
Chitin-Degrading Protein CBP49 Is a Key Virulence Factor in American Foulbrood of Honey Bees
American Foulbrood and its etiological agent, Paenibacillus larvae, pose a serious threat to global honey bee health. So far, molecular mechanisms of host-microbe interactions are poorly understood in this system and no key virulence factor for the entire species has been identified. Here, we demonstrate that P. larvae expresses a chitin-binding and -degrading protein PlCBP49 harboring one module that belongs to the auxiliary activity 10 (AA10) family of lytic polysaccharide monooxygenases (LPMOs). We provide evidence that PlCBP49 degrades chitin via a metal ion-dependent, oxidative mechanism, as already described for other members of the AA10 enzyme family. Using P. larvae mutants lacking PlCBP49 expression, we demonstrate that PlCBP49 is crucial for the degradation of the chitin-rich peritrophic matrix, a key step in pathogenesis of P. larvae infections. In the absence of PlCBP49 expression the peritrophic matrix remained nearly intact and about 95% of the infected larvae survived infection. This clearly indicated that PlCBP49 is a key virulence factor of P. larvae. These results constitute important progress in our understanding of both P. larvae pathogenesis and the biological role of LPMOs in entomopathogens. Furthermore, knowing PlCBP49 and its role in pathogenesis opens new possibilities to develop curative measures for this disease.
Published in the journal:
. PLoS Pathog 10(7): e32767. doi:10.1371/journal.ppat.1004284
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004284
Summary
American Foulbrood and its etiological agent, Paenibacillus larvae, pose a serious threat to global honey bee health. So far, molecular mechanisms of host-microbe interactions are poorly understood in this system and no key virulence factor for the entire species has been identified. Here, we demonstrate that P. larvae expresses a chitin-binding and -degrading protein PlCBP49 harboring one module that belongs to the auxiliary activity 10 (AA10) family of lytic polysaccharide monooxygenases (LPMOs). We provide evidence that PlCBP49 degrades chitin via a metal ion-dependent, oxidative mechanism, as already described for other members of the AA10 enzyme family. Using P. larvae mutants lacking PlCBP49 expression, we demonstrate that PlCBP49 is crucial for the degradation of the chitin-rich peritrophic matrix, a key step in pathogenesis of P. larvae infections. In the absence of PlCBP49 expression the peritrophic matrix remained nearly intact and about 95% of the infected larvae survived infection. This clearly indicated that PlCBP49 is a key virulence factor of P. larvae. These results constitute important progress in our understanding of both P. larvae pathogenesis and the biological role of LPMOs in entomopathogens. Furthermore, knowing PlCBP49 and its role in pathogenesis opens new possibilities to develop curative measures for this disease.
Introduction
Vertebrates and invertebrates alike need to protect their intestinal epithelia against various chemical, physical and biological challenges while the transport of nutrients and water must remain uninterrupted to aid in digestion. For this purpose, mucosal secretions line the digestive tract of vertebrates, while in most invertebrates the midgut epithelium is lined by the peritrophic membrane or peritrophic matrix (PM) [1], [2], an organized layer made of secreted chitin and (glyco)proteins. Chitin, an insoluble linear β-(1,4)-linked polymer of N-acetyl-D-glucosamine (GlcNAc), is considered the major structural component of the PM where it forms chitin fibrils. These fibrils are held together by chitin-binding proteins, while the interstitial spaces are filled by glycans (for a recent review see [3]). The resulting lattice acts as a molecular sieve with a large range of pore sizes. The functions attributed to the PM include (i) compartmentalization of digestive processes, (ii) protection from ingested xenobiotics, and (iii) acting as mechanical barrier against abrasive foodstuffs and pathogens (for a recent review see [2]). The latter function makes the PM a first-line defense against ingested pathogens and, hence, an important part of the invertebrates' complex system to combat infections. Accordingly, insect-pathogenic bacteria infectious per os must breach the PM before they can attack and invade or cross the epithelium. To this end, hydrolytic enzymes such as proteases and, most importantly, chitin-degrading proteins enabling PM degradation are produced and secreted by these pathogens.
Most of the chitin-degrading proteins are chitinases belonging to the family of glycosyl hydrolases (GH). To date, 133 different GH families classified on the basis of sequence similarities and forming 14 clans (GH-A – GH-N) of related families based on similarities in protein folds can be found in the Carbohydrate Active Enzymes (CAZy) database [4]. Another family of bacterial chitin-degrading proteins comprises proteins with a carbohydrate-binding module (CBM) and belong to the auxiliary activities 10 (AA10) family (formerly chitin binding module 33 (CBM33) family) of lytic polysaccharide monooxygenases (LPMOs) as defined in the Carbohydrate Active Enzymes (CAZy) database [4]. These proteins were originally thought to lack any catalytic activity of their own [4], although they had been shown to be involved in chitin degradation [5]. However, it was recently demonstrated that CBP21, an AA10 (formerly CBM33) family member expressed by the Gram-negative soil bacterium Serratia marcescens [6], [7], as well as EfCBM33A, expressed by the Gram-positive, opportunistic pathogen Enterococcus faecalis, are capable of degrading crystalline chitin via a novel, copper-dependent, oxidative enzymatic mechanism [8]–[10]. LPMOs comprise only two families, (i) the AA10 (formerly CBM33) family with bacterial, viral, and some eukaryotic members and (ii) the AA9 (formerly glycoside hydrolase 61 (GH61)) family with only fungal members so far. Both families are monooxygenases and target recalcitrant polysaccharides.
Bacterial chitin-degrading proteins are produced mainly to meet nutritional needs of the bacteria because chitin-degradation products, once transported into the bacterial cell, can be used as carbon sources [11], [12]. Because, in addition, many insect pathogens need to overcome chitin-containing structures (e.g., cuticula or peritrophic membranes) to enter a host and establish an infection, degradation of the PM by bacterial pathogens might be a process serving two purposes: nutrition and invasion.
Paenibacillus larvae is a bacterial pathogen of honey bee larvae which causes a notifiable disease called American Foulbrood (AFB) [13]. AFB is a highly contagious disease and is fatal for the entire colony when detected in too late a stage of disease. However, since most authorities consider burning of diseased colonies and contaminated hive material the only workable control measure, diseased colonies are inevitably lost in most cases. Hence, AFB causes considerable economic losses in apiculture worldwide. The etiological agent, P. larvae, is a Gram-positive, rod-shaped bacterium forming tenacious spores under adverse environmental conditions like lack of nutrients. These spores are the infectious form of P. larvae and they initiate a fatal infectious process in bee larvae once ingested with contaminated larval food. Honey bee larvae become less susceptible to infection with increasing age and as soon as two days after egg hatching they can be considered “resistant” (for recent reviews: [14]–[16]). This phenomenon has been attributed to the growing PM already in the early days of AFB research [17], [18]. Recently, we demonstrated that the honey bee larval gut is lined by a chitin-containing PM which is degraded during P. larvae infection [19], confirming earlier results showing that the PM is the first barrier the bacteria have to overcome when trying to breach the epithelium and to enter the haemocoel [20]. Proteases and chitin-degrading proteins are most likely involved in this process. For P. larvae it is long since known that an impressive number of proteases is expressed and secreted [21], [22]. These proteases, although poorly characterized, have already been implicated as virulence factors as they might aid in degrading the PM, breaching the epithelium and converting larval into bacterial biomass [23]–[25]. In contrast, little is known so far about the nature and expression of P. larvae chitin-degrading proteins and their role in the pathogenesis of AFB. Recently, the genomes of representatives of two P. larvae genotypes, ERIC I and ERIC II, were sequenced, annotated and used for comparative genome analysis [26]. Surprisingly, no complete, uninterrupted and, hence, putatively functional gene coding for a classical chitinase could be detected in any of the genomes [26] posing the intriguing question: how is the described chitin-degradation by P. larvae during infection [19] achieved? We are now answering this question by describing the identification and functional characterization of P. larvae PlCBP49, a novel member of the AA10 (formerly CBM33) family of chitin-binding and –degrading LPMOs. PlCBP49 was confirmed in the genome and secretome of P. larvae. Chitin-affinity purified PlCBP49 was used to demonstrate chitinolytic activity both on soluble and insoluble substrates as well as to confirm that chitin degradation was metal ion-dependent and involved an oxidative step. Furthermore, we studied the biological role of PlCBP49 in P. larvae infected larvae and were able to demonstrate that PlCBP49 is involved in PM degradation during infection and is a key virulence factor of P. larvae.
Results
P. larvae expresses chitin-binding and -degrading proteins
Recently, degradation of the larval midgut PM by P. larvae was demonstrated to be a key step in the pathogenesis of P. larvae infections [19]. In order to identify proteins which might be involved in this process, we isolated and analyzed chitin-binding proteins from P. larvae culture supernatants using chitin-coated beads. SDS-PAGE analysis of the chitin-binding fractions revealed two chitin-binding proteins (CBP) migrating around 60 kD (CBP60) and 49 kD (PlCBP49I) secreted by ATCC9545 (P. larvae ERIC I), while only one band migrating around 49 kD (PlCBP49II) was visible in the supernatant of DSM25430 (P. larvae ERIC II) (Fig. 1A).
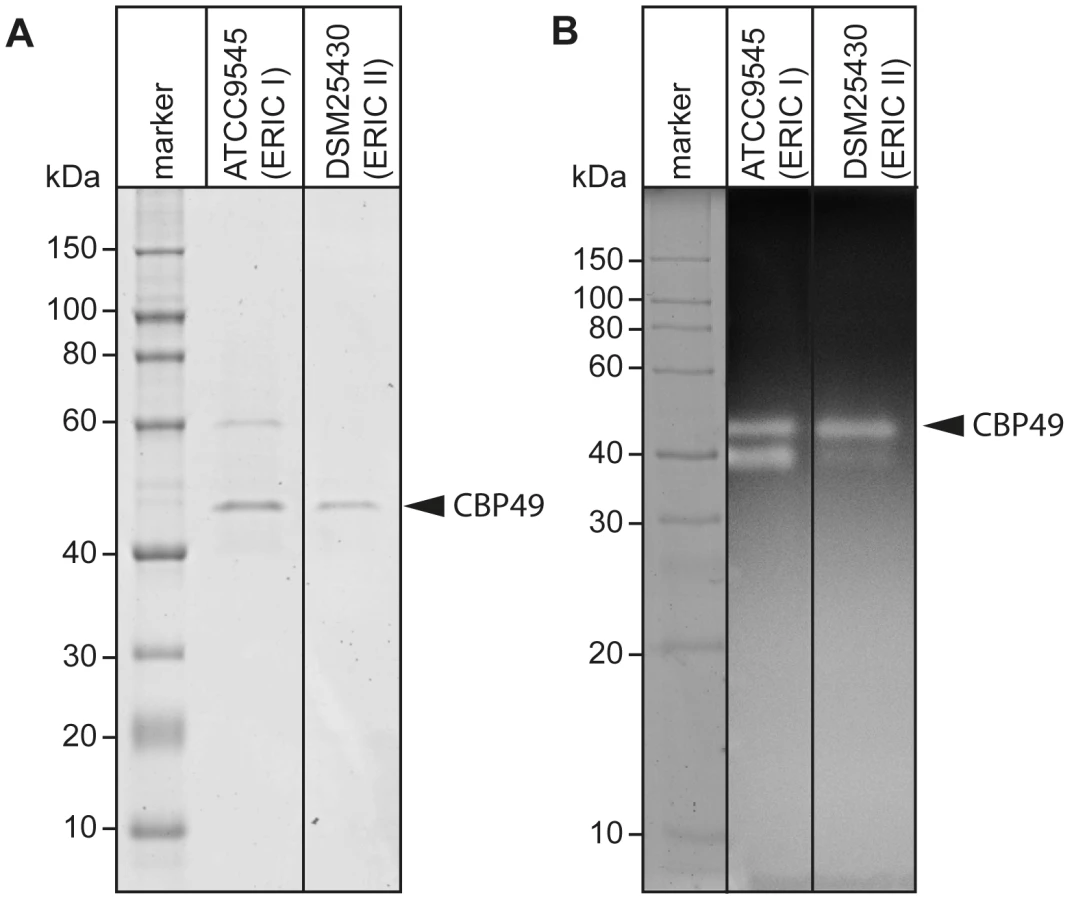
To determine the chitinolytic activity of the isolated proteins, the chitin-binding fractions were subjected to zymography performed with denaturing gels containing ethylene glycol chitin (EGC) as the substrate (Fig. 1B). The chitin binding fractions of both isolates produced a clear zone in the range of 49 kD suggesting that in both strains a protein migrating around 49 kD was able to degrade the chitin analogue EGC. We hypothesized that these bands corresponded to PlCBP49I and PlCBP49II identified in SDS-PAGE analysis of the chitin-binding fractions of ATCC9545 and DSM25430, respectively. In addition, chitinolytic activity could also be detected around 42 kD in the chitin-binding fraction of ATCC9545 and a similar activity was weakly but not reproducibly detectable in the chitin-binding fraction of DSM25430. These activities could not be related to any proteins detectable in the Coomassie-stained gels of the chitin-binding fractions hampering their further characterization. This phenomenon was not surprising because zymography is a highly sensitive method of detecting enzymatically active proteins even when present in such low amounts that they cannot be visualized with common staining procedures. In the chitin-binding fraction of ATCC9545, no chitinolytic activity could be observed around 60 kD indicating that CBP60, although binding to chitin, was not able to degrade EGC.
PlCBP49, the major chitin-degrading enzyme of P. larvae, is a member of the AA10 family of LPMOs
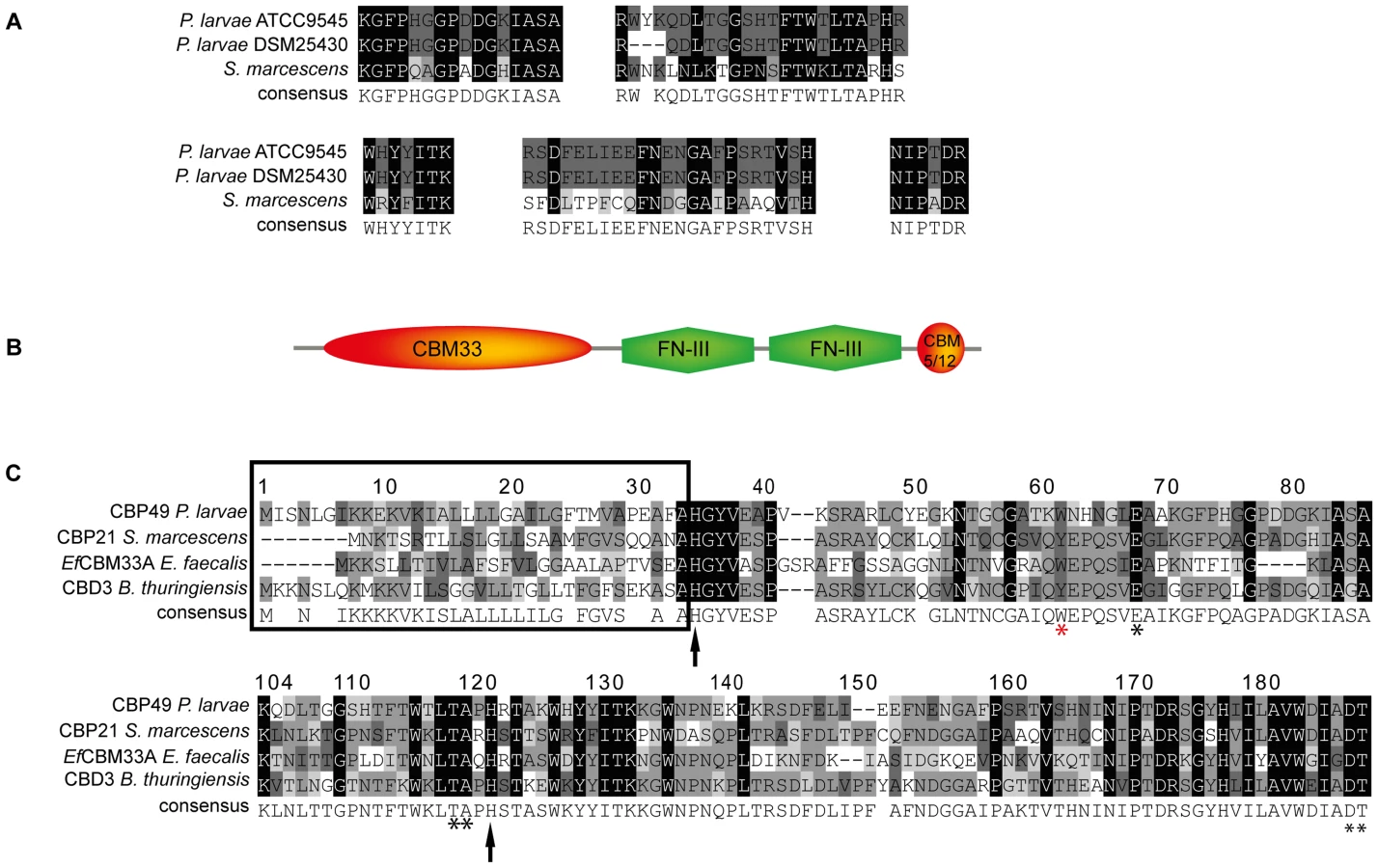
To further substantiate that PlCBP49I and PlCBP49II are not only chitin-binding but also chitin-degrading proteins, we determined their protein sequences via mass spectrometry analysis after separation of the bead-bound proteins by SDS-PAGE. Comparison of the obtained peptide sequence data suggested that PlCBP49I was identical to PlCBP49II (Fig. 2A). Protein BLAST analysis of the peptide sequences indicated that PlCBP49I and PlCBP49II were not classical chitinases but rather homologs of CBP21, a chitin-binding and –degrading protein of the chitinolytic bacterium S. marcescens belonging to the AA10 family of LPMOs [5], [8], [10] (Fig. 2A).
To analyze whether ATCC9545 and DSM25430 harbor a common gene putatively coding for PlCBP49 we determined the sequences of the corresponding genes cbp49I and cbp49II in ATCC9545 and DSM25430, respectively, by comparing the obtained peptide fragment sequences with the genomic sequences of P. larvae BRL 230010 [27] via TBLASTN analysis [28]. Nucleotide sequence analysis confirmed the presence of the same functional open reading frame (ORF) in both strains (Genbank accession numbers JX185746 for ATCC9545, JX185745 for DSM25430) indicating that both strains harbor the gene cbp49 coding for the protein PlCBP49 with 443 amino acids. Screening a collection of P. larvae field isolates for the presence of cbp49 confirmed the presence of this gene in all strains analyzed so far (data not shown).
Protein BLAST alignment of the translated genomic sequence of PlCBP49 followed by domain analysis of the deduced amino acid sequence using the Conserved Domain Database (CDD; NCBI) revealed a modular protein with an N-terminal domain homologous to AA10 (formerly CBM33) family members of LPMOs followed by two fibronectin type III-like domains, which are often found in bacterial glycosyl hydrolases, and a second small chitin-binding domain (CBM 5/12) at the C-terminus, which is also found in many different glycosyl hydrolase enzymes and presumed to have a carbohydrate binding function (Fig. 2B). Similar domain architecture has recently been described for several other members of the AA10 family of LPMOs [29]–[31]. Amino acid alignment of the AA10 domain of P. larvae PlCBP49 protein with other AA10 family members (Fig. 2C) identified an N-terminal signal sequence (framed) and two histidine residues (His35, His122; arrows) which are highly conserved in all proteins of the AA10 family and which are involved in coordinating copper [8], [10]. Some other conserved residues implicated in binding to chitin [5] were also present such as Tyr62, Glu68, Thr119, Ala120, Asp188, and Thr189 (asterisks) [8], [10] with Tyr62 in P. larvae (red asterisk) corresponding to Trp56 in CBP21 and Tyr54 in EfCBM33A [8], [10]. These results indicated that P. larvae PlCBP49 contains a domain characteristic for the AA10 family of LPMOs and suggested that PlCBP49 has LPMO activity.
PlCBP49 degrades chitin via a metal ion-dependent, oxidative mechanism
To further substantiate that P. larvae PlCBP49 is a member of the AA10 family of LPMOs we analyzed the chitin-degrading activity of PlCBP49 in greater detail by zymography in the presence of the metal chelator ethylenediaminetetraacetic acid (EDTA) and the di-oxygen mimic cyanide, which were both shown to be potential inhibitors of AA10 family members [8], [9]. P. larvae PlCBP49 activity was strongly inhibited in the presence of 20 mM EDTA and 2 mM potassium cyanide (KCN) (Fig. 3) supporting the involvement of divalent cations and the crucial role of the oxidative step in chitin degradation through PlCBP49. In contrast, presence of 20 mM caffeine, a competitive inhibitor of chitinases [32], did not inhibit chitin degradation (Fig. 3) suggesting that PlCBP49 indeed is not a classical chitinase.

Cbp49 gene disruption results in loss of both PlCBP49 expression and chitinolytic activity
To better analyze the chitin-degrading activity of PlCBP49, to functionally characterize this protein in the P. larvae genotypes ERIC I and ERIC II [13], [16], and to assess its role in the pathogenesis of AFB, P. larvae mutants deficient in the expression of PlCBP49 were constructed from ATCC9545 and DSM25430 as parent strains. The chitin-binding fractions of both mutant strains were analyzed by SDS-PAGE (Fig. 4A) and zymography (Fig. 4B) to demonstrate loss of PlCBP49 expression and of chitinolytic activity concomitant with cbp49 gene disruption. In both mutants, ATCC9545 Δcbp and DSM25430 Δcbp, the protein bands corresponding to PlCBP49 (Fig. 4A) as well as the chitinolytic activity migrating around 49 kDa (Fig. 4B) were missing in the corresponding chitin-binding fractions. In contrast, the chitinolytic activity visible around 42 kDa was unaffected (Fig. 4B) indicating that gene disruption of cbp49 had no downstream effects on the chitinolytic machinery detectable via zymography. These results clearly confirmed that the identified cbp49 gene encodes the identified protein PlCBP49 which in turn is responsible for the observed activity towards soluble chitin.

P. larvae PlCBP49 is involved in PM degradation
We recently showed that P. larvae is able to metabolize insoluble, colloidal chitin [19]. However, the data related to the chitin-degrading activity of PlCBP49 obtained so far were based on using EGC as a soluble substrate in zymograms. To further verify that degradation of insoluble recalcitrant polysaccharides is mediated by PlCBP49, as it is described for members of the AA10 family of LPMOs, we tested whether or not PlCBP49 might be able to degrade chitin-containing structures like an insect PM. To this aim, we used an Ussing chamber (Fig. 5A) to perform permeability assays with PMs which were isolated from S. frugiperda last instar larvae and subjected to the chitin bound fractions of the knock-out P. larvae strains (ATCC9545 Δcbp, DSM25430 Δcbp) and of the corresponding parent wild-type strains (ATCC9545, DSM25430). This comparative approach allows differences in PM permeabilization between mutant and wild-type bacteria to be linked with differences in PlCBP49 expression. Permeability of the PMs was measured as methylene blue (MB) efflux and was significantly higher after incubation with the chitin-binding fractions of the wild-type bacteria than after incubation with the chitin-binding fractions of the corresponding mutants (Fig. 5B). For P. larvae ATCC9545 MB efflux significantly (student's t-test, p-value = 0.0125) decreased from 0.0429±0.006 µg/ml/mm2/h in the presence of PlCBP49 expression (ATCC9545, mean values ± SEM) to 0.021±0.0016 µg/ml/mm2/h in the absence of PlCBP49 expression (ATCC9545 Δcbp, mean values ± SEM). Similar results were obtained for P. larvae DSM25430 (0.02418±0.002 µg/ml/mm2/h, mean values ± SEM) compared to P. larvae DSM25430 Δcbp (0.0094±0.0013 µg/ml/mm2/h, mean values ± SEM) which were also significantly different (student's t-test, p-value = 0.004). Remarkably, exposure of PMs to chitin-bound fractions of DSM25430 Δcbp resulted in PM permeability not significantly different from the negative control (student's t-test, p-value = 0.983) indicating that in the absence of PlCBP49 expression no PM degrading activity was active in these fractions. In contrast, chitin-bound fractions of ATCC9545 Δcbp resulted in PM permeability that was significantly higher than the negative control (student's t-test, p-value = 0.006) although also significantly reduced when compared to the effect achieved with ATCC9545 wild-type, meaning in the presence of PlCBP49 expression. These results indicated that the chitinolytic activity of PlCBP49 can act on chitin in its native crystalline form suggesting a role for PlCBP49 also in PM degradation observed during P. larvae infection of honey bee larvae [19].

PlCBP49 is an essential virulence factor of P. larvae
To test the proposed involvement of PlCBP49 in PM degradation in P. larvae infected honey bee larvae, we isolated (Fig. 6A) and stained PMs (Fig. 6B–D) from non-infected control larvae as well as from larvae infected with either wild-type P. larvae (ATCC9545) or mutant P. larvae (ATCC9545 Δcbp) in order to assess PM integrity. While control larvae at the age of six days after egg hatching contained intact PMs (Figs. 6A, B), the PM in ATCC9545-infected larvae of the same age appeared almost totally degraded with only small patches of stainable structures (Fig. 6C). In contrast, larvae of the same age infected with ATCC9545 Δcbp and, therefore, lacking the chitin-degrading activity of PlCBP49, did still contain an almost intact and stainable PM (Fig. 6D). This result indicated that PM degradation only occurs in the presence of PlCBP49 expression and, therefore, confirmed that PlCBP49 plays a key role in the degradation of the larval PM during infection.

PM degradation in P. larvae infected larvae has been shown to be a key step in AFB pathogenesis [19]. Therefore, we hypothesized that PlCBP49 involved in PM degradation in infected larvae should be a key virulence factor of this pathogen. To test this, we performed laboratory infection assays by feeding first instar honey bee larvae larval diet containing spores of the knock-out P. larvae strains (ATCC9545 Δcbp, DSM25430 Δcbp) and of the corresponding parent wild-type strains (ATCC9545, DSM25430). Comparing mortality in the groups infected with the wild-type and with the mutant bacteria revealed that the lack of PlCBP49-expression led to a significant reduction in mortality (Fig. 6E). While 500 cfu/ml of ATCC9545 resulted in a total mortality of 75.5%±5.0% (mean values ± SEM), the same spore concentration killed only 4.4%±1.1% (mean values ± SEM) in the group infected with the mutant strain ATCC9545 Δcbp (student's t-test, p-value = 0.0001). The results were similar for DSM25430 and DSM25430 Δcbp: 100 cfu/ml killed 77.7%±8.7% (mean values ± SEM) in the group infected with the wild-type DSM25430 and only 3.3%±1.9% (mean values ± SEM) died in the group infected with DSM25430 Δcbp (student's t-test, p-value = 0.0011). In other words, lack of PlCBP49 activity resulted in about 94% reduction in mortality for ATCC9545 and nearly 96% reduction in mortality for DSM25430. Therefore, the lack of PlCBP49 expression nearly abolished the virulence of both P. larvae strains. These results clearly indicated that PlCBP49 is a key virulence factor for both P. larvae genotypes.
Discussion
Attempts to understand the pathogenesis of P. larvae infections of honey bee larvae have been undertaken since the early days of AFB research. It was suggested that the PM acts as a protective shield against P. larvae infections because it might hamper vegetative bacteria in their attempts to cross the gut wall [17], [33]–[35]. Recently, we demonstrated that the PM indeed is the first barrier vegetative P. larvae bacteria have to overcome before attacking and breaching the epithelium [20]. In addition, we recently showed that P. larvae is able to metabolize colloidal chitin [19] and that the larval midgut PM contains chitin which is degraded during P. larvae infection, leading to loss of PM integrity [19]. We now describe the identification and functional analysis of P. larvae PlCBP49, a new member of the AA10 (formerly CBM33) family of chitinolytic LPMOs which were recently shown to be able to degrade crystalline chitin via a novel, oxidative mechanism [8]. We demonstrate that PlCBP49 is involved in PM degradation in infected honey bee larvae and that it is a key virulence factor for both P. larvae genotypes, as in the absence of PlCBP49 expression P. larvae virulence was almost lost (for a definition of the term virulence please see [36]).
When invading the infected larva, P. larvae faces two physical barriers: first the peritrophic membrane and second the epithelium [20]. Although the destruction of the epithelial cell layer by P. larvae is still poorly understood, the identification and characterization of PlCBP49 now allows the biochemical mechanism by which P. larvae penetrates and traverses the PM to be unravelled. Cbp49 encodes a chitin-binding and –degrading protein belonging to the AA10 (formerly CBM33) family of chitin-degrading LPMOs. The best analyzed member of this family is CBP21 expressed by S. marcescens, a Gram-negative soil bacterium described as an insect pathogen but which is also an opportunistic pathogen of mammals [6], [37], [38]. CBP21 was originally described as lacking any catalytic activity of its own despite its essential role in chitin-degradation [5]. However, it was recently shown to possess chitin-degrading activity [8] and it was demonstrated that CBP21 degrades crystalline chitin through a novel copper-dependent oxidative mechanisms [8], [10].
So far, the only function attributed to CBP21 is that of a food-scavenging enzyme allowing S. marcescens to use chitin as carbon source. However, S. marcescens is pathogenic to many invertebrates including insects where it causes intestinal infections [37]. Based on our results we now propose that the biological role of S. marcescens CBP21 is not only to enable these bacteria to use chitin as carbon source but also to allow them to degrade the protective PM as part of the pathogenic process when S. marcescens acts as insect pathogen. This assumption is further substantiated by recent findings showing that Drosophila melanogaster deficient in forming a proper PM due to a loss-of-function mutation in the gene dcy coding for Drosocrystallin, an integral component of D. melanogaster PM, are more susceptible to oral infection by S. marcescens than wild-type flies [39]. Furthermore, for EfCBM33A it was recently speculated that it is involved in host-microbe interactions based on the observed gene regulation [9].
PlCBP49 is 443 amino acids in length and in addition to the AA10 module also contained two fibronectin type III-like domains and another chitin-binding module, CBM5/12. The latter domain has often been found in proteins expressed by bacteria thriving in the digestive tract of invertebrates [40]. For instance, CBP50 expressed by B. thuringiensis has a similar domain architecture [30] and it was demonstrated that the FN III-like domains and the CBM5/12 domain are involved in substrate binding. Future experiments should address the question of the function of these domains for PlCBP49 activity. According to our results, PlCBP49 not only degraded insoluble chitin structures but also acted on EGC, a soluble analog of chitin. Further biochemical and molecular studies are needed to unravel the function of these additional modules in the context of LPMO activity and to analyze whether these modules are involved in cleavage of soluble chitin, a capability not yet described for other members of the AA10 family of LPMOs.
Normally, Bacilli and Paenibacilli express a wide range of classical chitinases. However, recent evidence suggests that the situation is totally different for P. larvae. When the entire genomes of two P. larvae strains representing the genotypes ERIC I (DSM25719) and ERIC II (DSM25430) were sequenced, manually curated, annotated, and searched for putative virulence factors, in both genomes only a pseudogene containing a chitinase A (GH18 family) N-terminal domain was identified. While the P. larvae genomes harbored more than 100 protease genes, genes coding for classical chitinases were missing [26]. These most recent in silico results further support our finding that PlCBP49 is a key chitin-degrading protein of P. larvae.
By using a chitin-binding activity based approach, we identified PlCBP49 as chitin-degrading protein and we demonstrated that PlCBP49 is able to degrade both soluble and insoluble chitin and is essential for PM degradation in vitro and in vivo. However, while in the Ussing chamber experiment gene disruption of cbp49 in DSM25430 resulted in loss of S. frugiperda PM degradation by DSM25430 Δcbp, the chitin bound fraction of ATCC9545 Δcbp still seemed to contain some activity affecting PM integrity. We speculate that this activity might be due to CBP60, present only in ATCC9545 and ATCC9545 Δcbp chitin bound fractions but absent in the corresponding DSM25430 fractions (Fig. 1). Although CBP60 is a chitin-binding protein, it did not show any chitin-degrading activity. However, CBP60 might have protease activity targeting non-chitin components of the PM like insect intestinal mucin (IIM) or other proteins thereby affecting PM integrity. More than hundred putatively functional protease genes were present in the P. larvae genomes [26] leaving abundant CBP60-candidates which might contribute to PM degradation in ATCC9545. Obviously, much further work is needed to unravel and understand the entire PM degrading system of ATCC9545, to identify CBP60, and to analyze whether it plays a role in PM degradation. Identification and characterization of PlCBP49 presented here is a first and important step towards this end.
Unfortunately, it was impossible to construct complementation mutants to provide the final control for the assays performed with mutant P. larvae. However, our data (i) on PM degradation during P. larvae infection [19], (ii) on the lack of classical chitinase genes in the genome of P. larvae [26], and (iii) the combination of in vitro and in vivo data presented here provide a convincing body of evidence for our interpretation of the role and relevance of PlCBP49 in AFB pathogenesis.
Weakening or even destroying the larval PM allows more ready access of bacteria or bacterial toxins to gut epithelial cells [3], [39], [41]. We recently described the identification of several putative toxin genes in the genome of P. larvae ERIC I [42] and the detailed analysis of two of these toxins, Plx1 and Plx2, which belong to the family of AB-toxins [43]–[45]. Binding of P. larvae AB-toxins to their cognate cell surface receptors of the midgut epithelial cells might be accomplished more easily or even only when the epithelium is no longer protected by a PM like already shown in other insect systems [3]. We, therefore, suggest that degradation of the PM is a prerequisite for P. larvae AB-toxins Plx1 and Plx2 to act on the epithelial cells.
We recently identified SplA, a P. larvae ERIC II-specific surface-layer (S-layer) protein which presumably mediates bacterial adhesion to midgut epithelial cells [46]. Direct adhesion of P. larvae ERIC II to host cells via SplA might only be possible if the cells are no longer protected by a PM. We, therefore, suggest that in the case of P. larvae ERIC II, degradation of the PM is a prerequisite for the bacteria to directly approach the epithelial layer.
Based on these scenarios we propose the following model for pathogenesis of P. larvae infections: during the initial, non-invasive phase of infection, degradation of the PM serves nutritional needs and enables P. larvae to use chitin as an additional carbon source. However, at some time point the PM is totally degraded and bacteria (e.g. via SplA for P. larvae ERIC II) and/or bacterial toxins (Plx1 and Plx2 for P. larvae ERIC I) can directly approach and act on the host cells leading to bacterial breaching of the epithelium and initiation of the invasive phase. According to this model, blocking PM degradation blocks the transition from the non-invasive phase to the invasive phase explaining the impressive loss of virulence connected with the inactivation of PlCBP49 activity. This makes PlCBP49 a key virulence factor of P. larvae.
Materials and Methods
Bacterial strains and growth conditions
Paenibacillus larvae strains ATCC9545 and DSM25430 representing the two P. larvae genotypes ERIC I and ERIC II [13], respectively, were used in this study. Strain ATCC9545 is the type reference strain and was obtained from the American Type Culture Collection (ATCC, USA) through U. Rdest (Biocenter Würzburg, Germany). Strain DSM25430 (Deutsche Sammlung von Mikroorganismen) corresponds to the field isolate 04-309 originating from an outbreak of American Foulbrood in Germany [47]. Both strains have been characterized in several previous studies [13], [42], [46], [48]–[50]. Non-manipulated P. larvae wild-type bacteria were cultivated either in MYPGP liquid broth or on Columbia sheep blood agar plates at 37°C as previously described [47], [51]. Manipulated knockout clones were cultivated on MYPGP-agar plates [52] supplemented with 5 µg/ml chloramphenicol and incubated at 37°C for 2–3 d as previously described [46].
Escherichia coli DH5α cells (Invitrogen) transformed with plasmids pTT_wsfA243 [53] or pTT_cbp573 (see below) were cultivated in selective Luria Bertani (LB) media (agar and broth) supplemented with 30 µg/ml chloramphenicol. Plasmid DNA was prepared following the manufacturer's protocols (QIAprep Spin Miniprep kit, Qiagen). Concentration and purity of the plasmid preparations were analyzed by photometric analysis (Nanodrop) and agarose gelelectrophoresis.
Preparation of spore suspensions for exposure bioassays and determination of spore concentrations by cultivating serial dilutions on Columbia sheep blood agar plates were performed as described previously [13], [48], [49].
Protein purification and analysis
For small-scale affinity purification of P. larvae proteins exhibiting a chitin-binding domain (CBD), P. larvae was grown under the conditions described above and the culture supernatant was collected in the stationary phase by centrifugation. Aliquots (50 µl) of magnetic beads coated with chitin (chitin magnetic beads, New England Biolabs) were washed twice with CBD Column Binding Buffer (500 mM NaCl, 20 mM Tris-HCl, 1 mM EDTA, 0.05% Triton X-100; pH 8.0) at 25°C. An aliquot of filtered (2 µm-filter, Roth) P. larvae culture supernatant (500 µl) was incubated with an aliquot of pre-washed beads for 1 h at 4°C with mild agitation. Beads were magnetically collected and subsequently washed three times with CBD buffer, resuspended in non-reducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer [54], heated for 5 min at 96°C to release the chitin-binding proteins which were then separated by SDS–PAGE and analyzed after staining with Coomassie Brilliant Blue.
Chitin-degrading activity was analyzed by zymography in 10% SDS-PA gels containing 0.1% ethylene glycol chitin (EGC) according to Trudel and Asselin [55]. Briefly, chitin-bound fractions were mixed with non-reducing SDS-PAGE sample buffer [54], heated for 5 min at 96°C to release the chitin-bound proteins and subjected to SDS-PAGE. Subsequently, gels were incubated with mild agitation at 37°C for 2 h in sodium acetate buffer (NaAc buffer; pH 5) containing 1%Triton X-100. Gels were stained for 5 min with 0.01% calcofluor in 0.5 M Tris-HCl (pH 9) and washed four times for 15 min with deionised water. Gel analysis was performed under UV-light. For analyzing the reaction mechanism of PlCBP49, zymography was also performed with NaAc buffer supplemented with 2 mM potassium cyanide (KCN), 20 mM ethylenediaminetetraacetic acid (EDTA), or 20 mM caffeine to determine the conditions inhibiting chitin-degrading activity of PlCBP49.
Determination of the protein and nucleic acid sequence of PlCBP49
In order to determine the protein sequence of PlCBP49I and PlCBP49II, chitin-binding fractions of ATCC9545 and DSM25430 were separated via SDS-PAGE. Coomassie stained bands of both, PlCBP49I and PlCBP49II were excised from the gel and analyzed by mass spectrometry (Alphalyse, Denmark) as already described [50]. The provided protein sequence analysis service included reduction and alkylation of cysteine residues, digestion of the proteins with trypsin, extraction and micro-purification of the obtained peptides using C18 ziptips followed by peptide mapping via MALDI-ToF and partial peptide sequencing via MALDI-ToF/ToF. Proteins were identified on the basis of peptide masses and sequence information by using the in-house databases of Alphalyse (Denmark) and the NCBI database. The obtained peptide sequences of PlCBP49I and PlCBP49II were compared to each other using the protein alignment tool of Vector NTI (Invitrogen).
To identify the genetic information belonging to the sequenced peptides in P. larvae, obtained peptide fragment sequences were compared by TBLASTN analysis [28] with the sequence of P. larvae BRL 230010 [27]. The best hit was with the sequence ZP_09067740.1 annotated as chitin-binding protein. The corresponding ORF was highly homologous to a hypothetical protein of Bacillus cereus (EJQ09528.1; E-value 4e-161) and chitin-binding domain 3 protein of Bacillus thuringiensis serovar berliner (ZP_04102491.1; E-value 4e-161). In order to determine the genomic sequence of PlCBP49 in P. larvae ATCC9545 and P. larvae DSM25430, we selected a primer pair amplifying the complete predicted ORF of PlCBP49 (cbp_F and cbp_R, Table 1) and a second primer pair located up and downstream of the predicted ORF (cbp_up and cbp_down, Table 1). The obtained amplicons for both genotypes were sequenced (Eurofins, Germany) and sequences were aligned using the DNA alignment tool of Vector NTI (Invitrogen). PCR-analysis with primer pair cbp_F and cbp_R (Table 1) of several field isolates of P. larvae ERIC I and ERIC II confirmed the presence of the PlCBP49-gene in all strains analyzed so far (data not shown).

Disruption of the gene coding for PlCBP49 in P. larvae
The PlCBP49-gene in the genomes of P. larvae ATCC9545 and P. larvae DSM25430 was disrupted via a recently described strategy [46], [56] using vector pTT_wsfA243 containing the bacterial mobile group II intron LI.LtrB sequence [46], [53] for constructing a targetron vector for targeted intron insertion at position 573/574 from the start codon of cbp49 in P. larvae determined as optimal insertion site by a computer algorithm provided by the manufacturer (http://www.sigma-genosys.com/targetron). Retargeting of the LI.LtrB targetron of vector pTT_wsfA243 prior to transformation into P. larvae was performed following the manufacturer's protocol (Sigma) and essentially as already described for disruption of the P. larvae S-layer gene splA [46]. In brief, primers IBS_cbp_573, EBS1d_cbp_573, and EBS2_cbp_573 (Table 1) designed by a computer algorithm provided by the manufacturer (see above) were used for modification of the LI.LtrB targetron to generate a specific cbp49 targetron. Replacement of the wsfA243 targetron in pTT_wsfA243 by the cbp49 targetron gave rise to the new vector pTT_cbp573 which was subsequently transformed into E. coli DH5α cells for plasmid replication and preparation.
For creation of P. larvae knockout mutants, electrocompetent P. larvae ATCC9545 and P. larvae DSM25430 cells were prepared as described [57] and 1 µg of plasmid pTT_cbp573 was transformed by electroporation as recently established [58]. Positive clones were selected on MYPGP-agar containing 5 µg/ml chloramphenicol. Successful insertion of the cbp49-specific retargeted intron (915 bp) into the P. larvae target gene cbp49 was demonstrated by PCR-analysis of the corresponding genomic regions in both knockout-strains designated P. larvae ATCC9545 Δcbp and P. larvae DSM25430 Δcbp using primers cbp_F and cbp_R flanking the cbp49 intron insertion position 573/574 of the ORF (Table 1) followed by sequencing the obtained PCR products. The mutant amplicons carrying the insertion migrated at 2259 bp whereas the wild-type amplicons had the expected size of 1344 bp (Fig. 7A). Further analyses of the mutant strains in comparison to their respective parent wild type strains were performed as already described [46] and did not reveal any differences in germination, sporulation, and growth in liquid broth (Fig. 7B,C) which otherwise might have influenced functional analyses. Clones P. larvae ATCC9545 Δcbp and P. larvae DSM25430 Δcbp were further analyzed for the absence of PlCBP49 in the chitin-binding fractions of the secretomes via SDS-PAGE and zymography.

Isolation of larval peritrophic membrane
Non-infected control larvae as well as larvae infected with wild-type strain ATCC9545 or with PlCBP49 deficient strain ATCC9545 Δcbp were reared in 24-well plates as described below. Larvae at 6 d of age were immobilized for 5 min on ice. Larvae were placed under the binocular and opened longitudinally. Fat body was carefully separated from larval midgut, discarded and midgut was washed with 1XPBS (phosphate buffered saline). Clean midgut was opened lengthways and peritrophic membrane was carefully pulled out. 1 ml of 1XPBS was dispensed per well in a 24-well plate and each PM corresponding to different larvae was disposed in a different well. Afterwards, clean PMs were extended on a microscope slide, dried out and stained with a methylene blue-basic fuchsine staining technique essentially as previously described [19].
Permeability tests
Changes in PM permeability were measured using an Ussing chamber (CHM8; World Precision Instruments, Stevenage, United Kingdom) and methylene blue (MB) as described before [59]. Spodoptera frugiperda was employed as a model insect since the required PM area necessary for performing permeability experiments could not be obtained from honey bee larvae. S. frugiperda PMs were isolated from actively feeding last-instar larvae. Each larva was anesthetized shortly on ice; midgut was extracted and longwise opened. PM was carefully pulled out, disposed on a cotton film and opened. The film was cut again to adjust PM containing area and gently washed with 1XPBS to remove food content. Cotton film was then assembled in an Ussing chamber. A suitably sized piece of the PM was used to cover the hole (12.6 mm2) in the Ussing chamber separating the two compartments. The ectoperitrophic side of the PM faced the compartment filled up with 400 µl of 0.2 mg/ml methylene blue solution, and the endoperitrophic side faced the second compartment with 400 µl of 1XPBS. Optical density at 661 nm (OD661) was measured at the beginning of the experiment and after 30 min to ensure the integrity of the PM. To test the activity of PlCBP49 against the PM, 100 µl of the buffer solution in the endoperitrophic compartment was replaced with an equivalent volume of chitin binding fraction of each P. larvae strain (ATCC9545, ATCC9545 Δcbp, DSM25430 and DSM25430 Δcbp). After additional 2 h of incubation the solutions in both compartments were recovered, and the concentration of MB was calculated based on the OD661 nm measures. The MB flux was expressed as µg of dye that passed through the 12.6-mm2 portion of mounted PM in 1 h. The flux measured at 30 min was subtracted from the final flux, to normalize the initial permeability of the PMs. Negative controls were performed with mock incubated chitin beads to rule out any influence of the beads or the buffers used on the PM. At least three independent replicates were performed for each data collection. Data represent mean values ± SEM. Data were statistically analyzed by student's t-test.
Exposure bioassays
In order to analyze the functional role of P. larvae PlCBP49 during pathogenesis of P. larvae infections, honeybee larvae reared in vitro were experimentally infected with the P. larvae knockout strains ATCC9545 Δcbp and DSM25430 Δcbp as well as with the corresponding P. larvae wild-type strains ATCC9545 and DSM25430. These exposure bioassays were performed essentially as previously described [20], [46], [48]. Briefly, spore suspensions with a defined concentration of colony forming units (cfu) were prepared from each of the four strains to be tested. First-instar larvae were collected from different Apis mellifera colonies of the institute's apiary. Larvae were reared in 24-well plates and larval diet (66% royal jelly (v/v), 33% glucose (w/v) and 33% fructose (w/v)) was fed ad libitum. During the first 24 h, larval diet of the infection groups was contaminated with spores to achieve infection. Subsequently, larvae were fed with normal larval diet and fresh larval diet was provided every day. Mock infected control larvae fed with normal larval diet during the whole experiment were used as internal quality control of the exposure bioassays. Only assays with less than 15% mortality in the control groups were considered valid. Spore concentrations were identical for the corresponding strains (500 cfu/ml for both ATCC9545 and ATCC9545 Δcbp; 100 cfu/ml for both DSM25430 and DSM25430 Δcbp). The final spore concentrations corresponded to the LC80 of the wild-type strains and resulted in 75.5%±5.0% and 77.8%±8.7% mortality in the larvae infected with P. larvae ATCC9545 and P. larvae DSM25430, respectively, corroborating previous findings [13], [48]. Taking the LC80 ensured that observation of both decrease and increase in mortality would be possible. Larval health status and mortality were monitored daily over 15 d. Larvae were only considered to have died from AFB if they contained a high number of vegetative P. larvae bacteria after overnight-cultivation of larval remains on CSA plates. P. larvae infection was never detected in control animals or in surviving pupae at day 15 post-infection of any of the infection groups. However, vegetative P. larvae entrapped in non-degraded PM could be demonstrated in engorged larvae of the P. larvae knockout mutant infected groups shortly before defecation. Cbp49 knockout stability (presence of the targetron insertion) in P. larvae cultivated from larval remains of the knock-out-infected groups was confirmed by PCR with the gene specific primer pair cbp_F and cbp_R (Table 1), flanking the intron insertion site 573/574. PCR amplicons were analyzed by gel electrophoresis on a 1% agarose gel, stained with ethidium bromide and visualised by UV light.
Total mortality of the P. larvae wild-type strains and the P. larvae knockout mutants was calculated for each replicate as the proportion of larvae that died from AFB compared to the total number of exposed larvae. For each strain, three biological replicates were performed with thirty larvae each. Data represent mean values ± SEM. Data were statistically analyzed by student's t-test.
Zdroje
1. RichardsAG, RichardsPA (1977) The peritrophic membranes of insects. Annu Rev Entomol 22 : 219–240.
2. TerraWR (2001) The origin and functions of the insect peritrophic membrane and peritrophic gel. Arch Insect Biochem Physiol 47 : 47–61.
3. HegedusD, ErlandsonM, GillottC, ToprakU (2009) New insights into peritrophic matrix synthesis, architecture, and function. Annu Rev Entomol 54 : 285–302.
4. CantarelBLea (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37: D233–238.
5. Vaaje-KolstadG, HornSJ, van AaltenDMF, SynstadB, EijsinkVGH (2005) The non-catalytic chitin-binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J Biol Chem 280 : 28492–28497.
6. GrimontPAD, GrimontF (1978) The genus Serratia. Annu Rev Microbiol 32 : 221–248.
7. JulianGS, BullaLAJr, SharpeES, AdamsGL (1973) Bacteria, spirochetes, and rickettsia as insecticides. Ann NY Acad Sci 217 : 65–75.
8. Vaaje-KolstadG, WesterengB, HornSJ, LiuZ, HZ, et al. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330 : 219–222.
9. Vaaje-KolstadG, BøhleLA, GåseidnesS, DalhusB, BjøråsM, et al. (2012) Characterization of the chitinolytic machinery of Enterococcus faecalis V583 and high-resolution structure of its oxidative CBM33 enzyme. J Mol Biol 416 : 239–254.
10. AachmannFL, SørlieM, Skjåk-BrækG, EijsinkVGH, Vaaje-KolstadG (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc Natl Acad Sci USA 109 : 18779–18784.
11. BolognesiR, TerraWR, FerreiraC (2008) Peritrophic membrane role in enhancing digestive efficiency:Theoretical and experimental models. J Insect Physiol 54 : 1413–1422.
12. BhattacharyaD, NagpureA, GuptaRK (2007) Bacterial Chitinases: Properties and Potential. Crit Rev Biotechnol 27 : 21–28.
13. GenerschE, ForsgrenE, PentikäinenJ, AshiralievaA, RauchS, et al. (2006) Reclassification of Paenibacillus larvae subsp. pulvifaciens and Paenibacillus larvae subsp. larvae as Paenibacillus larvae without subspecies differentiation. Int J Syst Evol Microbiol 56 : 501–511.
14. GenerschE (2007) Paenibacillus larvae and American foulbrood in honeybees. Berl Münch Tierärztl Wschr 120 : 26–33.
15. GenerschE (2008) Paenibacillus larvae and American foulbrood – long since known and still surprising. J Verbr Lebensm 3 : 429–434.
16. GenerschE (2010) American Foulbrood in honeybees and its causative agent, Paenibacillus larvae. J Invertebr Pathol 103: S10–S19.
17. DavidsonEW (1970) Ultrastructure of perithrophic membrane development in larvae of the worker honey bee (Apis mellifera). J Invertebr Pathol 15 : 451–454.
18. DavidsonEW (1973) Ultrastructure of American foulbrood disease pathogenesis in larvae of the worker honey bee, Apis mellifera. J Invertebr Pathol 21 : 53–61.
19. Garcia-GonzalezE, GenerschE (2013) Honey bee larval peritrophic matrix degradation during infection with Paenibacillus larvae, the aetiological agent of American foulbrood of honey bees, is a key step in pathogenesis. Environ Microbiol 15 : 2894–2901.
20. YueD, NordhoffM, WielerLH, GenerschE (2008) Fluorescence in situ-hybridization (FISH) analysis of the interactions between honeybee larvae and Paenibacillus larvae, the causative agent of American foulbrood of honeybees (Apis mellifera). Environ Microbiol 10 : 1612–1620.
21. HolstEC (1946) A simple field test for American foulbrood. Amer Bee 86 : 34.
22. HolstEC, SturtevantAP (1940) Relation of proteolytic enzymes to phase of life cycle of Bacillus larvae, and two new culture media for this organism. J Bacteriol 40 : 723–731.
23. DancerBN, ChantawannakulP (1997) The proteases of American foulbrood scales. J Invertebr Pathol 70 : 79–87.
24. AntúnezK, AnidoM, SchlappG, EvansJD, ZuninoP (2009) Characterization of secreted proteases of Paenibacillus larvae, potential virulence factors involved in honeybee larval infection. J Invertebr Pathol 102 : 129–132.
25. AntunezK, ArredondoD, AnidoM, ZuninoP (2011) Metalloprotease production by Paenibacillus larvae during the infection of honeybee larvae. Microbiology SGM 157 : 1474–1480.
26. DjukicM, BrzuszkiewiczE, FünfhausA, VossJ, GollnowK, et al. (2014) How to kill the honey bee larva: Genomic potential and virulence mechanisms of Paenibacillus larvae. PLoS ONE 9: e90914.
27. QinX, EvansJD, AronsteinKA, MurrayKD, WeinstockGM (2006) Genome sequence of the honey bee pathogens Paenibacillus larvae and Ascosphaera apis. Insect Mol Biol 15 : 715–718.
28. AltschulSF, MaddenTL, SchäfferAA, ZhangJ, ZhangZ, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 : 3389–33402.
29. ManjeetK, PurushothamP, NeerajaC, PodileAR (2013) Bacterial chitin binding proteins show differential substrate binding and synergy with chitinases. Microbiol Res 168 : 461–468.
30. MehmoodMA, XiaoX, HafeezFY, GaiYB, WangFP (2011) Molecular characterization of the modular chitin binding protein Cbp50 from Bacillus thuringiensis serovar konkukian. Anton Leeuw Int J G 100 : 445–453.
31. MoserF, IrwinD, ChenS, WilsonDB (2008) Regulation and characterization of Thermobifida fusca carbohydrate-binding module proteins E7 and E8. Biotechnol Bioeng 100 : 1066–1077.
32. RaoFV, AndersenOA, VoraKA, DeMartinoJA, van AaltenDMF (2005) Methylxanthine drugs are chitinase inhibitors: Investigation of inhibition and binding modes. Chem Biol 12 : 973–980.
33. BamrickJ-F (1967) Resistance to American foulbrood in honey bees. VI. Spore germination in larvae of different ages. J Invertebr Pathol 9 : 30–34.
34. JaeckelS (1930) Zur pathologischen Anatomie der Biene Apis mellifica L. während der Metamorphose bei bösartiger Faulbrut. Archiv für Bienenkunde 11 : 41–92.
35. BamrickJ-F (1964) Resistance to American foulbrood in honey bees. V. Comparative pathogenesis in resistant and susceptible larvae. J Insect Pathol 6 : 284–304.
36. Shapiro-IlanDI, FuxaJR, LaceyLA, OnstadDW, KayaHK (2005) Definitions of pathogenicity and virulence in invertebrate pathology. J Invertebr Pathol 88 : 1–7.
37. FlygC, KenneK, BomanHG (1980) Insect pathogenic properties of Serratia marcescens: Phageresistant mutants with a decreased resistance to Cecropia immunity and a decreased virulence to Drosophila. J Gen Microbiol 120 : 173–181.
38. HejaziA, FalkinerFR (1997) Serratia marcescens. J Med Microbiol 46 : 903–912.
39. KuraishiT, BinggeliO, OpotaO, BuchonN, LemaitreB (2011) Genetic evidence for a protective role of the peritrophic matrix against intestinal bacterial infection in Drosophila melanogaster. Proc Natl Acad Sci USA 108 : 15966–15971.
40. NakjangS, NdehDA, WipatA, BolamDN, HirtRP (2012) A novel extracellular metallopeptidase domain shared by animal host-assocaited mutualistic and pathogenic microbes. PLoS ONE 7: e30287.
41. HayakawaT, ShitomiY, MiyamotoK, HoriH (2004) GalNAc pretreatment inhibits trapping of Bacillus thuringiensis Cry1Ac on the peritrophic membrane of Bombyx mori. FEBS Lett 576 : 331–335.
42. FünfhausA, AshiralievaA, BorrissR, GenerschE (2009) Use of suppression subtractive hybridization to identify genetic differences between differentially virulent genotypes of Paenibacillus larvae, the etiological agent of American Foulbrood of honeybees. Environ Microbiol Rep 1 : 240–250.
43. FünfhausA, PoppingaL, GenerschE (2013) Identification and characterization of two novel toxins expressed by the lethal honey bee pathogen Paenibacillus larvae, the causative agent of American foulbrood. Environ Microbiol 15 : 2951–2965.
44. BarthH, AktoriesK, PopoffMR, StilesBG (2004) Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol Mol Biol Rev 68 : 373–402.
45. HolbournKP, ShoneCC, AcharyaKR (2006) A family of killer toxins: Exploring the mechanism of ADP-ribosylating toxins. FEBS J 273 : 4579–4593.
46. PoppingaL, JaneschB, FünfhausA, SekotG, Garcia-GonzalezE, et al. (2012) Identification and functional analysis of the S-layer protein SplA of Paenibacillus larvae, the causative agent of American Foulbrood of honey bees. PLoS Path 8: e1002716.
47. GenerschE, OttenC (2003) The use of repetitive element PCR fingerprinting (rep-PCR) for genetic subtyping of German field isolates of Paenibacillus larvae subsp. larvae. Apidologie 34 : 195–206.
48. GenerschE, AshiralievaA, FriesI (2005) Strain - and genotype-specific differences in virulence of Paenibacillus larvae subsp. larvae, the causative agent of American foulbrood disease in honey bees. Appl Environ Microbiol 71 : 7551–7555.
49. RauchS, AshiralievaA, HedtkeK, GenerschE (2009) Negative correlation between individual-insect-level virulence and colony-level virulence of Paenibacillus larvae, the etiological agent of American foulbrood of honeybees. Appl Environ Microbiol 75 : 3344–3347.
50. FünfhausA, GenerschE (2012) Proteome analysis of Paenibacillus larvae reveals the existence of a putative S-layer protein. Environ Microbiol Rep 4 : 194–202.
51. NeuendorfS, HedtkeK, TangenG, GenerschE (2004) Biochemical characterization of different genotypes of Paenibacillus larvae subsp. larvae, a honey bee bacterial pathogen. Microbiology 150 : 2381–2390.
52. DingmanDW, StahlyDP (1983) Medium promoting sporulation of Bacillus larvae and metabolism of medium components. Appl Environ Microbiol 46 : 860–869.
53. ZarschlerK, JaneschB, PabstM, AltmannF, MessnerP, et al. (2010) Protein tyrosine O-glycosylation—A rather unexplored prokaryotic glycosylation system. Glycobiology 20 : 787–798.
54. LaemmliUK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 : 680–685.
55. TrudelJ, AsselinA (1989) Detection of chitinase activity after polyacrylamide gel electrophoresis. Anal Biochem 178 : 362–366.
56. ZarschlerK, JaneschB, ZayniS, SchäfferC, MessnerP (2009) Construction of a gene knockout system for application in Paenibacillus alvei CCM 2051T, exemplified by the S-layer glycan biosynthesis initiation enzyme WsfP. Appl Environ Microbiol 75 : 3077–3085.
57. MurrayKD, AronsteinKA (2008) Transformation of the Gram-positive honey bee pathogen, Paenibacillus larvae, by electroporation. J Microbiol Meth 75 : 325–328.
58. PoppingaL, GenerschE (2012) Heterologous expression of green fluorescent protein in Paenibacillus larvae, the causative agent of American Foulbrood of honey bees. J Appl Microbiol 112 : 430–435.
59. JakubowskaAK, CacciaS, GordonKH, FerréJ, HerreroS (2010) Downregulation of a chitin deacetylase-like protein in response to baculovirus infection and its application for improving baculovirus infectivity. J Virol 84 : 2547–2555.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Molecular and Cellular Mechanisms of KSHV Oncogenesis of Kaposi's Sarcoma Associated with HIV/AIDS
- Holobiont–Holobiont Interactions: Redefining Host–Parasite Interactions
- BCKDH: The Missing Link in Apicomplexan Mitochondrial Metabolism Is Required for Full Virulence of and
- Helminth Infections, Type-2 Immune Response, and Metabolic Syndrome
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy