
Genetic Analysis of a Novel Tubulin Mutation That Redirects Synaptic Vesicle Targeting and Causes Neurite Degeneration in
Axons and dendrites are two classes of neuronal process that differ in their functions and molecular compositions. Proteins important for synaptic functions are mostly synthesized in the cell body and sorted differentially into the axon or dendrites. Microtubules in the axon and dendrite maintain their structural integrity and regulate polarized protein transport into these compartments. We identified a novel α-tubulin mutation in C. elegans that caused mistargeting of synaptic vesicles and induced progressive neurite swelling, which resulted in late-onset neurodegeneration. We showed that this tubulin mutation weakened microtubule network and abnormally increased microtubule affinity for dynein, a motor protein responsible for cargo sorting to the dendrite. This enhanced microtubule-dynein affinity is due to augmented negative charge at the carboxyl terminus of α-tubulin. Neurite swelling and neurodegeneration could be ameliorated by reduced physical activity, suggesting that recurrent mechanical strain from muscle contraction jeopardized neurite integrity in the long run. Mutations in α - and β-tubulins are found in human neurological diseases; our findings therefore contribute to understanding the pathogenic mechanism of human neurological diseases associated with tubulin mutations.
Published in the journal:
. PLoS Genet 10(11): e32767. doi:10.1371/journal.pgen.1004715
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004715
Summary
Axons and dendrites are two classes of neuronal process that differ in their functions and molecular compositions. Proteins important for synaptic functions are mostly synthesized in the cell body and sorted differentially into the axon or dendrites. Microtubules in the axon and dendrite maintain their structural integrity and regulate polarized protein transport into these compartments. We identified a novel α-tubulin mutation in C. elegans that caused mistargeting of synaptic vesicles and induced progressive neurite swelling, which resulted in late-onset neurodegeneration. We showed that this tubulin mutation weakened microtubule network and abnormally increased microtubule affinity for dynein, a motor protein responsible for cargo sorting to the dendrite. This enhanced microtubule-dynein affinity is due to augmented negative charge at the carboxyl terminus of α-tubulin. Neurite swelling and neurodegeneration could be ameliorated by reduced physical activity, suggesting that recurrent mechanical strain from muscle contraction jeopardized neurite integrity in the long run. Mutations in α - and β-tubulins are found in human neurological diseases; our findings therefore contribute to understanding the pathogenic mechanism of human neurological diseases associated with tubulin mutations.
Introduction
Microtubule and molecular motors mediate polarized transport of neuronal proteins to either axons or dendrites [1]. Microtubules are oriented uniformly with their plus ends towards the distal end of the axon, which facilitates kinesin-dependent targeting of presynaptic proteins [2]. By contrast, targeting of postsynaptic molecules to the dendrite, such as glutamate receptors, requires the minus end-oriented dynein motors [3], consistent with the fact that many microtubules in the dendrites orient the minus end distally [4]. Mislocalization of presynaptic proteins to the dendrite occurs when this polarized pattern of axon-dendritic microtubule arrays is disrupted [5], [6], when kinesin function is compromised [7], [8], or when dynein activity is inadvertently increased [3], [7], [8]. Synaptic vesicle (SV) precursors are generated in the neuronal cell body and transported to the synapses by the unidirectional motor Kinesin 3/KIF1A [1]. On the other hand, the dynein motor complex mediates retrograde SV transport in the axon [1], [9]. Since SVs are cargos for both KIF1A and dynein, it is intriguing that they are exclusively targeted to the axon and prevented from entering the dendrites.
Previous biochemical and structural studies suggest that kinesin and dynein share an overlapping binding region at the C-terminus of α-tubulin [10]. The N-terminus of the H12 helix of the α-tubulin contains a stretch of absolutely conserved acidic residues (414EEGE, equivalent to 415EEGE in the yeast α-tubulin) and interacts with ATP-bound KIF1A [11]. A recent study on dynein structures also implicates this region in the interaction between microtubule and the microtubule-binding domain (MTBD) of dynein [12], although validation of this model in the context of in vivo, eukaryotic system is still lacking. Mutations of any of the three glutamic acids in the yeast α-tubulin to alanine dramatically reduced the frequency of kinesin binding to the microtubules [13]. Mutations of several conserved, acidic residues in the H12 helix of β-tubulin (E410, E412, D417) to alanine similarly reduced microtubule affinity for kinesins. Interestingly, E410K, D417H and D417N in the human β-tubulin TUBB3, among other point mutations, had been found in patients with congenital neurological syndrome with ophthalmoparesis and peripheral neuropathy [14]. In particular, TUBB3(E410K) and TUBB3(D417H), but not other disease-related TUBB3 mutations, were shown to impair the bindings of Kinesin 1/KIF5, Kinesin 3/KIF1A and KIF21 when expressed in cultured mammalian neurons [15]. The interaction between dynein and microtubule was not affected by these TUBB3 mutants [15]. These studies established the critical importance of negative charge in the H12 helix of the α - and β-tubulins in mediating microtubule-kinesin interaction, but the molecular mechanisms governing microtubule-dynein interaction and its physiological significance remain unexplored.
Here we describe a novel mutation of G416 in the α-tubulin MEC-12 of C. elegans to glutamic acid (G416E). Homologous mutations at this site of α-tubulin had not been reported in human diseases or tested in genetic model organisms. In C. elegans, mec-12 is highly expressed in the six touch receptor neurons that detect gentle mechanical stimulation on the worm cuticle [16], [17]. MEC-12 and the touch neuron-specific β-tubulin MEC-7 are required to form the unusual, 15-protofilament giant microtubules in these neurons [16]. These giant microtubules had been implicated in the transduction of mechanosensation, although the mechanisms remain enigmatic [18]. We show that this gain-of-function G416E mutation redirects SVs to non-axon compartment in the C. elegans mechanosensory neuron PLM, and it does so by increasing microtubule affinity for dynein.
Results
The gm379 mutation disrupted synaptic vesicle transport and caused synaptic vesicle mistargeting
In wild-type C. elegans, the bilaterally symmetric touch receptor neurons ALM and PLM develop a single anterior process that forms synapses in the nerve ring and in the ventral nerve cord, respectively (Figure 1A). Touch neuron synapses are enriched in RAB-3-(+) synaptic vesicles (SVs), the active zone protein SYD-2/Liprin-α, and mitochondria (Figure 1B, 1D, and S1) [19], [20]. The PLM neurons also have a short posterior process that does not form synapses. In an EMS mutagenesis screen (see Materials and Methods), we identified gm379, a mutant with prominent SV phenotypes in the touch neurons (Figure 1B′-1G′). gm379 animals lacked RAB-3-(+) SVs at the touch neuron synapses, and instead SVs accumulated in the neuronal soma (Figure 1B′-1E′, 1H). We refer to these as SV transport defects for the rest of the paper. Surprisingly, SVs were also redirected to the PLM posterior process, a phenotype that we call SV mistargeting (Figure 1E′, 1I). These results were confirmed using two other SV reporters, jsIs37(Pmec-7::SNB-1::GFP) that marked the SV membrane protein synaptobrevin/SNB-1, and jsIs219(Psng-1::SNG-1::GFP) labeling another SV protein synaptogyrin/SNG-1, with GFP (Figure 1F-1G′). These ectopic SVs showed very limited motility, and many were stationary (Figure 1J). We followed gm379 mutants through development, and confirmed that SV transport defects and mistargeting were present at early larval stages and progressively worsened (Figure 1H). The transport and targeting of synaptic active zone protein SYD-2 was affected to a much milder degree, and surprisingly, SYD-2 failed to mistarget to the PLM posterior process (Figure S1). The dissociation in the mutant phenotypes of SV and active zone proteins indicates that the gm379 mutation caused relatively specific defects in SV targeting rather than generally impaired axon transport or induced ectopic synapse formation.

The gm379 mutant developed microtubule disorganization, neurite swelling and degeneration of the touch neurons
In addition to SV transport defects, the gm379 mutant touch neurons had progressive neurite swelling and misshapen soma (Figure 2A and S2A). Neurite defects evolved from small beadings at early larval stages into triangular-shaped swellings in L4 and adult animals, and mitochondria were frequently found to be present at the swellings (Figure 2A, 2B and S2A). These swellings were dynamic in morphology, as movements often induced reversible buckling of the neurite and changed the width and height of the swellings (Figure 2C and Video S1), which was similar to what had been described earlier for the tubulin acetyltransferase mutant mec-17 [21]. Neurite buckling or swelling were never seen in the wild type even under maximal muscle contraction induced by levamisole. This observation suggests that the gm379 mutation rendered the touch neurite susceptible to deformation under mechanical strain, a phenotype that was also seen when the membrane skeleton protein UNC-70/β-spectrin was lost [22]. We therefore test whether genetic paralysis of the animals suppresses neurite defects of the gm379 mutant. Mutation in the muscle myosin gene unc-54 almost completely paralyzed the animals, and it significantly reduced the number of neurite swellings in the gm379 mutant touch neurons (Figure 2D and 2E). This result implies that the gm379 mutation compromises the ability of the touch neurites to cope with mechanical stress.

We were curious whether massive neurite swellings in the gm379 mutant predispose touch neurons to degeneration. We first performed longitudinal imaging of individual ALM and PLM neurons through adulthood, and found that touch neurites in the gm379 mutant underwent progressive disorganization (Figure 2F). Touch neuron degeneration, characterized by swelling of neuronal soma, neurite interruption and extensive beadings, began to emerge in the gm379 mutant at D9 and progressively increased (Figure 2G and 2H). Touch neuron degeneration was extremely rare in the wild type at comparable age (Figure 2G and 2H) [23]. Interestingly, touch neuron degeneration of the gm379 mutant was suppressed by the unc-54 mutation (Figure 2G and 2H). These results indicate that recurrent mechanical strain imposed on the touch neurons during locomotion is an important precipitating factor for late-onset neurodegeneration in the gm379 mutant.
Under serial thin-section electron microscopy, we found that the characteristic 15-protofilament microtubules of C. elegans touch neurons were preserved in the gm379 mutant, including those in the PLM posterior process (Figure 3A-C, S2B-D). Mitochondria could be found where touch neuron processes swelled abnormally (Figure 3B), consistent with our light microscopic observation (Figure 2B). In longitudinal sections, in contrast to the wild type, where neuronal microtubules formed long straight bundles, touch neuron microtubules in the gm379 animals curved focally at sites of organelle accumulation (Figure 3D). We observed bending and splitting of neuronal microtubule bundles at sites of mitochondria accumulation in focal axonal swellings (Figure 3D4). These ultrastructural studies suggest that the microtubule network of the touch neurons is abnormal in the gm379 mutant.
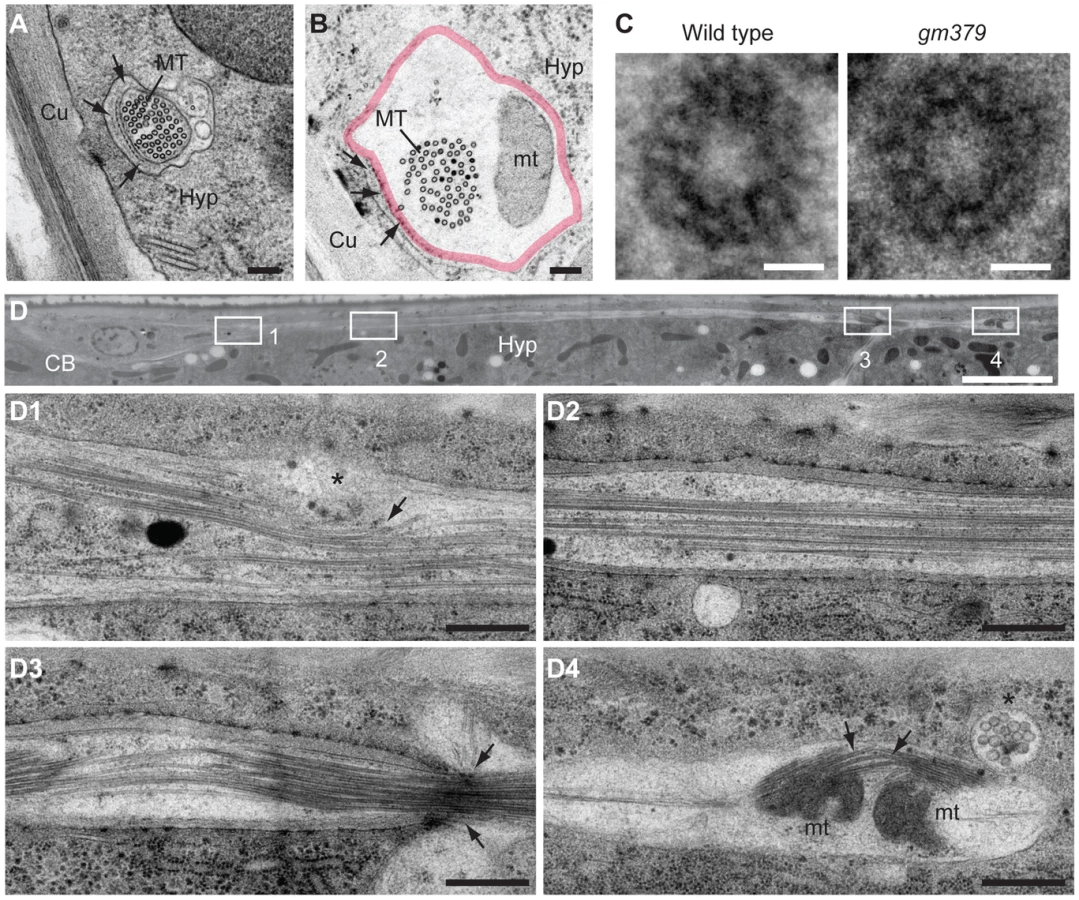
gm379 is a neomorphic allele of the α-Tubulin mec-12
We cloned gm379 by single nucleotide polymorphism (SNP) mapping, complementation test and DNA sequencing, and found that it contained a missense mutation of the touch neuron-specific α-tubulin mec-12 that alters an absolutely conserved C-terminal glycine residue to glutamate (G416E) [19], [17] (Figure 4A and 4B). Interestingly, gm379 animals were touch-insensitive (30% touch-sensitive, compared to wild type, 89%; and mec-12(e1607) null, 12%, n>35). We found another intron mutation in gm379 that was distant to exon-intron junctions (nucleotide 828 of unspliced transcript, G to A mutation). Two null mutants of mec-12, e1607 and tm5083, also had SV transport defects, but not SV mistargeting in the PLM or axon swelling in the touch neurons (Figure 4C, S3). RNAi against mec-12 in the gm379 mutant almost completely abolished axon swelling or SV mistargeting, indicating that these two phenotypes were neomorphic (Figure 4C, 4D). Expression of the MEC-12(G416E) mutant tubulin in the mec-12 null mutants recapitulated the SV mistargeting phenotypes of the mec-12(gm379) mutant (67% of transgenic animals showed SV mistargeting, n = 21 v.s. 6.5% of array-loss siblings showing SV mistargeting, n = 77), confirming that SV mistargeting and neurite swelling were indeed caused by the mec-12(gm379) rather than other unidentified mutations in the background.

To gain further insights into the genetic nature of the mec-12 phenotypes, we analyzed various heterozygous mec-12 mutants as well as trans-heterozygotes between these alleles (Figure 4E). Heterozygous animals containing the e1607, tm5083 or gm379 mutations all showed moderate SV transport defects that were less severe than the homozygous animals. These observations suggest that loss of mec-12 functions causes semi-dominant SV transport defects due to haploinsufficiency. Heterozygous gm379 mutants did not display neurite swelling or SV mistargeting. By contrast, mec-12(gm379)/mec-12(e1607) and mec-12(gm379)/mec-12(tm5083) trans-heterozygous mutants had SV mistargeting without neurite swelling, implying that the presence of wild-type MEC-12 somehow prevents MEC-12(G416E) from mistargeting SVs or disrupting microtubule organization. In conclusion, our genetic analysis indicates that gm379 causes both neomorphic (neurite swelling and SV mistargeting) and semi-dominant loss-of-function (SV transport defects) phenotypes of mec-12.
SV mistargeting in gm379 mutant was independent of microtubule polarity, tubulin posttranslational modifications or UNC-104/KIF1A
Stable microtubules formed in the gm379 touch neurons, based on the EM data and the preserved lysine 40 (K40) acetylation of α-tubulin [17], [24], [25] (Figure S4A). Moreover, a mec-7/β-tubulin null mutation completely suppressed neurite swelling and dramatically reduced SV mistargeting of mec-12(gm379) (Figure S5), suggesting that SV mistargeting and neurite swelling phenotypes require intact microtubules. Labeling touch neuron microtubules with the plus end-binding protein EBP-2::GFP showed that in the wild type, microtubules oriented plus-end distally in the anterior PLM process (Figure S6) [26]. In the PLM posterior process, microtubules showed mixed polarity (Figure S6). These patterns of microtubule polarity were preserved in the mec-12(gm379) mutant (Figure S6).
A recent study reported that mutations in the tubulin acetyltransferase mec-17 caused neurite degeneration with SV mislocalization in the touch neurons [27]. To test whether altered microtubule posttranslational modifications are responsible for SV mistargeting in the mec-12(gm379) mutant, we performed immunostaining experiments, but did not observe gross difference in microtubule acetylation or tyrosination in the touch neurons between the wild type and the mec-12(gm379) mutant (Figure S4A). Tubulin polyglutamylation signal was restricted to the amphid and phasmid sensory cilia as in the wild type, and was not ectopically expressed in the mutant touch neurons (Figure S4B). Moreover, we performed feeding RNAi to knock down the tubulin polyglutamylase ttll-4, the tubulin deglutamylase ccpp-6, and a few genes (ttll-5, ttll-12, ttll-15 and ccpp-1) that bear sequence homology to human tubulin amino acid ligases (Wormbase at http://www.wormbase.org) [28], [29], in both wild type and the mec-12(gm379) mutant. None of these RNAi resulted in SV mistargeting in the wild type or suppressed SV mistargeting in the mec-12(gm379) animals. Based on these results, we conclude that SV mistargeting in the mec-12(gm379) is not a consequence of altered microtubule posttranslational modifications.
It is possible that the interaction between KIF1A and microtubule was altered by the G416E mutation. We found that the strong loss-of-function unc-104(rh43)/KIF1A mutation caused completely penetrant SV transport defects and, surprisingly, low percentage of SV mistargeting in the PLM (Figure 5A and 5B). Moreover, this unc-104 mutation enhanced SV mistargeting of mec-12(gm379) rather than suppressing the phenotype, with more SVs mistargeted to the PLM posterior process and distributed more distally (Figure 5C, 5D). This result suggests that SV mistargeting in the mec-12(gm379) mutant is not caused by aberrant UNC-104 activity. Furthermore, overexpression of UNC-104 significantly rescued SV transport defects and mistargeting in the mec-12(gm379) mutant, with more SVs reaching synapses in the nerve ring or entering the anterior ALM and PLM processes (Figure 5E-5H, Figure S7A, S7B). While these data are consistent with the interpretation that UNC-104 activity was reduced in the mec-12(gm379) mutant, resulting in severe SV transport defects, they also indicate that SV mistargeting in the mutant requires the activity of an unknown molecule.

SV mistargeting in the gm379 mutant requires DHC-1/Dynein
We wondered whether increased activity of the minus end motor dynein is responsible for SV targeting to the PLM posterior process in the mutant, based on the presence of minus end-out microtubules in the PLM posterior process and the unc-104 effects. dhc-1 encodes the heavy chain for cytoplasmic dynein in C. elegans [30]. If enhanced dynein activity is responsible for SV mistargeting in the mutant, elimination of dynein function should suppress it. We could observe SV mistargeted to the PLM posterior process as early as 2-3 fold embryos, before the animal hatched. With the available dhc-1 mutant alleles, it was not possible to lose DHC-1 functions at such early stages without compromising animals' viability. To eliminate DHC-1 functions as early as possible, and to circumvent lethality due to widespread DHC-1 loss, we specifically knocked down dhc-1 in the touch neurons, but not in other somatic tissues, by simultaneously expressing sense and antisense dhc-1 from the mec-7 promoter, which we named transgenic dhc-1 RNAi. Strikingly, transgenic dhc-1 RNAi significantly suppressed SV mistargeting of the mec-12(gm379) mutant, with about one third of the transgenic animals completely devoid of mistargeted SVs (Figure 6A, 6B). This result was confirmed by another independently generated dhc-1 RNAi array (Figure S8A). Transgenic dhc-1 RNAi also significantly reduced SV mistargeting in the unc-104; mec-12(gm379) mutant (Figure 6C). In the wild type, transgenic dhc-1 RNAi had little effects on the intensity of GFP::RAB-3 or SNB-1::GFP in the PLM soma or synapses (Figure S8B). These data indicate that SV mistargeting in the mec-12(gm379) mutant is mediated by the dynein motor. The neurite swelling phenotypes of the mutant, by contrast, were not changed by dhc-1 RNAi, suggesting that SV mistargeting and neurite defects are mechanistically distinct.

We noted that eliminating DHC-1 functions in the mec-12(gm379) mutant significantly restored synaptic targeting and anterograde transport of SVs, with concomitant reduction of SV accumulation in the touch neuron soma (Figure 6A and 6D). These effects were similar to those caused by excess UNC-104 (Figure 5E-H, S7A, S7B). Indeed, synaptic targeting and anterograde transport of SVs were completely abolished in the dhc-1; unc-104; mec-12(gm379) triple mutant, suggesting that the phenotypic rescue caused by the dhc-1 mutation requires UNC-104 (Figure 6D). Although significantly increased compared to those in the mec-12(gm379) mutant, SV signals in the nerve ring synapses of the dhc-1; mec-12(gm379) were still much weaker than the wild type. Together with the severe reduction of presynaptic SVs in the mec-12(gm379) mutant, these observations suggest that UNC-104 was not able to display a full range of activity on the mutant microtubule scaffolds.
MEC-12(G416E) microtubules had increased affinity for dynein
To test whether the MEC-12(G416E) mutant microtubules have increased affinity for dynein, we performed microtubule sedimentation in transgenic strains expressing MEC-12 and MEC-7 pan-neuronally, and assayed for the amount of DHC-1 or UNC-104 associated with microtubules by blotting with anti-DHC-1 and anti-UNC-104 antibodies, respectively. The presence of MEC-12-containing, stable microtubules was verified by detecting 6-11B-1 antibody immunoreactivity in neurons other than the touch receptors (). After normalization to tubulin, the amount of DHC-1 co-sedimented with MEC-12(G416E)-containing microtubules dramatically increased, compared to that co-sedimented with wild-type microtubules (Figure 6E). In support of this conclusion, we found that GFP::DHC-1, expressed from the low-copy integrated transgene orIs17(Pdhc-1::gfp::dhc-1), which drives DHC-1 expression from the endogenous promoter, was accumulated in the PLM posterior process in the mec-12(gm379), but not in the wild type (Figure 6F). Together these results indicate that mutant microtubules have increased affinity for DHC-1. To our surprise, we did not detect a change in the amount of UNC-104 co-sedimented with mutant microtubules (Figure 6E) or a change of UNC-104 protein localization in the PLM neuron. This implies that UNC-104 can still associate with the mutant microtubules, although its function is somehow disrupted.
Negative charges at the H12 helix of MEC-12 instruct SV targeting
The aforementioned data indicate that G416 of MEC-12 plays a critical role in determining the relative affinity of microtubules for dynein. To further decipher the mechanisms that instruct microtubule-dynein affinity, we systemically replaced G416 with acidic (aspartic acid/D) or basic (lysine/K, arginine/R) residues, as well as alanine (A) and glutamine (Q), the latter being similar to glutamic acid in side chain length but did not carry charges (Figure 7A, 7B). These MEC-12 species were expressed in the touch neurons of the mec-12(e1607) null mutant. SV mistargeting was seen only with the expression MEC-12(G416D), but not other G416 substitutions (Figure 7A, 7B). These results suggest that SV mistargeting in the gm379 mutant was caused by the increased negative charges at the EEGE cluster of MEC-12.

Is the arrangement of the acidic residues important for dynein affinity of microtubules? To answer this question, we moved the glycine to residue 414, 415 or 417, with reciprocal glutamic acid substitution at 416 (E414G/G416E, E415G/G416E, G416E/E417G, referred as GEEE, EGEE and EEEG, respectively; Figure 7A, 7B), so that the arrangement, but not the sum, of negative charges was altered, and asked whether this manipulation affects SV targeting. While expression of MEC-12(GEEE) or MEC-12(EGEE) did not result in significant SV mistargeting, expression of MEC-12(EEEG) triggered SV mistargeting in about 30% of the mec-12(e1607) animals (Figure 7A, 7B). These observations indicate that SV targeting critically depends on the magnitude and the spatial arrangement of negative charges in the EEGE cluster of the H12 helix.
Mutation that disrupted intramolecular salt bridge in DHC-1 enhanced DHC-1 affinity with microtubules
Previous structural studies suggest that dynein binds the H12 helix of α-tubulin [10], [12]. Redwine et al. proposed that E3378 and R3382 of the dynein microtubule-binding domain (MTBD) form intramolecular salt bridge, and upon approaching the tubulin dimer, negative charges of the α-tubulin H12 disrupt this salt bridge by attracting R3382, which carries positive charges (Figure 8C). An E3378K mutation of MTBD disrupted this intramolecular salt bridge and increased both the affinity and the run length of dynein on the microtubules [12]. We speculate that the E3378K mutation facilitates the electrostatic interaction between MTBD and the negative charges of the α-tubulin H12 domain. To test this, we expressed and purified a fragment of C. elegans DHC-1 MTBD, and showed that this MTBD precipitated with microtubules synthesized from purified bovine tubulin in the in vitro sedimentation experiment (Figure 8A and S9). Moreover, D3323K mutation, which is equivalent to E3378K mutation of the yeast dynein MTBD, enhanced MTBD-microtubule interaction across a range of tested concentrations (Figure 8A, 8B and S9). This result supports our hypothesis that exaggerated microtubule affinity for dynein critically depends on the electrostatic interaction between the EEGE-containing H12 helix of α-tubulin and the dynein MTBD (Figure 8C).

Discussion
In the present study, we characterized a gain-of-function mec-12/tubulin mutant that displayed synaptic vesicle mistargeting and neurite swelling phenotypes, which were absent in the mec-12 null mutant. In this mutant, single amino acid substitution augments microtubule-dynein interaction, thereby mistargeting synaptic vesicles to non-axon compartments. This observation extends previous structural studies on the charged cluster of the α-tubulin H12 helix and provides a biological context for such charge-based coupling between microtubule and dynein. The neurite swelling and degeneration phenotypes could bear important implications for human neurological diseases associated with missense tubulin mutations, as discussed below.
Previous structural studies suggest that dynein binds the H12 helix of α-tubulin, and the interaction between dynein and microtubule is not as strong as that between kinesins and microtubule [10], [12]. One advantage for this suboptimal dynein-microtubule affinity is flexibility and dynamic range for dynein processivity on the microtubule scaffolds. The hypothesis that dynein-microtubule interaction is not optimized also suggests that it is possible to further enhance dynein-microtubule affinity by mutating residues at this interface. Here, we show that this is indeed the case: by increasing negative charges of the α-tubulin H12 helix or augmenting positive charges of the dynein MTBD, microtubule-dynein interaction was strengthened. More importantly, we show that this aberrantly high affinity of microtubule for dynein redistributed synaptic vesicles to ectopic compartments in the neurons. We speculate that the suboptimal affinity of microtubule for dynein prevents initial SV entry into the dendrite, yet allows for retrograde SV transport within the axon. The G416E mutation of MEC-12 increased microtubule affinity for dynein probably at the expense of dynein processivity: we found that motility of the mistargeted SVs in the PLM posterior process was profoundly compromised (Figure 1J). Consistent with this view, Redwine et al. showed that E3378K mutation of MTBD caused about 40% reduction in the velocity of dynein movements on microtubules. By contrast, none of the G416 substitutions restored the UNC-104/KIF1A-dependent, anterograde SV transport in the mec-12(e1607) null mutant. This is also consistent with the observation by Redwine et al. that microtubule-kinesin interaction was molecularly optimized, therefore changes to the tubulin residues at the microtubule-kinesin interface will only compromise but not compensate for or even further improve this interaction.
Rather than merely serving as a track, mounting evidence indicates that microtubules play an active role in directing axon transport [2]. One such example is the various posttranslational modifications on microtubules, which regulate axon transport by restricting different kinesin motors to discrete subcellular compartments [31], [32]. The intrinsic organization of microtubule is another critical factor regulating polarized axon transport. UNC-33 and UNC-44, the C. elegans homologs for the Collapsin Response Mediator Protein 2 (CRMP2) and neuronal ankyrin, respectively [33], [34], regulate axon transport by maintaining uniform microtubule polarity in the axon and the dendrite [5]. In C. elegans head neurons, microtubules are uniformly oriented with their minus-ends towards the distal of the dendrite. In the unc-33 and the unc-44 mutants, dendritic microtubules showed mixed polarity, which resulted in aberrant sorting of axonal proteins into the dendrite [5]. In these mutants, the dendritic localization of SVs requires UNC-104. Of note, mistargeting of axonal proteins in the unc-33 and unc-44 mutants occurred to the SVs and multiple active zone components. By contrast, SYD-2 was not mistargeted in the mec-12(gm379) mutant, and we found that SV transport in the touch neurons was not affected in unc-33 and unc-44 mutants. These observations indicate that while perturbation to the gross architecture of microtubule causes extensive mistargeting of multiple axonal cargos, changes at a restricted, yet functionally critical site of microtubule could lead to targeting defects of specific axonal proteins.
Many tubulin mutations associated with human diseases are point mutations that alter protein function rather than eliminating protein products [35]–[37]. Mutant tubulins are still incorporated into microtubule polymers; phenotypes presumably arise from altered microtubule dynamics or disrupted interactions with molecular motors or microtubule-associated proteins [14], [15], [38]. The G416E mutation of MEC-12 caused extensive neurite swellings that eventually led to degeneration of the touch neurons. Ultrastructurally, there were unbundling of microtubules and accumulation of mitochondria at focal neurite swellings. Interestingly, these neurite defects were independent of dynein activity, but could be suppressed by genetic paralysis of locomotion. In light of two recent studies that showed touch neurite buckling or swelling when unc-70/β-spectrin or mec-17/tubulin acetyltransferase was mutated [21], [22], this suggests that microtubules confer resistance of touch neurites to deformation imposed by constant muscle activity. When this structural resilience is compromised, touch neurons are likely to degenerate presumably in a wear-and-tear fashion induced by the animal's constant movements. Supporting this notion, commissural axons in unc-70 mutants also increasingly break as the animals grow and move, and paralysis of the animals prevents axon interruption [39]. Unlike the mec-17 mutant, in which defective tubulin acetylation was associated with abnormal microtubule protofilament number, tubulin acetylation and microtubule protofilaments were unaffected in the mec-12(gm379) mutant. We hypothesize that the G416E substitution may alter the association of microtubule-binding proteins with microtubules, changing the stability or structure of microtubule lattice and rendering the neurites susceptible to mechanical strain. It would be interesting to test whether any of the tubulin mutations found in human diseases generates similar axon defects and could also be ameliorated by reduced activity of neighboring musculature. The molecular mechanism by which G416E mutant microtubules cause axon degeneration awaits future investigation.
Materials and Methods
C. elegans mutants, transgenes and RNAi
Strains were cultured as described [40]. The following alleles were used in this study: N2 (Bristol strain), CB4856, LG I: dhc-1(or283ts), unc-54(e190); LG II: unc-104(rh43); LG III: mec-12(e1607) (a gift from Martin Chalfie, Columbia University), mec-12(tm5083), mec-12(gm379); LG V: sid-1(pk3321); him-5(e1490); LG X: mec-7(ok2152). Transgenes used in the current study are: jsIs37(Pmec-7::SNB-1::GFP)/IV, jsIs219(Psng-1::SNG-1::GFP)/II, jsIs821(Pmec-7::GFP::RAB-3)/X, jsIs973(Pmec-7:mRFP)/III, jsIs1111(Pmec-4::UNC-104::GFP), jsIs1238(Pmec-7::SYD-2::GFP) (jsIs821, jsIs973, jsIs1111, and jsIs1238 are gifts from Michael Nonet, Washington University), juIs76(Punc-25::GFP)/II, Punc-17::RFP/V (a gift from Joshua Kaplan, Massachusetts General Hospital), orIs17[Pdhc-1::GFP::DHC-1, unc-119(+)] (a gift from Bruce Bowerman, University of Oregon), otIs118(Punc-33::GFP)/IV, uIs71(Pmec-18::SID-1, Pmyo-2::mCherry), zdIs5[Pmec-4::GFP, lin-15(+)]/I, twnEx8(Pmec-7::TOMM20::mCherry, Pmyo-2::gfp) (“Pmec-7::mito::mCherry”), twnEx40(Pmec-7::GFP::EBP-2, Pdpy-30::dsRed), twnEx42[Pmec-7::dhc-1(RNAi), Pmyo-2::GFP], twnEx55[Pmec-7::MEC-12(G416E, E417G), Pdpy-30:: NLS::dsRed], twnEx73[Pmec-7::MEC-12(G416E) 5 ng/µl, Pdpy-30::NLS::dsRed], twnEx74[Pmec-7::MEC-12(G416D), Pdpy-30:: NLS::dsRed], twnEx75[Pmec-7::MEC-12(G416Q), Pdpy-30:: NLS::dsRed], twnEx76[Pmec-7::MEC-12(G416A), Pdpy-30:: NLS::dsRed], twnEx77[Pmec-7::MEC-12(G416K), Pdpy-30:: NLS::dsRed], twnEx78[Pmec-7::MEC-12(G416R), Pdpy-30:: NLS::dsRed], twnEx79[Pmec-7::MEC-12(E414G, G416E), Pdpy-30:: NLS::dsRed], twnEx80[Pmec-7::MEC-12(E415G, G416E), Pdpy-30:: NLS::dsRed], twnEx88[Pmec-7::MEC-12(G416E) 10 ng/µl, Pdpy-30::NLS::dsRed], twnEx89[Pmec-7::dhc-1(RNAi), Pdpy-30:: NLS::dsRed], twnEx98[Punc-119::MEC-12, Punc-119::MEC-7, unc-119(+)], twnEx99[Punc-119::MEC-12(G416E), Punc-119::MEC-7, unc-119(+)], Ex(Punc-104::UNC-104::GFP), Ex(Punc-104::UNC-104::mRFP) (both from Oliver Wagner, National Tsing-Hua University, Taiwan). mec-12(e1607) is a G to A point mutation at nucleotide 430 of mec-12 cDNA, resulting in glycine to serine mutation at amino acid 144. Germ line transformation was performed by microinjection of purified DNA of interest as described [41].
EMS screen identification of gm379
Initially, an EMS mutagenesis screen was performed in the zdIs5; cwn-1(ok546) animals to identify mutations that cause synthetic polarity defects of the touch neurons [42]–[44]. While causing no defects in neuronal polarity, gm379 was recovered due to its prominent axonal defects. The cwn-1 mutation was then removed from the mutant. The SV transport defects, SV mistargeting and axonal swellings were all independent of the cwn-1(ok546) mutation in the background, and the cwn-1 mutant displayed none of the gm379 phenotypes.
Single nucleotide polymorphism (SNP) mapping
Single nucleotide polymorphism mapping was performed as described [45]. zdIs5 was included in this mapping to assist the identification of the homozygous gm379 mutants. In brief, male animals of the Hawaiian strain CB4856 were crossed to zdIs5; gm379, and gm379 homozygotes were later recovered from F2 progeny. F3 animals from individually cloned F2 animals were washed off plates and genomic DNA extracted by proteinase K treatment. To map gm379, we selected 48 SNPs from the 5 autosomes and the sex chromosome, and semi-quantitatively determined the ratio of N2/Hawaiian SNP for each locus using the restriction enzyme DraI. Our SNP mapping located gm379 to a region between -12 and +7 MU of Chromosome III, a region that contains the mec-12 locus.
RNAi experiments
Feeding RNAi was performed as described [46], with 1 mM IPTG pre-induction for 2 hours. For touch neuron-specific RNAi, we used jsIs973(Pmec-7::mRFP) mec-12(gm379); sid-1(pk3321); jsIs821(Pmec-7::GFP::RAB-3); uIs71(Pmec-18::SID-1, Pmyo-2::mCherry) animals [47]. The only RNAi-sensitive cells in the sid-1; uIs71 background are the six touch receptor neurons. Five L4 animals were placed on the RNAi plates and cultured at 20°C. The F1 progeny of were then transferred to another freshly prepared RNAi plates at L4, and their progeny (F2) scored for axon and synaptic vesicle phenotypes. Each RNAi experiment was repeated three times to confirm the results. Efficiency of feeding RNAi against neuronal genes in this genotype was confirmed by mec-12 RNAi, which resulted in 55% animals losing the PLM branch (n = 22), with control RNAi having no effects (0%, n = 27). This penetrance was similar to what was observed in the mec-12(e1607) null mutant (58%, n = 60). Additional RNAi control included mec-7 and rho-1 and all showed results comparable with mutant analysis.
Molecular biology and plasmid construction
Cloning and construction of plasmids were performed with standard molecular biology techniques. All expression constructs in the twnEx series transgenes were in the pPD95.77 Fire vector backbone, which contains the unc-54 3′-UTR for optimized expression in C. elegans. Primer sequence information is available upon request.
Scoring of ALM and PLM axon defects
Neurite swelling of ALM and PLM was scored in live animals with the integrated GFP reporter zdIs5(Pmec-4::GFP), which is expressed in the six mechanosensory neurons: ALMs, PLMs, AVM and PVM. Beading is defined as oval or round swelling along primary axons. Neurite swelling is defined as triangular protrusion or looping of axonal membrane. Neurodegeneration is define as swelling and round-up of the neuronal soma with neurite interruption, thinning and large beading formation. To characterize the evolution of neurite defects in mutants, wild type and gm379 animals were synchronized by hatching and arresting early L1 in M9 at 20°C. Animals were then allowed to feed on regular E. coli plates with axon morphology scored at different time points (6 hr, 12 hr, 24 hr, 36 hr, 48 hr, and 60 hr post hatching) that correspond to distinct larval and adult stages. Because unc-54 animals are defective in egg laying and die from progeny hatched inside their bodies, 5-fluoro-2′-deoxyuridine (FUdR) was added to the plate at the final concentration of 50 µM to stop progeny production. FUdR was applied to the mec-12(gm379) mutant and the wild type in experiments where unc-54 was also tested.
Synaptic vesicle scoring
Synaptic vesicles in the touch neurons were visualized with the integrated GFP reporter jsIs821(Pmec-7::GFP::RAB-3) which labels synaptic vesicles in the six touch neurons. The authenticity of synaptic vesicle defects in the gm379 mutant was confirmed with another GFP reporter, jsIs37(Pmec-7::SNB-1::GFP). Touch neurons were simultaneously labeled by the RFP reporter jsIs973(Pmec7::mRFP). Animals were synchronized and jsIs821-labeled synaptic vesicles quantified at distinct developmental stages. Images were acquired using the 63x Carl Zeiss Apochromat objective and the Zeiss AxioImager M2 imaging system. Because the posterior PLM process was very thin (less than 0.5 µm, see Figure S2B and S2C), we did not take confocal z-axis image stacks for pixel quantification. Pixel density was derived using ImageJ by quantifying total pixel number divided by the area marked by the neuronal marker jsIs973. We excluded the neuronal nucleus when quantifying pixel density of the soma. For Figure 5G, because the UNC-104-overexpression array carries mCherry fused to UNC-104, which may complicate determination of neurite area by the jsIs973 marker, we decided to quantify total pixel number of fluorescence on the entire PLM posterior process. Mistargerted SVs often formed GFP::RAB-3 aggregates of variable size, and we therefore did not quantify GFP punctum number or individual punctual intensity. The length of the PLM posterior process was measured by the software Axio Vision Rel. 4.8. The distance of synaptic vesicle distribution was determined as the fraction of the PLM posterior process marked with synaptic vesicle GFP. All image quantification was done blind to avoid bias.
Electron microscopy
Worms were high pressure frozen in either a Bal-Tec HPM 010 (Bal-Tec AG, Liechenstein) or Leica HMP 100 (Leica Microsystems, Vienna) high pressure freezer and freeze substituted in 1% osmium tetroxide and 0.1% uranyl acetate in acetone over a period of 2 hours by the SQFS method of McDonald and Webb [45]. Infiltration of Epon epoxy resin was carried out by 15 minute incubations in 25, 50, and 75% acetone-resin mixtures on a rocker, then three 15 minute incubations in pure resin. Polymerization of resin was for 2 hours in a 100°C oven. Sections of 70 nm thickness were post-stained with 2% uranyl acetate in 70% methanol for 4 minutes and lead citrate (Reynolds, 1963) for 2 minutes. Images were viewed on a Tecnai 12 (FEI Inc., Hillsboro, OR, USA) transmission electron microscope operating at 120 kV, and images recorded with a Gatan Ultrascan 1000 CCD camera (Gatan Inc., Pleasanton, CA, USA). Some high magnification views of microtubule were taken out of focus in order to highlight protofilament patterns [48], [49].
Microtubule polarity by EBP-2::GFP dynamic imaging
EBP-2 comets were barely visible in wild type touch neurons, which could be attributed to the very stable microtubule structures in these cells. Therefore we devised an assay in which low-dose (0.125 mM) colchicine was applied to the worms to generate a moderate level of microtubule perturbation. L2 worms with twnEx40(Pmec-7::EBP-2::GFP, Pmyo-2::GFP) were grown on colchicine-containing NGM plates for 8 hours, picked off the plates and imaged one hour later. Under such treatment, a significant percentage of touch neurons displayed variable degree of microtubule growth with EBP-2 comets. Imaging acquisition was performed with the Zeiss AxioImager M2 imaging system.
Immunostaining
Worm immunostaining was performed as described [50]. Briefly, mixed-stage animals were flash-frozen in liquid nitrogen and fixed in 2% paraformaldehyde on ice for at least 4 hours, permeabilized by Tris-Triton, and subjected to series of reduction and oxidation by sequential β-mercaptoethanol, dithiothreitol (DTT) and hydrogen peroxide treatment in 1% borate base buffer, and stained with primary antibodies in PBST-A. The following primary antibodies were used in this study: 6-11B-1 (mouse monoclonal anti-K40 acetylated α-tubulin, 1∶500, Santa Cruz Biotech), GT335 (mouse monoclonal anti-polyglutamylated tubulin, 1∶100, Enzo Life Sciences), rabbit polyclonal anti-detyrosinated tubulin (1∶200, Millipore), YL1/2 (rat monoclonal anti-tyrosinated tubulin, 1∶200, Santa Cruz Biotech), and rabbit polyclonal anti-GFP (1∶250, Santa Cruz Biotech). Secondary antibodies are goat anti-rabbit, goat anti-rat or goat anti-mouse IgG conjugated with Alexa488 or Alexa568 used at 1∶100 (Molecular Probes). Animals were counterstained with DAPI at 1∶1000 diluation in 2% n-propylgallate (NPG) and observed with the Zeiss AxioImager M2 imaging system. For fluorescence confocal microscopy, L4 to young adult hermaphrodite animals were anesthetized with 1% sodium azide, mounted on agar pad, and observed under Zeiss LSM700 confocal imaging system.
Purification of DHC-1 MTBD
A fragment of the microtubule-binding domain (MTBD, amino acids 3207-3372) of C. elegans DHC-1 was cloned into the KpnI site of the pET30α vector with the primers: 5′ GGTACCCTCGCAGAGCAGCTGAAG 3′ (forward) and 5′ GGTACCTTATTCCTGGGTCTTCTTTGCAGC 3′ (reverse), and tagged at the N-terminus with 6xHis. Expression in E. coli was induced by 0.5 mM IPTG at 16°C for 4 hours, for avoiding protein aggregate formation in subsequent steps of purification. Bacterial pellet was collected by centrifugation and resuspended in the lysis buffer containing 50 mM NaH2PO4, 500 mM NaCl, 10 mM imidazole, 0.1% lysozyme, protease inhibitor cocktail and were homogenized by sonication. Cell extract was centrifuged at 15,000 g for 30 min at 4°C and MTBD was purified by passing the supernatant through HisPur™ Ni-NTA resin (Thermo Fisher Scientific, Walthem, USA). Purified MTBD was dialyzed in HEPES buffer (80 mM HEPES pH 7.0, 2 mM MgCl2, 0.5 EGTA) for microtubule binding protein spin-down assay.
Microtubule sedimentation assay with worm protein extract
The microtubule sedimentation assay was performed as described with modifications [51]. In brief, unc-119; twnEx98[Punc-119::MEC-12, Punc-119::MEC-7, unc-119(+)] and unc-119; twnEx99[Punc-119::MEC-12(G416E), Punc-119::MEC-7, unc-119(+)] transgenic animals were grown to gravid adults on standard NGM plates. Animals were collected by washing and centrifugation in 0.1 M PIPES (pH 6.94), 4.0 mM MgCl2, 5 mM EGTA, 0.1 mM EDTA, 0.9 M glycerol, 1 mM PMSF, and 1 mM DTT (PMEG) at 4°C and resuspension in cold PMEG with protease inhibitor cocktail. Worms were then manually homogenized and centrifuged at 20,000xg for 45 min and the pellet discarded. The supernatant was centrifuged at 150,000xg for 60 min and the pellet discarded. The translucent supernatant was then supplemented with 2 mM GTP, 10 pM taxol, 1 U/ml hexokinase, 50 mM glucose, and 25–50 nm AMP-PNP and incubated on ice for 90 min for microtubule polymerization. Microtubule polymers were sedimented through 20% sucrose cushion made in PMEG with10 pM taxol by centrifugation at 20,000xg for 90 min. The pellet was resuspended in 1 ml PMEG containing 10 nM taxol and 50 mM NaCl. Microtubules and associated proteins were pelleted at 20,000xg for 40 min and dissolved in water for further analysis.
In vitro microtubule binding protein spin-down assay
In vitro microtubule sedimentation assay was performed by using microtubule binding protein spin-down assay kit (Cytoskeleton, Denver, USA). In brief, microtubules were synthesized from purified bovine tubulins and incubated with purified MTBD at room temperature for 30 minutes. Microtubule-associated proteins were pelleted in sucrose cushion by centrifugation at 20000xg, resuspended and analyzed by SDS-PAGE. Coomassie blue-stained signals of tubulins and MTBD were quantified using ImageJ. The signal intensity of MTBD was normalized to respective tubulin signals.
Western blot analysis
Protein lysate from microtubule sedimentation assay was boiled with SDS lysis buffer and separated by SDS-PAGE (6% acrylamide). Proteins were transferred to a nitrocellulose membrane and probed with mouse anti-UNC-104 monoclonal antibody (1∶60, a gift from Dr. S. Koushika) [52], rabbit anti-DHC-1 polyclonal antibody (1∶200, a gift from Dr. P. Gonczy) [30] or 6-11B-1 (1∶1000, Santa Cruz Biotech) for acetylated α-tubulin, followed by HRP-based chemiluminescence detection. Signal intensity was quantified using ImageJ. All experiments were repeated at least three times. To quantify the amount of DHC-1 or UNC-104 co-sedimented with microtubules, the pixel intensity of individual bands was first quantified using ImageJ. We first normalized the amount of bound DHC-1 or UNC-104 relative to sedimented microtubules in respective experiments. Next, we normalized the DHC-1/microtubule values to the averaged UNC-104/microtubule value, and data from five independent experiments were expressed as a fold change relative to the UNC-104/microtubule ratio.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HirokawaN, NiwaS, TanakaY (2010) Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68 : 610–638.
2. CondeC, CaceresA (2009) Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci 10 : 319–332.
3. KapiteinLC, SchlagerMA, KuijpersM, WulfPS, van SpronsenM, et al. (2010) Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr Biol 20 : 290–299.
4. RollsMM (2011) Neuronal polarity in Drosophila: sorting out axons and dendrites. Dev Neurobiol 71 : 419–429.
5. ManiarTA, KaplanM, WangGJ, ShenK, WeiL, et al. (2011) UNC-33 (CRMP) and ankyrin organize microtubules and localize kinesin to polarize axon-dendrite sorting. Nat Neurosci 15 : 48–56.
6. YanJ, ChaoDL, TobaS, KoyasakoK, YasunagaT, et al. (2013) Kinesin-1 regulates dendrite microtubule polarity in Caenorhabditis elegans. Elife 2: e00133.
7. OuCY, PoonVY, MaederCI, WatanabeS, LehrmanEK, et al. (2010) Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell 141 : 846–858.
8. GoodwinPR, SasakiJM, JuoP (2012) Cyclin-dependent kinase 5 regulates the polarized trafficking of neuropeptide-containing dense-core vesicles in Caenorhabditis elegans motor neurons. J Neurosci 32 : 8158–8172.
9. KoushikaSP, SchaeferAM, VincentR, WillisJH, BowermanB, et al. (2004) Mutations in Caenorhabditis elegans cytoplasmic dynein components reveal specificity of neuronal retrograde cargo. J Neurosci 24 : 3907–3916.
10. MizunoN, TobaS, EdamatsuM, Watai-NishiiJ, HirokawaN, et al. (2004) Dynein and kinesin share an overlapping microtubule-binding site. EMBO J 23 : 2459–2467.
11. KikkawaM, HirokawaN (2006) High-resolution cryo-EM maps show the nucleotide binding pocket of KIF1A in open and closed conformations. EMBO J 25 : 4187–4194.
12. RedwineWB, Hernandez-LopezR, ZouS, HuangJ, Reck-PetersonSL, et al. (2012) Structural basis for microtubule binding and release by dynein. Science 337 : 1532–1536.
13. UchimuraS, OguchiY, HachikuboY, IshiwataS, MutoE (2010) Key residues on microtubule responsible for activation of kinesin ATPase. EMBO J 29 : 1167–1175.
14. TischfieldMA, BarisHN, WuC, RudolphG, Van MaldergemL, et al. (2010) Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140 : 74–87.
15. NiwaS, TakahashiH, HirokawaN (2013) beta-Tubulin mutations that cause severe neuropathies disrupt axonal transport. EMBO J 32 : 1352–1364.
16. Chalfie M.; SulstonJ (1981) Development Genetics of the Mechanosensory Neurons of C. elegans. Dev Biol 82 : 358–370.
17. FukushigeT, SiddiquiZK, ChouM, CulottiJG, GogoneaCB, et al. (1999) MEC-12, an alpha-tubulin required for touch sensitivity in C. elegans. J Cell Sci 112 : 395–403.
18. GoodmanMB (2006) Mechanosensation. WormBook 6 : 1–14.
19. Bounoutas A, Zheng Q, Nonet ML, Chalfie M (2009) mec-15 encodes an F-box protein required for touch receptor neuron mechanosensation, synapse formation and development. Genetics 183: : 607–617, 601SI-604SI.
20. ZhengQ, SchaeferAM, NonetML (2011) Regulation of C. elegans presynaptic differentiation and neurite branching via a novel signaling pathway initiated by SAM-10. Development 138 : 87–96.
21. TopalidouI, KellerC, KalebicN, NguyenKC, SomhegyiH, et al. (2012) Genetically separable functions of the MEC-17 tubulin acetyltransferase affect microtubule organization. Curr Biol 22 : 1057–1065.
22. KriegM, DunnAR, GoodmanMB (2014) Mechanical control of the sense of touch by beta-spectrin. Nat Cell Biol 16 : 224–233.
23. PanCL, PengCY, ChenCH, McIntireS (2011) Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc Natl Acad Sci U S A 108 : 9274–9279.
24. SiddiquiSS, AamodtE, RastinejadF, CulottiJ (1989) Anti-tubulin monoclonal antibodies that bind to specific neurons in Caenorhabditis elegans. J Neurosci 9 : 2963–2972.
25. SavageC, XueY, MitaniS, HallD, ZakharyR, et al. (1994) Mutations in the Caenorhabditis elegans beta-tubulin gene mec-7: effects on microtubule assembly and stability and on tubulin autoregulation. J Cell Sci 107 : 2165–2175.
26. Ghosh-RoyA, GoncharovA, JinY, ChisholmAD (2012) Kinesin-13 and tubulin posttranslational modifications regulate microtubule growth in axon regeneration. Dev Cell 23 : 716–728.
27. NeumannBH, MA (2014) Loss of MEC-17 Leads to Microtubule Instability and Axonal Degeneration. Cell Reports 6 : 93–103.
28. KimuraY, KurabeN, IkegamiK, TsutsumiK, KonishiY, et al. (2010) Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J Biol Chem 285 : 22936–22941.
29. O'HaganR, PiaseckiBP, SilvaM, PhirkeP, NguyenKC, et al. (2011) The tubulin deglutamylase CCPP-1 regulates the function and stability of sensory cilia in C. elegans. Curr Biol 21 : 1685–1694.
30. GonczyP, PichlerS, KirkhamM, HymanAA (1999) Cytoplasmic dynein is required for distinct aspects of MTOC positioning, including centrosome separation, in the one cell stage Caenorhabditis elegans embryo. J Cell Biol 147 : 135–150.
31. KonishiY, SetouM (2009) Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat Neurosci 12 : 559–567.
32. IkegamiK, HeierRL, TaruishiM, TakagiH, MukaiM, et al. (2007) Loss of alpha-tubulin polyglutamylation in ROSA22 mice is associated with abnormal targeting of KIF1A and modulated synaptic function. Proc Natl Acad Sci U S A 104 : 3213–3218.
33. GoshimaY, NakamuraF, StrittmatterP, StrittmatterSM (1995) Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 376 : 509–514.
34. OtsukaAJ, FrancoR, YangB, ShimKH, TangLZ, et al. (1995) An ankyrin-related gene (unc-44) is necessary for proper axonal guidance in Caenorhabditis elegans. J Cell Biol 129 : 1081–1092.
35. TischfieldMA, CederquistGY, GuptaMLJr, EngleEC (2011) Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr Opin Genet Dev 21 : 286–294.
36. SimonsC, WolfNI, McNeilN, CaldovicL, DevaneyJM, et al. (2013) A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 92 : 767–773.
37. LohmannK, WilcoxRA, WinklerS, RamirezA, RakovicA, et al. (2013) Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann Neurol 73 : 537–545.
38. KeaysDA, TianG, PoirierK, HuangGJ, SieboldC, et al. (2007) Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128 : 45–57.
39. HammarlundM, JorgensenEM, BastianiMJ (2007) Axons break in animals lacking beta-spectrin. J Cell Biol 176 : 269–275.
40. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94.
41. MelloCC, KramerJM, StinchcombD, AmbrosV (1991) Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10 : 3959–3970.
42. HilliardMA, BargmannCI (2006) Wnt signals and frizzled activity orient anterior-posterior axon outgrowth in C. elegans. Dev Cell 10 : 379–390.
43. PrasadBC, ClarkSG (2006) Wnt signaling establishes anteroposterior neuronal polarity and requires retromer in C. elegans. Development 133 : 1757–1766.
44. PanCL, BaumPD, GuM, JorgensenEM, ClarkSG, et al. (2008) C. elegans AP-2 and retromer control Wnt signaling by regulating mig-14/Wntless. Dev Cell 14 : 132–139.
45. DavisMW, HammarlundM, HarrachT, HullettP, OlsenS, et al. (2005) Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics 6 : 118.
46. KamathRS, Martinez-CamposM, ZipperlenP, FraserAG, AhringerJ (2001) Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2 RESEARCH0002.
47. CalixtoA, ChelurD, TopalidouI, ChenX, ChalfieM (2010) Enhanced neuronal RNAi in C. elegans using SID-1. Nat Methods 7 : 554–559.
48. McDonaldKL, WebbRI (2011) Freeze substitution in 3 hours or less. J Microsc 243 : 227–233.
49. ReynoldsES (1963) The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol 17 : 208–212.
50. FinneyM, RuvkunG (1990) The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell 63 : 895–905.
51. LyeRJ, PorterME, ScholeyJM, McIntoshJR (1987) Identification of a microtubule-based cytoplasmic motor in the nematode C. elegans. Cell 51 : 309–318.
52. KumarJ, ChoudharyBC, MetpallyR, ZhengQ, NonetML, et al. (2010) The Caenorhabditis elegans Kinesin-3 motor UNC-104/KIF1A is degraded upon loss of specific binding to cargo. PLoS Genet 6: e1001200.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- An RNA-Seq Screen of the Antenna Identifies a Transporter Necessary for Ammonia Detection
- Systematic Comparison of the Effects of Alpha-synuclein Mutations on Its Oligomerization and Aggregation
- Functional Diversity of Carbohydrate-Active Enzymes Enabling a Bacterium to Ferment Plant Biomass
- Regularized Machine Learning in the Genetic Prediction of Complex Traits
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy