seen in WT and PTH−/− mice (Figure 6A and 6B). We also measured serum Opn levels using an ELISA kit and were able to confirm significantly elevated serum Opn levels in Kl−/− mice, which were restored to normal levels in DKO mice (Figure 6C). Since we previously reported increased Opn expression in Fgf23−/− bones [17], we were interested in also examining their serum Opn levels and found that they were also significantly increased. In contrast, however, deletion of PTH from Fgf23−/− mice failed to normalize their serum Opn levels (Figure S2).

To further confirm the in vivo observations in Kl−/− and DKO mice calvarial osteoblasts from 2-day-old littermates were isolated. Cells were cultured in osteogenic medium for 2 weeks and RNA was isolated. qPCR analyses showed normal expression of Opn in osteoblasts of DKO mice (Figure 6D). These osteoblasts also exhibited normal mineralization as evaluated by Alizarin red staining while the mineralization of the osteoblasts from Kl−/− mice was markedly impaired (Figure 6E). We also evaluated the expression of Dmp1 and Matrix gla protein (Mgp) by in situ hybridization and/or qPCR. Both were significantly increased in Kl−/− mice, but rescued in DKO mice (Figure S3).
Infusion of PTH increases Opn levels and led to more severe mineralization defect
To further investigate the role of Opn in regulating the mineralization, we perfused PTH (1–34) peptides into the WT and Kl−/− mice using osmotic minipumps. After continuous infusion for 3 weeks, we observed that the serum Opn levels were significantly elevated in both WT and Kl−/− mice (Figure 7A). As expected, serum phosphate levels were significantly decreased in both kinds of mice (Figure S4). PTH infusion, as shown in Figure 7B–7D, did not change the bone volume in WT mice, but significantly decreased it in Kl−/− mice. More importantly, OV/BV (Figure 7E) and OS/BS (Figure 7F) of Kl−/− mice were further increased by the PTH infusion, while the mineralized bone volume (MdV/TV) (Figure 7G) was significantly decreased, indicating that extraneous PTH induces Opn and thereby worsens the skeletal mineralization defect in the Kl−/− mice. Continuous PTH infusion also increased osteoblast surface (Ob.S/BS), Oc.S/BS and N.Oc/B.Pm (Figure 7K–7M) in both WT and Kl−/− mice. Opn contains a RGD sequence, which is important for osteoclast attachment on the bone. Thus it is not surprising that infusion of PTH resulted in a significant elevation in serum CTX levels, indicating increased osteoclastic resorption (Figure 7N). In addition, PTH infusion significantly elevated serum PINP levels in both WT and Kl−/− mice (Figure 7O).

Discussion
This is the first study using a genetic mouse model with dual ablation of the Klotho and PTH genes. The results show that deletion of PTH from Kl−/− mice resulted in healthier mice with normalization of serum calcium levels and complete rescue of the skeletal phenotype, suggesting that PTH is a crucial contributor to the skeletal abnormalities caused by loss of Klotho function. More importantly, we found that deletion of the PTH completely rescued the mineralization defect in Kl−/− mice. Kl−/− mice, as well as Fgf23−/− mice, exhibit a severe mineralization defect in their bones despite excesses in serum calcium and phosphate when compared to WT mice. The underlying reason for this is largely unknown but FGF23 is recognized as an inhibitor of mineralization. Serum Fgf23 levels in Kl−/− mice are two thousand fold higher than in wild-type littermates. Wang et al [24] show that adenoviral overexpression of FGF23 in rat calvarial cells inhibits bone mineralization independent of its systemic effects on phosphate homeostasis. Our previous report also demonstrated that FGF23 treatment of primary calvarial osteoblasts from wild-type mice leads to an inhibition of mineralization [18]. However, FGF23 requires Klotho for its actions [9], [20], [25]. In the absence of Klotho, FGF23 has very low affinity to the FGFR1 and cannot induce signal transduction (phosphorylation) [9], [25], [26].
Another intriguing possibility is that Klotho may have a specific function in osteoblasts associated with PTH. Although Klotho has been widely accepted as the cofactor of FGF23 signaling [9], [26], [27], and Kl−/− and Fgf23−/− mice share very similar phenotypes [20], we show here that deletion of PTH fully rescues the mineralization defect in Kl−/− mice, but could not improve this defect in Fgf23−/− animals [23]. More recently, Klotho has been reported to be expressed in the osteoblastic cell linage [28]. In addition, previous studies have shown that Klotho does exert endogenous actions in calcium homeostasis and control of PTH secretion [13], [14] that are independent of FGF23. Similarly, Klotho directly mediates secretion of PTH through recruitment of Na+/K+ ATPase to the plasma membrane [14]. Recent studies also showed that Kl−/− mice have increased trabecular bone [29], [30]. In this study, we confirmed elevated bone volume in Kl−/− mice compared to the normal bone volume in Fgf23−/− mice. More importantly, cultured osteoblasts isolated from Kl−/− pups at the age of 2-days showed markedly impaired mineralization, suggesting that Klotho may play a specific role in osteoblasts. Moreover, we detected low Klotho expression by qPCR in both cultured osteoblasts and isolated cortical bone (Figure S5). An osteoblast-specific Klotho knockout mouse model may be required to dissect the role of Klotho during skeletogenesis.
To examine the bone mineralization defect in more detail, we determined the expression of Opn by in situ hybridization, immunohistochemistry, ELISA and quantitative PCR. Opn is a well-known mineralization inhibitor, and mice deficient in Opn show soft tissue calcification and premature bone mineralization [31], [32]. Our analyses showed that the increased amount of Opn detected in Kl−/− mice was normalized in DKO mice. PTH is known to be an important regulator of Opn and can induce the expression of Opn in MC3T3-E1 cells within 3 hours [33], [34]. Therefore, the complete ablation of PTH might be responsible for the normalization of the increased Opn levels in Kl−/− mice and subsequently rescue the mineralization defect. Furthermore, infusion of exogenous PTH into Kl−/− mice resulted in a significant elevation in Opn levels with a worsened mineralization defect. This further strengthens our hypothesis that PTH could regulate skeletal mineralization in Kl−/− mice via Opn.
Although studies suggest that phosphate could also regulate the expression of Opn [35]–[37], our previous study using Fgf23−/−/NaPi2a−/− mice suggested that the elevated expression of Opn in bones of Fgf23−/− mice is at least partially independent of systemic phosphate levels [18]. This was further supported by our observation in this study that PTH−/− and DKO mice had normal expression of Opn despite very high serum phosphate levels. In addition, we also found in this study that PTH infusion could increase the serum Opn, even while the serum phosphate levels were decreased (Figure S4). Moreover, serum calcium, phosphate and vitamin D levels in Fgf23−/−/PTH−/− and Kl−/−/PTH−/− are identical and can therefore not contribute to the regulation of skeletal mineralization and/or Opn levels in these mice.
In summary, the findings in this study demonstrate that genetic ablation of PTH resulted in healthier DKO mice. More importantly, deletion of PTH completely rescued the skeletal abnormalities, including the severe mineralization defect in Kl−/− mice, and this effect is very likely associated with normalized expression of Opn in DKO mice. Interestingly, we previously showed that deletion of PTH in Fgf23−/− mice could not rescue mineralization, implying an independent function of Klotho in bone. This study demonstrates that the activity of the low level of PTH remaining in Kl−/− mice contributes to the severe mineralization disorder and skeletal abnormalities caused by the loss of Klotho function. Moreover, we show that Klotho affects mineralization independently of its role as a co-factor for FGF23. Further analyses are needed to determine the independent roles of PTH and Klotho in mineral ion homeostasis and skeletal mineralization and their detailed molecular interactions, including those involving OPN.
Materials and Methods
Animals
Heterozygous - Kl+/− and PTH+/− animals were interbred to attain wild-type (WT), Kl−/−, Kl−/−/PTH−/− (double knockout, DKO) and PTH−/− animals for subsequent analyses. Routine PCR was used to genotype various mice as described previously [20], [38]. The total body weight of each mouse was measured weekly starting at 3 weeks after birth. All studies performed were approved by the Institutional Animal Care and Use Committee at the Harvard Medical School.
Biochemical analyses
Blood was obtained by puncturing the cheek pouch of animals. Total serum calcium and phosphorus levels were determined using Stanbio LiquiColor (Arsenazo III) and LiquiUV kits (Stanbio Laboratory, Boerne, TX), respectively. Serum concentrations of FGF23 and Opn were measured using commercial kits from Kainos Laboratories, Inc., (Tokyo, Japan), and R&D Systems, Inc. (Minneapolis, MN), respectively. The ELISA kits for 1,25(OH)2D, PINP and CTX were purchased from IDS (Fountain Hills, AZ).
Skeletal mineralization and bone mineral density
The mineralization pattern of the skeleton was analyzed by Alizarin red S and Alcian blue staining in 6 - week-old mice, as described by McLeod [39]. Femurs of all genotypes at 6 weeks of age were flushed and exposed to X-ray (20 kV, 5 seconds). As described previously [40], bone mineral density (BMD) and μCT analysis were performed by peripheral quantitative computerized tomography (pQCT) and by using a Scanco Medical μCT 35 system (Scanco), respectively.
Bone histology and histomorphometry
Processing of undecalcified bone specimens and cancellous bone histomorphometry in the distal femoral metaphysis were performed as described [40]. The area within 0.25 mm from the growth plate was excluded from the measurements.
Soft tissue calcifications
Von Kossa staining was performed using 5 µm paraffin sections of kidneys and lungs isolated from 9-week-old animals.
In situ hybridization
Complementary 35S-UTP-labeled riboprobe osteopontin (Opn) and dentin matrix protein 1 (Dmp1) were used for performing in situ hybridization on paraffin sections, as described previously [41].
Immunohistochemistry
Immunohistochemistry was performed using mouse Opn antibody (R&D, Minneapolis, MN) with a working concentration of 0.5 µg/ml overnight at 4°C. Tissue was stained with anti-goat HRP substrate and DAB (Vector, Burlingame, CA), and then counterstained with hematoxylin.
Mouse calvarial cell culture
Mouse calvarial cell culture was carried out as previously described [18]. Cells were treated with 50 mg/ml ascorbic acid and 10 mM β-glycerophosphate (βGP) to induce matrix mineralization. Total RNA isolation and Alizarin red S staining were performed 14 days after induction.
Quantitative real-time PCR
Total RNA was from cultured osteoblasts using Trizol reagents (Invitrogen) according to the manufacturer's protocol. For qRT-PCR, cDNA was prepared using QuantiTec reverse transcription kit (Qiagen) and analyzed with SYBR GreenMaster Mix (SABiosciences) in the iCycler (Bio-Rad) using specific primers designed for each targeted gene. Relative expression was calculated using the 2−ΔΔCt method by normalizing with Gapdh housekeeping gene expression, and presented as fold increase relative to control.
In vivo continuous PTH (1–34) infusion
50 µg per kilogram of body weight per day of human PTH 1–34 (Polypeptide Group, France) were delivered into 3-week-old animals for a 3-week period using implanted ALZET osmotic minipumps, Model-1004 (DURECT Corporation, Cupertino, CA). Animals of vehicle group were infused with an equal volume of sterile saline.
Statistics
Statistically significant differences between groups were evaluated by Student's t-test for comparison between two groups or by one-way analysis of variance (ANOVA) followed by Tukey's test for multiple comparisons. And those between vehicle and PTH-infused groups were evaluated by Student's t-test. All values were expressed as mean ± SD. A p value of less than 0.05 was considered to be statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. RenkemaKYAlexanderRTBindelsRJHoenderopJG 2008 Calcium and phosphate homeostasis: concerted interplay of new regulators. Ann Med 40 82 91
2. DussoASBrownAJSlatopolskyE 2005 Vitamin D. Am J Physiol Renal Physiol 289 F8 28
3. RazzaqueMSLanskeB 2007 The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol 194 1 10
4. LanskeBRazzaqueMS 2007 Mineral metabolism and aging: the fibroblast growth factor 23 enigma. Curr Opin Nephrol Hypertens 16 311 318
5. LeeMPartridgeNC 2009 Parathyroid hormone signaling in bone and kidney. Curr Opin Nephrol Hypertens 18 298 302
6. LiSAWatanabeMYamadaHNagaiAKinutaM 2004 Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct Funct 29 91 99
7. MatsumuraYAizawaHShiraki-IidaTNagaiRKuro-oM 1998 Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun 242 626 630
8. Shiraki-IidaTAizawaHMatsumuraYSekineSIidaA 1998 Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett 424 6 10
9. UrakawaIYamazakiYShimadaTIijimaKHasegawaH 2006 Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature
10. SaitoHKusanoKKinosakiMItoHHirataM 2003 Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J Biol Chem 278 2206 2211
11. ShimadaTUrakawaIYamazakiYHasegawaHHinoR 2004 FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 314 409 414
12. ChangQHoefsSvan der KempAWTopalaCNBindelsRJ 2005 The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310 490 493
13. BjorklundPKrajisnikTAkerstromGWestinGLarssonTE 2008 Type I membrane klotho expression is decreased and inversely correlated to serum calcium in primary hyperparathyroidism. J Clin Endocrinol Metab 93 4152 4157
14. ImuraATsujiYMurataMMaedaRKubotaK 2007 alpha-Klotho as a regulator of calcium homeostasis. Science 316 1615 1618
15. Ben-DovIZGalitzerHLavi-MoshayoffVGoetzRKuro-oM 2007 The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117 4003 4008
16. KrajisnikTBjorklundPMarsellRLjunggrenOAkerstromG 2007 Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol 195 125 131
17. SitaraDRazzaqueMSSt-ArnaudRHuangWTaguchiT 2006 Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am J Pathol 169 2161 2170
18. SitaraDKimSRazzaqueMSBergwitzCTaguchiT 2008 Genetic evidence of serum phosphate-independent functions of FGF-23 on bone. PLoS Genet 4 e1000154 doi:10.1371/journal.pgen.1000154
19. RazzaqueMSSitaraDTaguchiTSt-ArnaudRLanskeB 2006 Premature ageing-like phenotype in fibroblast growth factor 23 null mice is a vitamin-D mediated process. The FASEB Journal 20 720 722
20. NakataniTSarrajBOhnishiMDensmoreMJTaguchiT 2009 In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. Faseb J 23 433 441
21. NakataniTOhnishiMRazzaqueMS 2009 Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. Faseb J 23 3702 3711
22. BrownsteinCAZhangJStillmanAEllisBTroianoN Increased bone volume and correction of HYP mouse hypophosphatemia in the Klotho/HYP mouse. Endocrinology 151 492 501
23. YuanQSitaraDSatoTDensmoreMSaitoH 2011 PTH Ablation Ameliorates the Anomalies of Fgf23-Deficient Mice by Suppressing the Elevated Vitamin D and Calcium Levels. Endocrinology 152 4053 4061
24. WangHYoshikoYYamamotoRMinamizakiTKozaiK 2008 Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res 23 939 948
25. KurosuHOgawaYMiyoshiMYamamotoMNandiA 2006 Regulation of fibroblast growth factor-23 signaling by Klotho. J Biol Chem
26. TomiyamaKMaedaRUrakawaIYamazakiYTanakaT 2010 Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc Natl Acad Sci U S A 107 1666 1671
27. RazzaqueMS 2009 The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol 5 611 619
28. RheeYBiviNFarrowELezcanoVPlotkinLI Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 49 636 643
29. LiuHFergussonMMCastilhoRMLiuJCaoL 2007 Augmented Wnt signaling in a mammalian model of accelerated aging. Science 317 803 806
30. BrownsteinCAZhangJStillmanAEllisBTroianoN 2010 Increased bone volume and correction of HYP mouse hypophosphatemia in the Klotho/HYP mouse. Endocrinology 151 492 501
31. LuoGDucyPMcKeeMDPineroGJLoyerE 1997 Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 386 78 81
32. SteitzSASpeerMYMcKeeMDLiawLAlmeidaM 2002 Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol 161 2035 2046
33. SuttamanatwongSFranceschiRTCarlsonAEGopalakrishnanR 2007 Regulation of matrix Gla protein by parathyroid hormone in MC3T3-E1 osteoblast-like cells involves protein kinase A and extracellular signal-regulated kinase pathways. J Cell Biochem 102 496 505
34. GopalakrishnanRSuttamanatwongSCarlsonAEFranceschiRT 2005 Role of matrix Gla protein in parathyroid hormone inhibition of osteoblast mineralization. Cells Tissues Organs 181 166 175
35. FosterBLNocitiFHJrSwansonECMatsa-DunnDBerryJE 2006 Regulation of cementoblast gene expression by inorganic phosphate in vitro. Calcif Tissue Int 78 103 112
36. JulienMKhoshniatSLacreusetteAGatiusMBozecA 2009 Phosphate-dependent regulation of MGP in osteoblasts: role of ERK1/2 and Fra-1. J Bone Miner Res 24 1856 1868
37. FatheraziSMatsa-DunnDFosterBLRutherfordRBSomermanMJ 2009 Phosphate regulates osteopontin gene transcription. J Dent Res 88 39 44
38. MiaoDHeBLanskeBBaiXYTongXK 2004 Skeletal abnormalities in Pth-null mice are influenced by dietary calcium. Endocrinology 145 2046 2053
39. McLeodMJ 1980 Differential staining of cartilage and bone in whole fetuses by alcian blue and alizarin red S. Teratology 22 299 301
40. YuanQSatoTDensmoreMSaitoHSchulerC 2011 Fgf23/Klotho signaling is not essential for the phosphaturic and anabolic functions of PTH. J Bone Miner Res 26 2026 2035
41. LanskeBDivietiPKovacsCSPirroALandisWJ 1998 The parathyroid hormone (PTH)/PTH-related peptide receptor mediates actions of both ligands in murine bone. Endocrinology 139 5194 5204
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Inactivation of a Novel FGF23 Regulator, FAM20C, Leads to Hypophosphatemic Rickets in Mice
- Genome-Wide Association of Pericardial Fat Identifies a Unique Locus for Ectopic Fat
- Slowing Replication in Preparation for Reduction
- An Essential Role for Katanin p80 and Microtubule Severing in Male Gamete Production
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy