
The Impact of Juvenile Coxsackievirus Infection on Cardiac Progenitor Cells and Postnatal Heart Development
Coxsackievirus B (CVB) is a significant human pathogen, causing myocarditis, aseptic meningitis and encephalitis. The lasting consequences of juvenile CVB infection on the developing host have yet to be adequately inspected. Here, we show that CVB efficiently infected juvenile cardiac progenitor cells both in culture and the young heart. Furthermore, we describe a mouse model of juvenile infection with a subclinical dose of CVB which showed no symptoms of disease into adulthood. However following physiological or pharmacologically-induced cardiac stress, juvenile-infected mice underwent cardiac hypertrophy and dilation indicative of progression to heart failure. These results suggest that mild CVB infection in the young host may impair the ability of the heart to adapt to increased load leading to pathological remodeling later in adult life.
Published in the journal:
. PLoS Pathog 10(7): e32767. doi:10.1371/journal.ppat.1004249
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004249
Summary
Coxsackievirus B (CVB) is a significant human pathogen, causing myocarditis, aseptic meningitis and encephalitis. The lasting consequences of juvenile CVB infection on the developing host have yet to be adequately inspected. Here, we show that CVB efficiently infected juvenile cardiac progenitor cells both in culture and the young heart. Furthermore, we describe a mouse model of juvenile infection with a subclinical dose of CVB which showed no symptoms of disease into adulthood. However following physiological or pharmacologically-induced cardiac stress, juvenile-infected mice underwent cardiac hypertrophy and dilation indicative of progression to heart failure. These results suggest that mild CVB infection in the young host may impair the ability of the heart to adapt to increased load leading to pathological remodeling later in adult life.
Introduction
Coxsackieviruses (CV) are common human pathogens that typically cause a self-limited infection and mild symptoms such as fever, rash, and upper-respiratory complications. Though CV can also cause severe inflammatory diseases including myocarditis, a disease that can lead to dilated cardiomyopathy [1], [2], [3], the manifestation of a cardiac disease phenotype has been documented to be extremely rare (∼5% of infected patients) [4]. Collapse and death of young individuals during exertion can result from catastrophic dysfunction of the electrical pathways in the heart associated with unrevealed CV infection [5], [6]. Additionally, 70–80% of individuals with end-stage idiopathic dilated cardiomyopathy have detectable levels of CV RNA in the myocardium without any history of antecedent viral myocarditis [7], [8], [9]. These findings raise the possibility that mild infection with CV can cause subtle but lasting injury, although it is unclear whether such enduring damage is immune-mediated or due to virus-mediated cytopathic effects. Previous studies suggest that coxsackievirus B3 (CVB3) may exhibit unique tropism for undifferentiated cells such as neural and hematopoietic progenitor cells thereby altering cell lineage commitment or diminishing their restorative capacity [10], [11], [12], [13], [14], [15], [16], [17]. Infection of progenitor cells may also enhance virus dissemination in a process referred to autophagosome-mediated exit without lysis (AWOL) [18], [19]. Based on these observations, we hypothesized that CVB3 also infects cardiac progenitor cells which might lead to long-term consequences for heart development and function. Of note, the mechanistic basis and causal link between juvenile CVB3 infection and adult-onset dilated cardiomyopathy has not been previously inspected.
The heart was previously viewed as a terminally differentiated organ predominantly comprised of a fixed number of non-renewable cardiomyocytes. Recently, a distinct population of resident cardiac progenitor cells (CPCs) was identified that challenged the notion of a heart without regenerative capacity. These CPCs were described as exhibiting large nuclei, scant cytoplasm, and hematopoietic markers such as CD117 (c-Kit) and Sca-1 [20], [21], [22], [23]. c-kit+ cells isolated from the heart and grown in culture are capable of differentiating into all four cell types of the cardiac lineage which include cardiomyocytes, smooth muscle cells, endothelial cells, and fibroblasts. CPCs also play a beneficial role in adult cardiac repair, and the local injection of isolated CPCs have been shown to preserve myocardial muscle mass and reduce scar formation after experimental myocardial infarction in mice [24].
Due to their role in cardiac development as well as in cardiac maintenance, a perturbation of the CPC pool due to infection or other environmental factors could conceivably have deleterious effects on the developing heart. Previously, Huang et al demonstrated that in mouse pups, exposure to the chemotherapeutic drug doxorubicin before postnatal day 21 resulted in a decreased number of CPCs due to early senescence. Although doxorubicin-treated animals exhibited normal cardiac function at 8 weeks of age, endurance exercise led to the development of pathological hypertrophy and fibrosis [25]. This model seems to mirror the late consequences of childhood doxorubicin-induced cardiotoxicity observed in humans which manifests in weakening of the myocardium, an attenuation of cardiac pump performance, and progression towards congestive heart failure. Since previous studies described preferential CVB3 infection of progenitor cells in the CNS [10], [11], [15], [16], [26] and bone marrow [17], we investigated the effect of CVB3 on CPCs in the developing heart. We established a mouse model of juvenile CVB3 infection and observed that not only did CVB3 productively infect CPCs, but infection caused a sustained reduction in the CPC pool into adulthood. We suggest that the exhaustion of CPCs within the heart predisposed the adult animal to stress-induced cardiac hypertrophy, left ventricular dilation, and the accumulation of fibrotic tissue – features considered to be the early stages of dilated cardiomyopathy. These results provide evidence that a mild CVB3 infection may predispose the heart to pathologic remodeling later in adult life.
Results
CVB3 infected cardiac-derived c-kit+ cells grown in culture
We previously determined that proliferating neural stem and progenitor cells grown in the subventricular zone of the neonatal brain or grown in culture were preferentially targeted by CVB3 [10], [11], [16], [14], [26]. Furthermore, CVB3 reduced the levels of neurogenesis and altered brain development in a neonatal model of viral infection [15]. To investigate if CVB3 infected cardiac progenitor cells (CPCs) in a similar fashion, c-kit+ cells were isolated from juvenile (Day 17 post-birth) or adult mice and cultured in chamber slides with complete CPC growth medium. Cultured cardiac derived c-kit+ cells were infected with eGFP-CVB3 at a multiplicity of infection (MOI) of 1 or 1000. The production of infectious virus was determined by plaque assay or viral protein expression (eGFP) utilizing fluorescence microscopy up to 10 days post-infection (PI) (Figure 1). Our results showed that juvenile c-kit+ cells expressed high levels of viral protein (eGFP) at low and high MOI (Figure 1A). Also, juvenile c-kit+ cells produced high amounts of infectious virus (Figure 1B). In contrast, adult c-kit+ cells failed to express detectable levels of viral protein or significant amounts of infectious virus. These differences are consistent with clinical studies suggesting that infants are much more susceptible to CVB3 infections than adults [27], [28], [29]. As the course of infection progressed, a majority of juvenile c-kit+ cells exhibited cytopathic effects, detached from the surface of the culture dish and fragmented into apoptotic bodies. Nevertheless, some juvenile c-kit+ cells survived initial infection and continued to proliferate in culture.

Colocalization of c-kit and virus protein expression in cultured CPCs infected with eGFP-CVB3 was identified by immunofluorescence microscopy (Figure 1C). Additionally, we observed a population of low eGFP-expressing juvenile c-kit+ cells which remained morphologically normal. At later time points, eGFP was not detected, and infected cultures developed a flattened morphology consistent with differentiation (data not shown). In contrast, adult c-kit+ cells produce little to no infectious virus and appeared resistant to infection. Both juvenile and adult c-kit+ cell populations expressed high levels of coxsackie and adenovirus receptor (CAR) (Figure 1D), suggesting that virus receptor levels were not responsible for the differential susceptibility to infection. These results parallel our findings regarding infection in the neonatal CNS whereby CAR expression is widespread, yet the tropism of the virus is specific for proliferating NPSCs early after infection [10], [11]. The presence of CAR on adult c-kit+ cells suggests that factors other than CAR expression may ultimately be responsible for increased susceptibility of juvenile CPCs to CVB3 infection.
Next, to determine if CVB3 infected CPCs in vivo, immunostaining was performed on the hearts of mouse pups infected with eGFP-CVB3 (107 pfu IP). Hearts were harvested 2 days PI and paraffin-embedded for histological analysis. Viral protein expression (eGFP) colocalized with c-kit and Sca-1, indicating infection of CPCs in the juvenile heart (Figure 1E and Figure 1F, respectively). Interestingly, viral protein expression was not observed in the surrounding myocardium, suggesting that CPCs were preferentially targeted for infection in the juvenile heart.
Establishing a mouse model of subclinical CVB3 infection to examine CPC alterations in the heart
To study sublethal juvenile infection, mice were infected with 107 pfu GFP-CVB3 at postnatal day 3 (Figure 2). This inoculum of virus was well-tolerated, and infected mouse pups showed no overt health abnormalities. Suckling and weight gain in infected pups was similar to the mock-infected control mice. Despite the absence of overt illness, a large amount of infectious virus and viral genomic RNA was detectable in the heart up to 9 days PI, confirming cardiac tropism of the virus (Figure 2A) as shown previously in the adult mouse model by other groups [30], [31], [32]. Also, a stepwise reduction in the ratio of positive to negative sense strand viral genome was observed over time in the heart (Figure 2B), similar to previous studies describing CVB3 infection in the neonatal CNS [33]. Viral titers in the heart were determined over time by standard plaque assay on HeLa cells and closely paralleled results using quantitative real time RT-PCR for positive sense viral genome (Figure 2C). No significant difference in the heart weight to body weight ratio (HW/BW) was observed between CVB3 and mock-infected animals at any time point (Figure 2D; two-way ANOVA). Immunofluorescence for CD3 and Iba-1 (T-cells and macrophages, respectively) revealed no significant immune cell infiltration following CVB3 infection in the heart of juvenile mice given a sublethal dose of virus (Figure 2E). At 2 days PI, hematoxylin-eosin (H&E) staining showed that both infected and mock-infected mice showed normal arrangement of myofibrils. Also, Masson's trichrome staining revealed no myocardial scarring during the acute phase of infection.

Increased Sca-1 expression in the infected juvenile myocardium
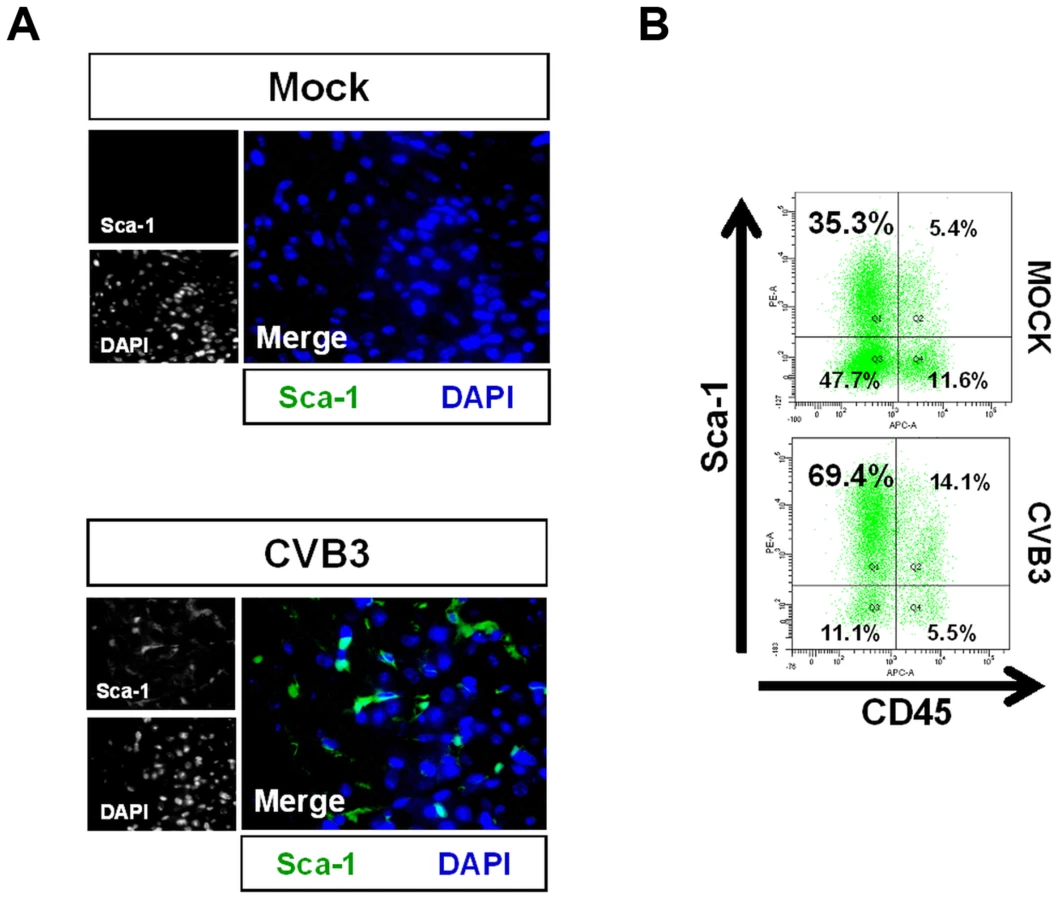
To further investigate the outcome of infected CPCs, immunohistochemistry was performed on the hearts of infected mouse pups using antibodies against Sca-1 and cell lineage markers at 2 days PI (Figure 3). c-kit+ progenitor cells are thought to represent the most primitive undifferentiated population, while Sca-1+ progenitor cells display a transcriptional profile closer to cardiomyocytes [34], [35]. We observed a large initial increase in the number of Sca-1+ cells in the heart compared to mock-infected control mice (Figure 3A). We determined if these Sca-1+ cells arose from resident cardiac progenitor cells, or were instead recruited from the peripheral blood in response to viral infection. Cell suspensions derived from enzymatically dissociated infected hearts were analyzed by flow cytometry for co-expression of Sca-1 and the hematopoietic cell marker CD45. Flow cytometric results confirmed a ∼two-fold increase in the percentage of Sca-1+ cells isolated from the heart at an early time point following infection compared to mock-infected control mice (83.5% versus 40.7%, respectively). Expansion of Sca-1+ cells in the heart following infection included both CD45+ and CD45− cell populations (Figure 3B). However, a greater percentage of Sca-1+ cells were largely negative for CD45 (69.4% versus 14.1%, respectively) or for the hematopoietic stem cell marker CD34 (31.8% versus 3.3%; data not shown), indicating the possible expansion of resident progenitor cells as opposed to the recruitment of progenitor cells having a peripheral blood origin. In combination with our histology results revealing an absence of immune infiltration, these results ruled out a significant hematopoietic cell response in the heart elicited by the virus at 2 days PI.
CVB3-infection induced premature differentiation of cardiac progenitor cells
Though the precise role of Sca-1+ cell expansion in the heart following CVB3 infection remains elusive, Sca-1 protein expression has previously been shown to function in suppressing cell cycle progression [36]. Recent literature suggests that in certain precursor cell types, Sca-1 expression remains at very low levels to prevent uncontrolled cell proliferation and premature senescence. As these cells enter a differentiation program, Sca-1 upregulation leads to complete withdrawal from the cell cycle which is a necessary prequel to differentiation [36], [37]. If Sca-1 plays a similar role as a late progenitor cell marker in CPCs [34], upregulation of Sca-1 could indicate a large increase in the number of progenitor cells which quickly undergo differentiation in response to CVB3 infection.
We next inspected early expansion of Sca-1+ cells in the heart following CVB3 infection by identifying expression levels of cell lineage markers which colocalized with Sca-1 staining. Infected hearts were observed by immunofluorescence microscopy for Sca-1 expression and the mature cardiac lineage markers for vascular smooth muscle cells, vascular endothelial cells, and cardiomyocytes (SM22, von Willebrand factor – vWF, and sarcomeric actin, respectively). Sca-1+ cells showed colocalization with SM22 and vWF (Figure 4A and Figure 4B, respectively). However, Sca-1 did not appear to co-localize with sarcomeric actin (Figure 4C), suggesting possible commitment of Sca-1+ cells to a vascular cell lineage in the heart following infection. Intriguingly, infected c-kit+ cells grown in culture exhibited a sharp increase in alpha-actin expression (Figure 4D; **p<0.01). Also, no increase in SM22 expression was observed in isolated c-kit+ cells following infection, indicating that CVB3 may induce preferential cardiomyocyte differentiation in CPCs grown in culture. These results contrast with increased SM22 expression levels in infected Sca-1+ cells found in the mouse heart. We hypothesize that the surrounding in vivo microenvironment may influence cell lineage commitment patterns following CVB3 infection in the heart.

Because many viruses including CVB3 are known to alter the cell cycle, heart sections were immunostained using antibodies against c-kit, Sca-1 and the proliferation marker, Ki67. A reduction in the number of Ki67+ cycling cells in hearts was observed in infected animals at 2 days PI relative to mock-infected controls (Figure 5A), and these results were quantified by ImageJ analysis (Figure 5B). Although we observed an early increase in the number of Sca-1+ cells in the heart early after infection suggesting an initial rapid expansion of progenitor cells, Sca-1+ cells predominantly lacked Ki67 expression by 2 days PI consistent with its proposed role as a cell cycle suppressor (Figure 5C). Immunostaining for both Ki67 and c-kit revealed a significant reduction in the number of proliferating c-kit+ cells in the hearts of infected pups (Figure 5D; **p<0.01).

Juvenile CVB3 infection depletes CPC pools in adult mice
CVB3 has been previously shown to induce caspase-3 dependent apoptosis in the heart and CNS [10]. To determine if CVB3 infection led to apoptosis and a permanent reduction in CPCs in the juvenile heart, sections from infected mice were immunostained using antibodies against c-kit and active caspase-3 (Figure 6). An increase in active caspase-3 (red) staining was observed in the infected heart, and active caspase-3 colocalized with some c-kit+ (green) cells (Figure 6A). Occasionally, infected c-kit+ cells in the heart showed characteristic apoptotic blebbing and pyknotic nuclei (Figure 6B). Quantification of active-caspase-3 staining indicated greater apoptosis in the infected heart at 2 day PI (Figure 6C; *p<0.05). Also, a greater number of TUNEL+ (ApopTag) cells per section were seen in infected hearts compared to those of mock-infected control mice at 5 days PI (Figure 6D). These results indicate that a significant number of cells in the infected juvenile heart which may include CPCs underwent virus-induced apoptosis.

We next determined if early CVB3 exposure resulted in a permanent decline in CPC numbers in the heart. Juvenile mice were infected with 107 pfu of eGFP-CVB3, and the number of c-kit+ cells was evaluated in heart tissue at 2, 5, 9, 28, and 77 days PI. At 2 and 5 days PI, we observed an increased trend in the number of c-kit+ cells although these results did not reach statistical significance. By 77 days PI, a statistically significant decrease in c-kit+ cells was observed in the infected heart (Figure 6E and Figure 6F; **p<0.001). Sca-1, a classical cardiac progenitor cell marker which signifies progression towards differentiation, increased transiently and then remained undetectable by 5 days PI (data not shown). The observed decline in c-kit+ CPCs persisted despite resolution of infection by 28 days PI as shown by plaque assay for infectious virus or by qPCR for viral RNA. These results coupled with our previous findings showing that CVB3 reduced the pool of self-renewing progenitor cells in part by triggering apoptosis or premature differentiation, and progenitor cell depletion continued into adulthood despite clearance of the virus and the absence of ongoing inflammation.
Subclinical CVB3 infection predisposed juvenile mice to beta adrenergic stress-induced heart failure later in life
Previous studies involving anthracycline-mediated cardiotoxicity suggested that juvenile exposure to the drug doxorubicin (DOX) was responsible for a profound effect on CPCs in mice, inducing their senescence and significantly reducing their number as time passed [25]. Though under basal conditions no physiological disparities or detectable cardiac deficits was observed when comparing DOX-treated mice to control animals, marked cardiomyopathy consisting of significantly increased heart mass, left ventricular dilation, and myocardial scarring was induced in DOX-treated mice following rigorous exercise via swimming. Sensitivity to stress-induced heart failure was attributed to the inability of DOX-treated animals to undergo vascular remodeling in response to the increased oxygen demand of the heart; a process which is known to be largely directed by paracrine signaling from CPCs.
Despite a reduction in the number of CPCs following CVB3 infection, juvenile-infected mice developed normally and were phenotypically indistinguishable from mock-infected control mice at 77 days PI. We determined if stress elicited pathologic remodeling in juvenile-infected adult mice administered with the beta adrenergic agonist, isoproterenol (ISO) [38]. At 91 days PI, juvenile-infected or mock-infected mice were treated with ISO for 10 days, treated with vehicle only, or given no treatment. Although mock-infected mice showed an increase in heart weight/tibia length ratios (HW/TL) following ISO treatment, juvenile-infected animals showed a significantly greater increase in heart mass than mock-infected mice (Figure 7A; *p<0.001). Transverse cross sections of heart tissue from ISO-treated mice infected at an early age showed enlarged left ventricles with thinning of the muscular wall (Figure 7B). Also, ISO-treated mice (CVB3-infected and mock-infected) developed fibrosis, as shown by Masson's Trichrome staining (Figure 7C). These observations supported our hypothesis that subclinical infection with CVB3 predisposed mice to greater pathologic remodeling in the heart. Of note, no infiltrating T cells were observed in CVB3 or mock-infected heart following ISO treatment (Figure 7D).

CVB3-mediated CPC loss impaired vascular remodeling during increased cardiac load
Cardiac vascular cells are highly dynamic and responsible for adaptive restructuring of the vascular network. Vascular remodeling is accomplished both through angiogenesis (branching of pre-existing blood vessels) and vasculogenesis (de novo blood vessel formation). These processes are critical for the heart to respond to various stimuli including stressors. With the induction of physiological stress, cardiac contractility increases, thereby increasing oxygen demand. Vascular remodeling is necessary to sufficiently perfuse the myocardium with oxygenated blood. Stem cells have been documented to be intimately involved in vascular remodeling through differentiation and release of paracrine factors such as vascular endothelial growth factor (VEGF) [25].
To examine if cardiac vasculature was affected by loss of progenitor cells due to prior CVB3 infection, we stained heart sections from ISO-treated animals for the blood vessel marker, CD31. In mice treated with vehicle alone, no significant differences in CD31+ blood vessel density was observed between infected and mock-infected animals (Figure 8A). As expected, treatment with ISO in mock-infected mice resulted in increased CD31 expression consistent with increased vascular remodeling in response to the increased workload imposed by beta adrenergic stress (*p<0.05). In contrast, no corresponding increase in CD31 expression was observed in juvenile-infected mice after ISO exposure (**p<0.01). These data indicate that under increased load, the heart remodels its vasculature to meet demand, but juvenile CVB3 infection impairs this compensatory function.

Because CPCs are known to release several factors including VEGF that stimulate blood vessel formation, we next sought to determine if the production of VEGF was impaired in the heart. Western blots of heart homogenates prepared from mock and juvenile-infected mice at 91 days PI were probed for VEGF expression levels. We found that VEGF expression was significantly lower in hearts of ISO-treated CVB3-infected mice compared to mock-infected control animals (Figure 8B; *p<0.05). Visual inspection of heart sections in mice treated with vehicle alone showed similar levels of CD31 staining between mock-infected and CVB3-infected mice treated with vehicle alone (Figure 8C and Figure 8D, respectively). In contrast, high levels of CD31 staining with associated vasculature was observed in ISO-treated mock-infected mice, particularly within regions of the heart showing signs of fibrosis outlined in purple (Figure 8E). In CVB3-infected mice following ISO treatment, lower levels of CD31 staining and less vasculature was observed in regions of the heart showing signs of fibrosis compared to ISO-treated mock-infected mice (Figure 8F).
The degree of fibrosis in heart sections was measured utilizing a Keyence microscope (BZ-9000) with brightfield capture in mock or CVB3-infected mice following treatment with ISO (Figure 9). CVB3-infected mice showed enlarged ventricles associated with greater amounts of fibrosis (blue signal) using the Keyence merge function. Fibrosis was more extensive in the hearts of CVB3-infected mice following ISO treatment compared to mock-infected mice, as quantified using the BZ-9000 Generation II Analyzer Keyence software (Figure 9B, Figure 9C and Figure 9D; *p<0.001). These results provide further evidence that a self-limited juvenile CVB3 infection may have lasting effects on the ability of adult mice to undergo compensatory vascular remodeling under cardiac stress.

Subclinical CVB3 infection predisposed juvenile mice to physiological stress-induced heart failure later in life
Intense swimming may be considered a natural “physiologic” stress. To determine if a natural stress could induce greater heart failure following infection, adult mice infected with CVB3 at an early age were subjected to exercise challenge which consisted of 90 minutes of daily continuous swimming for 14 consecutive days. Of note, mice previously infected with CVB3 did not swim as vigorously as the mock-infected control mice (data not shown). Following the swimming protocol, we observed a significant increase in heart weight/tibia length ratios (HW/TL) in infected mice compared to mock-infected control mice (Figure 10A; *p<0.05). Transverse cross sections from these hearts revealed enlarged left ventricles with thinning of the muscular wall (Figure 10B). Also, H&E staining showed the presence of myofibrillar disarray and Masson's trichrome staining revealed fibrosis in infected mice after the exercise challenge (Figure 10C), although no evidence of T cell infiltration was observed (Figure 10D). Taken together, these data suggest that a mild juvenile CVB3 infection can result in pathologic remodeling in adult mice after exercise challenge in the absence of significant inflammation.

Discussion
Coxsackieviruses (CV) are enteroviruses that most commonly cause a mild self-limited infection characterized by fever, upper-respiratory symptoms, and rash. In rare cases coxsackievirus B (CVB) infection can give rise to severe inflammatory diseases such as pancreatitis, meningo-encephalitis, and myocarditis. Studies of patients with end-stage idiopathic dilated cardiomyopathy have revealed detectable CVB RNA in the hearts of 70–80% of the cases, despite the absence of a history of viral myocarditis [8], [9]. This observation has raised the concern that mild CVB infection may contribute to long-term cardiac sequelae. Possible mechanisms have not been verified, although some studies have suggested autoimmune mechanisms which include superantigen expression, molecular mimicry, or bystander damage [39]. Dilated cardiomyopathy is the final common pathway of many pathologic processes, in which an enlarged ventricular cavity and thinned ventricular wall, results in poor contractility, low ejection fraction, and in many cases diastolic dysfunction due to fibrosis. These signs of disease may be preceded by many years of compensated cardiac hypertrophy characterized by thickening and enlargement of the ventricles before decompensation develops. While CVB-associated myocarditis is clearly linked to heart failure, the notion that mild CVB infection in childhood could predispose to heart failure in adults has not been previously examined. Based on our previous observation that neonatal exposure to the cardiotoxic anthracycline doxorubicin (DOX) resulted in late-onset heart failure through depletion of cardiac progenitor cells (CPCs), we hypothesized that juvenile CVB infection might reveal similar sequelae if progenitor cells in the heart were susceptible to infection similar to previous findings in the central nervous system and bone marrow [15], [17].
In this work, we established a model of mild juvenile coxsackievirus B3 (CVB3) infection which did not cause myocarditis or immune cell infiltration in the heart, but which triggered apoptosis or premature differentiation of CPCs, resulting in a significant and sustained reduction in the number of CPCs within the adult heart. Infectious virus and viral RNA were eventually cleared in the heart. We have not yet elucidated the immune mechanisms responsible for viral clearance from the heart in our juvenile mouse model. However, the limited CD3 staining in the heart observed at 2 and 77 days is consistent with previous studies demonstrating a lack of functional antiviral T cells following CVB3 infection [40], [41], and suggests that the type I interferon response may ultimately be responsible for controlling the viral infection [42]. We observed that after CVB3 exposure in vivo, CPCs showed a strong tendency to differentiate into vascular cells. Additionally, CVB3 has been previously shown to upregulate autophagy, a cellular process which may be essential during cellular differentiation [43]. Similar to DOX-induced senescence, CVB3-mediated premature differentiation reduced the number of CPCs available later in life. The role of CPCs in postnatal development and physiologic remodeling is poorly understood, but observations drawn from the juvenile DOX model suggest that CPCs are important for vascular development in the growing heart as well as in response to physiologic hypertrophy during exercise and in postinfarction remodeling [25]. The findings presented in this study support the hypothesis that a mild CVB3 infection early in development can impair the heart's ability to undergo physiologic remodeling, leading to dilated cardiomyopathy later in life.
Epidemiological studies have revealed an association between CVB3 infections and heart failure; however, a mechanistic link has not been found. To examine lasting consequences of CVB3 infection on the host, we infected three day-old pups with a sublethal dose of eGFP-CVB and examined hearts over time. We observed a significant decrease in the number of CPCs in the hearts of mice infected early in life. Additionally, after subjecting the mice to cardiac stress either via exercise or pharmacological induction, adult mice that had sustained juvenile CVB3 infection developed marked left ventricular hypertrophy and chamber dilation. To our knowledge, our study is the first model showing that mild neonatal CVB3 infection can predispose to pathologic remodeling in adult mice. We also observed premature differentiation of CPCs during acute infection and a sustained reduction in CPC numbers later in life despite an absence of detectable infectious virus or viral RNA in the hearts of adult animals. Also, no physiological abnormalities or cardiac damage were observed under basal conditions. Pathologic hypertrophy evidenced by left ventricular enlargement, chamber dilation, and histologic features of myofibrillar disarray and fibrosis was elicited only after adult mice were subjected to increased cardiac workload.
Our model of adult heart failure in juvenile-infected mice is shown in Figure 11. The propensity for pathologic alterations in the hearts of CVB3-infected mice may be due to an impairment of vascular remodeling in response to increased cardiac demand, a process which requires the participation of CPCs. Of note, endogenous c-kit+ cells were recently shown to substantially contribute to the generation of cardiac endothelial cells in the adult heart [44]. We did not observe differences in cardiac vascular density between mock and CVB-infected adult mice before ISO treatment. Our findings point to an impairment of compensatory remodeling rather than pre-existing deficiencies in vascular architecture. A similar phenomenon was observed following DOX-mediated CPC depletion [25]. Low dose DOX exposure in juvenile mice resulted in depletion of CPCs through early senescence. However, adult mice exhibited normal cardiac physiology and histologic findings. Stressors such as swimming exercise elicited pathologic remodeling, and experimental myocardial infarction resulted in more severe injury and poorer survival. The poor outcomes in DOX-treated mice were similarly attributed to an impaired capacity for vascular remodeling due to depleted CPCs. Stem cells have been shown to play a crucial role during vascular remodeling by stimulating angiogenesis through the release of paracrine factors, and differentiating to give rise to new blood vessels. We have previously shown that CVB3 infection of neural progenitor cells may have lasting effects on cell lineage commitment and development [45], [15]. We expect that progenitor cells in the heart surviving an early CVB3 infection may also alter CPC function and cell lineage commitment. In our studies here on the lasting consequences of juvenile CVB3 infection, a depletion or alteration of surviving CPCs which would normally participate in vasculogenesis, as well as reduced expression of the stimulatory paracrine factor VEGF in the heart likely impacted adaptive vascular rearrangement in response to cardiac stress.

These subtle cardiac alterations may go undetected under normal circumstances, but may emerge in the setting of increased cardiac demand, whether elicited by high-level exercise, chronic hypertension, or an ischemic event. Given the high prevalence of CV exposure in the population, many sedentary individuals may be asymptomatic. There are many unanswered questions that our animal model of juvenile CVB3 infection may help to address. For instance, does CVB3 infection affect stem cells at any age, or is there a high-risk window in early childhood? Anecdotally, we observed that CVB3 efficiently infected CPCs obtained from young mice (17 days post-birth), but only poorly infected CPCs obtained from adult animals. Defining the permissivity of CPCs may shed light on mechanism of pathogenesis and point to potential treatment strategies. Both permissive juvenile CPCs and non-permissive adult CPCs expressed high levels of coxsackie and adenovirus receptor (CAR). In addition, other cell types in the heart express high levels of CAR, including cardiomyocytes, without showing significant signs of infection. In contrast, some cells (B lymphocytes) lacking CAR expression are susceptible to infection [13]. The present study does not specifically define the basis for differential permissivity of cardiac progenitor cell (CPC) infection. Preferential juvenile CPC infection may be less dependent upon CAR expression and more likely determined by additional factors such as expression levels of type I interferon response genes, autophagy flux in progenitor cells, cellular differentiation, and the activation/proliferative status of these cells.
What is the fate of individual CPCs following CVB3 infection of the heart? The fate of individual CPCs infected with CVB3 would be difficult to determine without temporal cell tracking strategies. However taken as a population, our data suggest that CPCs vary in their permissivity to infection, both in terms of viral replication and cell death/survival. We have previously shown that neural progenitor cells grown in culture vary in their permissivity to infection based upon their differentiation status. CPCs grown in culture may follow a similar pattern of permissivity, although additional factors may also determine permissivity of CPCs in the heart tissue, including the host immune response.
These studies utilized a strain of CVB3 (Woodruff strain) that was obtained from a patient with myocarditis. Perhaps only some CVB3 strains pose a risk for CPC depletion and late cardiac sequelae. Due to the possible decades-long interval between childhood exposure to CV infection and development of cardiac symptomatology, challenges remain to prove causality in patients and to determine the age threshold for vulnerability. However, these studies raise concern that mild CVB3 infection in childhood may carry significant sequelae. Prevention of these late effects will depend upon introduction of a safe and effective vaccine and/or the development of rapid diagnosis and utilization of antiviral therapy in children exposed at a vulnerable age.
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the requirements pertaining to animal subjects protections within the Public Health Service Policy and USDA Animal Welfare Regulations. All experimental procedures with mice were approved by the San Diego State University Institutional Animal Care and Use Committee (Animal Protocol Form #10-05-013F), and all efforts were made to minimize suffering.
The generation of a recombinant CVB3 expressing the enhanced green fluorescent protein (eGFP–CVB3)
The generation of eGFP–CVB3 has been described previously [46]. Briefly, a DNA plasmid was obtained containing an infectious CVB3 clone [47] (pH 3 - kindly provided by Dr. Kirk Knowlton, University of California at San Diego). This DNA plasmid was engineered to contain an Sfi1 restriction site (pMKS1). The eGFP gene was amplified from an eGFP expression plasmid (Clontech, Palo Alto, CA) using eGFP primers with flanking Sfi1 sites. These PCR products were subsequently cloned into pMKS1. This construct was transfected into HeLa RW cells [47] (kindly provided by Dr. Rainer Wessely, University of California at San Diego) and lysates containing infectious virus were collected as viral stocks. Viral concentrations were measured via plaque assay. Viral stocks were prepared on HeLa cells and diluted in DMEM (Invitrogen, Gaithersburg, MD) before inoculation.
Mice and viral inoculations
BALB/c mouse breeding pairs were observed daily to ensure that pups were identified within 24 hours of birth. Three day-old pups were infected intraperitoneally (IP) with 1×107 pfu of eGFP–CVB3. At 2 to 10 days PI, animals were killed by hypothermia/CO2 asphyxiation, followed by immediate decapitation. Adult mice (4–11 weeks PI) were anesthetized with isoflurane and cervically dislocated. Hearts were removed and rinsed in cold PBS. For histology, hearts were fixed by immersion in 10% neutral-buffered formalin for 24 hours and then transferred to 70% ethanol for an additional 24 hours. The tissue was then paraffin-embedded.
Cardiac progenitor cell (CPC) isolation and infection
CPCs were isolated using the Millipore Cardiac Stem Cell Isolation Kit (Millipore cat# SCR061) according to the manufacturer's protocol. To determine CVB3 replication in cultured CPCs, cells were seeded in 6-well plates at a concentration of 104 cells/well. After 3 days, cells were infected with eGFP-CVB3 at MOI = 1.0 or 1000. Viral titers were measured in culture supernatants obtained each day following infection. For western blots, CPCs were seeded in 6-well plates at a concentration of 106 cells/well. After 3 days, cells were infected with GFP-CVB3 at MOI = 100. Three days PI, cells were lysed in RIPA buffer and protein lysates were used for western blot analysis.
Histological analysis
Paraffin-embedded hearts were cut into 4 µm thick sections. Sections were then deparaffinized with xylene and rehydrated in stepwise decreasing ethanol concentrations followed by PBS and distilled water. Antigen recovery was performed by boiling sections in sodium citrate buffer (10 mM sodium citrate+.05% Tween20, pH 6.0). For immunostaining, the following primary antibodies were used: anti-CD3 (1∶100, Biocare Med #CP215A), anti-Iba1 (1∶500, Wako #JNH4100), rabbit anti-GFP (1∶100, Invitrogen #A6455), goat anti-c-Kit (1∶40, R&D #AF1356), rat anti-Sca-1 (1∶25, Cedarlane cat #CL8934AP), mouse anti - Ki67 (1∶200, Vector #VP-K452), rabbit anti-SM22 (1∶2000, Abcam #ab14106), rabbit anti-Von Willbrand Factor (1∶100, Sigma #F3520), rabbit anti-CD31 (1∶100, Abcam #ab28364), mouse anti-sarcomeric actin (1∶100, Sigma #A2172). Perkin Elmer proprietary blocking solution (Perkin Elmer #NEL702001) was used for antibody dilution and tissue blocking. Apoptosis was detected using the ApopTag Red In Situ Apoptosis Detection Kit (Chemicon/Millipore, Billerica, MA) as described by the manufacturer.
Fibrosis quantitation
Transverse sections of paraffin-embedded hearts were stained with Masson's Trichrome. Whole heart sections were imaged with a Keyence microscope (BZ-9000) using brightfield capture. Multiple fields were captured using the 4× objective and stitched together using the Keyence Merge function. Fibrosis (collagen staining) was measured using the Single Extraction function of the Hybrid Cell Count software based on hue.
Whole heart dissociation for FACS analysis
Extracted hearts were placed in isolation buffer and excess blood was expressed by gentle compression with forceps. Hearts were then minced into ∼2 mm pieces in Millipore Isolation Buffer. Heart pieces were resuspended in Millipore Cardiac Tissue Dissociation Buffer and incubated at 37°C shaking at 200 rpm. After 45 minutes of incubation, tissue was mechanically dissociated for 5 minutes by repeated trituration with a wide-bore pipette tip. Tissues were then incubated for 15 more minutes and then mechanically dissociated for 5 minutes. Large tissue pieces were removed by filtering suspension through Millipore 100 um Steriflip unit. Cells were fixed in 10% formalin in PBS. For FACS analysis the following antibodies were used: PE-anti-Sca-1 (1∶100, BD #552108), FITC-anti-Sca-1 (1∶100, BD #557405), APC-anti-CD45 (1∶100, Caltag #MCD4505).
Exercise challenge
Ten to eleven weeks PI, neonatally-infected mice were subjected to exercise challenge which consisted of daily swimming in heated water tanks with a gentle pump-driven current to encourage active swimming. After each swimming session, animals were allowed to recover under a heating lamp. A seven day training period consisted of 5 min swimming on day 1, then increased daily (15, 30, 45, 60, 75 and finally 90 minutes). Following this initial week of training, mice underwent 90 min daily swimming exercise for 14 days. Upon completion of 14 days of full swimming (21 day total), hearts were excised for determination of heart weight (normalized to tibia length) and histology.
Beta-adrenergic stimulation
Isoproterenol hydrochloride was resuspended in sesame oil to prolong uptake. Thirteen weeks PI, juvenile-infected mice were injected intraperitoneally (IP) with 40 mg/kg/d isoproterenol hydrochloride daily for 10 days. Following the final day of injections, hearts were excised for determination of heart weight (normalized to tibia length) and histology.
Statistical analyses
Statistical analyses were carried out by Student's T-test using Excel software, and by two-way ANOVA using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). Significance was determined by a p-value of 0.05 or lower. Alternatively, statistical analyses were carried out by pairwise comparisons using Student's T-test with Bonferonni correction applied to control form multiple comparisons. Significance for Student's T-test with Bonferonni correction was determined by a p-value of 0.01 or lower.
Zdroje
1. CarthyCM, YangD, AndersonDR, WilsonJE, McManusBM (1997) Myocarditis as systemic disease: new perspectives on pathogenesis. Clin Exp Pharmacol Physiol 24 : 997–1003.
2. SoleMJ, LiuP (1993) Viral myocarditis: a paradigm for understanding the pathogenesis and treatment of dilated cardiomyopathy. J Am Coll Cardiol 22 : 99A–105A.
3. TamPE (2006) Coxsackievirus myocarditis: interplay between virus and host in the pathogenesis of heart disease. Viral Immunol 19 : 133–146.
4. GristNR, BellEJ (1969) Coxsackie viruses and the heart. Am Heart J 77 : 295–300.
5. WhittonJL, CornellCT, FeuerR (2005) Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol 3 : 765–776.
6. WardC (1978) Severe arrhythmias in Coxsackievirus B3 myopericarditis. Arch Dis Child 53 : 174–176.
7. FujiokaS, KitauraY, DeguchiH, ShimizuA, IsomuraT, et al. (2004) Evidence of viral infection in the myocardium of American and Japanese patients with idiopathic dilated cardiomyopathy. Am J Cardiol 94 : 602–605.
8. PetitjeanJ, KopeckaH, FreymuthF, LanglardJM, ScanuP, et al. (1992) Detection of enteroviruses in endomyocardial biopsy by molecular approach. J Med Virol 37 : 76–82.
9. ChiangFT, LinLI, TsengYZ, TsengCD, HsuKL, et al. (1992) Detection of enterovirus RNA in patients with idiopathic dilated cardiomyopathy by polymerase chain reaction. J Formos Med Assoc 91 : 569–574.
10. FeuerR, MenaI, PagariganRR, HarkinsS, HassettDE, et al. (2003) Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am J Pathol 163 : 1379–1393.
11. FeuerR, PagariganRR, HarkinsS, LiuF, HunzikerIP, et al. (2005) Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J Neurosci 25 : 2434–2444.
12. Tabor-GodwinJM, TsuengG, SayenMR, GottliebRA, FeuerR (2012) The role of autophagy during coxsackievirus infection of neural progenitor and stem cells. Autophagy 8 : 938–953.
13. FeuerR, WhittonJL (2008) Preferential coxsackievirus replication in proliferating/activated cells: implications for virus tropism, persistence, and pathogenesis. Curr Top Microbiol Immunol 323 : 149–173.
14. RhoadesRE, Tabor-GodwinJM, TsuengG, FeuerR (2011) Enterovirus infections of the central nervous system. Virology 411 : 288–305.
15. RullerCM, Tabor-GodwinJM, Van DerenDAJ, RobinsonSM, MaciejewskiS, et al. (2012) Neural stem cell depletion and CNS developmental defects after enteroviral infection. Am J Pathol 180 : 1107–1120.
16. TsuengG, Tabor-GodwinJM, GopalA, RullerCM, DelineS, et al. (2011) Coxsackievirus preferentially replicates and induces cytopathic effects in undifferentiated neural progenitor cells. J Virol 85 : 5718–5732.
17. AlthofN, WhittonJL (2012) Coxsackievirus B3 infects the bone marrow and diminishes the restorative capacity of erythroid and lymphoid progenitors. J Virol 87 : 2823–2834.
18. RobinsonSM, TsuengG, SinJ, MangaleV, RahawiS, et al. (2014) Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog 10: e1004045.
19. JacksonWT, GiddingsTHJr, TaylorMP, MulinyaweS, RabinovitchM, et al. (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 3(5): e156.
20. AnversaP, KajsturaJ, RotaM, LeriA (2013) Regenerating new heart with stem cells. J Clin Invest 123 : 62–70.
21. LiuJ, WangY, DuW, YuB (2013) Sca-1-Positive Cardiac Stem Cell migration in a Cardiac Infarction Model. Inflammation 36 : 738–749.
22. OhH, BradfuteSB, GallardoTD, NakamuraT, GaussinV, et al. (2003) Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A 100 : 12313–12318.
23. BeltramiAP, BarlucchiL, TorellaD, BakerM, LimanaF, et al. (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell %19 114 : 763–776.
24. FischerKM, CottageCT, WuW, DinS, GudeNA, et al. (2009) Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation 120 : 2077–2087.
25. HuangC, ZhangX, RamilJM, RikkaS, KimL, et al. (2010) Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater susceptibility to stress-induced myocardial injury in adult mice. Circulation 121 : 675–683.
26. PucciniJM, RullerCM, RobinsonSM, KnoppKA, BuchmeierMJ, et al. (2013) Distinct neural stem cell tropism, early immune activation, and choroid plexus pathology following coxsackievirus infection in the neonatal central nervous system. Lab Invest (In Press)..
27. ZhangY, SimpsonAA, LedfordRM, BatorCM, ChakravartyS, et al. (2004) Structural and virological studies of the stages of virus replication that are affected by antirhinovirus compounds. J Virol 78 : 11061–11069.
28. AnversaP, FitzpatrickD, ArganiS, CapassoJM (1991) Myocyte mitotic division in the aging mammalian rat heart. Circ Res 69 : 1159–1164.
29. PearceBD, SteffensenSC, PaolettiAD, HenriksenSJ, BuchmeierMJ (1996) Persistent dentate granule cell hyperexcitability after neonatal infection with lymphocytic choriomeningitis virus. J Neurosci 16 : 220–228.
30. WesselyR, KlingelK, KnowltonKU, KandolfR (2001) Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: implications for mortality and early viral replication. Circulation 103 : 756–761.
31. KimKS, HufnagelG, ChapmanNM, TracyS (2001) The group B coxsackieviruses and myocarditis. Rev Med Virol 11 : 355–368.
32. FeuerR, MenaI, PagariganRR, HassettDE, WhittonJL (2004) Coxsackievirus replication and the cell cycle: a potential regulatory mechanism for viral persistence/latency. Med Microbiol Immunol (Berl) 193 : 83–90.
33. FeuerR, RullerCM, AnN, Tabor-GodwinJM, RhoadesRE, et al. (2009) Viral persistence and chronic immunopathology in the adult central nervous system following Coxsackievirus infection during the neonatal period. J Virol 83 : 9356–9369.
34. DeyD, HanL, BauerM, SanadaF, OikonomopoulosA, et al. (2013) Dissecting the molecular relationship among various cardiogenic progenitor cells. Circ Res 112 : 1253–1262.
35. MagentaA, AvitabileD, PompilioG, CapogrossiMC (2013) c-kit-Positive cardiac progenitor cells: the heart of stemness. Circ Res 112 : 1202–1204.
36. EptingCL, LopezJE, ShenX, LiuL, BristowJ, et al. (2004) Stem cell antigen-1 is necessary for cell-cycle withdrawal and myoblast differentiation in C2C12 cells. J Cell Sci 117 : 6185–6195.
37. HolmesC, StanfordWL (2007) Concise review: stem cell antigen-1: expression, function, and enigma. Stem Cells 25 : 1339–1347.
38. GalindoCL, SkinnerMA, ErramiM, OlsonLD, WatsonDA, et al. (2009) Transcriptional profile of isoproterenol-induced cardiomyopathy and comparison to exercise-induced cardiac hypertrophy and human cardiac failure. BMC Physiol 9 : 23 doi:–10.1186/1472-6793-9-23.: 23–29
39. HorwitzMS, BradleyLM, HarbertsonJ, KrahlT, LeeJ, et al. (1998) Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med 4 : 781–785.
40. KemballCC, AlirezaeiM, FlynnCT, WoodMR, HarkinsS, et al. (2010) Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J Virol 84 : 12110–12124.
41. KemballCC, FlynnCT, HoskingMP, BottenJ, WhittonJL (2012) Wild-type coxsackievirus infection dramatically alters the abundance, heterogeneity, and immunostimulatory capacity of conventional dendritic cells in vivo. Virology 429 : 74–90.
42. AlthofN, HarkinsS, KemballCC, FlynnCT, AlirezaeiM, et al. (2014) In vivo ablation of type I interferon receptor from cardiomyocytes delays coxsackieviral clearance and accelerates myocardial disease. J Virol 88 : 5087–99.
43. GuanJL, SimonAK, PrescottM, MenendezJA, LiuF, et al. (2013) Autophagy in stem cells. Autophagy 9 : 830–49.
44. van BerloJH, KanisicakO, MailletM, VagnozziRJ, KarchJ, et al. (2014) c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509 : 337–341.
45. Tabor-GodwinJM, RullerCM, BagalsoN, AnN, PagariganRR, et al. (2010) A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J Neurosci 30 : 8676–8691.
46. FeuerR, MenaI, PagariganR, SlifkaMK, WhittonJL (2002) Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol 76 : 4430–4440.
47. KnowltonKU, JeonES, BerkleyN, WesselyR, HuberS (1996) A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J Virol 70 : 7811–7818.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Molecular and Cellular Mechanisms of KSHV Oncogenesis of Kaposi's Sarcoma Associated with HIV/AIDS
- Holobiont–Holobiont Interactions: Redefining Host–Parasite Interactions
- BCKDH: The Missing Link in Apicomplexan Mitochondrial Metabolism Is Required for Full Virulence of and
- Helminth Infections, Type-2 Immune Response, and Metabolic Syndrome
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy