
p53- and ERK7-Dependent Ribosome Surveillance Response Regulates Insulin-Like Peptide Secretion
Ribosome biogenesis is a major consumer of cellular energy and a rate-limiting process during cell growth. The ribosome biogenesis pathway is tightly connected with signaling pathways that regulate tissue growth. For example, nutrient-regulated signaling cues adjust the rate of ribosome biogenesis. On the other hand, the process of ribosome biogenesis is closely monitored by so-called surveillance mechanisms. The best-known ribosome surveillance factor is the transcription factor and tumor suppressor p53. In proliferating cells, activation of p53 upon disturbed ribosome biogenesis leads to cell cycle arrest and inhibition of proliferation. Here we show that ribosome surveillance not only regulates growth locally in proliferating cells, but is also coupled to hormonal growth control through regulation of insulin like peptide (dILPs) secretion. We observed that inhibition of ribosome biogenesis in the Drosophila insulin-producing cells generates a strong cell autonomous signal to inhibit dILP secretion. We identify two downstream effectors of this ribosome surveillance response by showing that p53 as well as an atypical MAP kinase ERK7 are mediators of the inhibition of dILP secretion. We also provide evidence that this ribosome surveillance mechanism contributes to nutrient-dependent regulation of dILP secretion.
Published in the journal:
. PLoS Genet 10(11): e32767. doi:10.1371/journal.pgen.1004764
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004764
Summary
Ribosome biogenesis is a major consumer of cellular energy and a rate-limiting process during cell growth. The ribosome biogenesis pathway is tightly connected with signaling pathways that regulate tissue growth. For example, nutrient-regulated signaling cues adjust the rate of ribosome biogenesis. On the other hand, the process of ribosome biogenesis is closely monitored by so-called surveillance mechanisms. The best-known ribosome surveillance factor is the transcription factor and tumor suppressor p53. In proliferating cells, activation of p53 upon disturbed ribosome biogenesis leads to cell cycle arrest and inhibition of proliferation. Here we show that ribosome surveillance not only regulates growth locally in proliferating cells, but is also coupled to hormonal growth control through regulation of insulin like peptide (dILPs) secretion. We observed that inhibition of ribosome biogenesis in the Drosophila insulin-producing cells generates a strong cell autonomous signal to inhibit dILP secretion. We identify two downstream effectors of this ribosome surveillance response by showing that p53 as well as an atypical MAP kinase ERK7 are mediators of the inhibition of dILP secretion. We also provide evidence that this ribosome surveillance mechanism contributes to nutrient-dependent regulation of dILP secretion.
Introduction
Animals coordinate tissue growth and body size with changing nutrient conditions and it is well established that insulin-like signalling has a key role in this process [1], [2]. In mammals, insulin-like signalling is mediated by insulin and insulin-like growth factors through their respective receptors. Drosophila possesses a single insulin-like receptor, which is activated by insulin-like peptides (dILPs). There are currently eight dILPs identified in Drosophila and several of them (e.g. dILP2, -3 and -5) are mainly expressed and secreted by a group of 14 median neurosecretory cells, also known as insulin-producing cells (IPCs) [3], [4]. These cells are critical in controlling organismal growth, as disturbance of IPC function leads to strongly reduced body size [4]. IPC function is coupled to nutrient status as starvation of Drosophila larvae leads to inhibition of dILP secretion [5]. Recent studies have demonstrated that fat body, the insect counterpart of adipose tissue and liver, senses nutrient status and regulates dILP secretion by hormonal mechanisms [5], [6]. Secreted fat body cytokine Unpaired 2 is regulated by dietary sugars and lipids and it controls a population of GABAergic neurons in the brain that project onto IPCs. Upd2 activates the JAK-STAT pathway in these neurons, which relieves the inhibitory effect of GABAergic neurons on IPCs, resulting in dILP secretion [6]. Moreover, the fat body senses amino acid levels and in turn secretes an unknown humoral signal, which facilitates dILP secretion from the IPCs [5]. Recently, it was shown that adiponectin receptor also regulates dILP secretion from the IPCs, but the corresponding ligand is not yet identified [7]. Beyond these examples, the regulation of IPC function in response to changing nutrient status is poorly understood. In particular, very little is known of how cell intrinsic pathways within the IPCs affect dILP secretion, and consequently organismal growth.
Ribosome biogenesis is a major consumer of cellular energy and a key determinant of cell growth capacity [8]. Ribosome biogenesis is tightly regulated by nutrient, growth factor and stress responsive signalling networks [1], [8]–[13]. As the ribosome biogenesis pathway is a major integrator of growth regulatory signals, its activity needs to be closely monitored. p53 serves as one of the ribosome biogenesis surveillance factors [14]. Impaired ribosome biogenesis activates p53, likely through several parallel mechanisms. For example, imbalance of ribosomal components allows ribosomal proteins RPL5 and RPL11 in complex with 5S rRNA to inhibit HDM2/MDM2-dependent degradation of p53 [15]–[17]. On the other hand, nucleolar Myb binding protein 1A (MYBBP1A) translocates to the nucleoplasm if rRNA production is inhibited [18]. Nucleoplasmic MYBBP1A increases p53 activity by promoting its tetramerization and acetylation [19]. In proliferating cells, activation of p53 through these mechanisms leads to cell cycle arrest [16]. However, the role of ribosome biogenesis surveillance in other physiological settings is poorly understood. This is a relevant question, as disturbed ribosome biogenesis is known to have highly tissue-specific consequences in vivo. In humans, mutations in ribosomal components or other ribosome biogenesis genes lead to genetic disorders called ribosomopathies [20]. They manifest, for example, as bone marrow failure, anemia, skin abnormalities, and pancreatic insufficiency [20]. In Drosophila, mutations that disturb ribosome biogenesis lead to so-called Minute phenotype, displaying impaired growth, poor fertility and short and thin bristles [21]. Inhibition of ribosome biogenesis specifically in the Drosophila fat body or prothoracic gland influences systemic growth by affecting hormonal regulation through insulin and ecdysone signalling axes, respectively [22]–[24]. Which signalling mechanisms mediate the distinct cell type-specific phenotypes of ribosome surveillance in vivo is, however, poorly understood.
Here we report that dILP secretion is tightly coupled to the activity of the ribosome biogenesis pathway within the IPCs. We show that the p53 dependent ribosomal surveillance response inhibits dILP secretion and identify atypical MAP kinase ERK7 as a downstream effector of the ribosome surveillance response in this setting. Furthermore, we provide evidence that both p53 and ERK7 activities within the IPCs contribute to regulation of dILP secretion in response to nutrient status. Thus, we propose that the p53 - and ERK7-dependent ribosome surveillance pathway serves as a local branch of the IPC-regulating nutrient sensing network, parallel to the humoral signals derived from the fat body.
Results
Kinome-wide screen identifies novel regulators of IPC function
While the role of Drosophila insulin-like peptides (dILPs) in systemic growth control is well established, it remains poorly understood how the secretion of dILPs is regulated in response to changing dietary conditions. To get a better insight into this question, we explored the cell autonomous signalling mechanisms that regulate dILP secretion, by performing a kinome-wide screen with RNAi expressed specifically in the IPCs. Impaired IPC function compromises tissue growth [4] and therefore we used body weight of emerging adults as a screening readout (Figure 1A, Table S1). To normalize variation between vials due to growth conditions, we determined the relative weight wherein the mean weight of the flies expressing both dILP2-Gal4 and RNAi was divided by the mean weight of control flies (not expressing the dILP2-Gal4) from the same vial. In total, 231 kinases were screened. All primary hits displaying >10% difference to the median relative weight were further analyzed by an independent RNAi line. Altogether 12 protein kinases showed significant body weight reduction in two independent RNAi lines (Figure 1B). All identified kinases have mammalian orthologs (Table 1). In addition to these 12 kinases identified here, we have earlier shown that atypical PKC is essential for IPC function [25]. One of the kinase hits was TOR (target of rapamycin). TOR kinase is present in two functionally distinct complexes, TOR complex 1 and 2 (TORC1 and TORC2). To explore which of the TOR complexes have a regulatory role in the IPCs, we depleted Raptor and Rictor, essential components of TORC1 and TORC2, respectively. Knockdown of Raptor in the IPCs significantly reduced total body weight, while depletion of Rictor had no significant impact (Figure 1C), showing that TORC1 regulates growth through the IPCs. However, as RNAi does not completely silence gene expression, we cannot rule out the involvement of TORC2.


To explore gene expression of three IPC-expressed dilp genes, namely dilp2, dilp3 and dilp5, we analyzed RNA from larval brains by quantitative RT-PCR (Figure 1D). In the case of most hits, expression of dilp2 and dilp5 remained nearly unchanged. In contrast, dilp3 expression was elevated in several samples. This is likely due to a feedback mechanism activated by reduced autocrine insulin signalling in the IPCs [26]. Only depletion of Tousled-like kinase (Tlk) significantly reduced dilp3 expression (Figure 1D).
Disturbed ribosome biogenesis in the IPCs inhibits dILP secretion
Secretion of dILPs is a key regulatory level in determining the activity of systemic insulin signalling [5]. dILP2 contributes to the total body weight [26] and its secretion can be assessed by monitoring its accumulation into the cell bodies of IPCs. Accumulation is observed when dILP2 secretion is inhibited upon starvation [5]; (Figure 2A, B). Of the hits, 5 kinases caused significant dILP2 accumulation upon knockdown. These include Cdk12, Adck (CG3608), Pkc98E (Figure S1), as well as two Rio kinases, Rio1 and Rio2 (Figure 2A, B). Rio1 and Rio2 belong to a group of atypical kinases and they have a conserved role in ribosome maturation [27]–[30]. As ribosome biogenesis is a process tightly coupled to nutrient sensing [1], [12], we chose to further explore the role of Rio kinases in dILP secretion. We confirmed that, similarly to starvation, IPC-specific depletion of Rio kinases led to elevated insulin-like receptor (inr) gene expression in the larva (Figure 2C), which is an established readout for reduced peripheral insulin signalling [31]. If the inhibition of dILP2 secretion was due to impaired ribosome biogenesis, we predicted that depletion of other ribosomal genes would phenocopy this effect. Indeed, IPC-specific knockdown of a number of other ribosomal components or genes involved in various steps of ribosome biogenesis led to significantly reduced body size (Figure 2D). To further confirm that the dILP2 secretion phenotypes of Rio kinases were due to impaired ribosome biogenesis, we depleted ribosomal protein Rpl35A and ribosome assembly factor Tsr1 (CG7338) in the IPCs and observed a similar inhibition of dILP2 secretion in both cases (Figure 2E, F). The dILP2-Gal4 driver we used [4] displays activity in the salivary glands, in addition to IPCs. Depletion of Rio2 by a salivary gland specific Sgs3-Gal4 driver [32] caused no growth reduction, ruling out the possibility of nonspecific effects through the salivary glands (Figure S2).

TORC1 signalling regulates ribosome biogenesis gene expression in Drosophila in a context-dependent manner. In S2 cells, expression of ribosome biogenesis genes is TORC1-dependent [33], [34], while in the context of whole larvae, TOR mutants do not display significantly reduced expression of ribosome biogenesis genes [35]. In accordance with the earlier observations in whole larvae, inhibition of Raptor in the IPCs did not lead to inhibited dILP2 secretion (Figure S3). Transcription factor dMyc is a conserved master regulator of ribosome biogenesis [13], [36]. It transcriptionally regulates the expression of rRNAs as well as ribosomal proteins [36]. In Drosophila larvae ribosome biogenesis genes that are inhibited upon starvation are targets of dMyc [35]. Therefore we tested whether dMyc activity in the IPCs has an impact on dILP2 secretion. Indeed, RNAi-mediated depletion of dMyc prominently inhibited dILP2 secretion (Figure 2G, H) and led to reduced body weight (Figure 2I).
We wanted to further explore whether inhibition of ribosome biogenesis affects the secretion of other dILPs in the IPCs. Similarly to dILP2, secretion of dILP5 was inhibited upon Rio2 knockdown (Figure 2J, K). Inhibition of systemic growth often manifests as prolonged larval development, in addition to reduced body size. Indeed, depletion of Rio2 in the IPCs led to significant delay in pupation kinetics (Figure 2L). In conclusion, inhibition of ribosome biogenesis in the IPCs triggers a response that blocks dILP secretion and consequently leads to slower larval development and reduced body size.
p53 mediates dILP2 accumulation upon inhibited ribosome biogenesis
How is the ribosome biogenesis pathway coupled to dILP secretion? We hypothesised that one of the ribosome biogenesis surveillance pathways might link secretion to the status of ribosome biogenesis in specialised secretory cells like the IPCs. As p53 is the best-established surveillance factor for the ribosome biogenesis pathway [14], [15], [37], we wanted to explore, whether it is involved in the regulation of dILP secretion. Indeed, overexpression of p53 in the IPCs revealed that p53 is sufficient to cause dILP2 accumulation (Figure 3A, B). To analyse the possible impact of transcriptional regulation, we measured dilp2 mRNA levels by quantitative RT-PCR. p53 overexpression caused modest downregulation of dilp2 mRNA levels (Figure S4), ruling out the possibility of transcriptional activation as a cause for p53-dependent dILP2 accumulation. To assess the contribution of p53-mediated dILP regulation on growth, we analyzed pupal volume, which is a sensitive means to measure changes in total body size [22]. In our hands, pupal volume data is consistent with the adult weight, but displays less random variation. In accordance with the immunofluorescence data suggesting inhibited dILP2 secretion, p53 overexpression in the IPCs strongly reduced the pupal volume (Figure 3C). In contrast, overexpression of p53 in the salivary glands caused no growth impairment (Figure S2). In sum, our data is consistent with the idea that p53 expression in the IPCs is sufficient to prevent dILP secretion and consequently inhibit tissue growth.
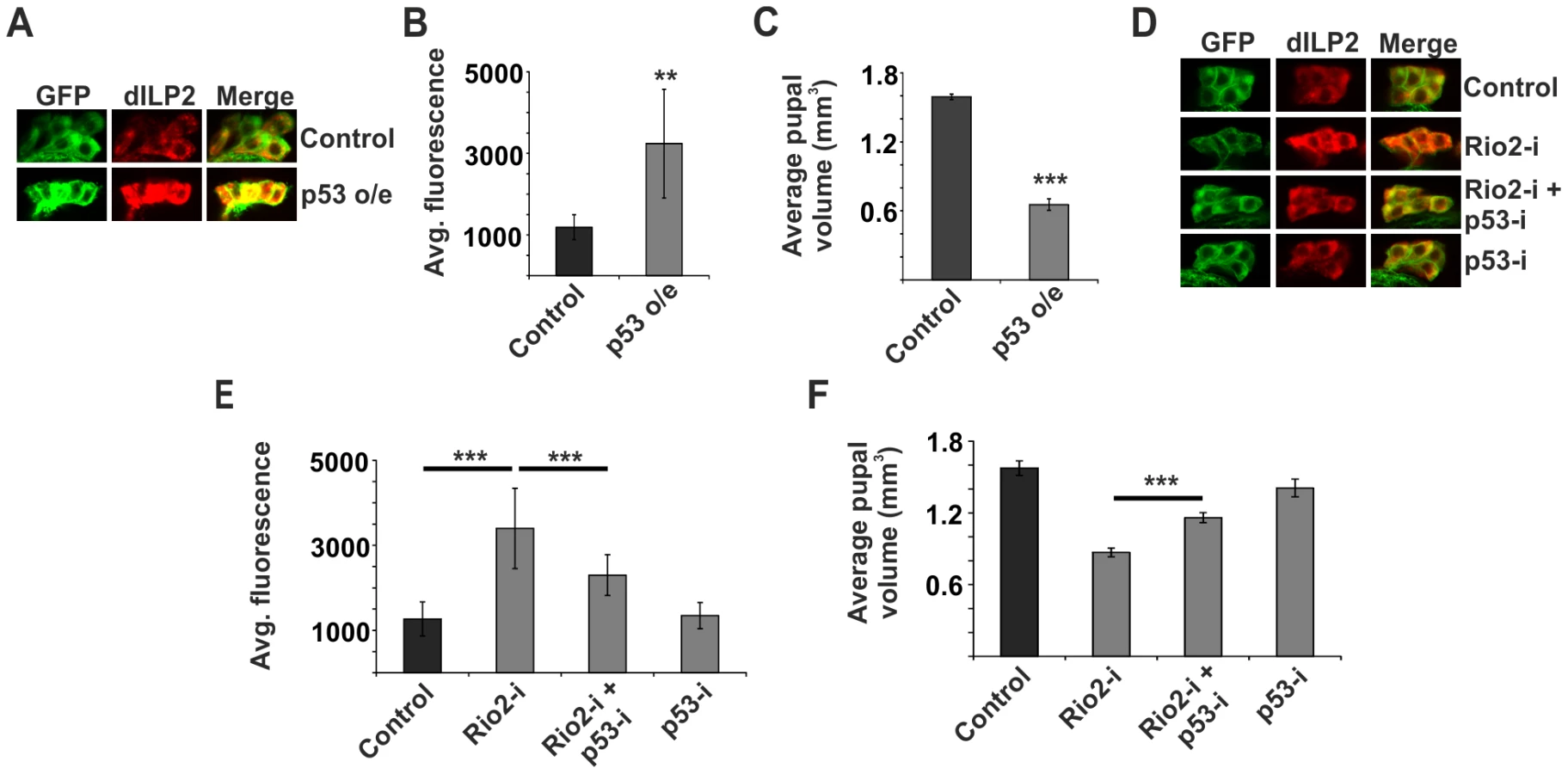
Next, we wanted to explore, whether the p53 would mediate the observed inhibition of dILP2 secretion upon disturbed ribosome biogenesis. This was the case, as depletion of p53 rescued the dILP2 accumulation upon Rio2 knockdown (Figure 3D, E) along with partial rescue of the growth impairment (Figure 3F). A similar rescue of growth was observed when p53 was depleted in combination with Rio1 (Figure S5). Moreover, a control RNAi targeting firefly Luciferase, but no endogenous genes, had no influence on the phenotypes of Rio2 RNAi in the IPCs (Figure S6), demonstrating that expression of an additional RNAi does not interfere with Rio2 RNAi activity.
ERK7 mediates the inhibition of dILP secretion upon impaired ribosome biogenesis
Identification of p53 as an essential inhibitor of dILP2 secretion implied that ribosome biogenesis regulates dILP secretion through an active surveillance mechanism, rather than through an indirect mechanism following reduced translation. To directly couple ribosome surveillance to secretion we turned our attention to known regulators of the secretory pathway. It was recently shown that an atypical MAP kinase called ERK7 (also known as ERK8 or MAPK15) inhibits secretion upon serum and amino acid starvation in Drosophila S2 cells [38]. ERK7 was shown to phosphorylate Sec16 and thus prevent its recruitment to ER exit sites, consequently preventing the export of the secretory cargo. Therefore, we wanted to explore, whether ERK7 is regulated in response to inhibited ribosome biogenesis. In order to do so, we used in situ hybridization of larval brain. erk7 expression was undetectable in fed control larvae, whereas overexpression of transgenic ERK7 by dILP2-Gal4 led to a strong IPC-specific signal (Figure S7). As a negative control we used an erk7 sense probe for the ERK7 overexpressing samples (Figure S7). After confirming the specificity of the in situ hybridization, we analysed erk7 expression upon disturbed ribosome biogenesis. Intriguingly, we observed that erk7 mRNA levels were clearly elevated in the IPCs following Rio2 depletion using dILP2-Gal4 (Figure 4A), implying that ERK7 is upregulated upon ribosome surveillance in the IPCs. As ribosome biogenesis is inhibited upon starvation [35], we hypothesized that starvation might also cause elevated expression of erk7, which indeed was the case (Figure 4A). Notably, in the context of brain tissue, the starvation-induced upregulation appeared specific to the IPCs, since erk7 mRNA remained undetectable in other brain areas. To confirm the regulation of erk7 mRNA by an independent method, we used quantitative RT-PCR on mRNA samples isolated from whole larvae. Disturbance of ribosome biogenesis by ubiquitous depletion of Rio1, Rio2 and Myc led to elevated erk7 expression (Figure 4B, C). Similarly, starvation caused significant upregulation of erk7 mRNA levels in the whole larval samples (Figure 4D). In sum, our data shows that erk7 gene expression is elevated upon impaired ribosome biogenesis and starvation, suggesting a possible role for ERK7 in the regulation of dILP secretion during these conditions.

To this end, we decided to explore the functional relationship between ribosome surveillance, ERK7 and dILP secretion in the IPCs. We analyzed transgenic flies that specifically overexpress ERK7 in the IPCs (Figure S7). Indeed, ERK7 overexpression led to prominent accumulation of dILP2 and dILP5 in the IPCs (Figure 5A–5D). Consistently, ERK7 overexpression in the IPCs, but not in salivary glands, led to reduced pupal volume (Figure 5E and Figure S2) as well as delayed pupation (Figure 5F). To rule out the possibility that the ERK7-dependent dILP accumulation was due to elevated transcription, we used quantitative RT-PCR to measure dilp2 and dilp5 mRNA levels, which remained unchanged upon ERK7 overexpression (Figure S8). Thus, we conclude that elevated ERK7 expression is sufficient to inhibit dILP secretion cell autonomously in the IPCs. We also performed genetic epistasis experiments to test if ERK7 is essential to inhibit dILP2 secretion upon disturbed ribosome biogenesis. Indeed, ERK7 RNAi efficiently suppressed the dILP2 accumulation caused by Rio2 knockdown (Figure 5G, H). Similar suppression was observed in the case of dILP2 accumulation following dMyc depletion (Figure S9). Consistent with the effects on dILP2 secretion, IPC-specific depletion of ERK7 partially suppressed the impaired growth upon knockdown of Rio2, while having no effect on body size in the wild type background (Figure 5I).

ERK7 acts downstream of p53 in nutrient responsive regulation of dILP2 secretion
As we had observed that both p53 and ERK7 were essential for the inhibition of dILP2 secretion upon impaired ribosome biogenesis, we wanted to explore whether they belong to the same pathway. We hypothesized that ERK7 might be a downstream effector of p53. Supporting this idea, we observed that overexpression of wild type p53 in the IPCs led to elevated expression of erk7 mRNA as detected by in situ hybridization (Figure 6A). To further test the possible relevance of the p53-mediated regulation of erk7 in the IPCs, we performed genetic epistasis experiments. Indeed, depletion of ERK7 efficiently suppressed the dILP2 accumulation upon p53 overexpression (Figure 6B, C). Consistently, ERK7 RNAi also partially suppressed the reduction of pupal volume caused by p53 overexpression (Figure 6D) along with the delay in pupation (Figure 6E). Notably, the suppression by ERK7 RNAi was only partial, leaving open the possibility for a parallel, ERK7-independent, mechanism. In sum, our data implies that p53 and ERK7 belong to the same pathway and ERK7 acts as a downstream effector of p53 in inhibiting dILP2 secretion.
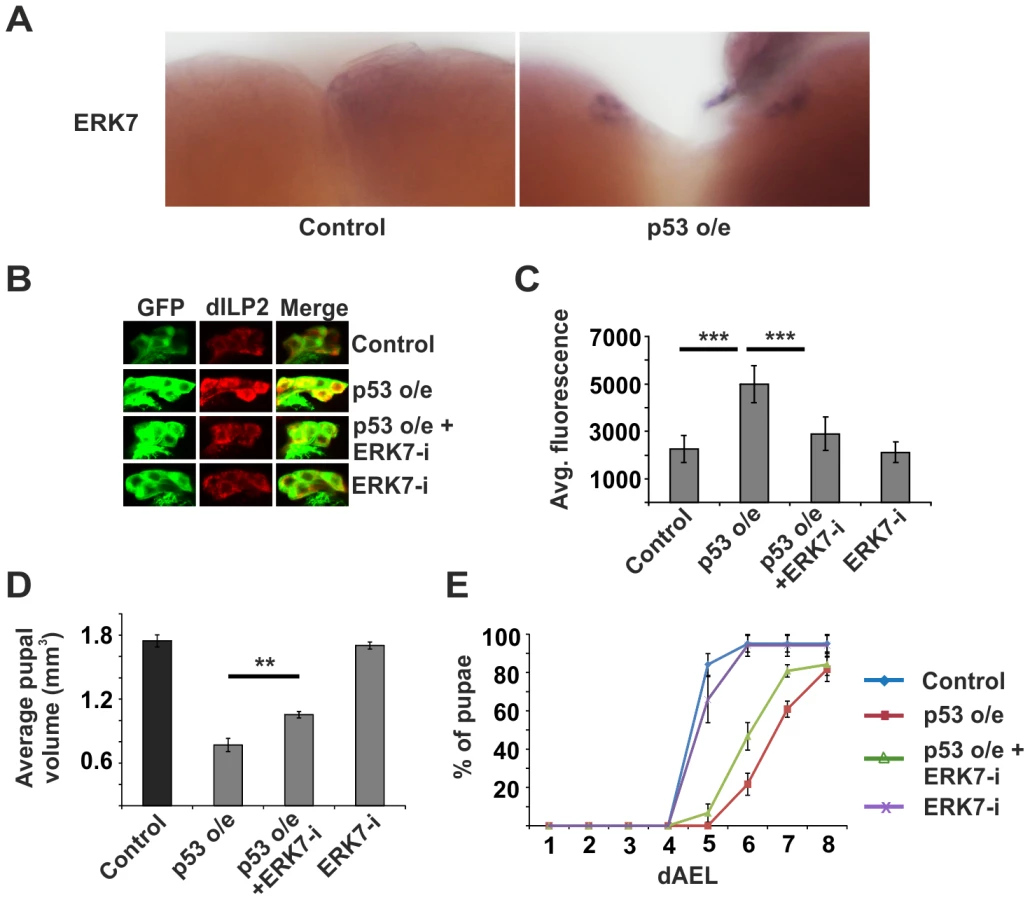
As ribosome biogenesis is dependent on nutrients [1], [12], [35], and erk7 expression was elevated upon starvation (Figure 4A, D), we postulated that perhaps the p53 - and ERK7-dependent ribosome surveillance response contributes to dILP accumulation in the IPCs upon starvation. This proved to be the case, as inhibition of either p53 (Figure 7A, B; Figure S10) or ERK7 (Figure 7C, D) in the IPCs partially reduced the accumulation of dILP2 following starvation of the larvae. While we cannot rule out the possibility that the knockdown of p53 and ERK7 affects dILP2 translation in this setting, the most parsimonious explanation is that p53 and ERK7 contribute to dILP2 secretion upon starvation.

Discussion
Here we report a novel cell-autonomous control mechanism for dILP secretion. Specifically, we conclude that i) inhibition of ribosome biogenesis in the IPCs at any level tested, including ribosomal gene expression (Myc), ribosome maturation (Rio1, Rio2, Tsr1) or by introducing imbalance of ribosomal components (Rpl35A), triggers a response to inhibit dILP secretion, ii) this inhibitory response is dependent on p53, a known surveillance factor for ribosome biogenesis, iii) a downstream effector of this ribosome surveillance pathway is protein kinase ERK7, as erk7 mRNA levels are elevated upon inhibited ribosome biogenesis and p53 activation and erk7 is essential to inhibit dILP secretion in both conditions, iv) the ribosome surveillance mechanism discovered here likely contributes to starvation-induced inhibition of dILP secretion. These findings significantly broaden the view about the regulatory functions of the ribosome surveillance pathways, which have been mainly explored at the level of proliferating cells. Ribosome biogenesis serves as the key determinant of cell autonomous growth control and it is finely tuned to match with the cellular nutrient and energy status [1], [12]. Coupling dILP secretion to the ribosome biogenesis pathway is an elegant mechanism for multicellular animals to synchronize the hormonal growth control with the local cell autonomous regulation of growth. Comparable to our finding in the IPCs, inhibition of ribosome biogenesis in the fat body leads to a block of dILP secretion in the IPCs through an unknown humoral mechanism [22], [24], [39]. Linking ribosome biogenesis to growth control through parallel mechanisms likely provides a robust regulatory network to tune down systemic growth signals whenever any region of the body experiences nutrient deprivation. This synchronization is likely important to maintain balanced growth throughout the spectrum of dietary conditions. Future studies should be aimed to explore the possible interrelationship between the fat body-derived signals and the cell-autonomous mechanism discovered here.
Our findings highlight that the p53-mediated ribosome surveillance pathway can serve highly cell type-specific functions in vivo. This is interesting when considering human ribosomopathies, genetic diseases caused by impaired ribosome biogenesis manifesting with a wide spectrum of tissue-specific defects. One of the ribosomopathies, Shwachman-Diamond syndrome (SDS), is manifested with a failure in pancreatic function [20]. A mouse model of SDS displays general preservation of ductal and endocrine compartments, but reduced amount of zymogen granules. Moreover, SDS mutant mice have reduced glucose tolerance, suggesting compromised endocrine function [40]. It will be interesting to learn whether p53 and ERK7 act as mediators of the secretion-related defects observed in SDS.
Compared to other members of the MAP kinase family, ERK7 has remained relatively poorly characterized [41]. Earlier studies in mammalian and Drosophila cells have shown that ERK7 protein levels are actively regulated at the level of protein degradation [38], [42]. In mammals, an increase in ERK7 levels leads to autophosphorylation and consequent activation [43], [44]. Consistent with the idea that ERK7 is mainly regulated through abundance, we observed that elevated ERK7 expression had a prominent impact in the function of IPCs. Interestingly, earlier studies have linked ERK7 function to growth regulation by showing that ERK7 protein is stabilized by serum and amino acid starvation [38]. Our data provides evidence that impaired ribosome biogenesis as well as starvation increases the expression of erk7 mRNA revealing a novel regulatory level for ERK7 function. In addition to the conditions explored here, ERK7 expression levels increase towards the end of larval development when growth is ceased [45]. It will be interesting to learn further how ERK7 expression is regulated and whether ERK7 has a function in tissue growth control beyond its role in the IPCs. Earlier studies in cell culture have shown evidence that ERK7 regulates cancer cell proliferation and autophagy [46], [47], suggesting that ERK7 may have a broader role in the regulation of tissue growth.
Materials and Methods
Fly stocks and husbandry
RNAi lines were from Vienna Drosophila RNAi Center (VDRC), Bloomington Drosophila Stock Center (BDSC) and National Institute of Genetics (NIG), Japan. Identities of RNAi lines and transgenes are listed in Table S2. Driver lines used: dILP2-Gal4, UAS-GFP [4] for all IPC specific experiments, Tub-G80TS; Tub-Gal4 (temperature sensitive ubiquitous driver) for ubiquitous expression of RNAi. Sgs3-Gal4 (BDSC#6870) was used for salivary gland specific experiments [32]. w1118 crossed to respective driver lines was used as control in all experiments. For generating UAS-ERK7-V5-His, ERK7 cDNA was amplified by PCR from pMT-ERK7-V5-HisB [38], cloned into KpnI site of the pUAST-attB vector and confirmed by sequencing. The transgene was directed to the attP landing site at 22A2. Transgenic flies were constructed by BestGene Inc.
Flies were grown on medium containing agar 0.6% (w/v), semolina 3.2% (w/v), malt 6.5% (w/v), dry baker's yeast 1.8% (w/v), propionic acid 0.7% (v/v) and Nipagin (methylparaben) 2.4% (v/v). For starvation, third instar larvae were placed on PBS, 1% sucrose, 0.4% agar for approximately 15 hours.
Drosophila relative weight determination
To normalize variation between vials due to growth conditions, we determined the relative weight. UAS-RNAi flies were crossed to dILP2-Gal4, UAS-GFP/CyO flies and progeny were allowed to develop at 29°C. Four days after adult emergence, male flies were weighed in groups (≥10 flies/group, N≥3), using a precision balance (Mettler). The mean weight of the flies expressing both dILP2-Gal4 and RNAi was divided by the mean weight of control flies (not expressing the dILP2-GAL4) from the same vial.
Measurement of pupal volume
UAS-RNAi flies were crossed to dILP2-Gal4, UAS-GFP/Cyo flies and allowed to lay eggs at +25°C. GFP positive L1 larvae were collected (30 per vial) 24 hours after egg deposition (AED) and grown at +29°C until pupation. Length and width of pupae were measured using ProgRes CapturePro 2.8.8 program (Jenoptik). Volumes of the pupae were calculated using the following formula: 4/3π(L/2)(W/2)2 (L, length; W, width).
Immunostainings and antibodies
Brains were dissected from 3rd instar wandering larvae, fixed in 4% formaldehyde-PBS for 30 min at RT and washed in PBT buffer (0.3% Triton X 100 in PBS). Following blocking for 2 h at RT in 5% bovine serum albumin (BSA)/PBT, samples were incubated with respective primary antibodies at 4°C o/n. Samples were washed three times (15 min each) with PBT and incubated with respective secondary antibodies for 2 h at RT. After three washes in PBT, brains were mounted in Vectashield Mounting Medium (Vector laboratories). Fluorescence images were acquired using a Leica TCS SP5 MP SMD FLIM confocal laser scanning microscope. Antibodies used: rat anti-dILP2 [5], rabbit anti-dILP2 [48], guinea pig anti-dILP2 (see below), rabbit anti-dILP5 (5), anti-rat Alexa fluor 633 (Invitrogen), anti-rabbit Alexa fluor 633 (Invitrogen) and anti-guinea pig Alexa fluor 633 (Invitrogen). Confocal images of each IPC cluster were taken using the same scan and laser power settings. Total signal from each cluster was quantified using ImageJ software (NIH). Representative images were cropped and processed identically for all the samples of every experiment.
dILP2 antibody generation
Two synthetic peptides (PHKRAMPGADSDLDA) and (AEVRRRTRQRQGI) corresponding to amino acid sequence of dILP2 from 49–63 and 102–114 amino acids respectively were produced by Storkbio Ltd. (Estonia). These peptides were mixed and used as immunogens to raise polyclonal antibodies in Guinea pigs by Storkbio Ltd.
RNA extraction
RNA extraction from larval brains: Equal numbers of L1 larvae were collected and grown at 29°C. 10 brains from third instar non-wandering larvae (approx. 85 hours AED) were dissected and RNA was extracted using Nucleospin RNA XS kit (Macherey-Nagel) according to the manufacturer's protocol. RNA extraction from whole larvae: equal numbers of L1 larvae were collected and grown at 29°C for 72 h prior to harvesting (96 h AED). ≥5 third instar larvae per sample in minimum of three replicates were homogenized and RNA was extracted using Nucleospin RNA II kit (Macherey-Nagel) according to the manufacturer's protocol.
Quantitative RT-PCR
RNA was reverse transcribed using RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific). qPCR was performed using Maxima SYBR Green qPCR Master Mix (2X) (Fermentas) with Light cycler 480 Real-Time PCR System (Roche). At least three biological replicates were used for each genotype and at least three technical replicates were used for each biological replicate. Primers for quantitative RT-PCR are presented as Table S3.
In situ hybridization
In situ hybridization was performed as described previously [49]. Briefly, antisense and sense probes were synthetized by T3/T7 RNA polymerase-dependent in vitro transcription reaction (Promega) in the presence of digoxigenin-UTP (Roche). Mid-3rd instar larval tissues were fixed using 4% formaldehyde and they were hybridized with the labeled probes for 16 h at 55°C. The hybridization was visualized by using alkaline phosphatase-conjugated anti-digoxigenin Fab fragments (1∶3,000; Roche) in the presence of NBT and BCIP substrates (Promega).
Primers for the erk7 probes were as follows:
5′-TAATACGACTCACTATAGGGCTCAAGAGCGACGCATTCAA-3′
5′-ATTAACCCTCACTAAAGGGATACTGGTCCACGTCGTAGCG-3′
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HietakangasV, CohenSM (2009) Regulation of tissue growth through nutrient sensing. Annu Rev Genet 43 : 389–410.
2. PartridgeL, AlicN, BjedovI, PiperMD (2011) Ageing in Drosophila: the role of the insulin/Igf and TOR signalling network. Exp Gerontol 46 : 376–381.
3. IkeyaT, GalicM, BelawatP, NairzK, HafenE (2002) Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr Biol 12 : 1293–1300.
4. RulifsonEJ, KimSK, NusseR (2002) Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science 296 : 1118–1120.
5. GeminardC, RulifsonEJ, LeopoldP (2009) Remote control of insulin secretion by fat cells in Drosophila. Cell Metab 10 : 199–207.
6. RajanA, PerrimonN (2012) Drosophila cytokine unpaired 2 regulates physiological homeostasis by remotely controlling insulin secretion. Cell 151 : 123–137.
7. KwakSJ, HongSH, BajracharyaR, YangSY, LeeKS, et al. (2013) Drosophila adiponectin receptor in insulin producing cells regulates glucose and lipid metabolism by controlling insulin secretion. PLoS One 8: e68641.
8. LempiainenH, ShoreD (2009) Growth control and ribosome biogenesis. Curr Opin Cell Biol 21 : 855–863.
9. SinghJ, TyersM (2009) A Rab escort protein integrates the secretion system with TOR signaling and ribosome biogenesis. Genes Dev 23 : 1944–1958.
10. LempiainenH, UotilaA, UrbanJ, DohnalI, AmmererG, et al. (2009) Sfp1 interaction with TORC1 and Mrs6 reveals feedback regulation on TOR signaling. Mol Cell 33 : 704–716.
11. JamesMJ, ZomerdijkJC (2004) Phosphatidylinositol 3-kinase and mTOR signaling pathways regulate RNA polymerase I transcription in response to IGF-1 and nutrients. J Biol Chem 279 : 8911–8918.
12. GrewalSS, EvansJR, EdgarBA (2007) Drosophila TIF-IA is required for ribosome synthesis and cell growth and is regulated by the TOR pathway. J Cell Biol 179 : 1105–1113.
13. GrewalSS, LiL, OrianA, EisenmanRN, EdgarBA (2005) Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol 7 : 295–302.
14. ChakrabortyA, UechiT, KenmochiN (2011) Guarding the ‘translation apparatus’: defective ribosome biogenesis and the p53 signaling pathway. Wiley Interdiscip Rev RNA 2 : 507–522.
15. LohrumMA, LudwigRL, KubbutatMH, HanlonM, VousdenKH (2003) Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3 : 577–587.
16. FumagalliS, IvanenkovVV, TengT, ThomasG (2012) Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev 26 : 1028–1040.
17. SloanKE, BohnsackMT, WatkinsNJ (2013) The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep 5 : 237–247.
18. KurodaT, MurayamaA, KatagiriN, OhtaYM, FujitaE, et al. (2011) RNA content in the nucleolus alters p53 acetylation via MYBBP1A. EMBO J 30 : 1054–1066.
19. OnoW, HayashiY, YokoyamaW, KurodaT, KishimotoH, et al. (2014) The nucleolar protein Myb-binding protein 1A (MYBBP1A) enhances p53 tetramerization and acetylation in response to nucleolar disruption. J Biol Chem 289 : 4928–4940.
20. TengT, ThomasG, MercerCA (2013) Growth control and ribosomopathies. Curr Opin Genet Dev 23 : 63–71.
21. MarygoldSJ, RooteJ, ReuterG, LambertssonA, AshburnerM, et al. (2007) The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol 8: R216.
22. DelanoueR, SlaidinaM, LeopoldP (2010) The steroid hormone ecdysone controls systemic growth by repressing dMyc function in Drosophila fat cells. Dev Cell 18 : 1012–1021.
23. LinJI, MitchellNC, KalcinaM, TchoubrievaE, StewartMJ, et al. (2011) Drosophila ribosomal protein mutants control tissue growth non-autonomously via effects on the prothoracic gland and ecdysone. PLoS Genet 7: e1002408.
24. MarshallL, RideoutEJ, GrewalSS (2012) Nutrient/TOR-dependent regulation of RNA polymerase III controls tissue and organismal growth in Drosophila. EMBO J 31 : 1916–1930.
25. JiuY, HasygarK, TangL, LiuY, HolmbergCI, et al. (2014) par-1, atypical pkc, and PP2A/B55 sur-6 are implicated in the regulation of exocyst-mediated membrane trafficking in Caenorhabditis elegans. G3 (Bethesda) 4 : 173–183.
26. GronkeS, ClarkeDF, BroughtonS, AndrewsTD, PartridgeL (2010) Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet 6: e1000857.
27. VanrobaysE, GelugneJP, GleizesPE, Caizergues-FerrerM (2003) Late cytoplasmic maturation of the small ribosomal subunit requires RIO proteins in Saccharomyces cerevisiae. Mol Cell Biol 23 : 2083–2095.
28. LaRonde-LeBlancN, WlodawerA (2005) A family portrait of the RIO kinases. J Biol Chem 280 : 37297–37300.
29. WidmannB, WandreyF, BadertscherL, WylerE, PfannstielJ, et al. (2012) The kinase activity of human Rio1 is required for final steps of cytoplasmic maturation of 40S subunits. Mol Biol Cell 23 : 22–35.
30. ZempI, WildT, O'DonohueMF, WandreyF, WidmannB, et al. (2009) Distinct cytoplasmic maturation steps of 40S ribosomal subunit precursors require hRio2. J Cell Biol 185 : 1167–1180.
31. PuigO, TjianR (2005) Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev 19 : 2435–2446.
32. CherbasL, HuX, ZhimulevI, BelyaevaE, CherbasP (2003) EcR isoforms in Drosophila: testing tissue-specific requirements by targeted blockade and rescue. Development 130 : 271–284.
33. GuertinDA, GunturKV, BellGW, ThoreenCC, SabatiniDM (2006) Functional genomics identifies TOR-regulated genes that control growth and division. Curr Biol 16 : 958–970.
34. TelemanAA, HietakangasV, SayadianAC, CohenSM (2008) Nutritional control of protein biosynthetic capacity by insulin via Myc in Drosophila. Cell Metab 7 : 21–32.
35. LiL, EdgarBA, GrewalSS (2010) Nutritional control of gene expression in Drosophila larvae via TOR, Myc and a novel cis-regulatory element. BMC Cell Biol 11 : 7.
36. van RiggelenJ, YetilA, FelsherDW (2010) MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer 10 : 301–309.
37. DeisenrothC, ZhangY (2010) Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 29 : 4253–4260.
38. ZacharogianniM, KondylisV, TangY, FarhanH, XanthakisD, et al. (2011) ERK7 is a negative regulator of protein secretion in response to amino-acid starvation by modulating Sec16 membrane association. EMBO J 30 : 3684–3700.
39. KillipLE, GrewalSS (2012) DREF is required for cell and organismal growth in Drosophila and functions downstream of the nutrition/TOR pathway. Dev Biol 371 : 191–202.
40. TourlakisME, ZhongJ, GandhiR, ZhangS, ChenL, et al. (2012) Deficiency of Sbds in the mouse pancreas leads to features of Shwachman-Diamond syndrome, with loss of zymogen granules. Gastroenterology 143 : 481–492.
41. CargnelloM, RouxPP (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75 : 50–83.
42. KuoWL, DukeCJ, AbeMK, KaplanEL, GomesS, et al. (2004) ERK7 expression and kinase activity is regulated by the ubiquitin-proteosome pathway. J Biol Chem 279 : 23073–23081.
43. AbeMK, KuoWL, HershensonMB, RosnerMR (1999) Extracellular signal-regulated kinase 7 (ERK7), a novel ERK with a C-terminal domain that regulates its activity, its cellular localization, and cell growth. Mol Cell Biol 19 : 1301–1312.
44. KlevernicIV, StaffordMJ, MorriceN, PeggieM, MortonS, et al. (2006) Characterization of the reversible phosphorylation and activation of ERK8. Biochem J 394 : 365–373.
45. GraveleyBR, BrooksAN, CarlsonJW, DuffMO, LandolinJM, et al. (2011) The developmental transcriptome of Drosophila melanogaster. Nature 471 : 473–479.
46. ColecchiaD, StrambiA, SanzoneS, IavaroneC, RossiM, et al. (2012) MAPK15/ERK8 stimulates autophagy by interacting with LC3 and GABARAP proteins. Autophagy 8 : 1724–1740.
47. XuYM, ZhuF, ChoYY, CarperA, PengC, et al. (2010) Extracellular signal-regulated kinase 8-mediated c-Jun phosphorylation increases tumorigenesis of human colon cancer. Cancer Res 70 : 3218–3227.
48. BaderR, Sarraf-ZadehL, PetersM, ModerauN, StockerH, et al. (2013) The IGFBP7 homolog Imp-L2 promotes insulin signaling in distinct neurons of the Drosophila brain. J Cell Sci 126 : 2571–2576.
49. MattilaJ, BremerA, AhonenL, KostiainenR, PuigO (2009) Drosophila FoxO regulates organism size and stress resistance through an adenylate cyclase. Mol Cell Biol 29 : 5357–5365.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- An RNA-Seq Screen of the Antenna Identifies a Transporter Necessary for Ammonia Detection
- Systematic Comparison of the Effects of Alpha-synuclein Mutations on Its Oligomerization and Aggregation
- Functional Diversity of Carbohydrate-Active Enzymes Enabling a Bacterium to Ferment Plant Biomass
- Regularized Machine Learning in the Genetic Prediction of Complex Traits
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy