
The Complex I Subunit Selectively Rescues Mutants through a Mechanism Independent of Mitophagy
Two genes linked to heritable forms of the neurodegenerative movement disorder Parkinson's disease (PD), PINK1 and parkin, play important roles in mitochondrial homeostasis through mechanisms which include the degradation of dysfunctional mitochondria, termed mitophagy, and the maintenance of complex I (CI) activity. Here we report the findings of an RNAi based screen in Drosophila cells for genes that may regulate the PINK1-Parkin pathway which identified NDUFA10 (ND42 in Drosophila), a subunit of CI. Using a well-established cellular system and in vivo Drosophila genetics, we demonstrate that while NDUFA10/ND42 only plays a minimal role in mitophagy, restoration of CI activity through overexpression of either ND42 or its co-chaperone sicily is able to substantially rescue behavioral deficits in pink1 mutants but not parkin mutants. Moreover, while parkin overexpression is known to rescue pink1 mutants, it apparently achieves this without restoring CI activity. These results suggest that increasing CI activity or promoting mitophagy can be beneficial in pink1 mutants, and further highlights separable functions of PINK1 and Parkin.
Published in the journal:
. PLoS Genet 10(11): e32767. doi:10.1371/journal.pgen.1004815
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004815
Summary
Two genes linked to heritable forms of the neurodegenerative movement disorder Parkinson's disease (PD), PINK1 and parkin, play important roles in mitochondrial homeostasis through mechanisms which include the degradation of dysfunctional mitochondria, termed mitophagy, and the maintenance of complex I (CI) activity. Here we report the findings of an RNAi based screen in Drosophila cells for genes that may regulate the PINK1-Parkin pathway which identified NDUFA10 (ND42 in Drosophila), a subunit of CI. Using a well-established cellular system and in vivo Drosophila genetics, we demonstrate that while NDUFA10/ND42 only plays a minimal role in mitophagy, restoration of CI activity through overexpression of either ND42 or its co-chaperone sicily is able to substantially rescue behavioral deficits in pink1 mutants but not parkin mutants. Moreover, while parkin overexpression is known to rescue pink1 mutants, it apparently achieves this without restoring CI activity. These results suggest that increasing CI activity or promoting mitophagy can be beneficial in pink1 mutants, and further highlights separable functions of PINK1 and Parkin.
Introduction
Parkinson's disease (PD) is the second most prevalent neurodegenerative disorder, the etiology of which remains unknown. Mitochondrial dysfunction, including complex I (CI) deficiency, are frequently observed in pathologic specimens. To elucidate the underlying molecular events, intensive research has focused on identifying the causative gene mutations for inherited forms of PD. An impressive array of disease-causing mutations have been found including autosomal recessive mutations in PARK6 which encodes PTEN-Induced Kinase 1 (PINK1), a mitochondrially targeted serine/threonine kinase, and PARK2, encoding the cytoplasmic E3 ubiquitin ligase Parkin [1], [2]. Previous work has shown that PINK1 and Parkin regulate several cellular processes that impinge on mitochondrial homeostasis [3]–[13], including the fission-fusion dynamics and trafficking of mitochondria [14]–[17], the degradation of damaged mitochondria [18]–[22], inter-organelle communication [23], and activity of CI of the electron transport chain [4], [8], [9], [24], [25]. However, the mechanisms by which PINK1 and Parkin affect these processes is incompletely understood.
In order to identify functional partners of PINK1 we performed a cell based RNAi screen to identify genes that either phenocopy or suppress PINK1 phenotypes. In Drosophila cells lacking pink1 or parkin steady state levels of Marf are increased causing excess fusion of the mitochondrial network [14], [17], [26]. This imbalance in mitochondrial fission-fusion at least partially contributes to the observed organismal phenotypes – enlarged and disrupted mitochondria, muscle degeneration, male sterility, and associated behavioral deficits and neurodegeneration – since they can be partially rescued by genetic interaction with fission (Drp1) or fusion (OPA1 and Marf) genes to restore the balance of normal mitochondrial morphology [14], [16]. We used this phenotype to screen for factors that impact on mitochondrial morphology and which may play a role in PINK1/Parkin function. Our RNAi screen identified multiple hits including a subunit of CI, ND42/NDUFA10. Given the link between CI deficiency and PD, we selected ND42/NDUFA10 for further characterization.
Knockdown of Drosophila ND42 phenocopied pink1 RNAi, causing excessive mitochondrial fusion. Genetic studies in Drosophila reveal that overexpression of ND42 or its co-chaperone sicily is sufficient to rescue behavioral deficits in pink1 mutants through restoration of CI activity. In contrast, overexpression of neither ND42 nor sicily rescued parkin mutant flies, and attenuation of the mammalian homolog, NDUFA10, in HeLa cells only modestly reduced mitophagy. Furthermore, NDUFA10 overexpression cannot restore Parkin translocation to mitochondria in the absence of PINK1, suggesting that ND42 selectively rescues pink1 mutants through a mechanism independent of mitophagy. Our study provides additional evidence in support of a role for PINK1 in CI activity, and further highlights separable functions of PINK1 and Parkin. Future characterization of the other factors from our screen promises to shed additional light on the functional roles of PINK1 and Parkin.
Results
An RNAi screen for suppressors or phenocopiers of pink1 RNAi-induced mitochondrial fusion
To identify new components in PINK1-Parkin pathway we performed an RNAi screen in Drosophila cells, using a subset library enriched for kinases and phosphatases, for genes that alter mitochondrial morphology (Figure 1A). In particular, we sought to identify genes whose knockdown could either phenocopy or suppress the pink1 RNAi-induced excess fusion. dsRNA treated cells were imaged live using MitoTracker Red to fluorescently label the mitochondrial network. The mitochondrial morphology in each image was scored as primarily falling into one of four categories based on control knockdown phenotypes (Figure 1B). Control (DsRed) dsRNA treated cells showed a typical mix of short-round and long-tubular mitochondria. This category was scored 2. Cells treated with dsRNA against the fly mitofusin homologue, Marf, caused a complete fragmentation of the mitochondrial network as expected and were scored 1. pink1 RNAi resulted in a tubular, highly interconnected network, as previously reported [14], [17], scoring 3. RNAi against the pro-fission factor Drp1 caused mitochondrial hyperfusion and peri-nuclear clumping. Being qualitatively different from the pink1 RNAi phenotype and an extreme result of hyperfusion this category scored 4.

The screen was performed on a library of selected genes mostly comprising kinases and phosphatases but with additional genes of interest. The effect on mitochondrial morphology was assessed in two backgrounds; one in a wild type background to identify manipulations that phenocopy pink1 RNAi tubulation, and another in a pink1 RNAi background to identify manipulations capable of suppressing the pink1 phenotype. The results from the two screens were cross-referenced to further refine the selection of possible hits (Figure 1C). The limits of the regions considered to phenocopy or suppress were defined by the mean ± standard deviation for pink1 or DsRed dsRNAs respectively. The gene targets whose dsRNAs either phenocopy or suppress pink1 RNAi are summarized in Table 1 and Table 2.

While there are several interesting candidates for further investigation in both suppressors and phenocopiers (see Discussion), a specific motivation for this work was to identify additional components of the PINK1-Parkin pathway that regulate mitochondrial dynamics and mitophagy. An attractive candidate that could fulfill this role would be a mitochondrially localized protein. Notably, we identified ND42, which encodes a subunit of CI of the electron transport chain, as a phenocopier; knockdown of ND42 caused excess tubulation similar to pink1 knockdown. Importantly, CI activity has been shown to be specifically affected in various models of pink1 deficiency, supporting ND42 as an attractive target for analysis in pink1 function.
ND42 knockdown specifically phenocopies pink1 RNAi induced mitochondrial hyperfusion
CI is a very large, multi-subunit complex comprising of around 44 subunits, consisting of a hydrophobic portion embedded in the inner membrane and a hydrophilic portion extending into the matrix [27], [28]. In order to assess the specific versus general effect of attenuating CI subunits on mitochondrial morphology, several additional subunits were analyzed. dsRNAs targeting 6 other subunits from different subdomains (α, β or λ) were generated and the effect of knockdown on mitochondrial morphology was assessed.
We confirmed that ND42 knockdown caused excess mitochondrial fusion in a WT background, indistinguishable from pink1 knockdown, but did not further enhance the pink1 phenotype (Figures 2A, 2B, and S1). In contrast none of the other selected subunits induced fusion; 4 subunits had no effect on morphology while 2 subunits caused fragmentation (Figures 2C and 2D). Further analysis of one of these subunits, CG7712, which did not perturb mitochondrial morphology, also did not modify the pink1 phenotype. These results support the specificity of the effect observed with ND42 knockdown.

ND42 overexpression can rescue pink1 but not parkin mutants
Since ND42 RNAi phenocopies loss of pink1 in cells we assessed whether loss of ND42 may phenocopy pink1 mutants in vivo. Drosophila mutant for pink1 exhibit characteristic locomotor deficits in climbing and flight, associated with degeneration of the musculature and profound disruption of mitochondria [3], [11]. In agreement with previous observations [29], we found that knockdown of ND42 in all tissues is lethal, consistent with the subunit playing a critical role in this essential metabolic enzyme.
Due to the essential nature of this gene, and lack of pink1 phenocopy, we did not further characterize ND42 loss-of-function. We hypothesized that, since pink1 mutants show CI deficiency, overexpressing ND42 may suppress pink1 phenotypes. While ND42 overexpression of two independent transgenes driven by the strong ubiquitous driver daughterless (da)-GAL4 had no effect on motor performance in a wild type background (Figures 3A and 3B), we found that expression of either transgene was able to significantly restore climbing and flight ability in pink1 mutants (Figures 3C and 3D). However, ND42 overexpression only partially restored flight muscle and mitochondrial integrity (Figure 3E), but was not able to improve the male sterility (Figure S2A).
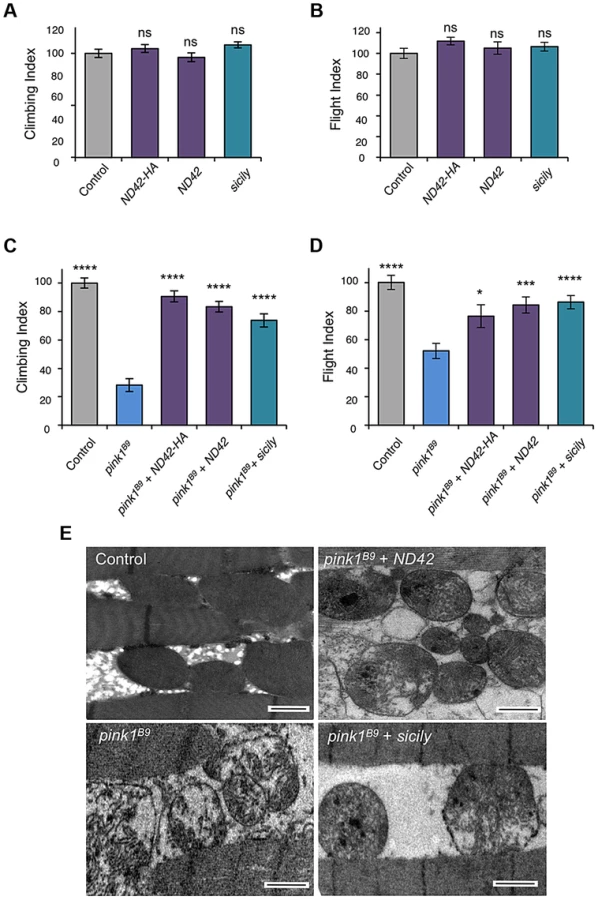
Genetic interaction studies in Drosophila have linked pink1 and parkin in a common pathway with pink1 acting upstream of parkin [3], [6], [11]. To further characterize the putative action of ND42/NDUFA10 in the PINK1-Parkin pathway, we tested whether ND42 overexpression could also rescue parkin mutants. Surprisingly, we found that overexpression of ND42 was not able to rescue any parkin phenotypes tested, including locomotor behaviors, muscle and mitochondrial integrity, and male sterility (Figures 4A–4C, and S2B).

Recently it was reported that Drosophila sicily acts as a co-chaperone to bind and stabilize ND42 in the cytoplasm, promoting its mitochondrial import and the formation of CI [29]. Supporting a potential role for sicily in pink1 function, we found that knockdown of sicily in Drosophila cells phenocopied pink1 mitochondrial hyperfusion (Figure S3). Since sicily promotes ND42 stability, and overexpression of ND42 can rescue pink1 mutant phenotypes, we hypothesized that sicily overexpression may also rescue pink1 mutants. Indeed, we found that sicily overexpression rescued pink1 mutant locomotor and mitochondrial phenotypes comparable to ND42 overexpression (Figures 3A–3E). Also, overexpression of sicily failed to rescue similar parkin mutant phenotypes (Figures 4A–4C), mirroring the effects of ND42. Together these results demonstrate a genetic interaction of ND42 and sicily with pink1 but not parkin.
NDUFA10 minimally affects Parkin translocation and mitophagy
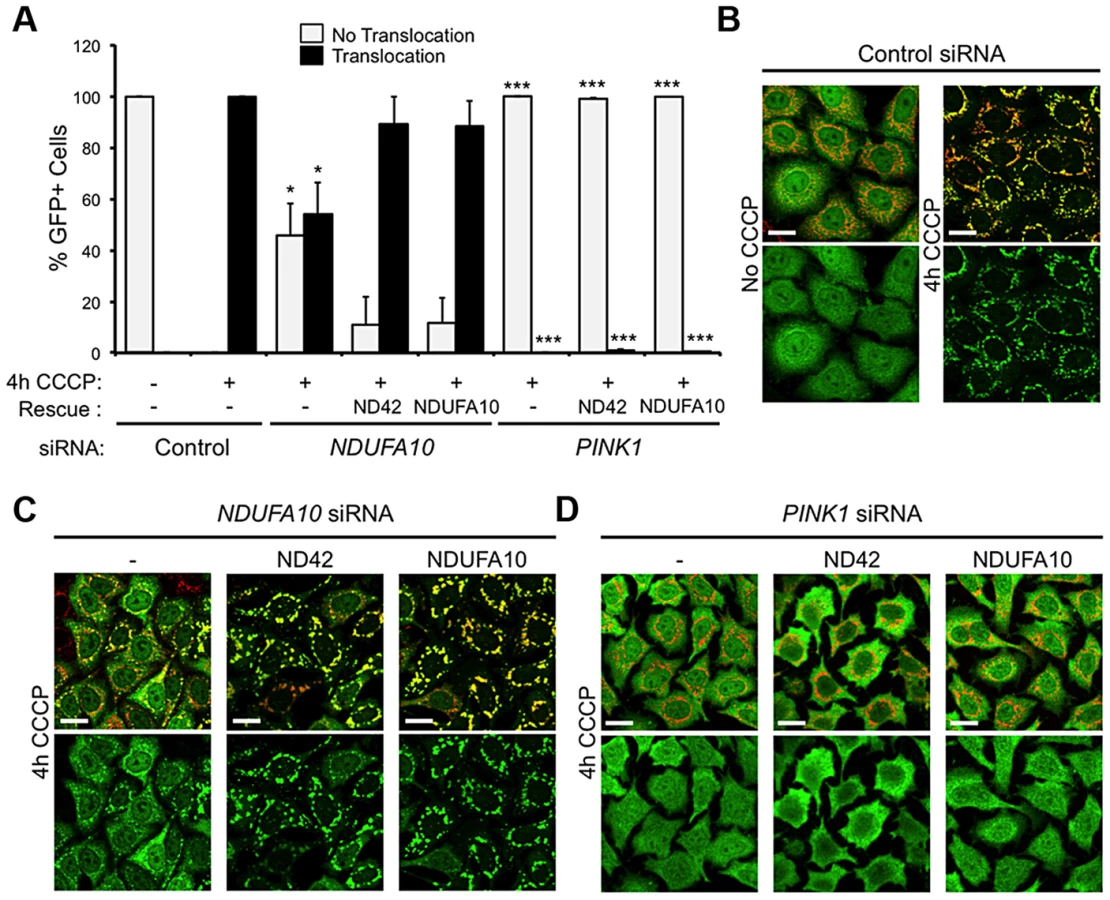
The fact that ND42 overexpression can rescue pink1 but not parkin mutants would be consistent with it acting in a common pathway between PINK1 and Parkin. An intensively studied field of PINK1-Parkin function is the autophagic turnover of mitochondria, termed mitophagy [30]. In HeLa cells overexpressing YFP-Parkin, depolarization of the mitochondrial membrane with the protonophore carbonyl cyanide 3-chlorophenylhydrazone (CCCP) causes a rapid stabilization of PINK1 on the outer mitochondrial membrane, which stimulates the re-distribution of cytoplasmic Parkin to co-localize with mitochondria (Figures 5A and 5B). Prolonged exposure to CCCP induces substantial degradation of the mitochondria (Figures 5C, 5D, and S4). These phenomena are almost completely abolished by PINK1 knockdown (Figures 5A–5D) [18]–[20], [22].

We next analyzed the effect of the human homolog of ND42, NDUFA10, on Parkin translocation and mitophagy. We found that NDUFA10 knockdown had a modest but significant effect on Parkin translocation, though clearly not as much as loss of PINK1 (Figures 5A, 5B, S1 and S5). Moreover, loss of NDUFA10 only very minimally reduced the degree of mitophagy (Figures 5C, 5D, and S6). We also found no effect of NDUFA10 knockdown on PINK1 stabilization following mitochondrial depolarization (Figure S7). These data indicate that NDUFA10 does not play a significant role in PINK1/Parkin mediated mitophagy and suggests that the in vivo rescue was unlikely via the mitophagy pathway.
To further exclude a role for mitophagy in the rescue of pink1 mutants, we assessed in vitro whether NDUFA10 overexpression could promote CCCP-induced Parkin translocation in the absence of PINK1. Encouragingly, re-expression of either NDUFA10 or ND42 almost completely restored Parkin translocation reduced by NDUFA10 knockdown (Figures 6A and 6C). However, when Parkin translocation was completely blocked by loss of PINK1, this was not rescued by expression of either NDUFA10 or ND42 (Figures 6A and 6D). These results support the idea that the rescue seen in vivo is unlikely due to activated mitophagy, raising the question of what mechanism is responsible.
ND42 overexpression restores CI activity in pink1 mutants
As loss of PINK1 has been reported to cause decreased CI activity [24], we reasoned that suppression of pink1 mutants by ND42 overexpression may be due to restoration of CI activity. As previously reported, we observed a CI deficiency in pink1 mutant flies, leading to decreased ATP production (Figures, 7A and 7B). We found that ND42 overexpression was indeed able to completely restore both CI activity and ATP levels in vivo (Figure 7A and 7B). Extending these analyses to parkin mutants, we saw a non-significant decrease in CI activity in parkin mutants that remained unchanged by ND42 overexpression (Figure 7C). Similarly, the significant depletion of ATP evident in parkin mutants was not rescued by ND42 overexpression (Figure 7D), consistent with a lack of phenotypic rescue by ND42 overexpression. Interestingly, we also found that sicily overexpression was able to completely restore CI activity in pink1 mutants (Figure 7A), while the increase in ATP levels was not significant (Figure 7B). Similar to ND42, sicily overexpression had no effect in parkin mutants (Figures 7C and 7D).

Analysis of ND42 Ser-250 phospho-variants in the rescue of pink1 mutant locomotion and CI activity
While this work was in preparation, Morais et al [25] reported that NDUFA10 lacked phosphorylation at serine-250 in the absence of PINK1, and that expression of phospho-mimetic NDUFA10/ND42 specifically reversed PINK1 deficits in various model systems, including restoring CI activity in mammalian systems and synaptic phenotypes in Drosophila pink1 mutants. Our preceding data concur that overexpression of ND42 can rescue some pink1 mutant phenotypes (and not others), but interestingly we found that this can be achieved with expression of the wild type version without a specific requirement for the phospho-mimetic.
To further explore the potential role of Ser-250 phosphorylation in these assays, we tested in parallel our existing lines (previously reported in [31]) and those of Morais et al [25] and of the yeast equivalent of CI, NDI1 [31]. As before, we found that expression of the previous WT transgene (designated ND42HB) significantly rescued pink1 climbing and flight defects (Figures 8A and 8B). We also found that the Morais et al. transgenes (designated ND42BDS) expressing either WT or phospho-mimetic (SD) also partially rescued climbing, albeit to a lesser extent (Figure 8A). The WT version of these lines did not statistically improve flight ability whereas the SD did provide a modest rescue (Figure 8B). We also found that the non-phosphorylatable version (SA) provided significant rescue of climbing, equivalent to the phospho-mimetic, but again did not rescue flight (Figure 8A and 8B). Notably, in these assays NDI1 expression significantly rescued climbing but not flight ability (Figure 8A and 8B), consistent with previous observations [31].

To better understand the relationship between the behavioral rescue and CI activity, and to assess functional efficacies of the various transgenic lines, we tested the ability of the phospho-variants transgenes to rescue the CI deficiency in pink1 mutants. We found that expression of all phospho-variants were able to fully restore CI activity in pink1 mutants (Fig. 8C). The degree of rescue was similar to that seen with the previous WT transgene (Fig. 7A), consistent with an equivalent level of expression between these lines (Fig. S1G). Interestingly, while we see the highest level of CI activity with the phospho-mimetic (SD), we also see a substantial rescue by the phopho-null (SA) in this in vitro assay.
Overexpression of parkin rescues pink1 mutants but does not restore CI activity
The ability of multiple transgenes that restore CI activity to at least partially rescue climbing behavior supports the idea that promoting CI activity is differentially beneficial in pink1 mutants but not parkin mutants and may hint at different underlying causes of pathogenicity. However, a puzzling aspect of this is the long-standing observations that parkin overexpression is sufficient to almost completely rescue many pink1 mutant phenotypes (Figures 9A and 9B, and [3], [11], [32], [33]). Since, to our knowledge, it had never been reported, we tested whether the rescue may be due to restoration of CI activity in pink1 mutants. Surprisingly, we found that parkin overexpression mildly improved ATP levels but did not restore CI function (Figures 9C and 9D). Hence, these genetic interactions further support independent and separable functions of PINK1 and Parkin.

Discussion
PINK1 and complex I function
PINK1 and Parkin have long been genetically linked in a common pathway that promotes mitochondrial homeostasis at least partly by directing the autophagic degradation of dysfunctional mitochondria as a mechanism of mitochondrial quality control. While this model potentially explains the occurrence of CI deficiency, oxidative stress, calcium dysregulation and elevated mtDNA mutations seen in patient tissues, and the age-related onset of PD [34], other models have been proposed to explain the pathological consequences of PINK1 and Parkin deficiency. Moreover, many mechanistic details by which the PINK1-Parkin pathway functions remain unexplained. To address these matters, we conducted an RNAi screen to identify genes whose loss-of-function either phenocopied or suppressed a pink1 RNAi phenotype. We have identified a number of genes that fulfill these criteria (discussed below) but focused our current investigation on ND42/NDUFA10 given the extensive literature implicating CI deficiency in PD pathogenesis and the fact that CI deficiency has previously been reported in PINK1 mutant models and patient samples.
Loss of ND42/NDUFA10 phenocopies the effect of pink1 loss on mitochondrial morphology in Drosophila cells, and ND42 overexpression rescues the pink1 mutant phenotypes. However, NDUFA10 knockdown has only modest effects on mitophagy, supporting a separate link between CI and PINK1 function. The simplest interpretation of these findings is that PINK1 normally regulates ND42/NDUFA10 abundance or activity through direct phosphorylation. Indeed, it was recently reported that NDUFA10 lacks phosphorylation at Ser-250 in Pink1-/- cells [25], although it remains to be determined whether PINK1 directly or indirectly regulates NDUFA10 phosphorylation. Moreover, it was reported that expression of a phospho-mimetic version of ND42/NDUFA10 specifically rescued phenotypes in multiple PINK1 deficient systems, while an S250A mutant version of ND42/NDUFA10 that is incapable of being phosphorylated was unable to confer rescue. Consistent with this we find that, from equivalent expression levels, the phospho-mimetic (SD) provides a slightly better phenotypic rescue than the other variants, and likewise promotes a higher CI activity. Nevertheless, our results also show that the non-phosphorylatable S250A version is still able to restore CI activity and significantly rescue the climbing deficit in pink1 mutant flies.
While further studies are needed to clarify the functional relationship between PINK1 and NDUFA10 in the regulation of CI, our findings provide further support mounting evidence that many manipulations that promote CI activity – overexpression of NDUFA10, sicily, heix, Ret, dNK, TRAP1 and NDI1, or treatment with vitamin K, deoxynucleosides or folic acid [25], [31], [35]–[38] – can rescue pink1 mutants, suggesting a more general defect underlies CI deficiency in loss of pink1. We hypothesize that the loss of CI activity in pink1 mutants may be due to a general de-stabilization of CI. Assembly is a particular challenge for such a large, multi-subunit complex and occurs in a stepwise process that is highly regulated by many factors [39]. Even its association with other ETC complexes in supercomplexes affects CI's stability [40]. There is evidence for reduced complex stability in pink1 mutants, though this may not be specific to CI [37], [41], [42]. One possibility is that PINK1 influences CI stability by directly promoting the assembly of CI, which may be regulated by NDUFA10.
The current findings also further support that the mechanism by which PINK1 influences CI activity appears to be separable from its well-characterized role in mitophagy, since, in agreement with some studies [24], [31] but in contrast to others [4], [8], [9], we do not find clear evidence of CI deficiency in parkin mutants flies. Moreover, it was unexpected to find that overexpression of parkin does not rescue the CI deficiency in pink1 mutants, because substantial previous work has shown that parkin overexpression rescues all of the other pink1 phenotypes, and because a prediction of the PINK1-parkin mitophagy pathway is that activation would trigger the selective removal of mitochondria deficient in CI activity. This finding suggests that CI deficiency alone cannot fully account for adult locomotor phenotypes seen in pink1 mutants. Further studies are needed to clarify full spectrum of cellular defects in pink1 and parkin mutants and their relative importance to the pathologic mechanism.
Consideration of other screen hits
The present screen analyzed the effect of ∼600 genes comprising mostly genes with homology to kinase or phosphatase domains. Other hits from this screen, identifying both phenocopiers and suppressors, could also be attractive candidates as potential new factors of pink1/Parkin function. Notably several hits play a role in lipid biology. This is particularly noteworthy in light of our recent report that another RNAi screen identified the master regulator of lipogenesis, SREBF1, to affect pink1/Parkin-mediated mitophagy [43].
The phenocopier Sphingosine kinase 1 and the suppressor easily shocked (encoding Drosophila Ethanolamine Kinase) are involved in phospholipid metabolic pathways. Sphingosine kinase catalyzes the production of sphingosine-1-phosphate (S1P), a key signaling molecule affecting cell growth and survival [44]. While S1P affects many cellular processes perhaps the most intriguing is its role in calcium mobilization from the endoplasmic reticulum (ER) [45] since Parkin was recently shown to promote ER-mitochondrial calcium transfer [23]. Interestingly, the breakdown of S1P generates phosphoethanolamine, the enzymatic product of ethanolamine kinase and a precursor metabolite of the key phospholipid phosphatidylethanolamine (PE). Loss of mitochondrial PE has been shown to affect mitochondrial morphology, oxidative phosphorylation and even the formation of complex I-containing supercomplexes [46]. The identification of four wheel drive (encoding Drosophila phosphatidylinositol 4-kinase beta), which catalyzes the formation of PI(4)P, is also intriguing since mutations in SYNJ1, which encodes PI(4,5)P2 phosphatase, were identified in families with early onset parkinsonism [47], [48].
Also related to lipid biology is Nocturnin although a direct link to mitochondria biology is less obvious. Nocturnin encodes a circadian deadenylase thought to be involved in the rhythmic regulation of gene expression by removal of polyA tails from mRNAs. Mice lacking Nocturnin are resistant to diet-induced obesity and hepatic steatosis [49], linking its function to lipid metabolism. Further studies will be needed to determine the extent to which lipids in general or specific lipids, and their regulated synthesis, impact on pink1/Parkin biology and regulation of mitochondrial dynamics and quality control. Nevertheless, this screen provides a resource for characterizing novel factors that regulate mitochondrial morphology.
Materials and Methods
Cell culture
Drosophila S2R+ cells were cultured in Schneider's medium (Gibco) containing 10% (vol/vol) heat-inactivated fetal bovine serum (Sigma), Penicillin 10 units/ml (Sigma) and Streptomycin 10 µg/ml (Sigma). Cells were maintained in a 25°C incubator. HeLa cells were cultured in DMEM GLUTAMAX media (Gibco) containing 10% (vol/vol) heat-inactivated fetal bovine serum (Sigma), Penicillin 10 units/ml (Sigma) and Streptomycin 10 µg/ml (Sigma). Cells were maintained in a 37°C incubator with 5% CO2. A stable transfected HeLa cell line expressing YFP-Parkin in pLVX-puro was cultured in DMEM GLUTAMAX media (Gibco) containing 10% (vol/vol) heat-inactivated fetal bovine serum (Sigma), Penicillin 10 units/ml (Sigma) and Streptomycin 10 µg/ml (Sigma). Cells were maintained in a 37°C incubator with 5% CO2.
RNAi screening and high-content microscopy
The kinome/phosphatome sub-library was generated based upon the second generation Drosophila dsRNA library (Heidelberg 2). Detailed information on amplicon targets is available online (http://rnai-screening-wiki.dkfz.de/signaling/wiki/display/rnaiwiki/Drosophila+RNAi+libraries). This sub-library was designed to contain all known and computed kinases and phosphatases, genes with some homology to these enzyme classes, but also some other genes of general interest. Library dsRNAs were plated at 250 ng in 5 µl of H2O into Perkin Elmer 384 well view plate (Product number: 6007470). Screen plates were arrayed with the inclusion of gaps to allow for the addition of user-specific controls. Here we added dsRNAs targeting Marf, Drp1, OPA1, Fis1, pink1 and parkin. These controls consistently all showed the expected results. For the pink1 RNAi background, 250 ng dsRNA against pink1 was added to each well prior to screening. A ‘double dose’ of dsRNA did not appear to affect mitochondrial morphology (see below). 15,000 Drosophila S2R+ cells were added to each well in 30 µl of Schneider's medium (Gibco) without FBS (Sigma). Plates were incubated at 25°C for 1 hour in which time dsRNAs are taken up by the cells. Following this incubation 30 µl of Schneider's medium (Gibco) containing 20% FBS (v/v) was added. The plates were then sealed and incubated at 25°C incubator for 4 days. Cells were stained with 100 nM Mitotracker Red (Invitrogen, M7512) and 20 µg/ml Hoechst 33342 (Invitrogen, H3570) for 15 minutes. Media was replaced and imaging was performed on live cells. Imaging was performed on an IN Cell Analyzer 1000 (GE Healthcare) automated microscope using a 40× air objective (Nikon, 0.60 NA).
Mitochondrial morphology analysis
Cells were prepared identically as for the high-throughput screening conditions, except for wild type where background 250 ng dsRNA targeting DsRed was added to each condition to mirror the ‘double dose’ of dsRNA in the pink1 background. Cells were imaged live under ambient conditions on a Deltavision RT deconvolution wide field microscope (Olympus, 100× objective, 1.4 NA) using 8 well µ-Slides (Ibidi), with 10 images taken per condition. Cells were scored for their gross mitochondrial morphology by eye with the scorer blinded to the experimental conditions. Where rotenone (20 µM) was used, cells were treated for 2 hours before mitochondrial morphology was analysed. A score would be assigned for a whole field of view and an average score would be calculated for the 10 images per condition. A score of 1 would be given to a field of view that had mainly fragmented mitochondria. A score of 2 would be given to a field of view that had a mainly wild type mitochondrial network with a mix of short-round and long-tubular mitochondria. A score of 3 would be given to a field of view that had mainly tubular mitochondria. A score of 4 would be given to a field of view that had mainly clumped mitochondria where the mitochondria had formed a single or few large peri-nuclear clusters.
RNAi reagents
Drosophila gene dsRNAs were generated using the MEGAscript T7 Kit (Ambion), using T7-flanked DNA amplicons from the library as template. Control dsRNA for Drosophila cell qRT-PCR analysis was a 782 bp sequence targeted against C. elegans gene R06F6.2 which has no ∼21mer homology within the Drosophila genome. siRNAs targeting human genes were obtained from the siGENOME SMARTpool collection (Dharmacon) as follows: control siRNA is Non-Targeting siRNA Pool #1 (product code; D-001206-13-20); PINK1 (product code; M-004030-02); NDUFA10 (product code; M-006752-00).
Quantitative real-time PCR
Total RNA from live cells was prepared from three replicates of each dsRNA treatment using RNeasy (Qiagen). RNA concentration was then determined spectrophotometrically. Once treated with DNase, total RNA was reverse-transcribed using RETROscript (Ambion) or Protoscript (NEB) according to the manufacturer's protocol. Quantitative PCR was performed using SYBR Green (Sigma) on a MyIQ real time PCR detection system (Bio-Rad). Each PCR included three technical replicates, which were repeated as three biological replicates. Levels for each transcript were normalized to a 18S rRNA (Drosophila: 18SrRNA; Human: RNA18S5) control by the 2-ΔΔCT method.
For Drosophila genes, primers used were:
18S - Forward: TCTAGCAATATGAGATTGAGCAATAAG
18S - Reverse: AATACACGTTGATACTTTCATTGTAGC
pink1 - Forward: GACGACCCTCGCACATAA
pink1 - Reverse: AACAGTCCGGAGATCCTACAG
ND42 - Forward: CGTTTCGATGTCCCGGAGCT
ND42 - Reverse: GTCTGCATTGTAGCCAGGAC
CG7712 - Forward: CGCAATGTGACCGACATCCG
CG7712 - Reverse: CGCATGATATGGCCTTCTG
For human genes, primers used were:
18S - Forward: CAGCCACCCGAGATTGAGCA
18S - Reverse: TAGTAGCGACGGGCGGTGTG
PINK1 - Forward: GCCGGACGCTGTTCCTCGTT
PINK1 - Reverse: TGGACACCTCTGGGGCCATC
NDUFA10 - Forward: GATCCGAGAAGCAATGATG
NDUFA10 - Reverse: TGGAGCGCTCCAACACAACA
Antibodies
The following primary antibodies were used, mouse anti-ATP5A (MS507, MitoSciences; 1∶2000), rabbit anti-GFP (ab6556, Abcam; 1∶5000). Secondary antibodies used were rabbit polyclonal anti-mouse IgG-H&L (DyLight 594, Invitrogen; 1∶5000) and goat anti-rabbit IgG (Alexa Fluor 488, Invitrogen; 1∶5000).
Parkin translocation and mitophagy assays
YFP-Parkin HeLa cells were reverse-transfected with siRNAs using DharmaFECT 1 (Dharmacon). For Parkin translocation, cells were incubated for 4 days then treated with 10 µM CCCP or equivalent volume of the solvent (EtOH) for 4 hours. For mitophagy, cells were incubated for 3 days then treated with 10 µM CCCP or equivalent volume of the solvent (EtOH) for 24 hours. Cells were fixed in ice-cold methanol for 10 minutes and washed in PBS. Mitochondrial staining was achieved by using anti-ATP5A antibody. Imaging was performed on an Olympus FV1000 confocal microscope (Olympus, 60× oil objective, 1.25 NA). For Parkin translocation, cells were scored for the accumulation of YFP-Parkin on mitochondria. For mitophagy, cells were scored for mitochondrial load based on having a normal load of mitochondria, few mitochondria or no mitochondria. At least 20 cells were scored per treatment and 3 biological replicates were performed.
PINK1 accumulation assay
HeLa cells were reverse-transfected with siRNAs using DharmaFECT 1. After 3 days cells were transfected with PINK1-EGFP using Effectene (Qiagen). After a further 1 day cells were treated with 10 µM CCCP or equivalent volume of the solvent (EtOH) for 1 hour before fixation with ice-cold methanol for 10 minutes and washed in PBS. Immunofluorescence was performed using anti-ATP5A and anti-GFP antibodies, and appropriate fluorescent secondary antibodies. Imaging was performed on an Olympus FV1000 confocal microscope. Cells were scored for the accumulation of PINK1-EGFP on mitochondria. At least 20 cells were scored per treatment and 3 biological replicates were performed.
TMRM assay
TMRM assay to measure mitochondrial membrane polarity was done as previously described [8]. Briefly, HeLa cells were reverse-transfected with siRNAs using DharmaFECT for 4 days. Cells were then treated with 10 µM CCCP or equivalent volume of the solvent (EtOH) for 1 hour. Cells were incubated with 50 nM TMRM (VTX668, Fisher) in PBS with 10 µM CCCP or equivalent volume of solvent for 30 minutes, then washed in PBS with 10 µM CCCP or equivalent volume of solvent 5 times. TMRM fluorescence was read on a spectrophotometer at 550 nm (Berthold technologies Mithras LB940). Triplicate readings were taken from 3 biological replicates.
Drosophila genetics
Drosophila were raised under standard conditions at 25°C on food consisting of agar, cornmeal and yeast. pink1B9 mutants [11] were provided by J. Chung (KAIST). park25 mutants, fertility tests, flight and climbing assays were performed as previously described [6], [50]. w1118 and da-GAL4 strains were obtained from the Bloomington Drosophila Stock Center (Bloomington, IN). UAS-ND42-RNAi lines (GD: 14444; KK: 101787) were obtained form the Vienna Drosophila Resource Centre [51]. UAS-ND42, UAS-ND42-HA and UAS-sicily have been described previously [29] and were a gift from H. Bellen (Baylor College of Medicine). The additional UAS-ND42 lines (WT, SA and SD) from Morais et al. [25] were provided by Patrik Verstreken.
Measurement of complex I activity
Mitochondria-enriched fractions were prepared from whole adult male flies (∼3 days old) with the indicated genotype (10 flies were used for each sample). Flies were gently crushed in chilled isolation buffer (250 mM sucrose, 10 mM Tris-HCl, pH 7.4, 0.15 mM MgCl2) using a plastic pestle homogenizer, then centrifuged twice at 500×g for 5 minutes at 4°C to remove debris. The supernatant was centrifuged 5000×g for 5 minutes at 4°C. The resulting pellet containing mitochondria was re-suspended in the isolation buffer or assay buffers. Complex I activity was measured using a modified method from Birch-Machin et al [52]. Briefly, samples were subjected to 3 cycles of freeze-thaw in liquid nitrogen. Complex I activity was determined by following the oxidation of NADH at 340 nm with a reference wavelength of 425 nm (ε = 6.22 mM−1 cm−1) at 30°C using a BMG Labtech FLUOStar plate reader. The assay buffer contained 25 mM KH2PO4, 5 mM MgCl2, (pH 7.2), 3 mM KCN, 2.5 mg per ml BSA, 50 µM ubiquinone, 2 µg/ml antimycin A and mitochondrial extract. The baseline was recorded for 5 minutes and the reaction was started with 125 µM NADH measured for 30 minutes, 15 µg/ml rotenone was added to inhibit the reaction and measured for 15 minutes. The results are expressed as µmol NADH oxidised/min/citrate synthase activity. Citrate synthase was measured by following the production of 5-thio-2-nitrobenzoate at 30°C using a BMG Labtech FLUOStar plate reader after samples had undergone 3 cycles of freeze-thaw in liquid nitrogen. The assay buffer was 100 mM Tris HCl (pH 8.0), 0.1 mM DTNB, 50 µM acetyl Coenzyme A, 0.1% Triton X-100 and mitochondrial extract per well. The baseline was recorded for 5 minutes at 412 nm, then the reaction was started by the addition of 0.5 mM oxaloacetate acid and the rate was recorded for 15 minutes.
ATP assays
Five male flies (3 days old) were homogenized in 100 µl of 6 M guanidine-HCl in extraction buffer (100 mM Tris, 4 mM EDTA, pH 7.8) to inhibit ATPases. Homogenized samples were subjected to rapid freezing in liquid nitrogen, followed by boiling for 3 min. Samples were cleared by centrifugation, and supernatant was diluted (1/100) with extraction buffer and mixed with a luminescent solution (CellTiter-Glo Luminescent Cell Viability Assay, Promega, Fitchburg, WI, USA). Luminescence was measured on a Varioskan™ Flash Multimode Reader (Thermo Scientific, Waltham, MA, USA). The relative ATP levels were calculated by dividing the luminescence by the total protein concentration, which was determined by the Bradford method.
Statistical analyses
For Parkin translocation, mitophagy and PINK1 stabilization, statistical significance was calculated by using Student's t-test on triplicate experiments comparing against control siRNA/dsRNAs of the equivalent experimental condition. Biochemical and behavioral assays in Drosophila were analyzed by one-way ANOVA with Bonferroni correction. Male fertility was analyzed by Chi-square test.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KitadaT, AsakawaS, HattoriN, MatsumineH, YamamuraY, et al. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392 : 605–608.
2. ValenteEM, Abou-SleimanPM, CaputoV, MuqitMM, HarveyK, et al. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304 : 1158–1160.
3. ClarkIE, DodsonMW, JiangC, CaoJH, HuhJR, et al. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441 : 1162–1166.
4. FlinnL, MortiboysH, VolkmannK, KosterRW, InghamPW, et al. (2009) Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 132 : 1613–1623.
5. GautierCA, KitadaT, ShenJ (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105 : 11364–11369.
6. GreeneJC, WhitworthAJ, KuoI, AndrewsLA, FeanyMB, et al. (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 100 : 4078–4083.
7. GrunewaldA, GeggME, TaanmanJW, KingRH, KockN, et al. (2009) Differential effects of PINK1 nonsense and missense mutations on mitochondrial function and morphology. Exp Neurol 219 : 266–273.
8. MortiboysH, ThomasKJ, KoopmanWJ, KlaffkeS, Abou-SleimanP, et al. (2008) Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol 64 : 555–565.
9. MuftuogluM, ElibolB, DalmizrakO, ErcanA, KulaksizG, et al. (2004) Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord 19 : 544–548.
10. PalacinoJJ, SagiD, GoldbergMS, KraussS, MotzC, et al. (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279 : 18614–18622.
11. ParkJ, LeeSB, LeeS, KimY, SongS, et al. (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441 : 1157–1161.
12. VedR, SahaS, WestlundB, PerierC, BurnamL, et al. (2005) Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem 280 : 42655–42668.
13. Wood-KaczmarA, GandhiS, YaoZ, AbramovAY, MiljanEA, et al. (2008) PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS One 3: e2455.
14. PooleAC, ThomasRE, AndrewsLA, McBrideHM, WhitworthAJ, et al. (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A 105 : 1638–1643.
15. WangX, WinterD, AshrafiG, SchleheJ, WongYL, et al. (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147 : 893–906.
16. YangY, OuyangY, YangL, BealMF, McQuibbanA, et al. (2008) Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA 105 : 7070–7075.
17. ZivianiE, TaoRN, WhitworthAJ (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A 107 : 5018–5023.
18. MatsudaN, SatoS, ShibaK, OkatsuK, SaishoK, et al. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189 : 211–221.
19. NarendraDP, JinSM, TanakaA, SuenDF, GautierCA, et al. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298.
20. GeislerS, HolmstromKM, SkujatD, FieselFC, RothfussOC, et al. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12 : 119–131.
21. NarendraD, TanakaA, SuenDF, YouleRJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183 : 795–803.
22. VincowES, MerrihewG, ThomasRE, ShulmanNJ, BeyerRP, et al. (2013) The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A 110 : 6400–6405.
23. CaliT, OttoliniD, NegroA, BriniM (2013) Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim Biophys Acta 1832 : 495–508.
24. MoraisV, VerstrekenP, RoethigA, SmetJ, SnellinxA, et al. (2009) Parkinson's disease mutations in PINK1 results in decreased Complex I activity and deficient synaptic function. EMBO Molecular Medicine 1 : 99–111.
25. MoraisVA, HaddadD, CraessaertsK, De BockPJ, SwertsJ, et al. (2014) PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344 : 203–207.
26. PooleAC, ThomasRE, YuS, VincowES, PallanckL (2010) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5: e10054.
27. JanssenRJ, NijtmansLG, van den HeuvelLP, SmeitinkJA (2006) Mitochondrial complex I: structure, function and pathology. J Inherit Metab Dis 29 : 499–515.
28. VinothkumarKR, ZhuJ, HirstJ (2014) Architecture of mammalian respiratory complex I. Nature. E-pub ahead of print doi:10.1038/nature13686
29. ZhangK, LiZ, JaiswalM, BayatV, XiongB, et al. (2013) The C8ORF38 homologue Sicily is a cytosolic chaperone for a mitochondrial complex I subunit. J Cell Biol 200 : 807–820.
30. YouleRJ, NarendraDP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12 : 9–14.
31. VilainS, EspositoG, HaddadD, SchaapO, DobrevaMP, et al. (2012) The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet 8: e1002456.
32. ExnerN, TreskeB, PaquetD, HolmstromK, SchieslingC, et al. (2007) Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 27 : 12413–12418.
33. YangY, GehrkeS, ImaiY, HuangZ, OuyangY, et al. (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA 103 : 10793–10798.
34. ExnerN, LutzAK, HaassC, WinklhoferKF (2012) Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J 31 : 3038–3062.
35. KleinP, Muller-RischartAK, MotoriE, SchonbauerC, SchnorrerF, et al. (2014) Ret rescues mitochondrial morphology and muscle degeneration of Drosophila Pink1 mutants. EMBO J 33 : 341–355.
36. VosM, EspositoG, EdirisingheJN, VilainS, HaddadDM, et al. (2012) Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 336 : 1306–1310.
37. TufiR, GandhiS, de CastroIP, LehmannS, AngelovaPR, et al. (2014) Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson's disease. Nat Cell Biol 16 : 157–166.
38. ZhangL, KarstenP, HammS, PogsonJH, Muller-RischartAK, et al. (2013) TRAP1 rescues PINK1 loss-of-function phenotypes. Hum Mol Genet 22 : 2829–2841.
39. LazarouM, ThorburnDR, RyanMT, McKenzieM (2009) Assembly of mitochondrial complex I and defects in disease. Biochim Biophys Acta 1793 : 78–88.
40. Acin-PerezR, Bayona-BafaluyMP, Fernandez-SilvaP, Moreno-LoshuertosR, Perez-MartosA, et al. (2004) Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell 13 : 805–815.
41. LiuW, Acin-PerezR, GeghmanKD, ManfrediG, LuB, et al. (2011) Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci U S A 108 : 12920–12924.
42. AmoT, SaikiS, SawayamaT, SatoS, HattoriN (2014) Detailed analysis of mitochondrial respiratory chain defects caused by loss of PINK1. Neurosci Lett 580 : 37–40.
43. IvattRM, Sanchez-MartinezA, GodenaVK, BrownS, ZivianiE, et al. (2014) Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc Natl Acad Sci U S A 111 : 8494–8499.
44. OliveraA, SpiegelS (2001) Sphingosine kinase: a mediator of vital cellular functions. Prostaglandins Other Lipid Mediat 64 : 123–134.
45. ChoiOH, KimJH, KinetJP (1996) Calcium mobilization via sphingosine kinase in signalling by the Fc epsilon RI antigen receptor. Nature 380 : 634–636.
46. TassevaG, BaiHD, DavidescuM, HaromyA, MichelakisE, et al. (2013) Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem 288 : 4158–4173.
47. KrebsCE, KarkheiranS, PowellJC, CaoM, MakarovV, et al. (2013) The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat 34 : 1200–1207.
48. QuadriM, FangM, PicilloM, OlgiatiS, BreedveldGJ, et al. (2013) Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat 34 : 1208–1215.
49. StubblefieldJJ, TerrienJ, GreenCB (2012) Nocturnin: at the crossroads of clocks and metabolism. Trends Endocrinol Metab 23 : 326–333.
50. TainLS, ChowdhuryRB, TaoRN, Plun-FavreauH, MoisoiN, et al. (2009) Drosophila HtrA2 is dispensable for apoptosis but acts downstream of PINK1 independently from Parkin. Cell Death Differ 16 : 1118–1125.
51. DietzlG, ChenD, SchnorrerF, SuKC, BarinovaY, et al. (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448 : 151–156.
52. Birch-MachinMA, BriggsHL, SaboridoAA, BindoffLA, TurnbullDM (1994) An evaluation of the measurement of the activities of complexes I-IV in the respiratory chain of human skeletal muscle mitochondria. Biochem Med Metab Biol 51 : 35–42.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- An RNA-Seq Screen of the Antenna Identifies a Transporter Necessary for Ammonia Detection
- Systematic Comparison of the Effects of Alpha-synuclein Mutations on Its Oligomerization and Aggregation
- Functional Diversity of Carbohydrate-Active Enzymes Enabling a Bacterium to Ferment Plant Biomass
- Regularized Machine Learning in the Genetic Prediction of Complex Traits
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy