
A Thermolabile Aldolase A Mutant Causes Fever-Induced Recurrent Rhabdomyolysis without Hemolytic Anemia
Aldolase A deficiency has been reported as a rare cause of hemolytic anemia occasionally associated with myopathy. We identified a deleterious homozygous mutation in the ALDOA gene in 3 siblings with episodic rhabdomyolysis without hemolytic anemia. Myoglobinuria was always triggered by febrile illnesses. We show that the underlying mechanism involves an exacerbation of aldolase A deficiency at high temperatures that affected myoblasts but not erythrocytes. The aldolase A deficiency was rescued by arginine supplementation in vitro but not by glycerol, betaine or benzylhydantoin, three other known chaperones, suggesting that arginine-mediated rescue operated by a mechanism other than protein chaperoning. Lipid droplets accumulated in patient myoblasts relative to control and this was increased by cytokines, and reduced by dexamethasone. Our results expand the clinical spectrum of aldolase A deficiency to isolated temperature-dependent rhabdomyolysis, and suggest that thermolability may be tissue specific. We also propose a treatment for this severe disease.
Published in the journal:
. PLoS Genet 10(11): e32767. doi:10.1371/journal.pgen.1004711
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004711
Summary
Aldolase A deficiency has been reported as a rare cause of hemolytic anemia occasionally associated with myopathy. We identified a deleterious homozygous mutation in the ALDOA gene in 3 siblings with episodic rhabdomyolysis without hemolytic anemia. Myoglobinuria was always triggered by febrile illnesses. We show that the underlying mechanism involves an exacerbation of aldolase A deficiency at high temperatures that affected myoblasts but not erythrocytes. The aldolase A deficiency was rescued by arginine supplementation in vitro but not by glycerol, betaine or benzylhydantoin, three other known chaperones, suggesting that arginine-mediated rescue operated by a mechanism other than protein chaperoning. Lipid droplets accumulated in patient myoblasts relative to control and this was increased by cytokines, and reduced by dexamethasone. Our results expand the clinical spectrum of aldolase A deficiency to isolated temperature-dependent rhabdomyolysis, and suggest that thermolability may be tissue specific. We also propose a treatment for this severe disease.
Abstract Summary
Using recent technical advances involving exome analysis, we identified a new missense mutation in the ALDOA gene, encoding a key enzyme in the glycolytic pathway. The patients presented with severe recurrent rhabdomyolysis without hemolytic anemia. The decrease of aldolase A activity in myoblasts was enhanced at high temperature and could explain the fever-induced rhabdomyolysis. By contrast, enzyme thermolability was not found in erythrocytes, possibly accounting for the unusual clinical phenotype of the patients. Enzyme thermolability was rescued by arginine supplementation in vitro but not by other chaperone compounds.
Introduction
Massive rhabdomyolysis is a life threatening condition and has been associated with mitochondrial fatty acid ß-oxidation defects (FAO) [1]–[3], LPIN1 mutations [4]–[6], as well as, rarely, with mitochondrial respiratory chain (RC) deficiency, dystrophinopathies and inborn errors of glycogenolysis and glycolysis [3], [7]. Among inherited defects of glycolysis, isolated rhabdomyolysis is not an usual presentation. Because most metabolic mechanisms of rhabdomyolysis are triggered by fever, differential diagnoses include myositis and viral infections for non recurrent cases. Metabolic work-up focuses on plasma carnitine and acylcarnitine profiles, urinary organic acids analysis, then sequencing of LPIN1 gene in young children. In older children, ischemic stress test can orient toward anaerobic glycolysis defects. In both young and older patients, skeletal muscle biopsy may be proposed for histological studies in cases of negative biochemical and molecular results. In spite of this wide range of investigations, the disease mechanism remains unknown in at least half of the recurrent cases [8].
In order to identify new etiologies of recurrent rhabdomyolysis in young children, we used exome sequencing in siblings suffering from severe episodes of rhabdomyolysis triggered by fever since age 2 months. This led us to identify a new phenotype of ALDOA mutations. The absence of hemolytic anemia was explained by tissue specific expression of protein thermolability. The occurrence of thermolability supports the contention that viral infections should remain a diagnosis of exclusion for rhabdomyolysis. Our results raise the possibility of medical therapy by arginine.
Results
Case report
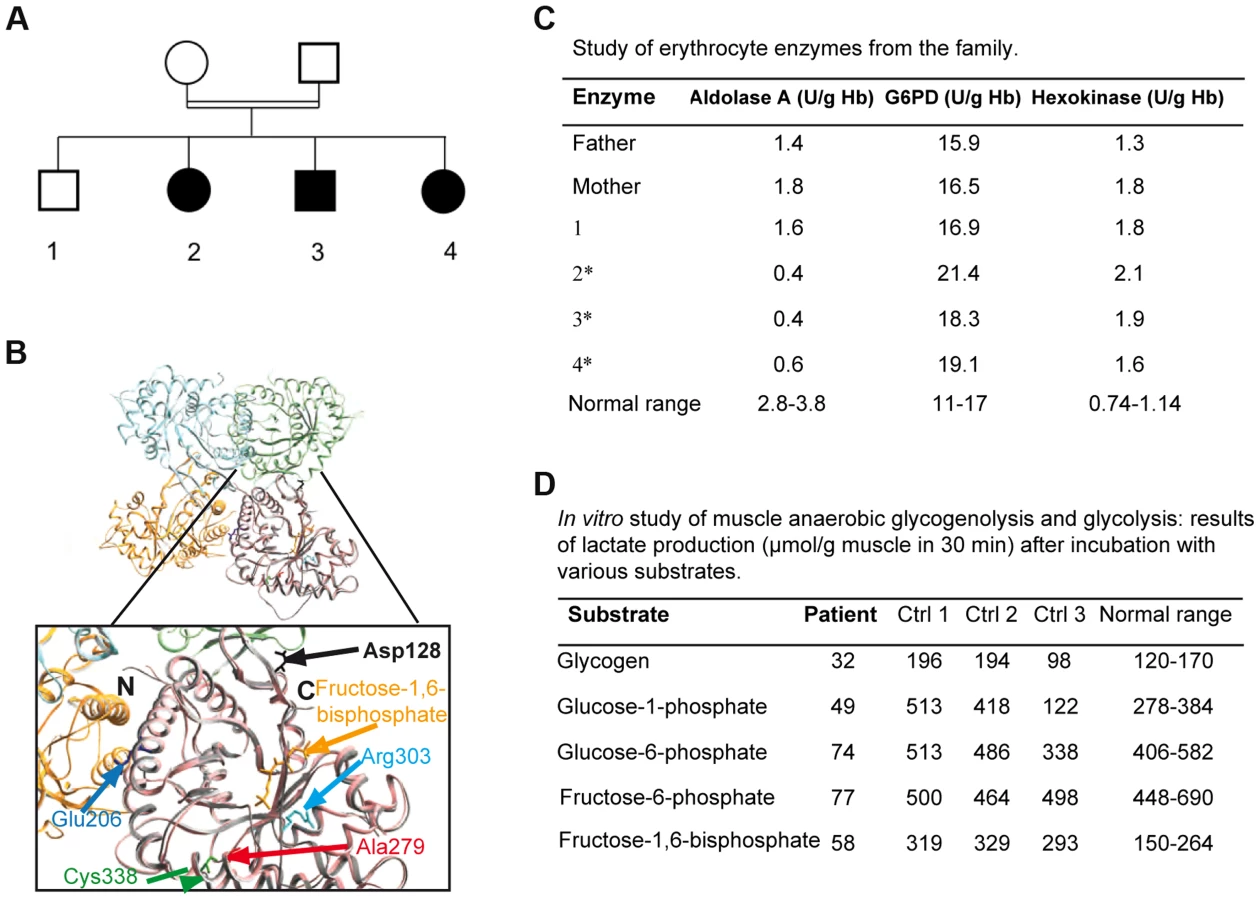
Three patients from a Moroccan consanguineous family (Figure 1A) suffered from recurrent episodes of rhabdomyolysis that required numerous hospitalizations from 2 months of age. These acute episodes were invariably triggered by febrile illnesses. The presenting symptoms were an inability to walk and myalgia. During the acute episodes, plasma creatine phosphokinase (CK) levels were variable, ranging from markedly elevated (peak levels: 180,000–450,000 U/L, N<150) with overt myoglobinuria to milder elevations (3,000 U/L). The following tests were normal: hemoglobin, hematocrit, mean corpuscular volume, plateletcount, reticulocyte count, bilirubin, haptoglobin, ferritin, Coombs' test, urea, creatinine, blood gasses, plasma lactate, carnitine, blood acylcarnitine profile, plasma amino acids, and urinary organic acids. Electromyography, brain MRI, abdominal ultrasonography, and echocardiography were also normal. CK levels ranged from normal (<150 U/L) to elevated (up to 1,800 U/L) in all 3 patients between acute episodes. The clinical examination and muscle tests performed 2 months after an episode of rhabdomyolysis were normal for each patient, at ages 9, 10, and 11 years respectively. Family history revealed neither chronic hemolytic anemia, nor episodes of jaundice or blood transfusions. Two patients suffered from learning disabilities and required a special school.
Molecular studies
Exome sequencing analysis pointed to 10 candidate genes harboring homozygous mutations (Table S1). The ALDOA gene was considered because all 3 affected patients harbored the homozygous mutation c.839 C>T (p.Ala279Val, NM_000034), whereas the healthy sibling and the parents were heterozygous. This gene was located in a homozygous region revealed by homozygosity mapping, between the polymorphic markers D16S3022 and D16S323. Polyphen software predicted that this change, located in a sequence conserved between species (Figure S1), was “probably damaging”. The homozygous mutation p. Ala279Val was localized near the previously described Cys338residue involved in a hematological and muscle phenotype[9] close to a “hinge” region that is critical for a conformational change in the C-terminus (Figure 1B). Prediction from seven combined softwares as implemented in iSTABLE [10] was consistent with A279V destabilizing the protein. Therefore, this mutation is likely involved in maintaining the correct spatial conformation of the enzyme (see below) [9], [11].
Morphological studies
All histological and cellular experiments were performed with the tissue and myoblasts from the same patient aged 10 (Patient 3, Figure 1C).
Microscopy showed an excessive lipid droplet (LD) accumulation, visualized with oil-red-O staining (Figure 2A), in the muscle biopsy from the patient (a) compared with control biopsies (b). However, the muscle tissue of the patient showed a well-preserved fascicular architecture with normal type 1 and type 2 fibers. In addition, the cytochrome c-oxidase and phosphorylase staining patterns were similar to controls.

Representative cultures of the patient under basal (a) or pro-inflammatory conditions (b) are shown in Figure 2B. Oil-red-O staining of the neutral lipids revealed massive accumulation of LDs after 24 hours of TNFα+IL-1ß stimulation (b), whereas no change in the size or the number of the LDs was observed at 40°C (Figure S2). In contrast, control myoblasts exhibited no LDs under basal condition and a moderate accumulation after TNFα+IL-1ß stimulation (Figure S3). Co-treatment with anti-inflammatory agents and particularly treatment with the synthetic glucocorticoid dexamethasone combined with TNFα+IL-1ß (c) or alone (d), dramatically decreased the number of LDs in patient myoblasts.
The effect of the ALDOA knockdown on LD accumulation was investigated in order to test if muscle abnormalities seen in patients result from ALDOA decrease. Transfection with a siRNA that knocked down ALDOA expression (as confirmed by RT-PCR, Figure 2C), induced a 48% increase in the number of LDs in the control myoblasts (Figure 2Da,b) and a 5.5% increase in the patient cells (Figure 2Dc,d).
Biochemical studies
ALDOA activity was dramatically decreased in patient erythrocytes (0.4 to 0.6 U/g Hb; control 4.6) (Figure 1C) and frozen skeletal muscle (55 nmol/h/mg; normal 581-5188) though to a lesser degree in myoblasts (1.08±0.4 U/µg protein; normal 2.4±0.025). Activity was approximately half the normal in the heterozygote (Figure 1C), supporting the pathogenic nature of the c.839 C>T mutation and the recessive inheritance of the disease. Studies of muscle anaerobic glycogenolysis and glycolysis in vitro revealed reduced lactate production, consistent with dysfunctional glycolysis (Figure 1D). The glucose-6-phosphate dehydrogenase and hexokinase activities were normal or elevated in all affected patients' erythrocytes (Figure 1C). Carbons from stable isotope labeled glutamine were incorporated to a lesser degree into Krebs cycle intermediates in the patient myoblasts relative to controls, consistent with ALDOA deficiency leading to decreased glycolytic flux (Figure S4).
We studied whether temperature or pro-inflammatory cytokines affected ALDOA expression. To this end, the patient and the control myoblasts were cultured at 37 or 40°C, under basal conditions or with the combination of TNFα+IL-1β. Myoblasts from the patient and the controls responded to pro-inflammatory stress by significant secretion of IL6, peaking at approximately 24 hours of TNFα+IL-1ß stimulation (20-fold). The level of ALDOA mRNA was unchanged in patient myoblasts after exposure to TNFα+IL-1β or 40°C (Figure 3A upper panel), whereas the corresponding protein level was reduced in patient myoblasts in basal conditions (0.6±0.09; control: 1.3±0.17) and was further abated at 40°C (barely detectable) compared to control (0.8±0.16) (Figure 3A, lower panel). In contrast, TNFα+IL-1β treatment did not affect the protein level (1.2±0.11 to 0.97±0.11 in control and 0.7±0.09 to 0.8±0.13 in patient myoblasts). Accordingly, aldolase activity in the patient myoblasts dramatically decreased after incubation from 25°C, 37°C through 40°C (residual activity 10% and 5% respectively), and to a lesser degree in the control myoblasts (residual activity 61% at 37°C and 43% at 40°C) (Figure 3B). TNFα+IL-1β treatment did not change the level of activity in the patient or the control myoblasts (Figure 3B). Interestingly, aldolase A activity in erythrocytes from the three patients was not modified by high temperature (Figure 3C). These results suggest that the mutant enzyme might be differentially destabilized in distinct tissues, i.e., only in myoblasts and not in erythrocytes.

Arginine supplementation significantly enhanced aldolase A activity in patient myoblasts compared to untreated cells (Figure 3D, upper panel); this effect may be explained by an increase of the protein level (Figure 3D, lower panel). By contrast, glycerol, benzylhydantoin and betaine did not modify aldolase activity and protein (Table S2), suggesting that arginine-mediated rescue did not operate as a protein chaperone. Arginine did not contribute carbons to Krebs cycle intermediates in 6-hr stable isotope labeling experiments in myoblasts (Figure S4), thus indicating that arginine did not represent a major source of compensatory fuel for energy production.
The respiratory chain activities were increased in the frozen skeletal muscle compared to control and similar in the myoblasts of the patient and the control (Table S3). Similarly, fatty acid oxidation (FAO) results were normal in patient myoblasts compared to control and were not affected by pro-inflammatory conditions (Table S4).
Discussion
Massive rhabdomyolysis is a life-threatening situation, yet a molecular mechanism is found in only half of the recurrent cases. Because glycolysis is the most important source of energy in erythrocytes and in some types of skeletal muscle fibers, inherited defects of glycolysis can cause hemolytic anemia or the combination of hemolytic anemia, neurologic abnormalities, and myopathy [12]. By exome sequencing and homozygosity mapping, we identified a new phenotype associated with aldolase A deficiency. The phenotype affects skeletal muscle with no hemolytic symptoms, while mild learning disabilities were identified in 2 of the 3 siblings.
Only 5 patients with aldolase A deficiency have been reported so far (Table 1) [9], [13]–[16]. All presented with non-spherocytic hemolytic anemia (OMIM #103850), a few had mental retardation [13], [15] and 2 also displayed rhabdomyolysis leading to death [9], [13]. Because of the involvement of ALDOA in skeletal tissue function and the observation of muscle symptoms in 2 previous cases with hemolytic anemia, we inferred that ALDOA mutations could be responsible for rhabdomyolysis in our family. In agreement with this, we showed the effect of the ALDOA knockdown on LD accumulation in patient myoblasts, and that they were enhanced by ALDOA siRNA in non-inflammatory conditions compared to control. LD accumulation was further enhanced in inflammatory conditions, consistent with the fact that intracellular accumulation of lipids is a common feature of proinflammatory stress [17], [18]. LD are dynamic organelles and provide key storage compartments that participate in the response to increased energetic demand [19] and in the regulation of cellular lipid metabolism, thus being involved in the delicate balance between triacylglycerol deposition and mobilization. LD accumulation as frequently observed in energetic diseases could result from a metabolic adaptation of the affected cells.

We were puzzled to find no evidence of hemolytic anemia in our patients although residual enzyme activity was 9% in erythrocytes and 45% in skeletal myoblasts. This may be related to the greater residual enzyme activity observed in our patients (approximately 10%) compared to previously reported cases of ALDOA deficiency (approximately 5%). Also, the hematological investigation was performed outside of an acute episode of decompensation. Of note, alternative explanations cannot be ruled out, including variable expressivity (degree of metabolic stress) or a selective resistance to thermolability in erythrocytes (cell type specific protein-protein interactions).
Because aldolase A is physiologically active as an homotetramer, the integrity of the quaternary structure was suggested to confer thermal stability to the enzyme [20] and was found to be vulnerable in erythrocytes [11], [16] as well as according to iSTABLE prediction [10]. However, thermolability of aldolase A was found only in patient myoblasts, not in erythrocytes, and it was observed at lower temperature than previously described [11], [13], [14], [16]. Red blood cells contribute to about 40% of the blood volume and are the first cellular structures to respond to increased reactive oxygen species (ROS) activity [21]. Because they do not have nucleus, they rely on pre-existing proteins for protection against ROS damage [22] and possess an extensive array of antioxidants [23]. Also, they develop a system of defenses that represent an excellent example of redox balance maintenance [24]. Interestingly, we showed that protein and catalytic breakdown can be reversed by arginine but not by glycerol, benzylhydantoin and betaine which all might act as a chaperone as recently proposed for unrelated metabolic disorders [25]–[27]. Also, another mechanism may be proposed concerning the effective role of L-arginine on aldolase A activity, namely its antioxidant function via increased NO formation and reduced release of superoxide.
Wen et al. [28] described the role of the inflammasome complex and IL-1β [29] in the Warburg effect of aerobic glycolysis, which was recently shown to be promoted by lipopolysaccharides. [30] Conversely, glucose metabolism plays a crucial role in IL-1β transcription because high glucose boosts the production of IL-1 β in pancreatic beta cells. [31] However, myoblasts from the patient did not exhibit altered or abnormal IL-1β secretion. Moreover, the levels of most cytokines measured (see above) were undetectable or very low in patients' plasma. Similar results were obtained for healthy donors.
Due to LD accumulation in our patient muscle cells, consistent with findings in a previous patient [13], and because adenosine triphosphate (ATP) has several origins, we also examined FAO metabolism and OXPHOS. Moreover, pro-inflammatory cytokines are known to down regulate lipid metabolism. [32] However, FAO and OXPHOS metabolism were normal compared to control myoblasts, and no change in patient myoblasts was observed after TNFα+IL-1β treatment. LD accumulation may result from anaplerotic dysfunction of the Krebs cycle. [33] Consistent with this possibility, we found reduced incorporation of carbon from stable isotope labeled glutamine (Figure S4). A number of secondary events may also play a role in muscle pain such as lipotoxicity [34] and osmotic and ionic modifications with consequences for ionic exchange. [35] The role of ALDOA as a scaffold protein that coordinates actin and microtubule networks can also be speculated to participate in the biogenesis of lipid droplet. [36], [37] [38] Aldolase A has been shown to bind to vacuolar-type H+-ATPase (V-ATPase) with ARNO (an ADP-ribosylation factor guanine nucleotide exchange factor). [39] Interestingly, ARNO has been recently found to restore/promote lipid droplet formation. [40] Therefore, attractive mechanisms may stem from putative aldolase-ARNO interacting properties.
In conclusion, aldolase A deficiency is a rare cause of severe myoglobinuria in early childhood, as a consequence of impaired generation of ATP to fuel muscle metabolism. Our study points to the crucial role of fever as the trigger of rhabdomyolysis in our patients. High temperature and a combination of pro-inflammatory cytokines, utilized to mimic inflammatory conditions, led to decreased aldolase A activity and LD accumulation, respectively, in both the patient myoblasts and to a lesser extent, in the control myoblasts.
Thermolability selectively found in myoblasts and not in erythrocytes of the patients plays a crucial role in the pathophysiology of this disease. Finally, we showed that arginine may be a useful therapy that enhances the enzymatic activity in patient cells, probably more by another role, i.e. its antioxidant effect than by a chaperone role, whereas inflammatory conditions enhanced LD accumulation and, therefore, lipotoxicity.
Materials and Methods
Molecular studies
To identify the causative mutations for rhabdomyolysis in this family, exome sequencing was performed in 1 child using previously described methods. [41] All variants were annotated using an in-house-developed annotation software system. The variants were classified as previously unidentified when they were absent from the control populations and from all datasets, including dbSNP129, the 1000 Genomes Project, and in-house exome data.
The coding regions and flanking splice sites of the ALDOA (NM_000034) gene were sequenced using genomic DNA prepared from leukocytes.
Real-time quantitative PCR (RT-qPCR) assays were performed using cDNAs from myoblasts. All experiments were conducted in triplicate using an ABI PRISM 7300 Sequence Detection System instrument with SYBR Green fluorescence dye (Applied Biosystems).
The protein concentrations in myoblasts were determined using the Bradford method (Sigma).Forty micrograms of protein were separated using denaturing PAGE and transferred to PVDF membranes. After probing with the suitable antibodies (ALDOA (sc-12059, Santa Cruz), α-tubulin (T9026, Sigma) or β−actin (sc-81178, Santa Cruz)), the signals were detected using the ECL kit (GE Healthcare).
Swiss-Pdb Viewer 3.7 (http://www.expasy.org/spdbv) was used to analyze the crystal structure of the human muscle fructose 1,6-bisphosphate aldolase complex (PDB code 4ALD). Prediction of protein stability changes was obtained from seven combined softwares as implemented in iSTABLE [10].
The ethics committee of the Necker Hospital approved the research proposal. Informed consent was obtained from the siblings' parents.
Morphologic and biochemical studies
Histological studies were performed on a skeletal muscle biopsy and myoblasts obtained from Patient3 left deltoid muscle (Figure 1A) as previously described. [42] Samples from 2age - and sex-matched controls were obtained. Myoblasts were subjected to various stress conditions mimicking those believed to trigger the episodes of rhabdomyolysis, including high temperature (40°C) [13] and pro-inflammatory cytokines TNFα+IL-1ß(10 ng/mlfor 24 hours; all from R&D Systems). [43]To evaluate the response of myoblasts to these stimuli,IL6/IL8 release into the culture medium was measured using an immunoradiometric assay kit (Immunotopics). [44] An inflammatory cytokine kit was used to determine the contents of 10 cytokines (BD, Bioscience) in blood plasma. The cytokine inhibitors anakinra (inhibitor of the IL-1β receptor, 1 µg/mL) and ab9635 (inhibitor of TNFα, 1 µg/mL), or the synthetic glucocorticoid dexamethasone (0.2 µM) were added to the culture medium 1 hour or 12 hours, respectively, prior to the 24 hour-incubation with TNFα+IL-1β.
The enzymatic activities in erythrocytes and myoblasts were determined according to the methods of the International Committee for Standardization in Hematology. [45] To study the effect of temperature, myoblast extracts and erythrocytes were incubated for 30 minutes at 4°C, 25°C, 37°C, 39°C,or 40°C before enzyme assay. In the presence of suitable cofactors (including ATP and NAD+) and substrates for multiple enzymes involved in glycolysis (Figure1D), we measured L-lactate formation after incubation of frozen homogenized muscle tissue at 37°C for 30 min under nitrogen gas according to a published protocol. [46]
Mitochondrial OXPHOS activities of the myoblasts were evaluated as previously described. [47] Fatty-acid oxidation (FAO) measurements were performed through the assay of deuterated C2 to C16 acylcarnitines generated by incubation of intact myoblasts with a pentadeuterated C16 fatty acid ([16-2H3, 15-2H2]-palmitate) according to a procedure used for the detection of β-oxidation defects. [48]
RNA-silencing (siRNA) experiments were performed on myoblasts using the jePRIME transfection reagent (Polyplus) according to the supplier's recommendations and 25 nM of siRNA targeting human AldoA(M-010375,Dharmacon). The non-targeting siRNA #2 (Dharmacon) was used as a negative control.
The effect of arginine supplementation was investigated in patients' myoblasts incubated with 2 mM of L-arginine hydrochloride (Sigma) for 10 days. Three other chaperons (sigma) were also tested, glycerol (100 mM) [25] for 10 days, betaine (10 and 50 mM) [26], [27] and benzylhydantoin (130 µM) for 3 days. Uniformly stable isotope labeled (U-13C5) glutamine or (U-13C6) arginine (Eurisotop, Saint-Aubin, France) were provided to myoblasts (1 mM) in the presence of glucose (2.5 mM) and incubated without serum for 6 hours. Krebs cycle intermediates and their isotopomers were measured by gas chromatography mass spectrometry (Scion TQ, Brüker) using standard derivation by BSTFA [N,O-Bis(trimethylsilyl)trifluoroacetamide] and 1% trimethylchlorosilane.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LaforetP, Vianey-SabanC (2010) Disorders of muscle lipid metabolism: diagnostic and therapeutic challenges. Neuromuscul Disord 20 : 693–700.
2. DiMauroS, GaroneC, NainiA (2010) Metabolic myopathies. Curr Rheumatoly Rep 12 : 386–393.
3. TeinI, DiMauroS, DeVivoDC (1990) Recurrent childhood myoglobinuria. Adv Pediatr 37 : 77–117.
4. ZehariaA, ShaagA, HoutkooperRH, HindiT, de LonlayP, et al. (2008) Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am J Hum Genet 83 : 489–494.
5. Michot C, Hubert L, Brivet M, De Meirleir L, Valayannopoulos V, et al. (2010 LPIN1 gene mutations: a major cause of severe rhabdomyolysis in early childhood. Hum Mutat 31: E1564–1573.
6. MichotC, HubertL, RomeroNB, GoudaA, MamouneA, et al. (2012) Study of LPIN1, LPIN2 and LPIN3 in rhabdomyolysis and exercice-induced myalgia. J Inherit Metab Dis 35(6): 1119–1128.
7. ToninP, LewisP, ServideiS, DiMauroS (1990) Metabolic causes of myoglobinuria. AnnNeurol 27 : 181–185.
8. OhkumaA, NoguchiS, MalicdanMC, FukudaT, et al. (2009) Clinical and genetic analysis of lipid storage myopathies. Muscle Nerve 39 : 333–342.
9. YaoDC, TolanDR, MurrayMF, HarrisDJ, DarrasBT, et al. (2004) Hemolytic anemia and severe rhabdomyolysis caused by compound heterozygous mutations of the gene for erythrocyte/muscle isozyme of aldolase, ALDOA(Arg303X/Cys338Tyr). Blood 103 : 2401–2403.
10. ChenCW, LinJ, ChuYW (2013) iStable: off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinformatics 14 Suppl 2S5.
11. TakahashiI, TakasakiY, HoriK (1989) Site-directed mutagenesis of human aldolase isozymes: the role of Cys-72 and Cys-338 residues of aldolase A and of the carboxy-terminal Tyr residues of aldolases A and B. J Biochem. 105 : 281–286.
12. van AdelBA, TarnopolskyMA (2009) Metabolic myopathies: update 2009. J Clin Neuromuscul Dis 10 : 97–121.
13. KreuderJ, BorkhardtA, ReppR, PekrunA, GottscheB, et al. (1996) Brief report: inherited metabolic myopathy and hemolysis due to a mutation in aldolase A. N Engl J Med. 334 : 1100–1104.
14. MiwaS, FujiiH, TaniK, TakahashiK, TakegawaS, et al. (1981) Two cases of red cell aldolase deficiency associated with hereditary hemolytic anemia in a Japanese family. Am J Hematol 11 : 425–437.
15. BeutlerE, ScottS, BishopA, MargolisN, MatsumotoF, et al. (1973) Red cell aldolase deficiency and hemolytic anemia: a new syndrome. Trans Assoc Am Physicians 86 : 154–166.
16. KishiH, MukaiT, HironoA, FujiiH, MiwaS, et al. (1987) Human aldolase A deficiency associated with a hemolytic anemia: thermolabile aldolase due to a single base mutation. ProcNatl Acad Sci U S A 84 : 8623–8627.
17. PachecoP, Vieira-de-AbreuA, GomesRN, Barbosa-LimaG, WermelingerLB, et al. (2007) Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene B4-synthesizing function of macrophage lipid bodies elicited by innate immune response. J Immunol 179 : 8500–8508.
18. GomesRN, FiguieredoRT, BozzaFA, PachecoP, AmancioRT, et al. (2006) Increased susceptibility to septic and endotoxic shock in monocyte chemoattractant protein 1/cc chemokine ligand 2-defiient mice correlates with reduced interleukin 10 and enhanced macrophage migration inhibitory factor production. Shock 26 : 457–463.
19. FareseRVJr, WalterTC (2009) Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 139 : 855–860.
20. BeerninkPT, TolanDR (1994) Subunit interface mutants of rabbit muscle aldolase form active dimers. Protein Sci 3 : 1383–1391.
21. TsantesAE, BonovasS, TravlouA, SitarasNM (2006) Redox imbalance, macrocytosis, and RBC homeostasis. Antioxid Redox Signal 8 : 1205–1216.
22. MarinkovicD, ZhangX, YalcinS, LucianoJP, BrugnaraC, et al. (2007) Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Invest 117 : 2133–2144.
23. MatesJM, Perez-GomezC, OlallaL, SeguraJM, BlancaM (2000) Allergy to drugs: antioxidant enzymic activities, lipid peroxidation and protein oxidative damage in human blood. Cell Biochem Funct 18 : 77–84.
24. CrawfordJH, IsbellTS, HuangZ, ShivaS, ChackoBK, et al. (2006) Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood 107 : 566–574.
25. BerendseK, EbberinkMS, IjlstL, Poll-TheBT, WandersRJ, et al. (2013) Arginine improves peroxisome functioning in cells from patients with a mild peroxisome biogenesis disorder. Orphanet J Rare Dis 8 : 138.
26. RothSD, SchuttrumpfJ, MilanovP, AbrissD, UngererC, et al. (2012) Chemical chaperones improve protein secretion and rescue mutant factor VIII in mice with hemophilia A. PLoS One. 7: e44505.
27. SenesiP, LuziL, MontesanoA, MazzocchiN, TerruzziI (2013) Betaine supplement enhances skeletal muscle differentiation in murine myoblasts via IGF-1 signaling activation. J Transl Med 11 : 174.
28. WenH, TingJP, O'NeillLA (2012) A role for the NLRP3 inflammasome in metabolic diseases—did Warburg miss inflammation? Nat Immunol 13 : 352–357.
29. ArendWP, PalmerG, GabayC (2008) IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 223 : 20–38.
30. KrawczykCM, HolowkaT, SunJ, BlagihJ, AmielE, et al. (2010) Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115 : 4742–4749.
31. MaedlerK, SergeevP, RisF, OberholzerJ, Joller-JemelkaHI, et al. (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110 : 851–860.
32. FeingoldKR, MoserA, PatzekSM, ShigenagaJK, GrunfeldC (2009) Infection decreases fatty acid oxidation and nuclear hormone receptors in the diaphragm. J Lipid Res 50 : 2055–2063.
33. BarronJT, KoppSJ, TowJ, ParrilloJE (1994) Fatty acid, tricarboxylic acid cycle metabolites, and energy metabolism in vascular smoth muscle. Am J Physiol 267: H764–769.
34. SharmaS, AdrogueJV, GolfmanL, UrayI, LemmJ, et al. (2004) Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18 : 1692–1700.
35. VissingJ, HallerRG (2012) Mechanisms of exertional fatigue in muscle glycogenoses. Neuromuscul Disord 22 Suppl 3S168–171.
36. WaingehVF, GustafsonCD, KozliakEI, LoweSL, KnullHR, et al. (2006) Glycolytic enzyme interactions with yeast and skeletal muscle F-actin. Biophys J 90 : 1371–1384.
37. Ritterson LewC, TolanDR (2012) Targeting of several glycolytic enzymes using RNA interference reveals aldolase affects cancer cell proliferation through a non-glycolytic mechanism. J Biol Chem 287 : 42554–42563.
38. RangarajanES, ParkH, FortinE, SyguschJ, IzardT (2010) Mechanism of aldolase control of sorting nexin 9 function in endocytosis. J Biol Chem 285 : 11983–11990.
39. MerkulovaM, Hurtado-LorenzoA, HosokawaH, ZhuangZ, BrownD, et al. (2011) Aldolase directly interacts with ARNO and modulates cell morphology and acidic vesicle distribution. Am J Physiol Cell Physiol 300: C1442–1455.
40. WilflnigF, ThiamAR, OlarteMJ, WangJ, BeckR, et al. (2014) Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife 3: e01607.
41. ByunM, AbhyankarA, LelargeV, PlancoulaineS, PalanduzA, et al. (2010) Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med 207 : 2307–2312.
42. DjouadiF, AubeyF, SchlemmerD, RuiterJP, WandersRJ, et al. (2005) Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum Mol Genet 14 : 2695–2703.
43. MichotC, MamouneA, VamecqJ, ViouMT, HsiehLS, et al. (2013) Combination of lipid metabolism alterations and their sensitivity to inflammatory cytokines in human lipin-1-deficient myoblasts. Biochim Biophys Acta 1832 : 2103–2114.
44. GallucciS, ProvenzanoC, MazzarelliP, ScuderiF, BartoccioniE (1998) Myoblasts produce IL-6 in response to inflammatory stimuli. Int Immunol 10 : 267–273.
45. BeutlerE, BlumeKG, KaplanJC, LohrGW, RamotB, et al. (1977) International Committee for Standardization in Haematology: recommended methods for red-cell enzyme analysis. Br J Haematol 35 : 331–340.
46. LayzerRB, RowlandLP, RanneyHM (1967) Muscle phosphofructokinase deficiency. Arch Neurol 17 : 512–523.
47. RustinP, ChretienD, BourgeronT, GerardB, RotigA, et al. (1994) Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta 228 : 35–51.
48. DesseinAF, FontaineM, DobbelaereD, Mention-MulliezK, Martin-PonthieuA, et al. (2009) Deuterated palmitate-driven acylcarnitine formation by whole-blood samples for a rapid diagnostic exploration of mitochondrial fatty acid oxidation disorders. Clin Chim Acta 406 : 23–26.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- An RNA-Seq Screen of the Antenna Identifies a Transporter Necessary for Ammonia Detection
- Systematic Comparison of the Effects of Alpha-synuclein Mutations on Its Oligomerization and Aggregation
- Functional Diversity of Carbohydrate-Active Enzymes Enabling a Bacterium to Ferment Plant Biomass
- Regularized Machine Learning in the Genetic Prediction of Complex Traits
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy