
Larger Mammalian Body Size Leads to Lower Retroviral Activity
Retroviruses have been invading mammalian genomes for over 100 million years, leaving traces known as endogenous retroviruses (ERVs). Early genome sequencing studies revealed a marked difference in the activity of retroviruses among species, with humans largely containing inactive lineages of ERVs, while the mouse contains numerous lineages of active ERVs. We explore the hypothesis that life history traits determine the activity of ERVs in mammalian genomes, and show that larger mammals have fewer ERV copies over recent evolutionary time (the last 10 million years) compared to smaller mammals. This association is determined by body size independently of any confounding variables. We build a mathematical model that shows that ERV abundance in genomes decreases with larger body size and increases with horizontal transmission. Retroviral integration can cause cancer, and our analysis suggests that larger bodied animals control ERV replication in order to postpone cancer until a post-reproductive age. This is in line with a long-standing observation that cancer rates do not fluctuate among mammals of different body size, a phenomenon known as Peto's paradox, and opens up the possibility that larger animals have evolved mechanisms to limit ERV activity.
Published in the journal:
. PLoS Pathog 10(7): e32767. doi:10.1371/journal.ppat.1004214
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004214
Summary
Retroviruses have been invading mammalian genomes for over 100 million years, leaving traces known as endogenous retroviruses (ERVs). Early genome sequencing studies revealed a marked difference in the activity of retroviruses among species, with humans largely containing inactive lineages of ERVs, while the mouse contains numerous lineages of active ERVs. We explore the hypothesis that life history traits determine the activity of ERVs in mammalian genomes, and show that larger mammals have fewer ERV copies over recent evolutionary time (the last 10 million years) compared to smaller mammals. This association is determined by body size independently of any confounding variables. We build a mathematical model that shows that ERV abundance in genomes decreases with larger body size and increases with horizontal transmission. Retroviral integration can cause cancer, and our analysis suggests that larger bodied animals control ERV replication in order to postpone cancer until a post-reproductive age. This is in line with a long-standing observation that cancer rates do not fluctuate among mammals of different body size, a phenomenon known as Peto's paradox, and opens up the possibility that larger animals have evolved mechanisms to limit ERV activity.
Introduction
Mammalian genomes contain large numbers of endogenous retroviruses (ERVs), derived from multiple independent germline invasions over evolutionary time. The human genome contains 31–40 such ERV invasions, termed ‘families’, each derived from a distinct ancestral exogenous retrovirus [1], [2]. These ERVs can continue proliferating after the initial germline invasion until they are inactivated, either through the acquisition of substitutions that occur at the host background level (∼10−3 per base per my) or by recombinational deletion [3], [4]. Most ERV families proliferate by reinfection, although some ERVs occasionally switch from reinfecting germline cells to an entirely intracellular life, and this switch can lead to an increase in the size of the ERV family [5]. As a result of these processes, ERVs have come to occupy ∼5–10% of their hosts' genomes [6], [7].
The fixation of a new ERV insertion is influenced by its fitness consequences to the host, and other population genetic parameters [8]; for example a neutral ERV could fix by drift, and a slightly deleterious insertion may hitchhike or fix during a population bottleneck [9]. A small number of ERVs have been exapted and have beneficial functions in their host [10]–[12], but the integration of retroviruses into or near host genes can have highly deleterious effects, as the consequent disruption or alteration of gene expression can lead to malignant transformation [13]. Furthermore, illegitimate recombination between ERVs at different loci can also have deleterious effects, as can the expression of viral proteins. The uncontrolled proliferation of ERVs would therefore be extremely detrimental to their host [14], and this process must be limited either by cessation of replication activity, or by host mediated suppression [15].Vertebrate genomes have evolved a range of responses that act at various stages of the viral life cycle to limit retroviral replication and its associated tumorigenic potential [16], [17].
The diversity and activity of ERVs across mammalian genomes has not been systematically assessed, and it remains unclear what factors have determined ERV abundance in their hosts. Mice and humans, the first two mammalian species to have their genomes sequenced, show strikingly different patterns of ERV activity – most human endogenous retroviruses are inactive, with a striking deceleration in activity over the last 25 million years [7]. In contrast, the mouse genome shows no sign of deceleration in ERV activity and a large number of murine ERVs are active and unfixed in the mouse population [6]. This difference is also reflected in the proportion of catalogued mutant alleles that are due to ERV insertions; ∼10% of mutant alleles can be attributed to ERVs in mice, whereas no such alleles can be attributed to ERVs in humans [18]. It has been suggested that the markedly different ERV activity in human and mouse genomes can be explained by systematic factors in the biology of these hosts [14]. We explore the hypothesis that differences in ERV activity across mammals are determined by differences in host life history, with smaller bodied animals expected to have higher levels of ERV activity. We compare body size with ERV numbers using data from a diverse set of 38 mammals in a multivariate analysis, controlling for confounding variables such as life history traits. We also explore the effect of body size and horizontal transmission on ERV dynamics through a mathematical model. Finally, we discuss our associations of body size and ERV replication in the light of evolutionary theory and cancer biology.
Results
Body size has a negative correlation with ERV abundance across mammals
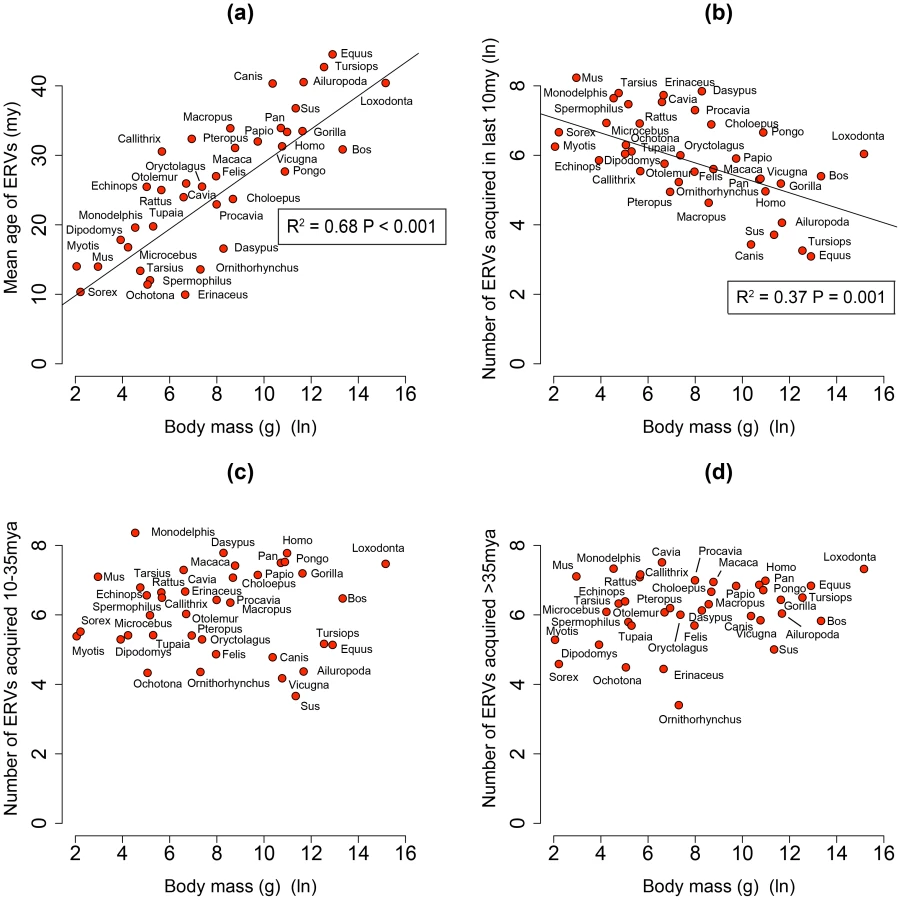
By analysing 38 mammalian genomes over approximately the last 10 my period we find a negative relationship between the number of integrated ERVs and body size (Figure 1b, Figure 2a). The correlation is robust if instead of present day body size we use the reconstructed body size at 5 million years ago (P = 0.0069, R2 = 0.31), and remains significant if we use a single substitution rate for all mammals (P = 0.01, R2 = 0.25). The correlation is dependent on the age of the integration in the genome and is no longer significant when we consider ERVs that are older than 10 my (Figure 1c, Figure 1d). If we exclude ERVs that belong to our previously defined megafamilies [5] with mean divergence <10%, namely the IAP family from Cavia Porcelus, the IAP family from Dipodomys Ordii, a Class I family from Felis catus, and the IAP and ERV-L families from Mus Musculus, the correlation remains (P = 0.0042, R2 = 0.30). If we split ERVs into their traditional classes, the correlation is significant only for the class II ERVs (Figure 3). There is no data to suggest systematic differences in the biology of retroviruses from different classes, given that the majority of ERVs are derived from extinct retroviral lineages. The three classes differ in their age distribution; class II ERVs are much younger (Figure 4), with the majority of insertions falling within the 10 my era that we use to define the young age category. Thus the observed relationship between body size and Class II ERVs is likely due to their recent replication and not some other difference in their biology such as higher pathogenicity.



The correlation of body size with ERV abundance is not confounded by another life history trait
Since life history traits are correlated with each other, it is possible that the apparent and inferred correlation of ERVs with body size could be confounded by another trait such as reproductive output (for which gestation period is a proxy) and timing (age at sexual maturity) [19]. The number of mates or the type of placenta might also influence ERV abundance via an increased risk of horizontal or vertical retroviral transmission, respectively. To clarify if number of sexual partners has played a role in determining the number of ERVs per genome, we use testis size as a proxy as it is known to correlate with the number of mates and the strength of sexual selection in mammals [20], [21]. To assess the effect of the placental type we modeled placental invasiveness as a semiquantitative parameter (i.e. marsupials = 1, epitheliochorial = 2, endotheliochorial = 3 and hemochorial = 4) [22]. We evaluated the correlation between ERV integration rate and potential confounders with multivariate models and standard stepwise forward model selection. We included in turn the following confounding variables; time to sexual maturity, gestation period, life span, testis size and placental invasiveness. Body size remained as the only significant variable confirming that it is the only significant predictor of ERV integration rate over the last 10 my (Table 1). The models remain significant when we account for phylogenetic non-independence [23], reconstruct ancestral mass and/or incorporate a body mass dependent substitution rate. Thus, unlike substitution rate [24], [25], ERV integration rate is not a result of shorter generation time. We do not find a significant correlation with testis size, either as an additional predictor variable (P = 0.3) with body size included in the model, or as an interaction term with body size (P = 0.2). Thus, the number of mates does not appear to have played a significant role on the number of ERVs per genome.

Another possible confounder is the effective population size of the host [26]: species with higher effective population sizes are expected to be more efficient at purging slightly deleterious mutations such as those incurred by ERV proliferation [27]. As a result, since larger bodied animals have smaller effective population sizes [19], [28], we would expect them to have more, not fewer ERVs. Thus, confounding due to effective population size would lead to a correlation in the opposite direction to what we observe, indicating that the observed correlation between body size and ERV numbers is robust against variations in effective population size.
Relationship between ERV abundance and body size can be explained by a mathematical model of retrovirus-host dynamics
To explore the possible mechanistic links between body size, integration rate and transmission route, we designed a mathematical model (Figure S1). We constructed a compartmental mathematical model using a system of ordinary differential equations to describe the epidemiological dynamics of exogenous and endogenous retroviral infections. There are two broad classes of individuals that need to be considered, the susceptible population and the infected population. In order to gain a more detailed picture of the latter compartment, namely to elucidate the inter-connected roles between exogenous and endogenous retroviral infections, we further distinguish three infected sub-populations: individuals infected with an exogenous retrovirus , those infected with a single integrated copy of the retrovirus through the process of endogenisation , and lastly, those infected with an endogenous retrovirus that has undergone amplification .
The overall level of retroviral activity is directly related to the copy number of endogenised retroviruses in the infected population. Since the vast majority of endogenous retrovirus present in the host population persists in the pool of AERV-infected individuals, the level of retroviral activity can be represented by the magnitude or size of this compartment. We first explore the roles of three key factors: body size , the rate of retroviral endogenisation which governs vertical transmission of the retrovirus, and the force of infection which determines the rate of horizontal transmission of the retrovirus. As shown in Figure S2 increased body size results in a lower number of individuals harbouring amplified endogenous retrovirus when this system reaches equilibrium, (Figure S2, upper plot), while the rate of retroviral endogenisation and the force of infection display the opposite relationship (Figure S2, lower plot).
Our model demonstrates that horizontal and vertical transmission are both crucial for the eventual endogenisation and amplification of the retrovirus. If there is no horizontal transmission (i.e. ), then the retrovirus cannot spread and persist in the population, even if there is a large initial pool of infected individuals (figure 5). Similarly, in the situation where no endogenisation events occur via vertical transmission (i.e. ), our model shows that infection can become endemic, but remains completely exogenous.

We explored the impact of body size on the structure of the population at equilibrium in relation to the extent of retroviral endogenisation and the force of infection (Figure 5) in more detail. The results in Figure 5 illustrate that higher rates of horizontal transmission, represented by the force of infection , lead to a higher proportion of AERV infections for a given body size, and furthermore, highlight our finding that larger body size is associated with a lower extent of AERV activity, with all other parameters fixed (Figure S2, lower plot). Furthermore, we observe from Figure 5 that for sufficiently high rates of horizontal transmission, the proportion of AERV infections plateaus with respect to increasing body size . In this case, larger body size is associated with a greater proportion of exogenous infections, as new (exogenous) infections would arise through horizontal transmissions at a faster rate than endogenisation via vertical transmissions. To explore the possibility that the number of horizontal transmissions confound the number of elements per genome, we tested if the number of families per genome is correlated with body size, and find no significant correlation (P = 0.15, R2 = 0.08).
Discussion
We identified 84,223 ERVs, of which 27,711 have integrated in the last 10 my across 38 species of mammal (Table 2). We find that the number of ERV integrations in mammals is negatively correlated to body size. This correlation can explain 37% of the variance in the number of ERV integrations over the past 10 my. We have controlled for confounding variables such as life history and sexual selection, and also confirmed robustness to variation in effective population size. Nevertheless body size can be influenced by other parameters, and it is possible that other factors (e.g. environmental, dietary) contribute to both body size and ERV abundance, thereby explaining part of the remaining variance; for example they might account for the residual variance of outliers (e.g. Dasypus Novemcinctus and Canis familiaris). Interestingly, Microcebus murinus, whose life history evolved rapidly due to its isolation in Madagascar [29], might be expected to be a significant outlier in the correlation, but is very close to the regression line. Perhaps the global distribution and geographic isolation of a species is another determinant of the variance in ERV abundance.

We also see that 68% of the variance observed in the mean age of ERV integrations in a genome (a proxy for recent replication) is explained by body size (Figure 1a), with the number of young (i.e. recently replicating) insertions correlating inversely with body size (Figure 1b) while the number of older insertions do not (Figure 1c, d). These observations suggest that body size limits the number of ERVs in a genome and that the presence of recently replicating ERVs is detrimental to their host. As ERVs accumulate host-induced mutations over time, their activity diminishes until they eventually become neutral. Our study suggests that ERVs that have been active within the last 10 million years could still have moderately deleterious effects, probably in the post reproductive age. Furthermore, the age of an ERV is a good proxy for its pathogenic potential, with pathogenicity decreasing over time. Some ERV families have retained replication capacity for millions of years; HERV-K (HML2) first invaded the primate genome >30 my yet has still been active up until at least ∼500,000 years ago [30], [31]. These recently active ERVs may retain some level of virulence, and therefore still have the potential for malignant transformation [13]. In line with this prediction of intermediate virulence, reconstructed ERVs [32] or recently established present day ERVs [33], [34] have low but detectable viral loads. The presence of pathogenic ERVs in a genome after such a long period of time may appear surprising. It could however be explained by analogy to models of the transmissibility of pathogens within the context of host-parasite co-evolutionary dynamics [35], [36]. Such models incorporate the effects of both transmissibility and virulence on the reproductive success of a parasite, and show that they do not necessarily evolve to be harmless; in some empirical datasets reproductive success is maximised at intermediate levels of both of these parameters [35]. In other words a pathogen can continue to be virulent despite selection imposed by the host for a more benign infection.
We have modelled the spread of retroviruses among hosts and within genomes, distinguishing between exogenous and endogenous retroviruses, and taking into account vertical and horizontal modes of transmission. A key aspect of our model is the assumption that the deleterious effects of a retrovirus in a genome scale with body size. The model shows that as body size increases, the proportion of individuals in the host population that carry ERVs drops (figure S2). Elevated rates of either endogenisation or horizontal transmission lead to higher ERV abundance and accelerate the rate at which ERV abundance increases with body size. For any given rate of horizontal transmission however the overall relationship between body size and ERV abundance is maintained (figure 5). In our model, the body size-associated pathogenic effect of an ERV in a genome is equivalent, whether it has been generated by vertical or horizontal transmission. Horizontal transmission of an ERV would require somatic expression and replication of the virus in order to propagate effectively, which may in turn increase the mortality of the host via a direct result of retroviral infection. Experimental evidence suggests that infections with replication competent retroviruses are more pathogenic when retroviral replication is high (e.g. HIV, or the recently endogenised Koala retrovirus [34], [37]). One way in which the pathogenicity of an ERV can be reduced while its replicative capacity is maintained is through epigenetic regulation in somatic cells. During genomic reprogramming of the germline, transposable elements are expressed and can replicate before being silenced [38], [39], resulting in lower levels of expression in somatic tissues and hence lower transmissibility. Thus, low levels of replication in somatic cells may be favorable for an ERV, enabling it to maximize its own success via vertical transmission while minimizing harm to the host. The association between ERV abundance and body size indicates that somatic replication cannot be completely suppressed and that the pathogenetic effects of ERVs cannot be dissociated from their copy number.
On a macroevolutionary timescale, ERV copy number will be determined both by the number of cross species transmissions and the subsequent proliferation of ERV families. The number of families per genome is orders of magnitude lower than the number of ERVs (mean number of families = 23, mean number of elements = 1073), and most ERVs within a genome come from a small number of families, the so-called superspreaders (or megafamilies) [5]. In line with this uneven distribution of ERVs among families, we do not see a correlation between the number of families within a genome and body size (P = NS, R2 = 0.08). Furthermore, ERVs that belong to megafamilies lack the env genome that is required for horizontal transmission, highlighting the importance of vertical transmission in determining ERV abundance, despite the ability of ERVs to cross species on timescales spanning millions of years.
Crucially, according to our model the selective cost of an ERV is determined by the body size of the host. Larger bodied animals would be expected to have a higher lifetime risk of cancer as a consequence of having both more dividing cells and longer lifespans. No such association is observed in nature, with relatively invariable risks of cancer in animals with differing body sizes, a phenomenon known as Peto's Paradox [40], [41]. Under our model, the risk of retrovirally induced cancer also scales similarly with body size. The observed negative correlation between body size and ERV integration rate suggests that larger mammals attain a lower ERV virulence cost per body size unit by reducing the number of ERVs in their genome. This should therefore enable them to postpone the onset of cancer until after their reproductive age.
Our results indicate that larger animals exert greater control over ERV proliferation. This could be due to the evolution of mechanisms capable of limiting retroviral activity and consequently limiting the incorporation of ERVs in the genome. Such mechanisms could involve the enhancement of innate or adaptive responses to retroviruses [16], [17], or perhaps epigenetic regulation [42] is more potent in larger mammals. An intriguing alternative is that the effect is indirect via an improved immune surveillance – some genes involved in pattern recognition for defence against pathogens such as viruses are also involved in controlling cancers [43]. Antiviral genes are the result of a continuous and ancient arms race between viruses and their hosts [15], and elucidating their roles in controlling cancer across animals of different body size could provide insights into cancer susceptibility.
Materials and Methods
ERV mining and dating of insertions
Our mining of the 38 mammal genomes has been described previously [5], [44], [45]. We estimated age based on the divergence from the most similar other ERV insertion in the same genome (“nearest neighbour”). We favour this approach over cruder metrics that are based on divergence from a consensus sequence, as it takes into account the phylogeny of the ERVs, and over approaches based on divergence between paired LTRs due to the variable quality of the genomes being analysed, most of which do not contain contigs that are long enough to include complete proviral elements. We first calculated nucleotide divergence from the most similar other ERV insertion in the same genome as described in Magiorkinis et al. [5], and then converted this to an integration date assuming a mean nucleotide substitution rate at neutral nuclear protein coding sites in mammals of 2.2×10−9 per site per year [46], and corrected for multiple hits using the Jukes-Cantor model. To calculate the average age of ERVs in each genome we took into account the known effect of body size on substitution rate by using a regression of rate against mass with slope of −0.09, i.e. log(adjusted rate) = 0.09×(log(mean mass)−log(mass))+log(unadjusted rate) [47]. We also repeat the correlation between body size and ERV number with a single substitution rate for all mammals.
Incorporating ancestral body mass
Using the data above we reconstructed ancestral body masses assuming a Brownian motion model of trait evolution as implemented in the package GEIGER in the R language [48]. This program returns the estimated body mass at nodes in the tree, and from these we calculated values at the mid-points of our time intervals (averaging where necessary). We then manually pruned our trees to this point and repeated the regression between number of ERVs and body mass for each time interval, taking the phylogeny into account (Table 2). Our regressions were performed with both present day body size and the reconstructed body size at the mid-point of our time intervals (e.g. body size at 5 million years ago for regression against activity during the last 10 million years).
Multivariate analysis
Life history traits correlate with each other; for example larger bodied animals tend to live longer and have smaller effective population size [19], [28]. Therefore body size could in principle be a surrogate measure of a different life history trait, as has been previously shown for substitution rate [24]. Mammalian life history data was taken from [49] and the phylogenetic tree from [50]. We collected the testis size for 24 out of 38 species in our study (Table S1). We used the Generalized Least Squares (GLS) approach as implemented by the Analysis of Phylogenetics and Evolution (APE) package [51] in R. We used standard model selection to identify significant confounders of ERV numbers per genome (Table 1).
A mathematical model of ERV persistence and evolution
Model (1) captures the fundamental dynamics of retroviral infections including the processes of retroviral endogenisation and amplification. The key interactions of the model are illustrated schematically in Figure S1.




In model (1), we consider both vertical and horizontal routes of transmission. We also distinguish between exogenous and endogenous retroviral infections. Whereas horizontal transmission can only lead to infection with an exogenous retrovirus (i.e. RV compartment), vertical transmission can result in retroviral endogenisation (i.e. ERV compartment) and subsequent amplification (i.e. AERV compartment).
There are two ways in which new (exogenous) retroviral infections may arise horizontally in an initially susceptible individual, either through contact with an individual infected with an exogenous retrovirus (i.e. RV compartment), or alternatively via exposure to an individual infected with an amplified endogenous retrovirus (i.e. AERV compartment). We assume that individuals infected with only a single integrated copy of the retrovirus (i.e. ERV compartment) are unable to transmit the infection horizontally between hosts. The force of infection is composed of two terms, , and thus reflects the dual modes of horizontal transmission. There are various different functional forms for the force of infection, and we choose the commonly used form , where and are the respective coefficients of infectious transmissibility for RV-infected and AERV-infected individuals, and N is the total population which is assumed to be constant.
A small proportion , where , of births from individuals who are infected by an exogenous retrovirus acquire an integrated endogenous copy of the retrovirus, thereby entering the ERV compartment. Meanwhile, individuals infected with an integrated endogenous retrovirus (in the ERV compartment) undergo retroviral amplification at a rate , which is dependent on body size . A consequence of retroviral amplification is a greater number of endogenous retroviruses, therefore the size of the compartment of individuals harbouring amplified, endogenised retroviruses is an indirect measure of the overall extent of retroviral activity. Births arising from infected individuals with amplified, endogenous retrovirus (i.e. AERV compartment) themselves harbour amplified, endogenous retroviruses.
To investigate the system without unnecessarily over-complicating the dynamical behaviour of the model, we consider a population that is maintained at a fixed size so that . The pool of susceptible individuals is maintained by the birth of new susceptible individuals and is encapsulated in the term in the equation, which includes new births of susceptibles from all other compartments as well as a term to balance the in - and out-flux of individuals in the system and ensure that the total population remains constant. We assume that background birth and death rates in each compartment are equal at a constant rate . Additional mortality due to the detrimental effects of amplified, endogenous retroviral infection, such as the development of cancer, is reflected in the parameter , which depends on body size . Excess mortality as a consequence of cancer is fed back into the susceptible pool so that, therefore the birth of susceptible individuals can be encapsulated by the term , where
The above discussion highlights an important trade-off between retroviral amplification , which is beneficial to the long-term persistence of the retrovirus, and increased mortality in excess of background death rates as a consequence of the detrimental effects associated with increased retroviral activity. These two factors both depend on body size , but in opposing ways. Whereas larger body size means increased retroviral amplification, it also results in greater mortality so that both and are increasing functions of . We therefore investigate the role of body size on the outcome of infection. Several additional parameters of significance are the force of infection as well as the rate of retroviral endogenisation and how varying body size can influence the dynamical behaviour of the infection according to model (1). For the former, we explore how body size can affect the system when differences between the force of infection of individuals infected with exogenous retrovirus (i.e. the compartment) versus those carrying amplified, endogenous retrovirus (i.e. the compartment) are taken into account. In terms of the latter, it is expected that a higher rate of endogenisation would result in a greater proportion of individuals with integrated endogenous retroviruses, and we are interested in determining the role of body size with respect to differences in endogenisation rates. Because we have assumed that the total population remains constant, it is sensible to investigate the dynamics of the model with respect to proportions of the total population rather than in terms of the sizes of each compartment.
Supporting Information
Zdroje
1. BannertN, KurthR (2006) The evolutionary dynamics of human endogenous retroviral families. Annu Rev Genomics Hum Genet 7 : 149–173.
2. Katzourakis A, Tristem M (2005) Phylogeny of human endogenous and exogenous retroviruses. In: Sverdlov ED, editor. Retroviruses and primate genome evolution. Austin, TX: Landes Bioscience. pp. 186–203.
3. KatzourakisA, PereiraV, TristemM (2007) Effects of recombination rate on human endogenous retrovirus fixation and persistence. J Virol 81 : 10712–10717.
4. BelshawR, WatsonJ, KatzourakisA, HoweA, Woolven-AllenJ, et al. (2007) Rate of recombinational deletion among human endogenous retroviruses. J Virol 81 : 9437–9442.
5. MagiorkinisG, GiffordRJ, KatzourakisA, De RanterJ, BelshawR (2012) Env-less endogenous retroviruses are genomic superspreaders. Proc Natl Acad Sci U S A 109 : 7385–7390.
6. WaterstonRH, Lindblad-TohK, BirneyE, RogersJ, AbrilJF, et al. (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420 : 520–562.
7. LanderES, LintonLM, BirrenB, NusbaumC, ZodyMC, et al. (2001) Initial sequencing and analysis of the human genome. Nature 409 : 860–921.
8. KimuraM (1962) On the probability of fixation of mutant genes in a population. Genetics 47 : 713–719.
9. OhtaT, GillespieJH (1996) Development of Neutral and Nearly Neutral Theories. Theor Popul Biol 49 : 128–142.
10. LavialleC, CornelisG, DupressoirA, EsnaultC, HeidmannO, et al. (2013) Paleovirology of “syncytins”, retroviral env genes exapted for a role in placentation. Philosophical transactions of the Royal Society of London Series B, Biological sciences
11. CornelisG, HeidmannO, Bernard-StoecklinS, ReynaudK, VeronG, et al. (2012) Ancestral capture of syncytin-Car1, a fusogenic endogenous retroviral envelope gene involved in placentation and conserved in Carnivora. Proceedings of the National Academy of Sciences of the United States of America 109: E432–441.
12. AswadA, KatzourakisA (2012) Paleovirology and virally derived immunity. Trends Ecol Evol 27 : 627–636.
13. JernP, CoffinJM (2008) Effects of retroviruses on host genome function. Annu Rev Genet 42 : 709–732.
14. KatzourakisA, RambautA, PybusOG (2005) The evolutionary dynamics of endogenous retroviruses. Trends Microbiol 13 : 463–468.
15. StoyeJP (2012) Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol 10 : 395–406.
16. DaughertyMD, MalikHS (2012) Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet 46 : 677–700.
17. DuggalNK, EmermanM (2012) Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol 12 : 687–695.
18. MaksakovaIA, RomanishMT, GagnierL, DunnCA, van de LagemaatLN, et al. (2006) Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet 2: e2.
19. BielbyJ, MaceGM, Bininda-EmondsOR, CardilloM, GittlemanJL, et al. (2007) The fast-slow continuum in mammalian life history: an empirical reevaluation. Am Nat 169 : 748–757.
20. HarcourtAH, HarveyPH, LarsonSG, ShortRV (1981) Testis weight, body weight and breeding system in primates. Nature 293 : 55–57.
21. SoulsburyCD (2010) Genetic patterns of paternity and testes size in mammals. PLoS One 5: e9581.
22. ElliotMG, CrespiBJ (2009) Phylogenetic evidence for early hemochorial placentation in eutheria. Placenta 30 : 949–967.
23. PagelM (1999) Inferring the historical patterns of biological evolution. Nature 401 : 877–884.
24. Wilson SayresMA, VendittiC, PagelM, MakovaKD (2011) Do variations in substitution rates and male mutation bias correlate with life-history traits? A study of 32 mammalian genomes. Evolution 65 : 2800–2815.
25. MartinAP, PalumbiSR (1993) Body size, metabolic rate, generation time, and the molecular clock. Proc Natl Acad Sci U S A 90 : 4087–4091.
26. LynchM, ConeryJS (2003) The origins of genome complexity. Science 302 : 1401–1404.
27. LynchM, GabrielW (1990) Mutational load and the survival of small populations. Evolution 44 : 1725–1737.
28. DamuthJ (1981) Population density and body size in mammals. Nature 290 : 699–700.
29. CatlettKK, SchwartzGT, GodfreyLR, JungersWL (2010) “Life history space”: a multivariate analysis of life history variation in extant and extinct Malagasy lemurs. Am J Phys Anthropol 142 : 391–404.
30. BelshawR, PereiraV, KatzourakisA, TalbotG, PacesJ, et al. (2004) Long-term reinfection of the human genome by endogenous retroviruses. Proc Natl Acad Sci U S A 101 : 4894–4899.
31. BelshawR, DawsonAL, Woolven-AllenJ, ReddingJ, BurtA, et al. (2005) Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity. J Virol 79 : 12507–12514.
32. DewannieuxM, HarperF, RichaudA, LetzelterC, RibetD, et al. (2006) Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res 16 : 1548–1556.
33. TarlintonRE, MeersJ, YoungPR (2006) Retroviral invasion of the koala genome. Nature 442 : 79–81.
34. TarlintonR, MeersJ, HangerJ, YoungP (2005) Real-time reverse transcriptase PCR for the endogenous koala retrovirus reveals an association between plasma viral load and neoplastic disease in koalas. J Gen Virol 86 : 783–787.
35. MayRM, AndersonRM (1983) Epidemiology and genetics in the coevolution of parasites and hosts. Proc R Soc Lond B Biol Sci 219 : 281–313.
36. AndersonRM, MayRM (1982) Coevolution of hosts and parasites. Parasitology 85(Pt 2): 411–426.
37. FraserC, LythgoeK, LeventhalGE, ShirreffG, HollingsworthTD, et al. (2014) Virulence and pathogenesis of HIV-1 infection: an evolutionary perspective. Science 343 : 1243727.
38. SlotkinRK, MartienssenR (2007) Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8 : 272–285.
39. CastanedaJ, GenzorP, BortvinA (2011) piRNAs, transposon silencing, and germline genome integrity. Mutat Res 714 : 95–104.
40. CaulinAF, MaleyCC (2011) Peto's Paradox: evolution's prescription for cancer prevention. Trends Ecol Evol 26 : 175–182.
41. PetoR, RoeFJ, LeePN, LevyL, ClackJ (1975) Cancer and ageing in mice and men. Br J Cancer 32 : 411–426.
42. BarbotW, DupressoirA, LazarV, HeidmannT (2002) Epigenetic regulation of an IAP retrotransposon in the aging mouse: progressive demethylation and de-silencing of the element by its repetitive induction. Nucleic acids research 30 : 2365–2373.
43. VollmerJ (2009) Autophagy links pattern recognition receptor to tumor cell apoptosis. Mol Therapy 17 : 1839–1841.
44. KatzourakisA, GiffordRJ (2010) Endogenous viral elements in animal genomes. PLoS Genet 6: e1001191.
45. KatzourakisA, GiffordRJ, TristemM, GilbertMT, PybusOG (2009) Macroevolution of complex retroviruses. Science 325 : 1512.
46. KumarS, SubramanianS (2002) Mutation rates in mammalian genomes. Proc Natl Acad Sci U S A 99 : 803–808.
47. WelchJJ, Bininda-EmondsOR, BromhamL (2008) Correlates of substitution rate variation in mammalian protein-coding sequences. BMC Evol Biol 8 : 53.
48. HarmonLJ, WeirJT, BrockCD, GlorRE, ChallengerW (2007) GEIGER: investigating evolutionary radiations. Bioinformatics 24 : 129–131.
49. JonesKE, BielbyJ, CardilloM, FritzSA, O'DellJ, et al. (2009) PanTHERIA: a species-level database of life history, ecology, and geography of extant and recently extinct mammals. Ecology 90 : 2648–2648.
50. Bininda-EmondsORP, CardilloM, JonesKE, MacPheeRDE, BeckRMD, et al. (2007) The delayed rise of present-day mammals. Nature 446 : 507–512.
51. ParadisE, ClaudeJ, StrimmerK (2004) APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20 : 289–290.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Molecular and Cellular Mechanisms of KSHV Oncogenesis of Kaposi's Sarcoma Associated with HIV/AIDS
- Holobiont–Holobiont Interactions: Redefining Host–Parasite Interactions
- BCKDH: The Missing Link in Apicomplexan Mitochondrial Metabolism Is Required for Full Virulence of and
- Helminth Infections, Type-2 Immune Response, and Metabolic Syndrome
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy