
The Toll-Like Receptor Gene Family Is Integrated into Human DNA Damage and p53 Networks
In recent years the functions that the p53 tumor suppressor plays in human biology have been greatly extended beyond “guardian of the genome.” Our studies of promoter response element sequences targeted by the p53 master regulatory transcription factor suggest a general role for this DNA damage and stress-responsive regulator in the control of human Toll-like receptor (TLR) gene expression. The TLR gene family mediates innate immunity to a wide variety of pathogenic threats through recognition of conserved pathogen-associated molecular motifs. Using primary human immune cells, we have examined expression of the entire TLR gene family following exposure to anti-cancer agents that induce the p53 network. Expression of all TLR genes, TLR1 to TLR10, in blood lymphocytes and alveolar macrophages from healthy volunteers can be induced by DNA metabolic stressors. However, there is considerable inter-individual variability. Most of the TLR genes respond to p53 via canonical as well as noncanonical promoter binding sites. Importantly, the integration of the TLR gene family into the p53 network is unique to primates, a recurrent theme raised for other gene families in our previous studies. Furthermore, a polymorphism in a TLR8 response element provides the first human example of a p53 target sequence specifically responsible for endogenous gene induction. These findings—demonstrating that the human innate immune system, including downstream induction of cytokines, can be modulated by DNA metabolic stress—have many implications for health and disease, as well as for understanding the evolution of damage and p53 responsive networks.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1001360
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001360
Summary
In recent years the functions that the p53 tumor suppressor plays in human biology have been greatly extended beyond “guardian of the genome.” Our studies of promoter response element sequences targeted by the p53 master regulatory transcription factor suggest a general role for this DNA damage and stress-responsive regulator in the control of human Toll-like receptor (TLR) gene expression. The TLR gene family mediates innate immunity to a wide variety of pathogenic threats through recognition of conserved pathogen-associated molecular motifs. Using primary human immune cells, we have examined expression of the entire TLR gene family following exposure to anti-cancer agents that induce the p53 network. Expression of all TLR genes, TLR1 to TLR10, in blood lymphocytes and alveolar macrophages from healthy volunteers can be induced by DNA metabolic stressors. However, there is considerable inter-individual variability. Most of the TLR genes respond to p53 via canonical as well as noncanonical promoter binding sites. Importantly, the integration of the TLR gene family into the p53 network is unique to primates, a recurrent theme raised for other gene families in our previous studies. Furthermore, a polymorphism in a TLR8 response element provides the first human example of a p53 target sequence specifically responsible for endogenous gene induction. These findings—demonstrating that the human innate immune system, including downstream induction of cytokines, can be modulated by DNA metabolic stress—have many implications for health and disease, as well as for understanding the evolution of damage and p53 responsive networks.
Introduction
The p53 master regulator is responsive to a variety of DNA metabolic stresses resulting in induction or repression of over 200 genes as well as several LINC - and micro-RNAs [1], [2]. In its role as tumor suppressor and “guardian of the genome” many of the target genes in humans influence cell cycle progression or apoptosis. Over the past decade the p53 network has been extended to transcriptional regulation of genes associated with a wide variety of biological functions including DNA repair, angiogenesis, cellular metabolism, autophagy, stem cell renewal, fertility, differentiation and cellular reprogramming. To better understand the broad role that p53 can play in human biology, we have pursued “functionality rules” for identifying target response element (REs) sequences where p53 can directly influence transactivation. Using in vivo transactivation systems based in yeast and human cells as well as binding in human cell extracts [3], [4], we recently found that functionality of the binding consensus RRRCWWGYYYnRRRCWWGYYY (R, pyrimidine; Y, pyrimidine; W, A or T; n, spacer of 0 to 13 bases) is greatest when there is at most a single base spacer and if “WW” is “AT.” Furthermore, we defined functionality for half-sites (RRRCWWGYYY) and found in cis synergy when another transcription factor, estrogen receptor, was bound nearby (for a description of noncanonical REs including half-sites see [3], [5] and summary in [6]). Based on these functionality rules, we found that the evolution of p53 control of at least one gene family, DNA metabolism and repair, is limited to primates [7]. Furthermore, we identified single nucleotide polymorphisms in REs that are predicted to modify responsiveness of genes to p53 mediated stress [8].
Our functionality studies of canonical and noncanonical promoter p53 REs suggested that p53 may have a role in human Toll-like receptor (TLR) expression. We had reported that a single nucleotide polymorphism (ChrX:12923681, rs3761624 A/G) in the promoter of the TLR8 gene creates an RE that can be targeted by p53 in yeast and cell line reporter systems [8] although we could not detect endogenous expression of the TLR8 gene. In addition, the TLR3 gene in epithelial cancer cell lines was found to be induced by p53 following exposure to 5-fluorouracil (5FU) [9].
Innate immunity is paramount to infection and tissue injury responses [10]. The TLR gene family mediates innate immunity to a wide variety of pathogenic threats. The TLRs recognize conserved exogenous pathogen-associated molecular patterns (PAMPs) and endogenous danger-associated molecular patterns (DAMPs) [11], while adaptive immune receptors ensure specific antigenic responses through clonal expansion. TLR function is associated with circulating immune cells such as monocytes and dendritic cells, tissue phagocytes that include alveolar macrophages and with nonimmune cells (such as epithelial cells of gut, skin, lung, etc.) that are exposed to environmental injury or pathogens. The roles of TLRs in mammalian biology are continuously expanding and they are now understood to function in such diverse processes as inflammation, cell differentiation and cell survival [11]. Altered TLR function is implicated in human diseases such as systemic lupus erythematosus, inflammatory bowel disease (IBD) and cancer [11], [12], and agonist/antagonist manipulations of the TLR system are being pursued to alleviate various diseases (reviewed in [13]).
Given the varied functions of TLRs, factors that regulate their expression are expected to shape immune responses [14]. However, there are few examples of TLR gene induction and these are limited to specific TLR-stimulus interactions [15], [16]. The TLR-dependent innate immune response is thus generally considered to be “hard-wired.” Although environmental stress can influence gene expression indirectly, e.g., through epigenetic mechanisms, to our knowledge there are no reports of TLR induction by environmental factors in primary human cells [15], [17]–[20].
These collected observations led us to investigate the responsiveness of TLR genes as a class to common DNA stressors. We have examined expression of the entire TLR gene family following exposure to anti-cancer agents that induce the p53 network in primary immune cells obtained directly from human subjects as well as the impact on downstream cytokines. Agents were applied to T-lymphocytes and alveolar macrophages ex vivo. Prior to this there were no reports that we are aware of addressing DNA damage-induced responses of any TLR genes in cells directly associated with innate immunity. As part of this study, we establish that in the evolution of the TLR responses, the p53-mediated expression of TLRs is unique to primates.
Results
DNA stress induces TLR gene family expression
We selected ionizing radiation (IR), 5FU and Doxorubicin (Doxo) because these agents represent a cross-section of DNA metabolic stressors, such as damage or replication inhibition, that may occur endogenously or environmentally and are well-known to activate p53 and its network of target genes. Furthermore, these agents are often employed in cancer treatments. To address TLR responses ex vivo in primary human immune cells, T-lymphocytes were expanded by phytohemagglutinin (PHA) stimulation of peripheral blood mononuclear cells (PBMC) freshly isolated from the blood of healthy human volunteers (see Figure S1A; for demographics of volunteers see Table 1); alternatively, T-lymphocytes were obtained from the PBMC fraction using anti-CD3 antibody-based purification as described in Figure S1B.

Presented in Figure 1 are the expression responses of the entire family of TLR genes in stimulated lymphocytes for 18 subjects (except where noted) following exposure to IR (4 Gray), 5FU (300 µM), and Doxo (0.3 µg/ml) (Figure 1A, 1B, and 1C, respectively). These responses are relative to no treatment controls and normalized to 18S ribosomal DNA. As additional controls, we also examined the expression of the beta-glucuronidase, GUSB, and actin genes (Figure S2) that are considered to be nonresponsive to chromosomal and/or p53 stress; they showed little variation after doxorubicin and nutlin exposure. The doses chosen were similar to therapeutic doses or doses commonly used in the literature. By way of comparison, we also examined the response of the cyclin-dependent kinase inhibitor gene p21WAF1 (CDKN1A), a prototypical p53 target gene induced by these agents in a variety of human cells. Notably, these results establish that expression of all TLRs can be responsive to DNA metabolic insults, even exceeding p21 induction. However, there is considerable variability in the individual responses between subjects, TLRs, and treatments as summarized in the “box and whiskers” presentation of Figure S2. For example, the IR induction of the ten TLR genes varies from 1 - to over 4-fold for each TLR except TLR3 (Figure 1 and Figure S2), which is not detected in the PHA-stimulated lymphocytes of most subjects. This agrees with the large differences in expression of IR-inducible genes between human lymphoblastoid cell lines [21]. The variability in gene expression among the 18 subjects can represent a continuum of responses (e.g., TLR4 expression after IR), or exhibit more of a binary induction pattern (e.g., TLR8 response after IR; see Figure 1). Specifically, for the TLR8 gene, approximately half the subjects respond strongly and half exhibit a much lower response, a finding that appears to be genetically determined [8], as discussed below. Different agents also elicit differential TLR gene expression responses. For example, TLR1 is responsive to IR by more than 2-fold in most subjects, but generally there is only a small level of induction in response to 5FU or Doxo.
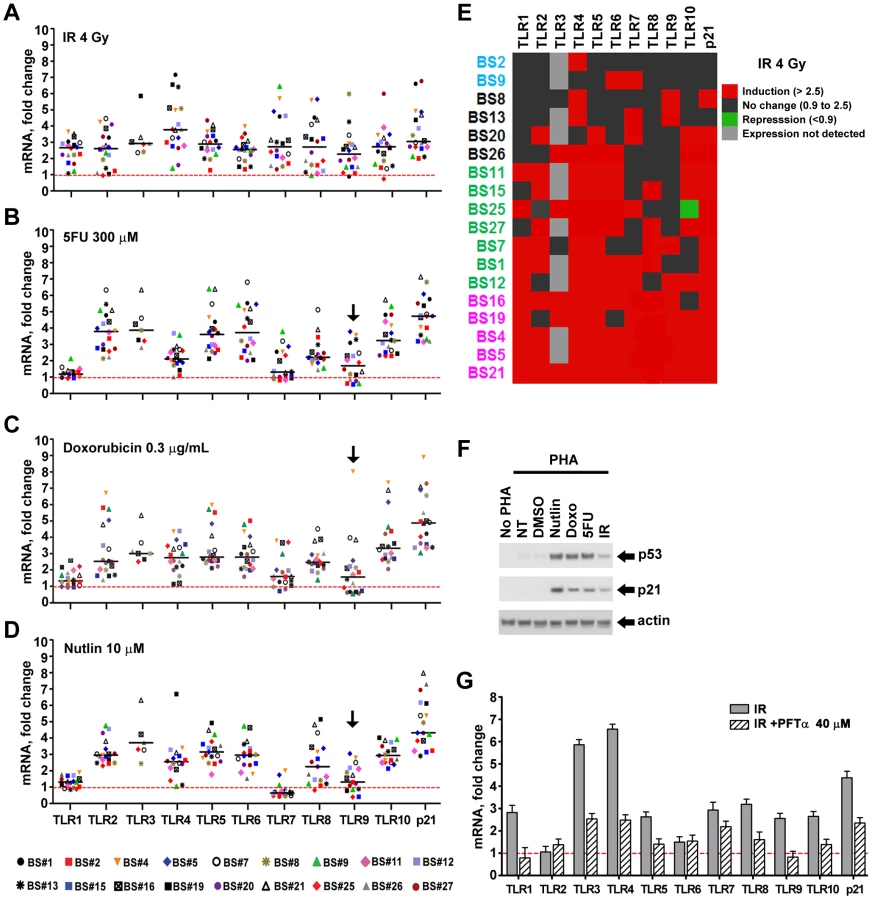
To better assess each TLR gene response across subjects, we portrayed the induction of TLRs for each subject in a format akin to a heat map, as described in Figure 1E for IR, where 2.5-fold induction is indicated in red and <2.5 fold in black (this value corresponds to the minimal p21WAF1 response for nearly all agents and subjects; see Figure S3 for other agents). Among three subjects (BS4, 5, 21), all TLRs were induced at least 2.5-fold by IR exposure, while only 1 (BS2), 2 (BS9) or 3 (BS8, 13) TLRs were induced in four others. There is no obvious pattern to the differences between TLRs or subjects for IR as well as for 5FU and Doxo, although it is clear that TLR1 is generally much less responsive to the last two agents (Figure 1, Figures S2 and S3). Consistent with TLR3 induction by DNA damage in cancer cell lines [9], its expression was induced in 5 out of 7 subjects; however, TLR3 gene expression was not detected in 11 subjects (Figure 1 and Figure S3). Even though all TLRs are responsive in at least one subject, only subjects BS4, 5 and 21 exhibited high responses for most TLRs for all agents tested (Figure S3).
We also addressed statistically the responsiveness of the population as a whole (18 subjects, Figure 1A, 1B, 1C, except TLR3 which was expressed only in 7 subjects) to IR, 5FU, and Doxo employing a t-test (see Material and Methods) to assess the ability of each agent to induce expression of each TLR gene in the population. As expected, induction of the p21 gene in the population was highly significant for all the treatments (p<0.0001). While there was variation between individuals, all the TLR genes in the population were responsive to IR (p<0.0001). A similar likelihood of responsiveness (p<0.0001) was observed for 5FU and Doxo treatment for all but the TLR1 and 7 genes (p<0.003) and TLR9 (p = 0.011 for 5FU and 0.053 for Doxo); however, there were substantial responses for the TLR 9 genes of several individuals. Collectively, these results suggest that multiple pathways may influence TLR expression after DNA damage, and these are specific to the mode of chromosomal stress.
p53 directly enhances TLR expression
Since p53 mediates many DNA damage responses and given our finding that all TLRs respond to at least one DNA metabolic disruptor, we sought to examine in more detail the ability of p53 to induce TLRs. PHA-stimulated lymphocytes were exposed to the p53 activator nutlin (10 µM) to increase p53 levels. Stabilization of p53 normally occurs through stress-induced post-translational modifications affecting both p53 and MDM2 [22]. Nutlin can directly prevent p53 destruction by interfering with the MDM2-p53 interaction [23]. As expected, all treatments activated the p53 pathway in the lymphocytes of all individuals as assessed by immunodetection of p53 and p21 proteins (Figure S4; Figure 1F is a representative example). As shown in Figure 1D, Figures S2 and S3, nearly all TLR genes in most subjects are induced over 2.5 fold after nutlin treatment, except for TLRs 1 and 7. The TLR1 gene much less responsive while the TLR7 gene is generally repressed by p53 induction. Interestingly, TLR7 is induced by the DNA damaging treatments, suggesting p53-independent induction mechanisms. While TLR3 is not detected in 11 subjects, it is induced by nutlin in 5 of the remaining 7 subjects. Although there was variation between individuals, we found that most of the TLR genes in the population were responsive to nutlin. There was no statistically significant induction of the TLR9 gene (p = 0.11); however, there was statistically significant repression of the TLR7 gene (p = 0.006). The expression of all the remaining TLR genes was significantly induced in the population (p<0.003).
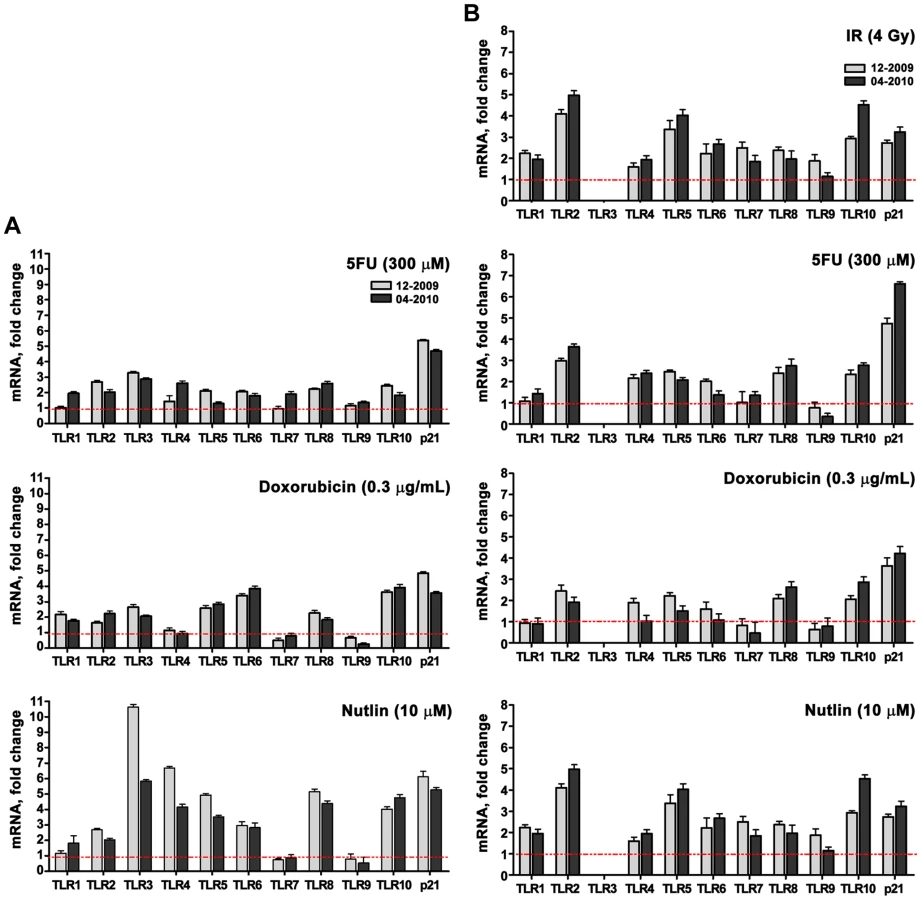
A role for p53 in TLR induction is further indicated by the finding that co-treatment with the p53 inhibitor pifithrin-alpha [24] represses the induction of TLRs by IR and other agents (Figure 1G for IR; Figure 2 for Doxo, 5FU and nutlin). Notably, samples taken on two occasions from the same individuals in a 4 month-interval, blind study revealed a striking consistency in TLR gene expression patterns (Figure 3), thereby excluding technical or temporal variables as significant sources of variability in findings.

The p53 inducibility of most TLRs led us to investigate if p53 could act directly on transcription. As discussed in the Introduction, the commonly accepted consensus target RE consists of two decamers composed of (RRRC A/T A/T GYYY) separated by up to 13 bases (reviewed in [25], [26]). We had established rules for predicting in vivo functionality of p53 REs including separation of <2 bases and dependence on a core CATG (summarized in [3], [6]). Among several potential p53REs identified by in silico search and bioinformatic tools, we found that each TLR has at least one p53 target sequence within ±5 kb of the transcription start site that is predicted to provide at least a weak-to-modest p53 responsiveness (Table 2 contains the sites predicted to have the greatest functional responses). As shown in Figure 4A, each of these target sequences (TLR7 was not examined because we did not find any p53-like binding sequences) could support p53-driven transcription of a luciferase reporter co-transfected, along with a p53 expression plasmid, into p53 null H1299 cells. For half of the TLRs, the induction levels were comparable to those obtained with the moderately responsive p53 target response element of AIP.


Based on these results, we examined whether the functional REs of Figure 4A are indeed targets of induced p53 using chromatin immunoprecipitation (ChIP) analysis. We chose a representative subject BS4 whose lymphocytes exhibited strong Doxo-induced expression (Figure 1) for all TLRs except TLR1 and TLR7. As shown in Figure 4B, there is clear binding following exposure of cells to Doxo at the established p53RE on p21WAF1 as well as at the TLR2, 4, 5, 6, 8, 9 and 10 sequences described in Figure 4A. Thus, all TLR genes are subject to DNA-damage associated transcriptional regulation and most are directly targeted by p53. (We also identified other p53-like sequences in the analyzed regions; however, these were predicted to be less functional than the sequences examined). Furthermore, a direct role for p53 in TLR gene expression was demonstrated using p53 null SaOS2 osteosarcoma cells that have p53 under the control of a tetracycline inducible promoter [27]. Expression of wild-type p53, but not the transcriptionally inactive G279E mutant protein, results in induction of all TLRs except TLR7 (TLR8 was not tested because it is not expressed by SaOS2 cells; Figure S5).
We also determined whether p53 induction of TLR genes by nutlin can lead to a corresponding increase in protein. (Nutlin was chosen because it typically led to the largest increase in p53, as shown in Figure S3.) Using western blot analysis with TLR specific antibodies (see Materials and Methods), the levels of TLR2 and TLR5 proteins were examined in the membrane fraction from stimulated lymphocytes of volunteers BS25 and B26 (sufficient cells were obtained from these subjects to enable the protein measurements; the TLR2 and TLR5 proteins were only detected in the membrane fraction as noted in the Material and Methods). Nutlin treatment resulted in a substantial increase in both proteins in the membrane fraction which corresponded well with induced expression of the TLR 2 and TLR5 genes, as described in Figure 5.

To address the generality of DNA damage-induced TLR expression and the role of p53, we also examined alveolar macrophages since they are well-known to play a pivotal role in pulmonary innate immunity and are susceptible to DNA damage from environmental exposures including cigarette smoke and particulate matter [28]–[30]. The cells were collected by bronchoalveolar lavage of healthy human subjects and treated ex vivo. Among 6 subjects, there were considerable differences between TLRs with little or no induction of TLR1 and TLR6 by Doxo or nutlin, as described in Figure 6, and a dramatic induction of TLR9 by Doxo, suggesting cell-specific factors that determine damage-TLR response profiles. In general, fewer of the TLR genes in the macrophages responded to Doxo and nutlin as compared to lymphocytes and the response levels were not as large.

Activation of p53 influences PAMP-induced TLR cytokine responses
Induction of cytokines is a prototypical functional response that is downstream of TLR activation [11], [31], [32], and increases in TLR expression can enhance PAMP or DAMP-induced signaling and innate-immune mediated effects. As shown in Figure 7, pretreatment of freshly isolated CD3+ T-lymphocytes (see Figure S1) with nutlin to induce TLR2 by p53 resulted in 2 - to 5-fold increased expression of interleukin IL-1 and IL-8 by the TLR2 ligand PAM3CSK4, suggesting that TLR induction by p53 can directly affect innate immune function. (The TLR2 induced cytokine response was examined because of our observation that T-lymphocytes from all subjects experience nutlin-induced TLR2 expression, as shown in Figure 1 and Figure S3). The magnitude of cytokine induction varied between subjects (see Figure S6), suggesting that other factors, besides p53-induced gene expression, affect innate immune responsiveness.

TLR8 promoter SNP determines nutlin and IR responsiveness
Our previous report [8] of a SNP in a potential p53 response element of the TLR8 promoter (AAACAT(G/A)TCa; see Table 1) provided a unique opportunity to directly assess the relationship between p53 and TLR expression. While we had established large differences in the potential p53-responsiveness of the two SNP sequences [8]; see Figure 4A for the “positive”, p53-responsive G-allele), their cellular impact could not be assessed because the TLR8 gene was not expressed in the human cell systems previously examined. Unlike the results with cell lines, we observed a dichotomous response in TLR8 expression in lymphocytes following IR and nutlin treatment (Figure 1), and in alveolar macrophages following nutlin exposure (Figure 6), with some subjects showing low TLR8 induction and others robust induction. We, therefore, determined which p53 response element alleles were present and their relationship to TLR8 induction. Since TLR8 is located on the X-chromosome, males carry only a single copy and females 2 copies (the specific alleles for each volunteer are described in Table 1). As shown in Figure 8 and Figure S3, the ability of nutlin and IR to induce TLR8 correlates well with the presence of the G-allele. The frequency of this allele in our study is 0.43 (13/28, which corresponds to the frequency in the general population (Table 1). Importantly, TLR8 induction was always high when only G-alleles were present (both alleles in females or the single allele in males) and absent if there were only A-alleles. However, among the 3 female subjects that were heterozygous for these alleles, only one responded poorly to both nutlin and IR. Possibly, the variability between female subjects heterozygous for the SNP is related to X chromosome inactivation.

While these results demonstrate differences in the impact of the polymorphic alleles, the differences do not extend to 5FU or Doxo, suggesting that other p53-related target sequences are responsible for the induction or involvement of additional p53-independent mechanisms (Figure 8 and Figure S3). Notably, these findings with TLR8 provide the first direct demonstration, to our knowledge, in humans of the ability of a specific RE in a p53 target gene to drive transcription. The SNP-associated differences in expression may provide opportunities to identify other factors in human primary tissue that determine the ability of specific REs to support p53-driven transcription [33], [34]. Although there are SNPs in the REs of TLR5 and TLR6, as described in Table 2, they are predicted to have no functional impact (see [6]–[8]).
Evolution of TLR gene family responsiveness to p53
Our results with primary human cells have been confirmed for DNA damage induction of TLRs (to be presented elsewhere) in several human cell lines including the previously reported TLR3 [9]. However, as shown in Figure S7, they do not extend to murine cells. The TLR responses to Doxo, 5FU and IR were found to be small in mouse peritoneal elicited macrophages, bone marrow-derived macrophages, and embryonic fibroblasts (MEFs; except for TLR9 in MEFs). The low induction level appears to be p53-independent based on results with nutlin and on the similar responses in p53-positive and -null MEFs. These results are consistent with the lack of sequence conservation between humans and rodents of functional p53 response elements in TLR genes (Figure S8) even though the coding sequences are well-conserved. Although we were able to identify p53 RE-related sequences in the vicinity of the transcription start site of several mouse TLRs, none were predicted to be functional p53 targets.
Discussion
Cellular stress and the inflammatory response are intricately linked pathways subject to endogenous and exogenous challenges. Here, we provide the first evidence that DNA stressors may also modulate inflammatory responses at a fundamental level in primary human cells, namely, by altering TLR expression. All members of the human TLR gene family tested (TLR1-10) are responsive to at least one disruptor of chromosome metabolism. While there are considerable variations between individuals, for each TLR gene there is a group of several subjects that is responsive to at least one of the stressors (5FU, doxorubicin, IR and/or nutlin). Note, for example, that there is at most a low level of TLR1 induction by 5FU and nutlin in the primary cells from 18 subjects and there is even a general repression of TLR7 by nutlin. We thus propose that, contrary to the paradigm that innate immunity is hard-wired, the TLR system in humans actually has a complex, robust responsiveness to environmental conditions that challenge the integrity of the genome. We establish that induction of TLRs by DNA damage is a class effect that in many cases can be mediated by p53 (including repression of TLR7) and prevented by the p53 inhibitor pifithrin. In support of these findings, several potential p53 binding sites were identified in the proximity of the transcription start sites for all TLRs, except TLR7. The new p53REs were identified using functionality rules to predict p53RE responsiveness [6], [7]. These were confirmed with reporter assays and by the binding of p53 in human lymphocytes following stress activation. In addition the functional TLR8 SNP in the p53 target response element directly confirms a role for p53. Interestingly, for TLR2 and TLR10, the most functional sites were predicted to be noncanonical, containing only a half–site p53RE (i.e., one decamer). Previously, the genes FLT1 [35] and RAP80 [36] were shown to be directly controlled by p53 through half-site REs and several other target genes have been identified that are predicted to be regulated by noncanonical p53 RE (summarized in [6]).
The inclusion of TLR genes expands the universe of genes and biological functions that fall within control of the p53 master regulator. The addition of the set of TLR genes identifies a biological gene niche regulated by the p53 master regulator that is distinct from rodents whose TLR genes appear to lack functional p53 REs. It is interesting that a similar niche was identified for all the DNA metabolic genes that are p53-responsive in humans in that none of them respond to p53 in rodents [7], [36]. p53 provides a rapid, integrated signaling mechanism for increasing or maintaining gene responses to acute and chronic DNA damage stress. In a more general sense, we suggest that the evolutionary inclusion of sets of genes with related functions may provide an efficient means of dealing with sudden, temporary challenges that can result from DNA damage and/or DNA damage itself may be a modulator of broader biological threats, as for the case of infection.
We speculate that there may be feedback loops that integrate this newly identified role for p53 in DNA damage and innate immune responses, as described in Figure 9. In this scheme, DNA damage from environmental agents or potentially from TLR-elicited reactive oxygen species (ROS) may amplify the responsiveness of the innate immune system by promoting TLR upregulation. p53 itself displays a variety of roles in mediating ROS signals [37], [38]. On the other hand, TLR upregulation may also sensitize tissues to maladaptive aseptic inflammation in the setting of environmental injury. For example, tissue injury is reported to induce inflammation through release of DAMPs that act upon TLR2, TLR4, and TLR9 [39], [40]. We speculate that, during tissue injury, upregulation/activation of TLRs may serve as a cell-fate counterbalance to p53-mediated pro-apoptotic responses by leading to activation of the pro-survival factor NF-κB [39]. This may be particularly relevant to cancer therapy, as stimulation of TLR5, 7, 8, and 9 have been shown to modify cellular radio - and chemoresistance [41], [42]. These findings demonstrating that p53 can increase an inflammatory response differ from the generally held view relating to the antagonistic affect of p53 on inflammation directed by NF-κB [41]. However, the mechanism here is quite different in that it involves the p53-mediated increase in a receptor that translates ligand interactions into cytokine responses. Our results may be particularly relevant to diseases in which variations in T-cell function can impact pathogenesis, such as autoimmune disease, asthma, and IBD. For example, recent reports suggest that intestinal inflammation can induce genotoxicity in circulating leukocytes [43], while increased DNA damage is also detected in lymphocytes obtained from rheumatoid arthritis patients [44]. The heightened pro-inflammatory status in such patients, as well as the common finding of systemic or multi-organ inflammation during exacerbations of autoimmune disease, might be mediated by circulating immune cells which have suffered DNA damage during passage through inflamed tissues.

Beyond the TLR gene and cell subset variability in response to DNA damage and p53 activation, we also demonstrate considerable inter-individual variation. This variability may be relevant to inter-individual differences in susceptibility to a wide spectrum of diseases and therapies. Our results suggest that various anti-cancer agents may yield different patterns of responses across TLRs and between subjects. Since TLR ligands are increasingly used as adjuvants for vaccines (TLR4 and TLR9) and cancer treatment (TLR3/7/9) (reviewed in [45]), the ability to detect and predict genetically determined inter-individual variability in TLR induction may prove a useful therapeutic tool. Future studies are warranted to determine whether single agents such as nutlin, or even factors that induce chromosome stress, may serve as useful immune adjuvants through manipulation of TLR expression in human subjects.
Materials and Methods
Lymphocyte isolation
Healthy adult volunteers were recruited to the NIEHS Clinical Research Unit and underwent phlebotomy. Subjects were excluded if they had a history of recent infection, were on anti-inflammatory medications, or tested positive for hepatitis B, C or HIV. Up to 300 ml of whole blood were withdrawn from an antecubital vein into citrated tubes. Lymphocytes were isolated using percoll (Sigma) and anti-CD3-coupled Magnetic Beads (Miltenyi Biotec) as per manufacturer's protocol. Cell purity was >98% after percoll/magnetic bead isolation based on flow cytometry. We maintained lymphocytes in RPMI supplemented with 10% FBS. For T cell stimulation, cells were activated with phytohemagglutinin-M (PHA, Invitrogen, 3% vol/vol) for 72 h. The total number of lymphocytes available per treatment conditions after PHA was typically around 10 million cells or less. Cells were treated starting at 48 h post PHA addition and cell cultures were harvested 24 h later. Freshly isolated CD3+ cells were treated with nutlin for 20 h or DMSO as a vehicle control, then washed and exposed to TLR1/2 ligand PAM3CSK4 (1 µg/ml) at 1×106 cells/ml. Total yield of CD3+ cells from a single subject was typically around 10–15 million cells or less. Protocol and procedures were approved by the NIEHS Institutional Review Board.
Alveolar macrophage isolation
Healthy, nonsmoking male volunteers, 18 to 40 yr of age, underwent fiberoptic bronchoscopy with lavage to procure alveolar macrophages. The screening procedures for each subject included a medical history, physical examination, and routine hematologic and biochemical tests. None of the subjects had a history of asthma, allergic rhinitis, chronic respiratory disease, or cardiac disease. Subjects were excluded from the study if they had suffered a recent acute respiratory illness and were asked to avoid exposure to air pollutants such as tobacco smoke and paint fumes. A fiberoptic bronchoscope was wedged into a segmental bronchus of the lingula. Six 50-ml aliquots of sterile saline were instilled and immediately aspirated. The procedure was repeated on the right middle lobe, again using 300 ml of saline. Samples were put on ice immediately after aspiration and centrifuged at 300 x g for 10 min at 4–8°C. Cells from all aliquots were pooled, washed twice with RPMI 1640, and re-suspended in RPMI 1640 at 1.0×106/mL. Total yield was typically around 10–15 million cells or less. Around 2.0×106 cells per well were seeded in a 12 well plate. After 2 hr, cells were washed twice with warm PBS and 2 ml of growing media was added to each well. Cells were then treated with nutlin or doxorubicin. Cells were harvested 24 hr post-treatment. The protocol and consent form were approved by the University of North Carolina School of Medicine Committee on the Protection of the Rights of Human Subjects. Prior to participation in the study, subjects were informed of the procedures and potential risks and each signed a statement of informed consent.
Cancer cell cultures
H1299 lung cancer cells (American Type Culture Collection) were routinely maintained following standard conditions and procedures for culturing mammalian cells. All cultures were incubated at 37°C with 5% CO2. p53 tetracycline inducible SaOS2 TET-off cell lines expressing the wild-type or the G279E mutant protein were cultured as described previously [27]. p53 expression was kept “off” by 2 mg/ml doxycycline (Clontech). To induce the p53 expression, cells were washed 3X with phosphate-buffered saline (PBS) and placed in medium lacking doxycyline during 24 h.
Reagents and treatment conditions
For drug treatment and p53 activation, cells were incubated with doxorubicin (Sigma 0.3 µg/mL), nutlin3 (Sigma, 10 µM) and 5-fluorouracil (Sigma, 300 µM). For ionizing radiation treatment, cells were irradiated at 1.56 Gy/min from a Shepherd cesium irradiator in PBS at room temperature at final dose of 4 Gy. Where indicated cells were also pretreated 2 h with p53 inhibitor pifithrin-alpha (Sigma, 40 µM).
RNA isolation and gene expression analysis
Total RNA was isolated by RNEasy kit (Qiagen). Real-time PCR was performed in triplicate with Taqman PCR Mix (Applied Biosystems) in the 7000 ABI sequence Detection System (Applied Biosystems). All human and mouse primers were purchased from Applied Biosystems (information available upon request). For PHA stimulated lymphocytes and alveolar macrophages expression of TLR genes was normalized to 18S ribosomal RNA gene while for freshly isolated CD3+ T cells, the glucuronidase-beta gene was used for normalization.
Luciferase reporter assays
Pairs of complimentary oligonucleotides for the desired p53RE from selected TLRs and containing restriction sites were cloned into the open reading frame of firefly luciferase pGL4.26 plasmid (Promega) previously double digested by Xho I/Kpn I restriction enzymes. The identity of the inserts was confirmed by DNA sequencing. Luciferase activity was measured 48 h after Fugene6 - mediated co-transfection of the TLR p53RE constructs in the presence of p53 (pC53-SN3) or empty vector pCMV NEO-BAM3 along with pRL-TK Renilla as a transfection efficiency control into p53 null H1299 cells, as previously described [5]. Forty-eight hours post-transfection extracts were prepared using the Dual Luciferase Assay System (Promega) following the manufacturer's protocol and luciferase activity was measured on a Victor Wallac multilabel plate reader (PerkinElmer). Relative luciferases activities for each construct was defined as the mean value of the firefly luciferase/Renilla luciferase rations obtained from 4 independent experiments performed in triplicate.
Immunoblot analysis
Whole cell extracts were quantified using the Bradford protein assay kit and gamma globulin as a reference standard (BioRad). For TLR protein detection, cellular pellets were subjected to subcellular protein fractionation (Thermo Scientific) following the manufacturer's instructions and protein was quantified using BCA protein assay kit (Thermo Scientific). For TLR western blot analysis ∼30 µg of total membrane fraction was used, while for the analysis of other proteins ∼25 µg of total cell extract were used. As expected, we did not detect TLR2 and TLR5 in the cytosolic fractions; therefore, those data are not included. Proteins were resolved on 4–12% BisTris NuPAGE and transferred to polyvinylidene difluoride membranes (Invitrogen) and were visualized with primary antibodies followed by horseradish peroxidase–conjugated goat anti–mouse or donkey anti-goat immunoglobulin (Santa Cruz Biotechnology) through the use of enhanced chemiluminescence reagents (Amersham Biotechnology). The primary antibodies used in these studies were against p53 (DO1, Santa Cruz Biotechnology,), p21 (SXM30, BD Biosciences Pharmigen) and Actin (C-11 Santa Cruz Biotechnology). The following is the list of TLR antibodies tested in this study in order to detect TLR protein expression in whole cell extracts as well as membrane and cytosol protein fractions: TLR8 ab24185 and TLR10 ab45088 from Abcam, Inc.; TLR1#2209, TLR2#2229, TLR7#2633 and TLR9#2254 from Cell Signaling. We also used a Toll-like receptor detection kit that includes antibodies for all human TLRs (TLR1 to TLR10 antibodies from ProSci, Inc., as well as TLR3-4H270 and TLR5-H1-27 antibodies from Santa Cruz Bitoechnology. The TLR3-IMG-315A, TLR4-IMG6370A antibodies were from IMGENEX. Only the TLR2 (Cell Signalling) and TLR5 (Santa Cruz) gave clear results. Attempts to detect other TLRs with these antibodies were unsuccessful and appear to be a general problem with TLR antibodies from our collective experience. One of the antibodies enabled us to detect induction of full length TLR4 by nutlin; however, those results are not presented due to the appearance in both untreated and treated samples of nonspecific bands.
Chromatin Immunoprecipitation (ChIP) assays
ChIP assays were done as previously described [35] using ChIP kits (Millipore). Approximately 40×106 PHA stimulated lymphocytes were used for each experimental sample. Cell lysates were sonicated using conditions that yield chromatin fragments 200–500 bp long. One microgram of DO-7 p53-specific monoclonal antibody (BD Biosciences Pharmigen) was used per ChIP assay. As a negative control, mouse Ig (Santa Cruz Biotechnology) was used. PCR amplifications were performed on immunoprecipitated chromatin using primers to amplify specific regions on the TLRs promoters (sequence information available upon request). The PCR cycles were as follows: initial 10 min Taq polymerase (Invitrogen) at 95°C followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The PCR products were then run on a 1.8% agarose gel.
Flow cytometry analysis
Cells were resuspended in 100 µl of PBS and incubated with 5 µl of fluorescent antibody per sample for 30 min, then washed and fixed with 0.5% paraformaldehyde. The fluorescence intensity was evaluated using a Becton Dickinson LSR II Flow Cytometer. All antibodies used for FACS were from BD Pharmigen.
Genotyping
SNPs were assessed by three different approaches. In RFLP assays, genomic DNA was extracted from Percoll-isolated lymphocytes by DNeasy kit (Qiagen). For the SNP in the TLR8 p53RE#6 (AGGCAAGATGAAACAT(G/C)TCA), the G-SNP creates a unique restriction cutting site for NspI (R CATG Y). PCR was performed with 100 ng of DNA, 50 pmol of each primer, 1.5 mM MgCl2, 1 µL 10× PCR buffer, and 0.0125 U of Taq (Invitrogen). After 10 min at 94°C, 35 cycles were repeated as follows: 94°C 30 s, 60°C 30 s, and 72°C 35 s; this cycling was followed by a final extension at 72°C for 10 min. The PCR product was digested with 5 U Nsp I (New England Biolabs, Ipswich, MA), at 37°C for 4 h. Since Nsp I recognizes the polymorphic sequence, a G allele is demonstrated by the presence of two fragments 109 and 69 bp in a gel. The A allele is revealed by the presence of a single 177 bp band. The following primers were used for amplifying the region containing the p53RE on TLR8 promoter region:
Forward: 5′TCATAACAAGGTGTTCCACAGTC-3′
Reverse: 5′-ATCTGGCCCTTTACAGAAAAAGTT-3′.
The status of this SNP was determined also by using a Taqman SNP genotyping assay. All primers were purchased from Applied Biosystems (Assay ID:C_27497635_10). For direct sequencing the region containing the p53RE was first PCR amplified using the following pairs of primers:
Forward: 5′-TTGAATTCCCTTAGGGTGTGA-3′,
Reverse: 5′-AAACTGCCTTCGATTATTATTATTACA-3′
This was followed by running the samples on a TEA-agarose gel. The expected product (397 bp) was cut out and cleaned using QIAquick gel extraction kit (QIAGEN). The sequencing reactions used Big dye (Applied Biosystems) per manufacturer recommendations and the following primers:
Foward: 5′TCATAACAAGGTGTTCCACAGTC-3
Reverse: 5′-ATCTGGCCCTTTACAGAAAAAGTT-3′
Mouse primary cell culture
p53+/+ and p53−/ − mouse embryonic fibroblasts (MEFs) were cultured in DMEM media and 10% of FBS. Female C57BL/6 mice were purchased from Jackson Laboratories. All experiments were performed in accordance with the Animal Welfare Act and the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals after review of the protocol by the Animal Care and Use Committee of the National Institute of Environmental Health Sciences. For murine peritoneal macrophage harvests, mice were injected i.p. with 2 ml of 4% Brewer's thioglycollate and euthanized 96 h later. The peritoneum was washed with 10 ml ice cold PBS three times. Cells were centrifuged (1,000x RPM, 6 minutes, 4°C) and washed twice with sterile PBS. Peritoneal exudate macrophages were resuspended in DMEM/0.1% FBS, counted, and plated at 2×106 cells/well in a 12-well plate. Cells were allowed to settle for 2 h (37°C/5% CO2) before replacing media with DMEM complimented with 10% FBS.
For bone marrow-derived macrophages (BMM), marrow was flushed from femoral and tibial bones using bone marrow media (DMEM/2 mM L-glutamine/10% L929-conditioned medium/10% FBS). Cells were spun down (2200x RPM, 5 min, 4°C), brought up in 1 ml sterile ACK buffer, incubated 4°C for 1 min after which 10 ml PBS was added. Cells were spun as above, resuspended in bone marrow medium, counted, and plated at 1×106 cells/well in a 12-well plate. Cells were cultured at 37°C and 10% CO2 in 2 ml bone marrow medium/well and fed on Day 5 with addition of 1 ml medium/well. Experiments were performed on Day 6. At 24 h post-treatment, cells were harvested for RNA extraction.
Statistical methods
To examine statistically whether the average mRNA fold-change at each locus in the population sampled differed from 1 for the various exposures, we applied one-sample Student's t tests to log-transformed values of mRNA fold change. The logarithmic transformation helps the data meet the distributional assumptions for the t test. This procedure, in effect, tests the null hypothesis that the geometric mean mRNA fold change at the locus is equal to 1 against the two-sided alternative that the geometric mean differs from 1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HeL
HeX
LoweSW
HannonGJ
2007 microRNAs join the p53 network–another piece in the tumour-suppression puzzle. Nat Rev Cancer 7 819 822
2. HuarteM
GuttmanM
FeldserD
GarberM
KoziolMJ
2010 A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142 409 419
3. JordanJJ
MenendezD
IngaA
NourredineM
BellD
2008 Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet 4 e1000104 doi:10.1371/journal.pgen.1000104
4. NoureddineMA
MenendezD
CampbellMR
BandeleOJ
HorvathMM
2009 Probing the functional impact of sequence variation on p53-DNA interactions using a novel microsphere assay for protein-DNA binding with human cell extracts. PLoS Genet 5 e1000462 doi:10.1371/journal.pgen.1000462
5. MenendezD
IngaA
ResnickMA
2010 Estrogen receptor acting in cis enhances WT and mutant p53 transactivation at canonical and noncanonical p53 target sequences. Proc Natl Acad Sci U S A 107 1500 1505
6. MenendezD
IngaA
ResnickMA
2009 The expanding universe of p53 targets. Nat Rev Cancer 9 724 737
7. JeggaAG
IngaA
MenendezD
AronowBJ
ResnickMA
2008 Functional evolution of the p53 regulatory network through its target response elements. Proc Natl Acad Sci U S A 105 944 949
8. TomsoDJ
IngaA
MenendezD
PittmanGS
CampbellMR
2005 Functionally distinct polymorphic sequences in the human genome that are targets for p53 transactivation. Proc Natl Acad Sci U S A 102 6431 6436
9. TauraM
EgumaA
SuicoMA
ShutoT
KogaT
2008 p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol Cell Biol 28 6557 6567
10. PalmNW
MedzhitovR
2009 Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227 221 233
11. BauerS
MullerT
HammS
2009 Pattern recognition by Toll-like receptors. Adv Exp Med Biol 653 15 34
12. GarantziotisS
HollingsworthJW
ZaasAK
SchwartzDA
2008 The effect of toll-like receptors and toll-like receptor genetics in human disease. Annu Rev Med 59 343 359
13. HennessyEJ
ParkerAE
O'NeillLA
2010 Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov 9 293 307
14. IwasakiA
MedzhitovR
2010 Regulation of adaptive immunity by the innate immune system. Science 327 291 295
15. ArmstrongL
MedfordAR
HunterKJ
UppingtonKM
MillarAB
2004 Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol 136 312 319
16. Komai-KomaM
JonesL
OggGS
XuD
LiewFY
2004 TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A 101 3029 3034
17. HarterL
MicaL
StockerR
TrentzO
KeelM
2004 Increased expression of toll-like receptor-2 and -4 on leukocytes from patients with sepsis. Shock 22 403 409
18. HayashiF
MeansTK
LusterAD
2003 Toll-like receptors stimulate human neutrophil function. Blood 102 2660 2669
19. Kurt-JonesEA
MandellL
WhitneyC
PadgettA
GosselinK
2002 Role of toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF enhances TLR2 expression and TLR2-mediated interleukin 8 responses in neutrophils. Blood 100 1860 1868
20. MuzioM
BosisioD
PolentaruttiN
D'AmicoG
StoppacciaroA
2000 Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol 164 5998 6004
21. CorreaCR
CheungVG
2004 Genetic variation in radiation-induced expression phenotypes. Am J Hum Genet 75 885 890
22. LevineAJ
1997 p53, the cellular gatekeeper for growth and division. Cell 88 323 331
23. VassilevLT
VuBT
GravesB
CarvajalD
PodlaskiF
2004 In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303 844 848
24. KomarovPG
KomarovaEA
KondratovRV
Christov-TselkovK
CoonJS
1999 A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285 1733 1737
25. el-DeiryWS
KernSE
PietenpolJA
KinzlerKW
VogelsteinB
1992 Definition of a consensus binding site for p53. Nat Genet 1 45 49
26. RileyT
SontagE
ChenP
LevineA
2008 Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9 402 412
27. MenendezD
IngaA
ResnickMA
2006 The biological impact of the human master regulator p53 can be altered by mutations that change the spectrum and expression of its target genes. Mol Cell Biol 26 2297 2308
28. DesleeG
WoodsJC
MooreC
ConradiSH
GieradaDS
2009 Oxidative damage to nucleic acids in severe emphysema. Chest 135 965 974
29. IshidaT
HironoY
YoshikawaK
HuteiY
MiyagawaM
2009 Inhibition of immunological function mediated DNA damage of alveolar macrophages caused by cigarette smoke in mice. Inhal Toxicol 21 1229 1235
30. MengZ
ZhangQ
2007 Damage effects of dust storm PM2.5 on DNA in alveolar macrophages and lung cells of rats. Food Chem Toxicol 45 1368 1374
31. PasareC
MedzhitovR
2005 Control of B-cell responses by Toll-like receptors. Nature 438 364 368
32. SchnareM
BartonGM
HoltAC
TakedaK
AkiraS
2001 Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2 947 950
33. EspinosaJM
VerdunRE
EmersonBM
2003 p53 functions through stress - and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell 12 1015 1027
34. GomesNP
EspinosaJM
2010 Differential regulation of p53 target genes: it's (core promoter) elementary. Genes Dev 24 111 114
35. MenendezD
KrysiakO
IngaA
KrysiakB
ResnickMA
2006 A SNP in the flt-1 promoter integrates the VEGF system into the p53 transcriptional network. Proc Natl Acad Sci U S A 103 1406 1411
36. YanJ
MenendezD
YangXP
ResnickMA
JettenAM
2009 A regulatory loop composed of RAP80-HDM2-p53 provides RAP80-enhanced p53 degradation by HDM2 in response to DNA damage. J Biol Chem 284 19280 19289
37. HussainSP
AmstadP
HeP
RoblesA
LupoldS
2004 p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res 64 2350 2356
38. LiuB
ChenY
St ClairDK
2008 ROS and p53: a versatile partnership. Free Radic Biol Med 44 1529 1535
39. JiangD
LiangJ
FanJ
YuS
ChenS
2005 Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11 1173 1179
40. ZhangQ
RaoofM
ChenY
SumiY
SursalT
2010 Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464 104 107
41. BurdelyaLG
KrivokrysenkoVI
TallantTC
StromE
GleibermanAS
2008 An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science 320 226 230
42. Cherfils-ViciniJ
PlatonovaS
GillardM
LauransL
ValidireP
2010 Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J Clin Invest 120 1285 1297
43. WestbrookAM
WeiB
BraunJ
SchiestlRH
2009 Intestinal mucosal inflammation leads to systemic genotoxicity in mice. Cancer Res 69 4827 4834
44. ShaoL
FujiiH
ColmegnaI
OishiH
GoronzyJJ
2009 Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med 206 1435 1449
45. KanzlerH
BarratFJ
HesselEM
CoffmanRL
2007 Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med 13 552 559
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy