
Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
Recent advances in DNA sequencing have enabled mapping of genes for monogenic traits in families with small pedigrees and even in unrelated cases. We report the identification of disease-causing mutations in a rare, severe, skeletal dysplasia, studying a family of two healthy unrelated parents and two affected children using whole-exome sequencing. The two affected daughters have clinical and radiographic features suggestive of anauxetic dysplasia (OMIM 607095), a rare form of dwarfism caused by mutations of RMRP. However, mutations of RMRP were excluded in this family by direct sequencing. Our studies identified two novel compound heterozygous loss-of-function mutations in POP1, which encodes a core component of the RNase mitochondrial RNA processing (RNase MRP) complex that directly interacts with the RMRP RNA domains that are affected in anauxetic dysplasia. We demonstrate that these mutations impair the integrity and activity of this complex and that they impair cell proliferation, providing likely molecular and cellular mechanisms by which POP1 mutations cause this severe skeletal dysplasia.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1002027
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002027
Summary
Recent advances in DNA sequencing have enabled mapping of genes for monogenic traits in families with small pedigrees and even in unrelated cases. We report the identification of disease-causing mutations in a rare, severe, skeletal dysplasia, studying a family of two healthy unrelated parents and two affected children using whole-exome sequencing. The two affected daughters have clinical and radiographic features suggestive of anauxetic dysplasia (OMIM 607095), a rare form of dwarfism caused by mutations of RMRP. However, mutations of RMRP were excluded in this family by direct sequencing. Our studies identified two novel compound heterozygous loss-of-function mutations in POP1, which encodes a core component of the RNase mitochondrial RNA processing (RNase MRP) complex that directly interacts with the RMRP RNA domains that are affected in anauxetic dysplasia. We demonstrate that these mutations impair the integrity and activity of this complex and that they impair cell proliferation, providing likely molecular and cellular mechanisms by which POP1 mutations cause this severe skeletal dysplasia.
Introduction
Skeletal dysplasias (SD) are a group of genetic disorders affecting skeletal development that cause deficiencies and deformities of the limbs and spine, dwarfism, or abnormal bone strength. SDs are usually inherited as dominant or recessive monogenic Mendelian traits or occur as a result of de novo mutations. Recent advanced in targeted whole-exome DNA re-sequencing have enabled several groups to identify of causative mutations underlying various Mendelian diseases, in which traditional linkage approaches were not feasible because of the paucity of familial cases in which to perform the mapping [1]–[7]. In this study we report the mapping of mutations causing a severe bone dysplasia in a family of unrelated unaffected parents with two affected siblings. The clinical and radiographic features of the affected siblings showed similarities to anauxetic dysplasia, an autosomal recessive spondylo-epi-metaphyseal dysplasia characterized by extremely short stature [8], [9]. Both siblings had severe growth retardation of prenatal onset, a bone dysplasia affecting the epiphyses and metaphyses of the long bones particularly in the lower limbs, and abnormalities of the spine including irregularly shaped vertebral bodies and marked cervical spine instability (Figure 1). Anauxetic dysplasia is caused by mutations in RMRP, an untranslated intronless gene, mutations of which also cause cartilage-hair hypoplasia (CHH, OMIM 250250), another severe form of dwarfism [10]. As RMRP mutations had been excluded in this family, we sought to identify the disease-causing variants by whole-exome sequencing.

Results/Discussion
We sequenced exomes of both parents and the affected siblings using the Nimblegen SeqCap Ez exome capture protocol and the Illumina Genome Analyser II paired-end sequencing method. High quality sequence reads were aligned to the human reference genome (UCSC assembly hg19); 90% of targeted bases had coverage of fourfold or higher; and 79% of targeted bases had coverage greater than tenfold. We then used SAMtools [11], Genome Analysis Toolkit (GATK) [12] and custom scripts to detect polymorphic sites in the four individual exome sequence data sets. Following quality filtering, we retained approximately 15,000 SNPs per sequenced exome (Table 1). The vast majority of these SNPs (>96%) were reported in the recent NCBI dbSNP 131 release, and were therefore excluded from further analysis as unlikely to cause this severe, rare, phenotype. Following the functional annotation of the remaining novel SNPs, we focused our analyses on a set of 483 unique novel coding non-synonymous SNPs that were detected in at least one sample.

Given the affected status of both siblings and unaffected status of the parents we considered autosomal recessive homozygous and autosomal recessive compound heterozygous as the two most likely inheritance models; as the parents are unrelated, a compound heterozygous model was considered most likely. All novel non-synonymous SNPs were analyzed assuming either a homozygous or a compound heterozygous model. We did not find any novel non-synonymous mutations that satisfied a recessive homozygous inheritance model. To be accepted as candidate mutations for a compound heterozygous inheritance model, we sought genes with at least two novel non-synonymous SNPs within the protein coding sequence in both siblings; additionally, the same alleles had to be present in the parents in a mutually exclusive manner. We identified four candidate genes (POP1, PKD1, MICALL2, and COL5A1) that fitted this model (Table S1).
To determine the gene likely to be causing the disease we analyzed the effects of the detected missense mutations on protein function using the SIFT algorithm [13]. In all but one gene the detected missense mutations were predicted to be tolerated in terms of effect upon protein function. The single remaining candidate gene carried two novel alleles – one creating a premature stop codon, and the other causing a missense mutation with predicted damaging effect on protein function, thus representing a strong candidate for the disease-causing gene in this family (Figure 2). Remarkably, we found that this gene encodes the human homolog of the yeast POP1 (processing of precursor RNA) protein, one of the core components of the RNase P and RNase MRP complexes [14], [15].

RNase P and RNase MRP are essential ribonucleoprotein complexes present in all eukaryotes [16]. Both enzymatic complexes have an evolutionarily conserved RNA component as a catalytic moiety. Several proteins essential for enzymatic activity form a constitutive part of RNAse P and RNase MRP complexes. POP1 is the largest constitutive component of both complexes (Figure S1) [15], [16]. Importantly, mutations in RMRP, which encodes the RNA moiety of the RNase MRP complex, cause anauxetic dysplasia and cartilage-hair hypoplasia [10].
To validate the identified POP1 mutations we re-sequenced genomic DNA fragments encompassing mutated alleles of the POP1 gene using Sanger sequencing, confirming that the two affected siblings carry both mutated alleles, while each parent carries only one allele (Figure 2A, 2B). While neither of these mutations is located within known protein domains (as determined by PFAM annotation), both amino acids affected by the mutations show high levels of evolutionary conservation, suggesting their importance for the functional integrity of POP1 and/or RNase P/RNase MRP complexes (Figure 2C; Figure S1). The p.Arg513Ter mutation is likely to be particularly damaging; the premature stop codon located in the exon 10 would trigger mRNA degradation via nonsense mediated decay mechanism. Alternatively (though less likely), p.Arg513Ter mutation would result in translation of a truncated protein lacking the evolutionarily conserved POPLD domain (Figure 2C). These results are consistent with findings in yeast. Xiao et al. [16] identified several point mutations in the yeast POP1 protein that affect either the stability or activity of RNase P and RNase MRP complexes. Importantly, some of the yeast mutations were found close to the mutations identified in our study, supporting our conclusion that these are likely to cause significant detrimental effect on RNase P and RNase RMP function.
The very low population frequency of the described skeletal dysplasia, and the essential roles of RNase P/RNase MRP enzymatic complexes in cellular functions, suggest that there is strong negative selection pressure on homozygous and compound heterozygous mutations with detrimental effects on the activity of these complexes. Indeed, analysis of mutations that ablate POP1 function in model organisms demonstrate that POP1 is essential for viability in yeast and Drosophila [14], [17]. In this context, it is likely that one or both detected POP1 mutations have only a partial effect on function of either RNAse P or RNase MRP complexes. This conclusion is consistent with the broad spectrum of mutations observed in RMRP in cases of anauxetic dysplasia and cartilage-hair hypoplasia and their correlation with the severity of the disease phenotype [18]. RMRP mutations implicated in anauxetic dysplasia and cartilage-hair hypoplasia have distinct clustering within the RMRP RNA molecule [18]. Importantly, POP1 directly interacts with the same RMRP RNA domains that are affected in anauxetic dysplasia (Figure S1) [18], [19]. This further strengthens the link between our findings and the molecular mechanisms underlying the clinical phenotype in anauxetic dysplasia and our patients.
To assess population frequency of the POP1 alleles we screened 186 DNA samples from healthy control subjects. For the NM_001145860.1:c.1573C>T (p.Arg513Ter) allele we used direct Sanger sequencing of PCR-amplified DNA fragments. To estimate the frequency of the second POP1 allele, NM_001145860.1:c.1748G>A (p.Gly583Glu), we used a restriction fragment length polymorphism (RFLP) assay designed to detect the loss of the BsaJ1 restriction site (CCNNGG) created by c.1748G>A (p.Gly583Glu) mutation. None of the 186 control samples contained either mutation. Further, these variants are not reported in public SNP databases including dbSNP 131. Therefore we conclude that both mutations represent novel rare alleles.
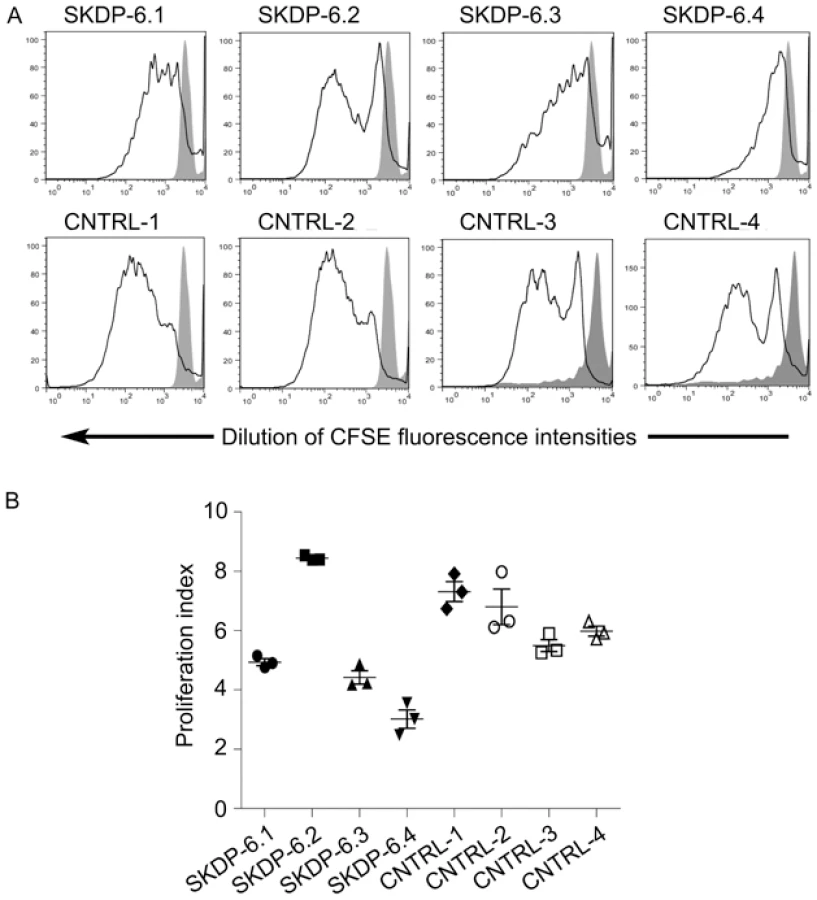
One of the cellular functions of the RNase RMP complex is processing of precursor 5.8S ribosomal RNA into the mature form [15]. To determine the impact of the mutated alleles on functional integrity and activity of the RNase P/MRP complexes, we measured relative abundance of the RMRP RNA and unprocessed pre-5.8S rRNA in the affected siblings, non-affected parents, and unrelated controls. We found that the relative abundance of RMRP RNA was markedly reduced in the affected individuals compared with non-affected parents and controls (Figure 3), indicating that the mutated alleles are likely to cause mis-assembly and destabilization of the RNase MRP complex. These results are consistent with the previous study in yeast demonstrating that mutations in POP1 affect integrity of the RNase MRP complex, resulting in destabilization of the complex and reduction of the abundance of NME1 RNA (the yeast homolog of human RMRP) [16]. We also found that affected individuals have higher abundance of the unprocessed pre-5.8S rRNA compared to non-affected parents and controls (Figure 3), consistent with the established role of RNase P in processing of rRNA [15] and demonstrating that activity of the RNAse MRP complex is impaired in the affected individuals. To assess the cellular impact of this molecular defect, we measured cell proliferation in stimulated peripheral blood mononuclear cells (Figure 4). These studies showed that, in comparison with healthy controls, the two affected cases had markedly diminished cell proliferation in response to proliferation stimulus, as evidenced by the reduced rate of dilution of CFSE fluorescence intensities (Figure 4A). Additionally, individual SKDP-6.1, carrying the c.1573C>T (p.Arg513Ter) allele, had noticeable reduction in cell proliferation rate, consistent with the predicted more damaging effect of this mutation (Figure 4). These findings also explain similarities with the anauxetic dysplasia phenotype, as individuals with this disease also have impaired RNase P/RNase MRP activity, and diminished cell turnover [10], [18].

In conclusion, we have identified mutations in POP1 underlying the skeletal dysplasia presented in this study. Involvement of POP1 in the skeletal dysplasia highlighted the existence of a shared molecular pathway in the etiology of skeletal dysplasias involving the RNAse P and RNase MRP complexes [10], [18] and raises the possibility of the involvement of other components of the pathway, or allelic variants of RMRP or POP1, in other as yet unmapped severe short-stature syndromes. Screening of RMRP-negative cases of anauxetic dysplasia or CHH, or patients with similar phenotypic features, may yield additional mutated alleles within POP1, and possibly in other components of the RNase MRP complex. We further hypothesize that the distinct skeletal phenotype associated with POP1 mutations may indicate that RNAse P and RNase MRP complexes may process other yet unidentified target RNAs with regulatory roles in specific developmental pathways such as osteogenesis. This study also provides an example of the feasibility of whole-exome re-sequencing approach in rare familiar monogenic diseases with small pedigrees.
Materials and Methods
Human Subjects
This study was approved by the University of Queensland ethics committee (Approval #2007001196). Written, informed, consent was obtained from all subjects or their respective guardians.
Construction of DNA Libraries and Sequencing
5 µg of human genomic DNA extracted from peripheral venous blood samples was used for preparation of each DNA sequencing library. The DNA libraries were prepared using Illumina's paired-end sequencing DNA sample preparation kit according to the manufacturer's protocol.
Whole-exome sequence capture was performed using “SeqCap EZ Exome Library v1.0” liquid phase sequence capture kit (Roche/Nimblegen) according to the manufacturer's recommendations. Sequence capture efficiency was assessed by quantitative real-time PCR using a standard set of control primers recommended in the sequence capture protocol. Individual DNA libraries were quality-checked and quantified on Agilent 2100 Bioanalyzer using DNA1000 kit, and DNA concentration adjusted to 10 nM.
Sequencing was performed on the Illumina Genome Analyzer II using a standard 56 cycle paired-end read sequencing protocol and Illumina's sequencing reagents according to the manufacturer's recommendations. Each library was sequenced individually on a single flow cell lane.
Analysis of Sequencing Data
Base calling and sequence reads quality assessment was performed using Illumina's Data Analysis Pipeline software v.1.6. Alignment of sequence reads to the reference human genome (hg19, UCSC assembly, February 2009) was performed using the Burrows-Wheeler alignment (BWA) tool [20]. Sequence alignment files conversions were performed using SAMtools [11]. Only high quality sequence reads with unique mapping positions to the reference human genome were used for calling SNPs and Indels. Identification of SNPs and Indels in the alignment files was performed using Genome Analysis Toolkit (GATK) [12]. Raw SNP calls were filtered using empirically derived cut-offs for the following GATK filter expressions: –filterExpression “QUAL <50.0 || AB>0.75 && DP>40 || QD<5.0 || HRun>5 || SB>−0.10” –filterName “StandardFilters” –filterExpression “DP<6” –filterName “LOW_DEPTH”, where QUAL – combined recalibrated quality score at the SNP position; AB – allele balance at the SNP position; DP – sequencing depth at the SNP position; QD – QUAL/DP ratio at the SNP position; HRun – maximal length of the homopolymer run; SB - strand bias at the SNP position. Known SNPs were obtained from the NCBI dbSNP build 131 (http://www.ncbi.nlm.nih.gov/SNP/). Prediction of the effect of non-synonymous amino acid substitutions on protein function was done using SIFT algorithm [13].
Sanger Sequencing
Sanger sequencing of the exons 10 and 12 of POP1 was performed on Genetic Analyzer 3130 (Applied Biosystems) using standard sequencing protocol for PCR-amplified DNA. Primers used for PCR amplification of POP1 DNA fragments and Sanger sequencing were as follows: POP1.10F 5′-CAATGGGAAGAGAGGGAATACATGTTT-3′; POP1.10R 5′-CTGGAGAGGTGTCAGAGAAAGAACTCTT-3′; POP1.12F 5′-CATCCTTGAGAAGGTGTCACTTAATTGTT-3′; POP1.12R 5′-CACCACTCAATTCCTCCTAAGTTGTACAT-3.
186 DNA samples from healthy white European controls were studied to determine the population frequency of the variants identified. The c.1573C>T (p.Arg513Ter) allele located within exon 10 of POP1 was tested by Sanger sequencing using POP1.10F and POP1.10R primers listed above. The c.1748G>A (p.Gly583Glu) allele located within exon 12 of POP1 was tested by a restriction fragment length polymorphism (RFLP) assay designed to screen for the loss of the BsaJ1 restriction site (CCNNGG) created by the mutation. Following PCR amplification of exon 12 DNA fragments with POP1.12F and POP1.12R primers, DNA fragments were digested with BsaJ1, separated by agarose gel electrophoresis, and visualised by ethidium bromide staining and UV transillumination.
RNA Isolation and cDNA Synthesis
Total RNA was extracted from peripheral blood mononuclear cell samples using TRIZOL reagent (Invitrogen) following the manufacturer's protocol. The RNA concentrations and purity were determined using the RNA 6000 Bioanalyzer kit (Agilent). For cDNA synthesis, 500 ng of total RNA was treated with DNase I (Invitrogen) and then utilized as a template for randomly primed reverse transcription using SuperScript III reverse transcriptase (Invitrogen), according to the manufacturer's instructions. The resulting cDNA was diluted 1∶25 (v/v) with nuclease-free water. A total of 5 µL of the cDNA was used as a template for quantitative real-time PCR.
Quantitative Real-Time PCR
Relative abundance of unprocessed 5.8S rRNA and RMRP transcripts in affected in non-affected individuals was determined by real-time PCR using SYBR Green RCR Master Mix (Applied Biosystems) according to manufacturer's protocol. The PCR amplification was performed on the ABI Prism 7900HT sequence-detection system (Applied Biosystems) in a final volume of 12 µL using standard cycling parameters (10 min, 95°C; 30 sec, 95°C; 30 sec 65°C; 30 sec 72°C, with the latter three steps repeated for 45 times). Primers for the real-time PCR reaction were designed using Vector NTI 11.0 software package (Invitrogen), following primer design guidelines given in the SYBR green PCR Master Mix and RT-PCR protocol (Applied Biosystems). The melting temperatures of all primers were between 65 and 71°C. All primers were purchased from the ITD (Glycon Australia Pty Ltd).
The primers used for detection of pre-5.8S rRNA were as follows: pre5.8S_F 5′-TGTGAAACCTTCCGACCCCTCT-3′; pre5.8S_R 5′-CGAGTGATCCACCGCTAAGAGTCGTA-3′.
The primers used for detection of the RMRP RNA (GenBank NR_003051) were as follows: RMRP_F 5′-AGGACTCTGTTCCTCCCCTTTCCGCCTA-3′, RMRP_R 5′-TGGAGTGGGAAGCGGGGAATGTCTA-3′.
The primers used for detection of human beta actin mRNA (ACTB, GenBank NM_001101) used as an internal normalization standard were as follows: ActF 5′-TCACCATTGGCAATGAGCGGTT-3′; ActR 5′-AGTTTCGTGGATGCCACAGGACT-3′.
The final optimized concentration of primers was 250 nM for all DNA amplicons. The absence of inter - and/or intramolecular duplex formation between primers was confirmed in a control real-time PCR reaction lacking template. Relative quantification was performed as described in ABI Prism 7900HT Sequence Detection System User Bulletin #2 (Applied Biosystems) according to Comparative CT Method. In brief, threshold cycle (CT) values of experimental samples were normalized to corresponding CT values of the ACTB mRNA control, and then quantified relative to the sample with the maximal CT value (calibrator). All real-time PCR reactions were done in five technical replicates, and results were confirmed in two independent experiments.
Cell Turnover Studies
Progressive dilution of the fluorescent dye CFSE during cell division was used as a proxy to assess cell division rates using flow cytometry [21]. PBMC from the affected children (SKDP 6.3 and 6.4), their parents (SKDP 6.1 and 6.2) and healthy controls were labelled with 2.5 µM CFSE and plated in triplicate at 5×105 cells/well in 48-well plates. PBMC were stimulated with PMA and Ionomycin or left unstimulated for seven days. Proliferation of PBMC in response to stimulation was assessed by examining CFSE dilution on a FACSCanto flow cytometer (BD). Comparative cell division rates were determined by calculating proliferation indices of cells undergoing 2 or more cell divisions using ModFit software (Verity Software House).
Accession Numbers
POP1 amino acid substitutions are given using human POP1 mRNA sequence and annotations with GenBank accession NM_001145860. Human beta actin mRNA, RMRP RNA, and rRNA polycistron sequences have GenBank accession numbers NM_001101, NR_003051, and U13369 respectively. NCBI and FlyBase accession numbers for Drosophila POP1 homolog are NP_572236 and FBgn0026702 respectively.
Web Resources
UCSC genome browser http://genome.ucsc.edu/; NCBI dbSNP http://www.ncbi.nlm.nih.gov/SNP/; Protein families database (PFAM) http://pfam.sanger.ac.uk/; SIFT website http://sift.jcvi.org/; GATK website http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit; Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/.
Supporting Information
Zdroje
1. NgSBTurnerEHRobertsonPDFlygareSDBighamAW 2009 Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461 272 276
2. HoischenAvan BonBWMGilissenCArtsPvan LierB 2010 De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nature Genetics 42 483 485
3. GilissenCArtsHHHoischenASpruijtLMansDA 2010 Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet 87 418 423
4. BilguvarKOzturkAKLouviAKwanKYChoiM 2010 Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467 207 210
5. WalshTShahinHElkan-MillerTLeeMKThorntonAM 2010 Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet 87 90 94
6. KrawitzPMSchweigerMRRodelspergerCMarcelisCKolschU 2010 Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nature Genetics 42 827 829
7. NgSBBuckinghamKJLeeCBighamAWTaborHK 2010 Exome sequencing identifies the cause of a mendelian disorder. Nature Genetics 42 30 35
8. MengerHMundlosSBeckerKSprangerJZabelB 1996 An unknown spondylo-meta-epiphyseal dysplasia in sibs with extreme short stature. Am J Med Genet 63 80 83
9. HornDRupprechtEKunzeJSprangerJ 2001 Anauxetic dysplasia, a spondylometaepiphyseal dysplasia with extreme dwarfism. J Med Genet 38 262 265
10. ThielCTHornDZabelBEkiciABSalinasK 2005 Severely incapacitating mutations in patients with extreme short stature identify RNA-processing endoribonuclease RMRP as an essential cell growth regulator. Am J Hum Genet 77 795 806
11. LiHHandsakerBWysokerAFennellTRuanJ 2009 The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 2078 2079
12. McKennaAHannaMBanksESivachenkoACibulskisK 2010 The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 1297 1303
13. KumarPHenikoffSNgPC 2009 Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4 1073 1081
14. LygerouZMitchellPPetfalskiESeraphinBTollerveyD 1994 The POP1 gene encodes a protein component common to the RNase MRP and RNase P ribonucleoproteins. Genes Dev 8 1423 1433
15. EsakovaOKrasilnikovAS 2010 Of proteins and RNA: the RNase P/MRP family. Rna 16 1725 1747
16. XiaoSHsiehJNugentRLCoughlinDJFierkeCA 2006 Functional characterization of the conserved amino acids in Pop1p, the largest common protein subunit of yeast RNases P and MRP. Rna 12 1023 1037
17. PeterASchottlerPWernerMBeinertNDoweG 2002 Mapping and identification of essential gene functions on the X chromosome of Drosophila. EMBO Rep 3 34 38
18. ThielCTMortierGKaitilaIReisARauchA 2007 Type and level of RMRP functional impairment predicts phenotype in the cartilage hair hypoplasia-anauxetic dysplasia spectrum. Am J Hum Genet 81 519 529
19. WeltingTJvan VenrooijWJPruijnGJ 2004 Mutual interactions between subunits of the human RNase MRP ribonucleoprotein complex. Nucleic Acids Res 32 2138 2146
20. LiHDurbinR 2009 Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 1754 1760
21. LyonsABParishCR 1994 Determination of lymphocyte division by flow cytometry. J Immunol Methods 171 131 137
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy