
Oncogenic Herpesvirus KSHV Hijacks BMP-Smad1-Id Signaling to Promote Tumorigenesis
Although KSHV exerts multiple mechanisms to promote cell survival by repressing TGF-β signaling, little is known whether KSHV manipulates BMP signaling and contributes to the pathogenesis of KSHV-induced malignancies. In the present study, we have identified Smad1 as a novel binding protein of LANA by tandem affinity purification. We demonstrated that LANA up-regulated Id transcription through BMP-Smad1-Id signaling pathway. Id proteins were significantly up-regulated in KSHV-transformed MM (KMM) cells, and were abundantly expressed in human KS lesions; therefore, they were probably relevant to the development of KS. Importantly, we have shown that Ids are required to maintain the oncogenic phenotype of KMM cells in vitro and in vivo. These findings illustrate a novel mechanism by which a tumor virus hijacks and converts a developmental pathway into an indispensable oncogenic pathway for tumorigenesis. Furthermore, we showed that BMP signaling inhibitors dramatically hampered the tumorigenicity of KMM cells in vitro and in vivo. Our results demonstrate that small inhibitors targeting BMP-Smad1-Id signaling pathway are promising candidates for the treatment of KS.
Published in the journal:
. PLoS Pathog 10(7): e32767. doi:10.1371/journal.ppat.1004253
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004253
Summary
Although KSHV exerts multiple mechanisms to promote cell survival by repressing TGF-β signaling, little is known whether KSHV manipulates BMP signaling and contributes to the pathogenesis of KSHV-induced malignancies. In the present study, we have identified Smad1 as a novel binding protein of LANA by tandem affinity purification. We demonstrated that LANA up-regulated Id transcription through BMP-Smad1-Id signaling pathway. Id proteins were significantly up-regulated in KSHV-transformed MM (KMM) cells, and were abundantly expressed in human KS lesions; therefore, they were probably relevant to the development of KS. Importantly, we have shown that Ids are required to maintain the oncogenic phenotype of KMM cells in vitro and in vivo. These findings illustrate a novel mechanism by which a tumor virus hijacks and converts a developmental pathway into an indispensable oncogenic pathway for tumorigenesis. Furthermore, we showed that BMP signaling inhibitors dramatically hampered the tumorigenicity of KMM cells in vitro and in vivo. Our results demonstrate that small inhibitors targeting BMP-Smad1-Id signaling pathway are promising candidates for the treatment of KS.
Introduction
Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiological agent of Kaposi's sarcoma (KS), which is the most common malignancy in AIDS patients [1]. The KSHV-infected proliferating spindle cells are the driving force of KS [2]. KSHV mainly displays latency in spindle cells. Viral latent genes were reported to promote cell proliferation and inhibit apoptosis through various mechanisms. In particular, latency-associated nuclear antigen (LANA), a multifunctional major viral latent protein, is responsible for maintaining viral episome, inhibiting viral reactivation, and promoting cell proliferation by targeting p53, pRb and GSK-3β, etc (reviewed in [3], [4]). We have also shown that LANA contributes to cell proliferation by promoting intracellular Notch (ICN) accumulation through inhibition of Sel10-mediated ICN degradation [5], [6].
Due to the lack of in vitro KSHV cellular transformation model and the lack of KS cell lines, the roles of KSHV-deregulated signaling pathways in KSHV-induced cellular transformation remain unclear. The recent development of a robust model of KSHV-induced cellular transformation and tumorigenesis has made this possible [7]. Specifically, KSHV can efficiently infect, immortalize and transform primary rat embryonic metanephric mesenchymal precursor (MM) cells. KSHV-transformed MM cells (KMM) efficiently induce tumors with virological and pathological features of KS. This work has paved a way for studying the intrinsic oncogenic pathways underlying the tumorigenesis driven by KSHV latent genes. Using this system, KSHV-encoded miRNAs and vCylin were recently demonstrated to play critical roles in KSHV-induced cellular transformation and tumorigenesis [8], [9].
Bone morphogenetic proteins (BMPs) belong to the transforming growth factor β (TGF-β) superfamily. BMP signaling pathways play critical roles in diverse developmental phases [10]. In recent years, BMP signaling pathways have increasingly been the focus in cancer research, since these developmental pathways are frequently disrupted in cancer [11]. BMP signaling pathways are involved in both promotion and inhibition of cancer progression depending on the context, which is similar to the TGF-β pathway [12].
Inhibitors of DNA-binding (Id) family are major downstream targets of BMP signaling, and belong to the helix-loop-helix (HLH) family of transcription factors. There are four known members of the Id family in vertebrates (called Id1, Id2, Id3 and Id4) [13]. Id proteins do not possess a basic DNA binding domain and functions as a dominant-negative regulator of basic HLH proteins [14]. Recent evidence has revealed that Id proteins, especially Id1, are able to promote cell proliferation and cell cycle progression. Moreover, up-regulation of Id1 has been found in many types of human cancers and its expression levels are also associated with advanced tumor stage. [15]. Id1 was once reported to be up-regulated in KSHV-infected endothelial cells and in KS tissues [16], however, the mechanism and implication of Id1 up-regulation remains unclear.
In this study, Smad1 was identified as a novel LANA-binding protein. LANA up-regulated Id expression through constitutively sustaining the activation of the BMP-Smad1-Id signaling pathway, and thus contributed to the oncogenicity of KMM cells in vitro and in vivo. These studies have identified a novel viral oncogenic signaling pathway, and our data indicate that small inhibitors targeting BMP-Smad1-Id signaling pathway could be promising candidates for the treatment of KS.
Results
LANA interacted with BMP-activated p-Smad1 in the nucleus
In order to explore the novel function of LANA, we utilized Strep-Flag (SF)-tag based tandem affinity purification (SF-TAP) method to identify novel LANA-binding proteins (Fig. 1A) [17]. Smad1, a critical transducer of BMP signaling [18], was one of the hit proteins co-purified by SF-LANA [19]. We confirmed that LANA physically interacted with Smad1 in 293T cells by reciprocal co-immunoprecipitation (Co-IP) (Fig. 1B, C). We further confirmed their interaction in KSHV-infected cells (Fig. S1). LANA is predominantly located in the nucleus [20], while Smad1 shuttles from cytosol to nucleus in complex with Smad4 resulting in the transcription of BMP target genes following phosphorylation at C terminus S463/465 (SXS motif) by type I BMP receptor [18]. To determine the compartment of LANA-Smad1 interaction, 293T cells were transfected with LANA and Smad1, then treated with BMP2 and harvested for cell fraction. Co-IP assay was performed with cytoplasmic fraction and nuclear fraction respectively. As expected, LANA-Smad1 interaction was only detected in the nuclear but not in cytoplasmic fraction (Fig. 1D). Moreover, Smad1 pulled-down by LANA was recognized by a p-Smad1/5/8 antibody (Fig. 1D). Since LANA did not bind to Smad5 (Fig. S1), these results suggested that LANA interacted with BMP-activated p-Smad1 in the nucleus.

We further mapped out the Smad1-binding domain of LANA. Smad1 could be pulled down by Myc-tagged full length LANA1–1162 and N-terminus LANA1–432, but not by C-terminus LANA762–1162, negative control Intracellular Notch1 (ICN) nor control vector (Fig. S1). Therefore, N-terminus LANA1–432 is responsible for Smad1-binding.
Next, we mapped out the LANA-binding domain of Smad1. Smad1 has highly conserved N - and C-terminal regions known as Mad homology (MH) 1 and MH2 domains, respectively, which are linked by a linker region with a highly variable structure [18]. HA-tagged full length Smad1, Smad1-C (Linker+MH2), Smad1-MH2, but not Smad1-N (MH1+Linker) were pulled down by LANA (Fig. 1E). Therefore, Smad1 MH2 domain is responsible for LANA-binding. To narrow down the LANA-binding domain within Smad1 MH2 domain, we constructed a series of MH2 truncation mutants, termed as MH2-N, MH2-M and MH2-C respectively. Deletion of neither C-terminus of MH2 (MH2-N) nor N-terminus of MH2 (MH2-C) totally abolished its binding to LANA while the center part of MH2 (MH2-M) retained LANA binding activity (Fig. 1F). Therefore MH2-M (Smad1308–407) was critical for LANA-binding.
We then asked whether LANA-Smad1 interaction depended on the phosphorylation of SXS motif of Smad1. The Smad1 mutant with the SXS motif deleted (ΔC3), inactivated (AVA) or constitutively activated (DVD) [21] bound to LANA as efficiently as the wild type Smad1 (Fig. 1G). The differences of the apparent molecular weight between the wild type Smad1 and Smad1 mutants in SDS-PAGE were due to tag sizes. These results indicated that the nuclear location but not the phosphorylation of Smad1 is the restriction factor for the LANA-Smad1 interaction.
LANA up-regulated Id1 transcription in a BMP-Smad1 dependent manner
BMP signaling regulates fundamental biological processes during embryonic development, postnatal development, as well as tumorigenesis [22]. The Smad1 MH2 domain is responsible for sensing BMP signaling, oligomer formation with other Smads, interaction with various DNA-binding proteins, and transcriptional activation of BMP downstream targets [18]. We wondered whether LANA modulated BMP-Smad1 signaling by regulating the expression and/or function of p-Smad1via interaction with Smad1-MH2 domain. To address this hypothesis, 293T cells were transiently transfected with LANA or a control vector and then treated with BMP2. 293T cells were harvested at different times and subjected to immunoblotting for the levels of p-Smad1 and BMP downstream target Id1. The levels of p-Smad1 activation and Id1 were normalized to their expression levels at 0 hour in two groups, respectively. Activation of p-Smad1 started to decline at 3 hours post BMP2 treatment and reached the basal level at 24 hours in the vector-transfected 293T cell while activation of p-Smad1 did not start to decline until 15 hours and continued to maintain at a relatively higher level at 24 hours in the LANA-transfected 293T cells (Fig. 2A, B). Therefore, BMP-induced p-Smad1 expression was significantly sustained by LANA (Fig. 2A, B). Consistent with these results, the induction of the canonical BMP downstream target Id1 was significantly potentiated in the LANA-transfected cells than the vector-transfected control cells by BMP2 (Fig. 2A, C).

Id1 was once reported to be up-regulated in KSHV-infected endothelial cells and in KS tissues; moreover, expression of LANA and vCyclin seemed to up-regulate Id1 expression in post-transcription level [16]. Since Id1 was well-recognized for its roles in tumorigenesis [13], [23], we sought to determine whether LANA up-regulated Id1 expression through the BMP-Smad1 pathway.
As previously reported, we showed that Id1 was up-regulated in KSHV-infected human primary endothelial cells (Fig. S2). However, LANA but no other KSHV latent genes significantly up-regulated Id1 expression in 293T cells (Fig. S3). Meanwhile, LANA did not obviously alter Id1 protein stability. These results indicated that LANA regulated Id1 expression mainly at transcription level (Fig. S3). Consistent with these results, Id1 transcription was up-regulated more than two fold in LANA-transfected 293T cells (Fig. 3A). Treatment with noggin, which inhibited BMP signaling, abolished LANA induction of Id1 expression (Fig. 3A); while treatment with BMP2 further enhanced LANA induction of Id1 expression (Fig. 3B). We then asked whether LANA was directly involved in Id1 transcription regulation. In a promoter reporter assay, LANA increased the activity of Id1 promoter reporter Id1-985, which contained a Smad1 binding site or BRE (BMP-responding element), but not that of the mutant reporter Id1-956 lacking the BRE [24] (Fig. 3C). Knock-down of Smad1 abolished LANA activation of the Id1-985 promoter reporter (Fig. 3D, Fig. S3). Therefore, LANA up-regulation of Id1 transcription depended on the BMP-Smad1 signaling pathway.
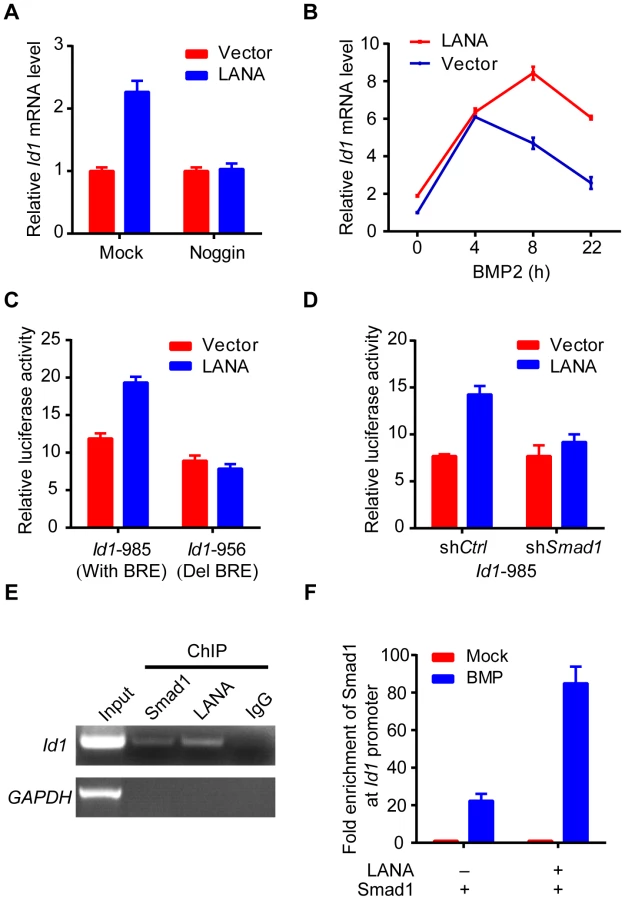
We showed that LANA was directly recruited to the Id1 promoter, together with Smad1 in ChIP-PCR assay (Fig. 3E). Moreover, LANA significantly enhanced the enrichment of Smad1 binding to the Id1 promoter after BMP2 treatment (Fig. 3F). Collectively, these results indicated that LANA promoted Smad1-mediated Id1 transcription activation through sustaining p-Smad1 expression, and probably facilitating and extending the loading of Smad1 on Id1 promoter.
Interestingly, we found that other Id family members, including Id2 and Id3 were also up-regulated in LANA-transfected 293T cells at both mRNA and protein levels (Fig. S4), whereas Id4 was not detected in our system. Furthermore, we showed that Id2 and Id3 were up-regulated in KSHV infected human primary endothelial cells (HUVECs) as Id1 (Fig. S5). Knockdown of Smad1 significantly impaired the expression of Id1, Id2 and Id3 in KSHV infected HUVECs, which suggested that Ids were mainly regulated by BMP-Smad1 pathway in those cells. We also showed that BMP signaling inhibitor Dorsomorphin dramatically repressed Id1, Id2 and Id3 in iSLK.219 cells (Fig. S5). Based on our data, we believed that LANA might generally up-regulated the transcription of Id family members through BMP-Smad1-Id signaling pathway in KSHV infected cells.
Ids were aberrantly expressed in KS tissues and in KSHV-transformed cells
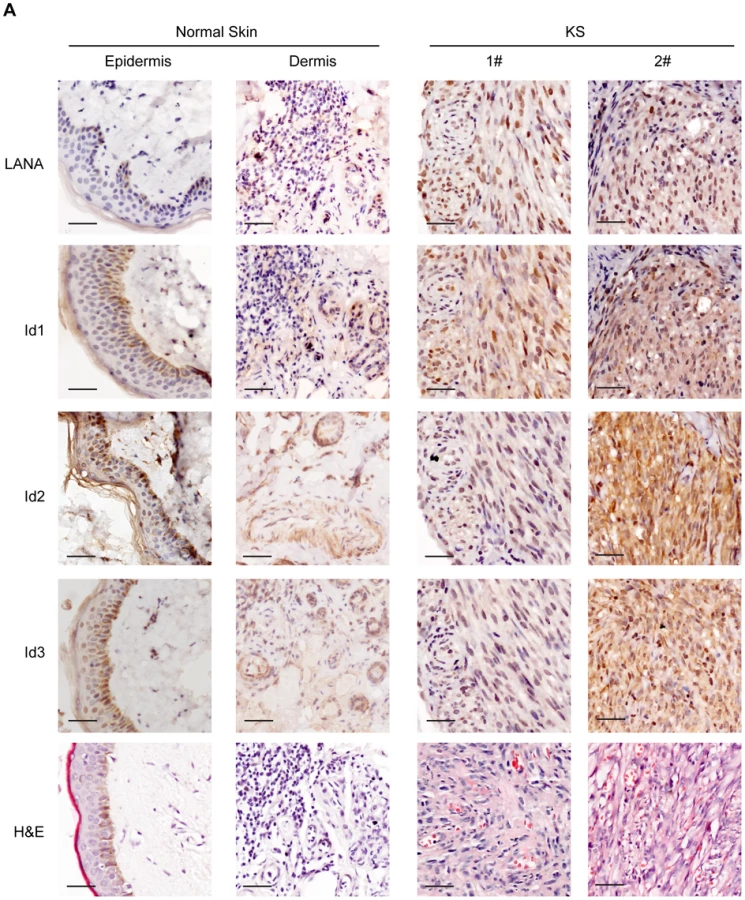
Since Ids were important oncogenic proteins, we sought to determine whether Ids were aberrantly expressed in KS tissues. We examined the expression of Id proteins and LANA in 10 cases of classical KS tissues and 5 cases of normal skin tissues by immunohistochemistry. As shown, there were weak to modest staining signals of Id1, Id2 and Id3 only in the basal cells of epidermis and around the hair follicle of dermis in normal skin tissues (Fig. 4). There was no LANA staining in any normal skin tissues (Fig. 4). In sharp contrast to normal skin tissues, there were strong staining signals of Id1, Id2 and Id3 in the spindle cells in the KS lesions. By staining for Ids and LANA in consecutive sections, positive signals of Ids were only observed in the spindle cells in KS lesions, which were also positive for LANA staining. No staining of Ids was observed in the adjacent tissues, which were negative for LANA (Fig. S6). Because of the small sample size, we were not able to perform a valid correlation analysis between Ids expression and the stage of the tumors. Nevertheless, our data suggested that Ids were aberrantly regulated in KS tumors and might be relevant to the development of KS. Because the p-Smad1 antibody was not suitable for immunohistochemical staining, we were not able to examine the expression of p-Smad1 in these KS lesions. Nevertheless, we showed that there was strong staining of Smad1 in the KS lesions but not in adjacent tissues (Fig. S6) indicating that BMP-Smad1-Id signaling might be involved in the aberrant expression of Ids in KS.
KSHV can efficiently infect and transform primary rat embryonic metanephric mesenchymal precursor (MM) cells [7]. KSHV-transformed MM cells (KMM) efficiently induce tumors with virological and pathological features of KS [7]. We asked whether Id family members were up-regulated by LANA in KMM cells. We detected significantly higher levels of Id1∼Id3 (about 3 fold) in KMM cells than in MM cells at both mRNA and protein levels (Fig. 5A, B). Knock-down of LANA dramatically suppressed the expression of Id1, Id2, and Id3 in KMM cells (Fig. 5C). These results indicated that LANA was responsible for the up-regulation of Ids in KMM cells.

Ids were indispensable driving forces for KSHV-induced tumorigenesis
Id proteins, especially Id1, are able to promote cell proliferation and cell cycle progression. To determine if LANA deregulation of the BMP-Smad1-Id pathway could contribute to KSHV-mediated tumorigenesis [7], we established KMM-shId1 cell lines with high Id1 knockdown efficiency and determined the effect on cellular transformation (Fig. 6A). Knock-down of Id1dramatically decreased the proliferation of KMM cells (Fig. 6B) and inhibited the formation of foci in culture (Fig. 6C, D), formation of colonies in soft agar (Fig. 6E, F), and induction of tumors in nude mice (Fig. 6G, H, I) In contrast, knockdown of Id1 in MM cells only slightly decreased the proliferation of MM cells (Fig. S7). We also established KMM-shId2 and KMM-shId3 cell lines (Fig. S8). Knockdown of Id2 and Id3 inhibited anchorage-independent growth of KMM cells in soft agar (Fig. S8). Moreover, knockdown of either LANA or Smad1 also severely impaired the anchorage-independent growth of KMM cells (Fig. S9). These results indicated that Ids were required for maintaining the oncogenic phenotype of KMM cells.

We further asked whether Ids were the driving force for KSHV-mediated cellular transformation. We overexpressed Id1 in MM cells, however, no direct cellular transformation was observed as expected (Fig. S10). Nevertheless, ectopic expression of Id1 in KMM cells (Fig. S11A) further accelerated cell proliferation (Fig. S11B), and increased the formation of foci in culture and formation of colonies in soft agar (Fig. S11C, D, E, F).
Collectively, our data provided evidence that LANA increased BMP-Smad1-Id signaling and this pathway was required for KSHV-induced tumorigenesis.
Targeting BMP-Smad1-Id pathway inhibited the growth of KSHV-induced tumors
Based on the above findings, we speculated that inhibitors of the BMP pathway might be potential therapeutic agents of KS. Dorsomorphin potently inhibits BMP-mediated Smad1/5/8 phosphorylation [25], while WSS25 disrupts the interaction between BMP and BMP receptor [26], [27]. Indeed, treatment with these two molecules dramatically inhibited BMP2-stimulated p-Smad1 expression (Fig. 7A), and inhibited the anchorage-independent cell growth of KMM cells in soft agar assay (Fig. 7B). Importantly, compared to MM cells, Dorsomorphin showed preferential toxicity to KMM cells (Fig. 7C), indicating Dorsomorphin selectively targeted KSHV-transformed cells. Furthermore, we found that Dorsomorphin dramatically inhibited the expression of Ids (Fig. 7D) while ectopic expression of Id1 significantly rescued Dorsomorphin induced G2/M arrest [28], cellular toxicity in KMM cells (Fig. 7E, F and Fig. S12), and partially rescued Dorsomorphin inhibition of anchorage-independent colony formation (Fig. 7G). These results indicated that Dorsomorphin mainly inhibited the oncogenicity of KMM cells through targeting the BMP-Smad1-Id pathway. To strengthen our conclusion, we showed that overexpression of Id1 significantly rescued Dorsomorphin-induced cellular toxicity in 293T cells in a dose-dependent manner (Fig. S13).

Finally, we determined the efficacy of Dorsomorphin in inhibiting in vivo tumor growth of KMM cells. We subcutaneously injected 1×106 KMM cells into BALB/c nude mice. When tumor volume reached about 50∼100 cm3, the nude mice were randomly divided into 2 two groups. One group was intraperitoneally injected with a single dose of Dorsomorphin at 10 mg/Kg [29] while the other group was injected with vehicle control. Impressively, single treatment with Dorsomorphin was sufficient to significantly inhibit the tumor growth of KMM cells (Fig. 7H). Immunohistochemical staining showed that Dorsomorphin inhibited Id1, Id2, Id3 and Ki67 expression and activated caspase 3 in the tumors (Fig. 7I). Interestingly, LANA remained positive in the Dorsomorphin-treated tumors (Fig. 7I), suggesting that the antitumor activity of Dorsomorphin was not dependent on the inhibition of KSHV infection and replication in KMM cells.
Discussion
Our results showed that KSHV LANA interacted with BMP-activated p-Smad1 in the nucleus, sustained p-Smad1 expression, and facilitated its loading on the Id promoter leading to aberrant expression of Ids, which were indispensable driving forces for KSHV-induced tumorigenesis. Thus, KSHV hijacks and converts a developmental pathway into an oncogenic pathway, which is essential for KSHV-induced transformation. Furthermore, our results have shown that small inhibitors targeting BMP-Smad1-Id signaling pathway may serve as potential candidates for the treatment of KS (summary in Fig. 8).

Although KSHV exerts multiple mechanisms to promote cell survival by repressing TGF-β signaling [30]–[32], little is known whether KSHV manipulates BMP signaling and contribute to the pathogenesis of KSHV-induced malignancies. Previously, KSHV lytic gene K5 was reported to inhibit BMP signaling by down-regulating BMPR-II through ubiquitination-mediated degradation [33]. However, KSHV is predominantly maintained in the latent state of replication in KS spindle tumor cells and KMM cells, in which K5 is usually not expressed. In this context, we believe that KSHV hijacks the BMP-Smad1-Id pathway to promote tumorigenesis.
We previously reported that Smad1 was not detected in PEL cells [31] and LANA did not interact with Smad5. It is unlikely that LANA is involved in Id regulation in PEL cells. We showed that Id1∼3 were expressed in KSHV-positive BCBL1 and BC3 cells at levels similar to KSHV-negative BJAB cells (Fig. S14). Since BJAB was not an ideal control for BCBL1 and BC3 cells, we further compared the expression of Ids in BJAB and KSHV stably transfected BJAB (KSHV-BJAB) cells. We found that Id2 and Id3, but not Id1 was decreased by about 50% in KSHV-BJAB cells compared to BJAB cells (Fig. S14). Since Id2 and Id3 but not Id1 were reported to be down-regulated in the vFLIP-transfected cells [34], vFLIP might be the main viral gene that regulates the expression of Ids in PEL cells.
Since Id1∼3 are significantly up-regulated in KS lesions compared to adjacent tissue and normal skin and in KMM cells compared to MM cells, up-regulation of Ids by LANA through LANA-Smad1-Id signaling is likely the principal mechanism that KSHV regulates the expression of Ids in KS tumor cells and in KSHV-transformed KMM cells. Interestingly, Id1 and Id3 were induced by EBV latent protein LMP1 [35], [36]. LMP1 inactivates the function of Foxo3a leading to up-regulation of Id1. Id1 increased cell proliferation and conferred resistance to TGFβ-mediated cell cycle arrest in nasopharyngeal epithelial cells [37]. Therefore, Id proteins may serve as conserved targets for oncogenic herpesviruses.
Ids inhibit apoptosis and promote cell proliferation through distinct mechanisms [13]. For example, Id1 had been shown to inhibit E-protein and Ets-protein-mediated activation of the p16/INK4a [38], [39]. Id2 has been found to reverse cellular growth inhibition by the retinoblastoma protein (pRb) through direct interaction with pRb [40]. How individual Ids promote KSHV-mediated oncogenesis remain to be further clarified. Our data showed that LANA up-regulated BMP-Smad1-Id signaling was required but not sufficient for KSHV-induced tumorigenesis. It is likely that additional oncogenic signaling pathways are involved in KSHV-induced cellular transformation and tumorigenesis. Discovery of these additional pathways could help better understanding of how KSHV induces tumorigenesis.
Dorsomorphin is known to potently inhibit the expression of Ids through suppressing BMP-induced Smad1 phosphorylation. Our results showed that Dorsomorphin dramatically inhibited the growth of KMM cells in vitro and tumor growth in vivo. Even though Dorsomorphin might also target other kinases [41], our results showed that Id1 was capable of rescuing Dorsomorphin-induced G2/M arrest and cellular toxicity in KMM cells. Therefore, we have demonstrated that, by targeting the KSHV-deregulated BMP-Smad1-Id pathway, Dorsomorphin inhibits KSHV-induced tumorigenesis. Dorsomorphin might be a promising lead compound for KS therapy.
Currently, it is still unknown how LANA sustains p-Smad1 activation through their interaction. In the basal state, Smad1 constantly shuttles between cytoplasm and nucleus through its N-terminal nuclear localization signal (NLS) motif and C-terminal nuclear export signal (NES) [42]. Upon activation by BMP, the C-terminal of Smad1, which is phosphorylated at SXS motif, undergoes conformation change, and creates an acidic knob to form a trimer with the homologous MH2 domain of another Smad1 molecule and MH2 domain of Smad4 [43]. The ligand-induced phosphorylation promotes the accumulation of the hetero-oligomer in the nucleus by inhibiting the nuclear export and enhancing its import [42]. In the nucleus, Smad1/Smad4 complexes bind to other co-transcription factors and initiate target gene transcription. The signal undergoes rapid termination through dephosphorylation in its C-terminal SXS motif by PPM1A [44] and/or SCP (small C-terminal domain phosphatase) family of nuclear phosphatases [45] or degradation via polyubiquitylation and proteasome-mediated degradation by Smurf1/2 [46] or CHIP [47]. In this study, we have demonstrated the interaction between the N-terminal of LANA and MH2 domain of Smad1. This interaction may have several effects on sustaining the activated Smad1 in the nucleus: 1) it may stabilize the heteromeric complex between the phosphorylated Smad1 and the common mediator Smad4, thus masking the NES motif; 2) it may protect Smad1 from dephosphorylation caused by PPM1A or SCPs; 3) it may disrupt the rapid Smad1 turnover via Ubiquitin-Proteasome Pathway mediated by Smurf1/2 or CHIP. Thus, LANA facilitates the loading of functional p-Smad1 on the Id promoter and ultimately leads to aberrant expression of Ids. However, additional works are required to confirm any of these speculations.
In a summary, our study has revealed that BMP-Smad1-Id signaling pathway is positively regulated by LANA and serves as an intrinsic oncogenic pathway of KSHV-induced tumorigenesis. More importantly, we have shown that the BMP-Smad1-Id pathway is a potential therapeutic target for KS.
Materials and Methods
Ethics statement
The clinical section of the research was reviewed and ethically approved by the Institutional Ethics Committee of the First Teaching Hospital of Xinjiang Medical University (Urumqi, 127 Xinjiang, China; Study protocol # 20082012). Written informed consent was obtained from all participants, and all samples were anonymized. All participants were adults.
The animal experiments were approved by the Institutional Animal Care and Use Committee of the Institut Pasteur of Shanghai, Chinese Academy of Sciences (Animal protocol # A2013010). All animal care and use protocols were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People's Republic of China.
Cell lines, plasmids, antibodies and reagents
Rat embryonic metanephric mesenchymal precursor cells (MM cells), KSHV-transformed MM cells (KMM), 293T cells were maintained in DMEM (HyClone) supplemented with 10% fetal bovine serum (HyClone). HUVEC was maintained in EGM (Lonza). KMM/shsmad1, KMM/shId1, KMM/shLANA, and KMM/shControl, KMM/Id1, KMM/Vector, 293T/shSmad1, 293T/shControl, 293T/SF-LANA and 293T/SF-Puro cell lines were established by infection of indicated lentivirus according to the manufacturer's instructions (System Bioscience).
LANA truncation plasmids were previously reported [48]. pCDH-SF-LANA was constructed by sub-cloned full-length LANA into pCDH-SF-EF1-Puro by EcoRI and BamHI sites. Reporter plasmids pGL3-Id1-985 and pGL3-Id1-956 were constructed as previously reported [24]. HA-Smad1and Flag-Smad1, were provided by Dr. Naihe Jing (Shanghai Institutions of Biological Sciences). ShLANA was previously reported [49]. shId1, shId2, shId3 and shSmad1 were constructed in pLKO.1 using the following targeting sequence: Id1 (AAGGTCACATTTCGTGCTTCT); Id2 (CAGCACGTCATCGATTATATC); Id3 (GTGATCTCCAAGGACAAGAGG) ; Smad1(CGGTTGCTTATGAGGAACCAA). Truncated or SXS motif mutated Smad1 plasmids were constructed by cloning the indicated sequence into pCMV-HA vector using the following primers:
Smad1-N (F: 5′-CGCGTCGACAATGAATGTGACAAGTTTATT-3′, R: 5′-CGCCTCGAGTTAAGCAACCGCCTGAACATCTC-3′);
Smad1-C (F: 5′-CGCGTCGACAATGCCTGTACTTCCTCCTGTGCT-3′, R: 5′-CGCCTCGAGTTAAGATACAGATGAAATAGGAT-3′);
Smad1-MH2 (F: 5′-CGCGTCGACAATGTATGAGGAACCAAAACACTG-3′, R: 5′-CGCCTCGAGTTAAGATACAGATGAAATAGGAT-3′);
Smad1-DVD (F: 5′-CGCGTCGACAATGAATGTGACAAGTTTATT-3′, R: 5′-CGCCTCGAGTTAATCTACATCTGAAATAGGATTA-3′);
Smad1-AVA (F: 5′-CGCGTCGACAATGAATGTGACAAGTTTATT-3′, R: 5′-CGCCTCGAGTTAAGCTACAGCTGAAATAGGATT-3′);
Smad1-ΔC3 (F: 5′-CGCGTCGACAATGAATGTGACAAGTTTATT-3′, R: 5′-CGCCTCGAGTTAAGCTACAGCTGAAATAGGATT-3′).
Expression plasmids of truncated Smad1-MH2 were constructed by cloning the indicated sequence into pEGFP vector using the following primers:
MH2-N (F: 5′-cgcAGATCTTATGAGGAACCAAAACACTG-3′, R: 5′-cgcGGATCCTTAATGATGGTAGTTGCAGTTCC-3′)
MH2-M (F: 5′-cgcAGATCTCGTTTCTGCCTTGGGCTGCT-3′, R: 5′-cgcGGATCCTTATGTAAGCTCATAGACTGTCTCA-3′)
MH2-C (F: 5′-cgcAGATCTGGATTTCATCCTACTACTGTTTGC-3′, R: 5′-cgcGGATCCTTAAGATACAGATGAAATAGG-3′)
MH2-F (F: 5′-cgcAGATCTTATGAGGAACCAAAACACTG-3′, R: 5′-cgcGGATCCTTAAGATACAGATGAAATAGG-3′).
The antibodies and reagents were used as follows: anti-LANA (1B5, prepared in our lab), anti-Smad1 (Santa cruz, sc-7965x), anti-pSmad1/5/8 (Cell signaling technology, #9511), anti-Id1 (Santa cruz, sc-488), anti-Id2 (Santa cruz, sc-489), anti-Id3(Santa cruz, sc-490), anti-Ki67 (Novocastra, NCL-Ki67p), anti-cleaved Caspase-3 (Cell signaling technology, #9661). Anti-Flag M2 affinity gel (Sigma, A2220), Strep-Tactin sepharose (IBA, 2-1201-010), desthiobiotin (IBA, 2-1000-001), BMP2 (Sigma, B3555), Cycloheximide (Sigma, C1988), Dorsomorphin (Sigma, P5499) and WSS25 were kindly provided by Dr. Kan Ding from Shanghai Institute of Materia Medica [26].
Tandem affinity purification (TAP)
TAP of SF-LANA was done as previously described [19]. Briefly, 293T-SF-LANA or 293T-SF-Puro cells were harvested and subjected to nuclear extraction as previously described [50]. Dialyzed nuclear extract was loaded into a column of prewashed Strep-Tactin Superflow (0.5 ml bed volume, IBA). The column was washed with 10 bed volume of Buffer W (50 mM Tris pH 7.9, 100 mM KCl, 10% Glycerol, 0.2 mM EDTA, 0.5 mM DTT, 0.1% Triton-X100, 0.2 mM PMSF) and eluted with 3 bed volume of Buffer E (Buffer W containing 2.5 mM D-desthiobiotin). The elute was then subjected to second round of affinity purification by anti-Flag M2 affinity gel for 2 hours at 4°C. The beads were washed with Buffer W for 5 times and eluted with 3×Flag Peptide in Buffer W. The elute was monitored by SDS-PAGE and subjected to mass spectrometry.
Coimmunoprecipitation (co-IP) and immunoblotting
Cells were lysed in radio immunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.6], 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 0.1 mM PMSF, 1×phosphatase inhibitors [Phospho-Stop, Roche]) for 1 h on ice with brief vortexing every 10 min. The lysate were incubated with antibody or affinity beads as indicated overnight at 4°C. The immunoprecipitations were separated by SDS-PAGE and analyzed by immunoblotting. For cytoplasmic protein and nuclear protein fractionation, cells were harvested and extracted as described [51].
Real-time RT-PCR
Cells were collected and lysed in Trizol buffer (Life technology), and RNA was isolated according to the manufacturer's instructions. Reverse transcription was performed with a cDNA Reverse Transcription Kit (Toyobo). Real-time RT-PCR was performed with a SYBR green Master Mix kit (Toyobo). Relative mRNA levels were normalized to Actin and calculated by ΔΔCT method. The primers were listed below:
Id1 (F: 5′-CTGCTCTACGACATGAACGG-3′, R: 5′-GAAGGTCCCTGATGTAGTCGAT-3′);
Id2 (F: 5′-GCTATACAACATGAACGACTGCT-3′, R: 5′-AATAGTGGGATGCGAGTCCAG-3′);
Id3 (F: 5′-GAGAGGCACTCAGCTTAGCC-3′, R: 5′-TCCTTTTGTCGTTGGAGATGAC-3′);
Actin (F: 5′-GCACGGCATCGTCACCAACT-3′, R: 5′-CATCTTCTCGCGGTTGGCCT-3′).
Chromatin immunoprecipitation assay (ChIP)
Chromatin immunoprecipitation (ChIP) was performed as previously described. Briefly, 5 µg correspondent antibody (anti-HA mAb, anti-Flag-mAb) or control mouse immunoglobulin (IgG) was added into each group of lysate at 4°C overnight. Then 50 µl proteinA/G beads, which had been precleared with binding buffer containing 0.2 mg of salmon sperm DNA per ml for 6 h, were added into each sample at 4°C for 2 h for immunoprecipitation. To extract the DNA fragment, TE buffer with 1% SDS and proteinase K (Beyotime) was added to the washed precipitates. After incubation at 65°C for at least 6 h, the eluted solution was subjected to DNA extraction kit (Bio-Dev). Specific primers used for chromatin immunoprecipitation (ChIP) DNA amplification matched the Id1 promoter region were: Id1-F: 5′-CAGTTTGTCGTCTCCATG-3′; Id1-R: 5′-TCTGTGTCAGCGTCTGAA-3′; GAPDH-F: 5′-TACTAGCGGTTTTACGGGCG-3′; GAPDH-R: 5′-TCGAACAGGAGGAGCAGAGAGCGA-3′.
Functional assay
MTT assay for cell proliferation or toxicity was conducted according to the manufacturer's instructions (Beyotime). For cell proliferation, 1000 cells were seeded per well in 96-well plates as indicated; for toxicity, 4000 cells were seeded per well in 96-well plates with DMEM containing Dorsomorphin of indicated concentrations.
Cell cycle assay was conducted according to manufacturer's instructions (Beyotime). KMM-Vector and KMM-Id1 cells were treated with DMSO or 5 mM Dorsomorphin for 48 hours. Then the cells were harvested and subjected to PI staining and cell cycle analysis by Mod Fit software.
Soft agar assay: Six-well plates were covered with a bottom layer of 1% agar (Invitrogen) in DMEM containing 10% FBS. Then 10000 cells were prepared in DMEM containing 10% FBS and 0.4% agar and seeded onto the solidified bottom layer. After two weeks of cell culture, colonies were photographed by microscopy and stained with 0.005% crystal violet. The number of colonies was analyzed by Quantity One.
Colony formation assay: 1000 cells were prepared in DMEM containing 10% FBS and seeded in six-well plates. After two weeks of cell culture, colonies were photographed by microscopy and stained with 0.005% crystal violet. The number of colonies was analyzed by Quantity One.
Histopathology and immunohistochemistry (IHC) analysis
The clinical tissue specimens from 10 patients with KS were collected from Xinjiang province, northwestern of China. The clinical section of the research was reviewed and ethically approved by the Institutional Ethics Committee of the First Teaching Hospital of Xinjiang Medical University (Urumqi, 127 Xinjiang, China; Study protocol # 20082012). The expression of LANA, Id1, Id2, Id3, Smad1, Ki67, and activated caspase 3 were analyzed by IHC as described [52].
Xenograft experiment
1×106 KMM-shCtrl cells or KMM-shId1 cells were subcutaneously injected into BALB/c Nude mice. There were 5 mice in each group. The size of tumor was measured every 3 day. Tumor volume was calculated by the formula: (length×width2)/2. Nude mice were sacrificed at the same time when the size of tumors in shCtrl group reaches about 2000 mm3. In another xenograft assay with drug treatment, 1×106 KMM cells were subcutaneously injected into BALB/c Nude mice. When tumor volume reached about 50∼100 cm3, nude mice were divided into 2 two groups randomly. There were 5 mice in each group. One group was intraperitoneal injection with a single dose of Dorsomorphin (10 mg/Kg), the other group was injected with vehicle. Tumor volume was monitored daily and calculated by the formula: (length×width2)/2. These animal experiments were approved by the Institutional Animal Care and Use Committee of the Institut Pasteur of Shanghai, Chinese Academy of Sciences (Animal protocol # A2013010).
Statistical analysis
Data were analyzed by Student's t test. P<0.05 was considered to be significant (two tailed). Error bars represent standard error of mean (s.e.m.).
Accession numbers
Gene IDs:
BMP2 : 650
Smad1 : 4086
Smad5 : 4090
Id1 : 3397
Id2 : 3398
Id3 : 3399
LANA: 4961527
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GanemD (2010) KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120 : 939–949.
2. MesriEA, CesarmanE, BoshoffC (2010) Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer 10 : 707–719.
3. BallestasME, KayeKM (2011) The latency-associated nuclear antigen, a multifunctional protein central to Kaposi's sarcoma-associated herpesvirus latency. Future Microbiol 6 : 1399–1413.
4. VermaSC, LanK, RobertsonE (2007) Structure and function of latency-associated nuclear antigen. Curr Top Microbiol Immunol 312 : 101–136.
5. LanK, VermaSC, MurakamiM, BajajB, KaulR, et al. (2007) Kaposi's sarcoma herpesvirus-encoded latency-associated nuclear antigen stabilizes intracellular activated Notch by targeting the Sel10 protein. Proc Natl Acad Sci U S A 104 : 16287–16292.
6. LanK, ChoudhuriT, MurakamiM, KuppersDA, RobertsonES (2006) Intracellular activated Notch1 is critical for proliferation of Kaposi's sarcoma-associated herpesvirus-associated B-lymphoma cell lines in vitro. J Virol 80 : 6411–6419.
7. JonesT, YeF, BedollaR, HuangY, MengJ, et al. (2012) Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. J Clin Invest 122 : 1076–1081.
8. JonesT, Ramos da SilvaS, BedollaR, YeF, ZhouF, et al. (2014) Viral cyclin promotes KSHV-induced cellular transformation and tumorigenesis by overriding contact inhibition. Cell Cycle 13 : 845–858.
9. MoodyR, ZhuY, HuangY, CuiX, JonesT, et al. (2013) KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog 9: e1003857.
10. HoganBL (1996) Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev 10 : 1580–1594.
11. KelleherFC, FennellyD, RaffertyM (2006) Common critical pathways in embryogenesis and cancer. Acta Oncol 45 : 375–388.
12. AlarmoEL, KallioniemiA (2010) Bone morphogenetic proteins in breast cancer: dual role in tumourigenesis? Endocr Relat Cancer 17: R123–139.
13. PerkJ, IavaroneA, BenezraR (2005) Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer 5 : 603–614.
14. SikderHA, DevlinMK, DunlapS, RyuB, AlaniRM (2003) Id proteins in cell growth and tumorigenesis. Cancer Cell 3 : 525–530.
15. WongYC, WangX, LingMT (2004) Id-1 expression and cell survival. Apoptosis 9 : 279–289.
16. TangJ, GordonGM, MullerMG, DahiyaM, ForemanKE (2003) Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen induces expression of the helix-loop-helix protein Id-1 in human endothelial cells. J Virol 77 : 5975–5984.
17. GloecknerCJ, BoldtK, SchumacherA, RoepmanR, UeffingM (2007) A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics 7 : 4228–4234.
18. MiyazonoK, KamiyaY, MorikawaM (2010) Bone morphogenetic protein receptors and signal transduction. J Biochem 147 : 35–51.
19. SunR, LiangD, GaoY, LanK (2014) Kaposi's Sarcoma-Associated Herpesvirus-Encoded LANA Interacts with Host KAP1 to Facilitate Establishment of Viral Latency. J Virol 88 : 7331–7344.
20. GaoSJ, KingsleyL, HooverDR, SpiraTJ, RinaldoCR, et al. (1996) Seroconversion to antibodies against Kaposi's sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi's sarcoma. N Engl J Med 335 : 233–241.
21. WangL, LiuYT, HaoR, ChenL, ChangZ, et al. (2011) Molecular mechanism of the negative regulation of Smad1/5 protein by carboxyl terminus of Hsc70-interacting protein (CHIP). J Biol Chem 286 : 15883–15894.
22. Blanco CalvoM, Bolos FernandezV, Medina VillaamilV, Aparicio GallegoG, Diaz PradoS, et al. (2009) Biology of BMP signalling and cancer. Clin Transl Oncol 11 : 126–137.
23. HollnagelA, OehlmannV, HeymerJ, RutherU, NordheimA (1999) Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J Biol Chem 274 : 19838–19845.
24. KatagiriT, ImadaM, YanaiT, SudaT, TakahashiN, et al. (2002) Identification of a BMP-responsive element in Id1, the gene for inhibition of myogenesis. Genes Cells 7 : 949–960.
25. YuPB, HongCC, SachidanandanC, BabittJL, DengDY, et al. (2008) Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol 4 : 33–41.
26. QiuH, YangB, PeiZC, ZhangZ, DingK (2010) WSS25 inhibits growth of xenografted hepatocellular cancer cells in nude mice by disrupting angiogenesis via blocking bone morphogenetic protein (BMP)/Smad/Id1 signaling. J Biol Chem 285 : 32638–32646.
27. QiuH, TangW, TongX, DingK, ZuoJ (2007) Structure elucidation and sulfated derivatives preparation of two alpha-D-glucans from Gastrodia elata Bl. and their anti-dengue virus bioactivities. Carbohydr Res 342 : 2230–2236.
28. VucicevicL, MisirkicM, JanjetovicK, Harhaji-TrajkovicL, PricaM, et al. (2009) AMP-activated protein kinase-dependent and -independent mechanisms underlying in vitro antiglioma action of compound C. Biochem Pharmacol 77 : 1684–1693.
29. RiosM, ForetzM, ViolletB, PrietoA, FragaM, et al. (2013) AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res 73 : 2628–2638.
30. LeiX, ZhuY, JonesT, BaiZ, HuangY, et al. (2012) A Kaposi's sarcoma-associated herpesvirus microRNA and its variants target the transforming growth factor beta pathway to promote cell survival. J Virol 86 : 11698–11711.
31. LiuY, SunR, LinX, LiangD, DengQ, et al. (2012) Kaposi's sarcoma-associated herpesvirus-encoded microRNA miR-K12-11 attenuates transforming growth factor beta signaling through suppression of SMAD5. J Virol 86 : 1372–1381.
32. Di BartoloDL, CannonM, LiuYF, RenneR, ChadburnA, et al. (2008) KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood 111 : 4731–4740.
33. DurringtonHJ, UptonPD, HoerS, BonameJ, DunmoreBJ, et al. (2010) Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type II. J Biol Chem 285 : 37641–37649.
34. WangYF, WangLY, LiYL, ShyuHW, ChiouYH, et al. (2010) Human herpesvirus 8 viral FLICE-inhibitory protein retards cell proliferation via downregulation of Id2 and Id3 expression. Mol Cell Biochem 343 : 83–89.
35. EverlyDNJr, MainouBA, Raab-TraubN (2004) Induction of Id1 and Id3 by latent membrane protein 1 of Epstein-Barr virus and regulation of p27/Kip and cyclin-dependent kinase 2 in rodent fibroblast transformation. J Virol 78 : 13470–13478.
36. LiHM, ZhuangZH, WangQ, PangJC, WangXH, et al. (2004) Epstein-Barr virus latent membrane protein 1 (LMP1) upregulates Id1 expression in nasopharyngeal epithelial cells. Oncogene 23 : 4488–4494.
37. LoAK, DawsonCW, LoKW, YuY, YoungLS (2010) Upregulation of Id1 by Epstein-Barr virus-encoded LMP1 confers resistance to TGFbeta-mediated growth inhibition. Mol Cancer 9 : 155.
38. AlaniRM, YoungAZ, ShifflettCB (2001) Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proc Natl Acad Sci U S A 98 : 7812–7816.
39. OhtaniN, ZebedeeZ, HuotTJ, StinsonJA, SugimotoM, et al. (2001) Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 409 : 1067–1070.
40. LasorellaA, UoT, IavaroneA (2001) Id proteins at the cross-road of development and cancer. Oncogene 20 : 8326–8333.
41. VogtJ, TraynorR, SapkotaGP (2011) The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell Signal 23 : 1831–1842.
42. XiaoZ, WatsonN, RodriguezC, LodishHF (2001) Nucleocytoplasmic shuttling of Smad1 conferred by its nuclear localization and nuclear export signals. J Biol Chem 276 : 39404–39410.
43. QinBY, ChackoBM, LamSS, de CaesteckerMP, CorreiaJJ, et al. (2001) Structural basis of Smad1 activation by receptor kinase phosphorylation. Mol Cell 8 : 1303–1312.
44. DuanX, LiangYY, FengXH, LinX (2006) Protein serine/threonine phosphatase PPM1A dephosphorylates Smad1 in the bone morphogenetic protein signaling pathway. J Biol Chem 281 : 36526–36532.
45. KnockaertM, SapkotaG, AlarconC, MassagueJ, BrivanlouAH (2006) Unique players in the BMP pathway: small C-terminal domain phosphatases dephosphorylate Smad1 to attenuate BMP signaling. Proc Natl Acad Sci U S A 103 : 11940–11945.
46. ZhangY, ChangC, GehlingDJ, Hemmati-BrivanlouA, DerynckR (2001) Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci U S A 98 : 974–979.
47. LiL, XinH, XuX, HuangM, ZhangX, et al. (2004) CHIP mediates degradation of Smad proteins and potentially regulates Smad-induced transcription. Mol Cell Biol 24 : 856–864.
48. LanK, KuppersDA, VermaSC, RobertsonES (2004) Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J Virol 78 : 6585–6594.
49. LiX, LiangD, LinX, RobertsonES, LanK (2011) Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen reduces interleukin-8 expression in endothelial cells and impairs neutrophil chemotaxis by degrading nuclear p65. J Virol 85 : 8606–8615.
50. QiHH, OngusahaPP, MyllyharjuJ, ChengD, PakkanenO, et al. (2008) Prolyl 4-hydroxylation regulates Argonaute 2 stability. Nature 455 : 421–424.
51. LiuY, CaoY, LiangD, GaoY, XiaT, et al. (2008) Kaposi's sarcoma-associated herpesvirus RTA activates the processivity factor ORF59 through interaction with RBP-Jkappa and a cis-acting RTA responsive element. Virology 380 : 264–275.
52. FengY, WangY, WangZ, FangZ, LiF, et al. (2012) The CRTC1-NEDD9 signaling axis mediates lung cancer progression caused by LKB1 loss. Cancer Res 72 : 6502–6511.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Molecular and Cellular Mechanisms of KSHV Oncogenesis of Kaposi's Sarcoma Associated with HIV/AIDS
- Holobiont–Holobiont Interactions: Redefining Host–Parasite Interactions
- BCKDH: The Missing Link in Apicomplexan Mitochondrial Metabolism Is Required for Full Virulence of and
- Helminth Infections, Type-2 Immune Response, and Metabolic Syndrome
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy