
Diverse Roles and Interactions of the SWI/SNF Chromatin Remodeling Complex Revealed Using Global Approaches
A systems understanding of nuclear organization and events is critical for determining how cells divide, differentiate, and respond to stimuli and for identifying the causes of diseases. Chromatin remodeling complexes such as SWI/SNF have been implicated in a wide variety of cellular processes including gene expression, nuclear organization, centromere function, and chromosomal stability, and mutations in SWI/SNF components have been linked to several types of cancer. To better understand the biological processes in which chromatin remodeling proteins participate, we globally mapped binding regions for several components of the SWI/SNF complex throughout the human genome using ChIP-Seq. SWI/SNF components were found to lie near regulatory elements integral to transcription (e.g. 5′ ends, RNA Polymerases II and III, and enhancers) as well as regions critical for chromosome organization (e.g. CTCF, lamins, and DNA replication origins). Interestingly we also find that certain configurations of SWI/SNF subunits are associated with transcripts that have higher levels of expression, whereas other configurations of SWI/SNF factors are associated with transcripts that have lower levels of expression. To further elucidate the association of SWI/SNF subunits with each other as well as with other nuclear proteins, we also analyzed SWI/SNF immunoprecipitated complexes by mass spectrometry. Individual SWI/SNF factors are associated with their own family members, as well as with cellular constituents such as nuclear matrix proteins, key transcription factors, and centromere components, implying a ubiquitous role in gene regulation and nuclear function. We find an overrepresentation of both SWI/SNF-associated regions and proteins in cell cycle and chromosome organization. Taken together the results from our ChIP and immunoprecipitation experiments suggest that SWI/SNF facilitates gene regulation and genome function more broadly and through a greater diversity of interactions than previously appreciated.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1002008
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002008
Summary
A systems understanding of nuclear organization and events is critical for determining how cells divide, differentiate, and respond to stimuli and for identifying the causes of diseases. Chromatin remodeling complexes such as SWI/SNF have been implicated in a wide variety of cellular processes including gene expression, nuclear organization, centromere function, and chromosomal stability, and mutations in SWI/SNF components have been linked to several types of cancer. To better understand the biological processes in which chromatin remodeling proteins participate, we globally mapped binding regions for several components of the SWI/SNF complex throughout the human genome using ChIP-Seq. SWI/SNF components were found to lie near regulatory elements integral to transcription (e.g. 5′ ends, RNA Polymerases II and III, and enhancers) as well as regions critical for chromosome organization (e.g. CTCF, lamins, and DNA replication origins). Interestingly we also find that certain configurations of SWI/SNF subunits are associated with transcripts that have higher levels of expression, whereas other configurations of SWI/SNF factors are associated with transcripts that have lower levels of expression. To further elucidate the association of SWI/SNF subunits with each other as well as with other nuclear proteins, we also analyzed SWI/SNF immunoprecipitated complexes by mass spectrometry. Individual SWI/SNF factors are associated with their own family members, as well as with cellular constituents such as nuclear matrix proteins, key transcription factors, and centromere components, implying a ubiquitous role in gene regulation and nuclear function. We find an overrepresentation of both SWI/SNF-associated regions and proteins in cell cycle and chromosome organization. Taken together the results from our ChIP and immunoprecipitation experiments suggest that SWI/SNF facilitates gene regulation and genome function more broadly and through a greater diversity of interactions than previously appreciated.
Introduction
Chromosomes undergo a wide variety of dynamic processes including transcription, replication, repair and packaging. Each of these activities requires the recruitment and congregation of a particular set of factors and chromosomal elements. For example visualization of nascent mRNA in HeLa cells has led to a model of transcription units being clustered into “factories” thereby facilitating optimal engagement of RNA Polymerase II (Pol II) and coordination with other crucial holoenzyme complexes [1]–[3]. In addition to RNA Pol II and transcription factors, transcriptional assemblages include proteins critical to regulating chromatin. The accessibility of nuclear proteins to DNA is often controlled by ATP-dependent chromatin remodeling complexes, which are thought to play a role in a number of different cellular transactions by reshaping the epigenetic landscape.
The SWI/SNF (switch/sucrose nonfermentable) chromatin remodeling proteins were first discovered in Saccharomyces cerevisiae as components of a 2 MDa complex that repositions nucleosomes for vital tasks such as transcriptional control, DNA repair, recombination and chromosome segregation [4], [5]. Mammalian SWI/SNF is comprised of approximately ten subunits and the combinations of these subunits, some of which have multiple isoforms, enable multiple varieties of SWI/SNF complexes to exist both within a given cell and across cell types [6]. Among these subunits either of the two ATPases, Brg1 or Brm, is sufficient to remodel nucleosome arrays in vitro, however maximal nucleosome remodeling activity is achieved when the SWI/SNF subunits BAF155, BAF170 and Ini1 are present in a 2∶1 stoichiometry relative to Brg1 [7]. Whereas the ATPases have an obvious catalytic function, the roles of the other SWI/SNF subunits are largely obscure. Several reports indicate that BAF155 and BAF170 provide scaffolding functions for other SWI/SNF subunits as well as regulating their protein levels [8], [9]. SWI/SNF also contains β-actin and the actin-related protein BAF53, suggesting a possible bridge to nuclear organization or signal transduction, e.g. through phosphatidylinositol signaling [10], [11]. Phosphatidylinositol 4,5-bisphosphate has been shown to bind to Brg1 and promote binding to actin filaments [12]. Mutations resulting in loss of Ini1 function are associated with rare but aggressive pediatric cancers [13], [14]. The SWI/SNF subunits Brg1 [15] and ARID1A [16]–[18] are likewise thought to have tumor suppressor roles based on mutations recovered from other tumor types. Curiously, Ini1 alone has a unique and largely undefined role in HIV-1 infection that includes binding of Ini1 to HIV-1 integrase and the cytoplasmic export of Ini1 and its incorporation into HIV-1 particles [19]–[21].
The role of SWI/SNF components in cancer and tumor suppression is poorly understood despite extensive study. Detailed investigations of individual loci have implicated SWI/SNF in various transcriptional pathways including the cell cycle and p53 signaling [22], insulin signaling [23], and TGFβ signaling [24], as well as signaling through several different nuclear hormone receptors [25]. Although in vitro experiments and single-gene studies have been informative and have laid the foundation for understanding chromatin remodeling, a global analysis of targets of SWI/SNF is expected to yield a more extensive view into the biological roles of SWI/SNF components and their involvement in human disease.
In this study we present two complementary global analyses of SWI/SNF subunits to provide a more systematic view of SWI/SNF functions. First we performed ChIP-Seq with the ubiquitous SWI/SNF components Ini1, BAF155, BAF170 as well as the Brg1 ATPase. Second, in a parallel set of studies we performed mass spectrometry identification of proteins that co-immunoprecipitate with SWI/SNF components. Using our ChIP-Seq results the resulting chromosomal locations were integrated with published annotations to yield a more complete understanding of SWI/SNF on a genome-wide scale. We find SWI/SNF components frequently occupy transcription start sites (TSSs), enhancers, CTCF regions and many regions occupied by Pol II. Further analyses of the SWI/SNF regions we identified by ChIP-Seq reveals that SWI/SNF factors target genes and signaling pathways involved in cell proliferation and cancer. Our investigation of SWI/SNF protein interactions detected not only the expected co-occurrences of individual SWI/SNF factors with each other but also with cellular components such as nuclear matrix proteins, key transcription factors and centromere proteins implying a ubiquitous role in gene regulation and nuclear function. We find an overrepresentation of both SWI/SNF-associated chromosomal regions and proteins in cell cycle and chromosome organization. Collectively our results suggest that SWI/SNF is at the nexus of multiple signal transduction pathways, essential chromosomal functions and nuclear organization.
Results
Genome-wide mapping of SWI/SNF subunits reveals many different co-associations
We identified the targets of four SWI/SNF components, Ini1 (SMARCB1), Brg1 (SMARCA4), BAF155 (SMARCC1) and BAF170 (SMARCC2), using ChIP-Seq. Chromatin complexes were isolated from HeLa S3 nuclei following independent immunoprecipitations with antibodies for each factor. Each of these antibodies was characterized by both immunoblot and mass spectrometry analyses (see Materials and Methods). Reads that mapped uniquely to the genome were retained (29–33 million reads per data set; Table 1) and significant binding regions were identified using the PeakSeq program with q-value<0.05 [26]. The peaks were compared to a similarly-sized data set of uniquely mapped ChIP DNA reads obtained from control immunoprecipitation experiments using normal IgG (i.e. a control serum that is not directed to any known antigens). Using this approach we identified many Ini1-, Brg1-, BAF155 - and BAF170-associated regions (Table 1).

The majority of SWI/SNF binding occurs near (±2.5 kb) protein-coding genes, a distribution that is significant relative to a random target list (p<1×10−16; Genome Structure Correction (GSC) test [27]; see Materials and Methods). Several examples of SWI/SNF positioning relative to genic regions are shown in Figure 1 and Figure S1. In order to further examine SWI/SNF locations with respect to gene-rich and gene-poor regions we obtained a set of histone H3K27me3 domains that were identified in HeLa cells (Table S1; [28]) because this chromatin mark often occurs in gene-poor and repressed (i.e. heterochromatin) regions. Although most SWI/SNF-binding occurs outside H3K27me3 domains, we observed that SWI/SNF is occasionally found in heterochromatin regions, as shown in Figure 2. In this example a 7.5 Mb heterochromatin region on Chr16 contains a single gene, the neuronal cadherin CDH8, that is repressed and lacks RNA Pol II, however several SWI/SNF binding regions are found nearby.


We have performed considerable analyses of the targets for the individual SWI/SNF factors, particularly with respect to elements representing several major classes of genomic features including promoters (Ensembl protein-coding genes), RNA Pol II sites [26], CTCF sites [28], and predicted enhancers [29]. All of these features were identified in HeLa cells (Table 1, Tables S1 and S2; see Materials and Methods). In comparisons between the individual target lists for Ini1, Brg1, BAF155 and BAF170 with promoters, RNA Pol II sites, CTCF sites and enhancers we found that each SWI/SNF factor is significantly overrepresented for each of these major classes of genomic elements (p<1×10−16, GSC test, see Materials and Methods). To arrive at a single unified and more conservative list of SWI/SNF locations, we first took the union of all regions for Ini1, BAF155, BAF170 and Brg1, resulting in 69,658 SWI/SNF regions. We then trimmed this list to a high-confidence set of 49,555 sites by eliminating those regions where either only a single SWI/SNF subunit was present or that those regions that did not co-occur with either promoters, RNA Pol II sites, CTCF sites or predicted enhancers. We used this list of 49,555 SWI/SNF regions for all subsequent analyses unless otherwise noted (Table S3). The four major classes of genomic features mentioned above were overrepresented in both the 69,658 SWI/SNF regions as well as the more conservative list 49,555 SWI/SNF regions (p<1×10−16, GSC test).
We next examined the configurations of our 49,555 SWI/SNF regions (Figure 3A and Table 2). Ini1, BAF155 and BAF170 have been described as forming a ‘core’ based on their ability to stimulate remodeling activity of the Brg1 ATPase in reconstitution experiments [7]. Among our data 30,310 regions (61%) have two or more SWI/SNF components and 9,760 regions (20%) contain the core of Ini1, BAF155 and BAF170; for the purposes of this study we call this the ‘core set’. Among putative complexes comprised of two or more SWI/SNF subunits, we observed BAF155 was the subunit most common to each binding region. Only 770 SWI/SNF subunit co-occurrences were recovered that lacked BAF155 as compared to 6,467 for BAF170 and 14,824 for Ini1. This finding is consistent with several previous studies showing that BAF155 is important for SWI/SNF complex stability [8], [9]. BAF155 may increase the stability of the complex during assembly, or BAF155 may be easier to detect by ChIP.


Genome-wide locations of SWI/SNF components suggest diverse roles in gene regulation
One of the primary functions of chromatin remodeling complexes is to assist in gene regulation. Among the SWI/SNF regions in our high-confidence union set of 49,555 sites, 29% correspond to the 5′ ends of protein-coding Ensembl genes, 40% correspond to Pol II sites, 17% correspond to CTCF sites and 43% correspond to predicted enhancer regions (Figure 3B; Table 3). The various combinations of these four elements account for a total of 90% of the SWI/SNF union regions; 4,800 (10%) of the SWI/SNF regions are unclassified using the above elements. Similar trends were observed for the 9,760 SWI/SNF “core” regions where Ini1, BAF155 and BAF170 all co-occur (Table 3). None of these four particular SWI/SNF subunits or any combinations thereof exhibited a differential preference for one type of element (Table S4).

There are some differences between the SWI/SNF core and union regions. The SWI/SNF core regions are overrepresented for RNA Pol II (p<9.9×10−16; hypergeometric test) and 5′ ends (p<6.5×10−211; hypergeometric test) relative to all of the SWI/SNF high-confidence union regions; however the SWI/SNF high-confidence union regions are overrepresented for enhancer regions relative to the Ini1-BAF155-BAF170 core (p<2.4×10−67; hypergeometric test). Neither the SWI/SNF core nor the high-confidence union regions were over - or underrepresented for CTCF sites relative to each other (p>0.05; hypergeometric test).
Enhancers are often characterized by long-range interactions. We examined the locations of SWI/SNF binding regions in the 150 kb CIITA region where numerous chromosomal looping interactions have been mapped at high resolution in HeLa cells using 3C (Chromosome Conformation Capture). Brg1 has been previously mapped at several sites in this locus in these cells [30]. Superimposition of these 3C data on our SWI/SNF ChIP-Seq data (Figure 4) reveals that all six of the 3C interacting regions in the CIITA locus (−50 kb, −16 kb, −8 kb, pIV, +40 kb and +59 kb) are bound by SWI/SNF components. Moreover certain individual SWI/SNF component binding regions that appeared initially as orphans may now be seen as part of a complete complex when joined with a distal element. For example Ini1 at pIV when joined with BAF155 and BAF170 regions at the −16 kb element forms a SWI/SNF core. Thus in the CTIIA locus SWI/SNF regions are often associated with 3C regions and many of the regions bound by individual factors may in fact be part of entire SWI/SNF complexes inside the nucleus.

Overall our ChIP-Seq results are summarized in Table 1, Table 2, Table 3, and Figure 3 and indicate that SWI/SNF likely contributes to gene regulation through many different avenues in light of its binding to promoters, enhancers and CTCF sites. Furthermore SWI/SNF may facilitate looping interactions among these various elements as it has been shown in vitro that SWI/SNF can interact simultaneously with multiple DNA sites and generate loops between them [31]. Interestingly we found a slightly higher presence of the SWI/SNF core at TSSs and with Pol II than the SWI/SNF union regions with these elements (Table 3). Thus a complete core of Ini1, BAF155 and BAF170 may be required for effective promoter function whereas only a subset of these factors may be required for enhancer function. Alternatively a full SWI/SNF core may be more difficult to recover from a single enhancer element as compared to a more compact promoter region due to the enhancer's presumed interaction with many different distal elements.
RNA polymerases are extensively colocalized with SWI/SNF
As detailed above SWI/SNF regions are enriched for Pol II. To explore the prevalence of SWI/SNF with transcriptional machinery we asked whether the converse would also be true, namely if regions bound by RNA polymerases are enriched for SWI/SNF. Indeed Pol II regions are enriched for SWI/SNF binding regions (p<1×10−16, GSC test). Although Pol II overlaps extensively with SWI/SNF it differs from SWI/SNF in its concordance with CTCF and enhancer regions (Table S5). Pol II regions lacking SWI/SNF show a five-fold decrease in CTCF sites and a two-fold decrease in enhancer regions as compared to those Pol II regions containing SWI/SNF.
We further compared our SWI/SNF regions with binding intervals identified for RNA polymerase III (Pol III), which in addition to transcribing tRNA and other non-protein coding RNAs has an emerging role in the formation of boundary elements [32], [33]. Pol III localization data were obtained from published ChIP-Seq studies using HeLa cells ([34], [35]; Tables S1 and S6) and constitute 478 known and novel Pol III-associated regions. Pol II is often associated with Pol III (Table 4; reviewed in [32]. Therefore we examined whether SWI/SNF was associated with Pol III binding regions independently of Pol II. Of the 478 Pol III regions, 253 Pol III intervals lack Pol II and among these 39% (98/253) contain one or more SWI/SNF components. These results suggest that SWI/SNF association with Pol III can occur independently of Pol II.
Overall 65% (309/478) of Pol III regions and 84% (19,541/23,320) of Pol II regions have at least one SWI/SNF factor associated with them. The Ini1-BAF155-BAF170 core is found at 41% (195/478) of Pol III regions and 52% (12,079/23,320) of Pol II regions. From the colocalizations of SWI/SNF, Pol II and Pol III we see that there is substantial overlap among these factors yet each of these factors also has distinct characteristics.
SWI/SNF components bind near many expressed regions
SWI/SNF is known to act as both an activator and repressor of transcription [36]. We examined the locations of four SWI/SNF components relative to transcribed regions in HeLa S3 cells using the RNA-Seq data of Morin et al. [37], Ini1, Brg1, BAF155 and/or BAF170 are present at or near the 5′ ends (±2.5 kb) of 71 to 92% of active protein-coding genes. As noted above, SWI/SNF occupancy in promoters is similar to that of Pol II and each of the factors is individually enriched in promoter regions (p<1×10−16, GSC test). Although the majority of Ini1, Brg1, BAF155 and BAF170 target genes are expressed, an appreciable fraction of gene targets have little or no detectable mRNA in HeLa cells. A closer examination of the union regions where a SWI/SNF component is located in the promoter of an inactive gene reveals that 58% (2,063/3,565) of these promoters are co-associated with Pol II suggesting transcriptional stalling (reviewed in [38], [39].
Considering that SWI/SNF components bind near many expressed regions and that SWI/SNF factors occur in a multitude of configurations (Figure 3 and Table S3), we examined transcript expression levels for all possible combinations of Ini1, Brg1, BAF155 and BAF170 occurrences. Using the RNA-Seq data of Morin et al. [37], we examined transcript expression levels corresponding to each of these configurations (Figure 5). We see that the highest levels of transcription are associated with the following four configurations: 1) the complete core of Ini1, BAF155 and BAF170; 2) the complete core plus Brg1; 3) Ini1 and BAF155 only and 4) Brg1, BAF155 and BAF170. Although BAF155 is the subunit that is common to all of the configurations associated with the highest levels of transcription, it does not appear to be the sole driver of transcriptional activity. Compared against each other, all three components of the core complex taken individually have nearly indistinguishable profiles. Despite the involvement of Brg1 in two of the four configurations with the highest expression levels, most other configurations involving Brg1 are restricted to profiles associated with the lowest expression levels. One inference from these data is that certain combinations of SWI/SNF subunits are likely synergistic in promoting transcription whereas other combinations may be inhibitory or unstable.

We also examined SWI/SNF occurrences relative to 48,403 non-canonical small RNAs from HeLa cells (≤156 bp; Table S1) where most (83%; p<1×10−16, GSC test) of these small RNAs are near protein-coding genes [40]. Approximately one third (30%) of this entire small RNA set is within 1 kb of a target from our high-confidence union list of 49,555 SWI/SNF regions. The incidence of small RNA-SWI/SNF co-associated regions was nearly equivalent in protein-coding genes and intergenic regions. From this we surmise that SWI/SNF may contribute to gene regulation of a variety of transcripts, many of which are newly annotated and of unknown function.
SWI/SNF targets genes involved in nuclear function and cancer pathways
Prior research has shown that a variety of signaling cascades are linked to SWI/SNF [25]. To gain further insights into potential actions of SWI/SNF components we examined the underlying Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) designations of their gene targets to determine significantly overrepresented annotations and pathways (Table 5 and Table S8). SWI/SNF gene targets were associated with ‘Pathways in cancer’ and several specific cancers types, e.g. chronic myeloid leukemia and pancreatic cancer. A number of signaling pathways and cellular processes that are “hallmarks of cancer” [41] were also overrepresented among the gene targets of Ini1, Brg1, BAF155 and BAF170. These include the Wnt, ErbB, p53, MAPK, and insulin signaling pathways, and processes endemic to oncogenesis and cancer progression such as DNA repair, the cell cycle and apoptosis. From these analyses we surmise the recruitment of SWI/SNF components is likely to influence the molecular basis of cancer through several potential mechanisms.

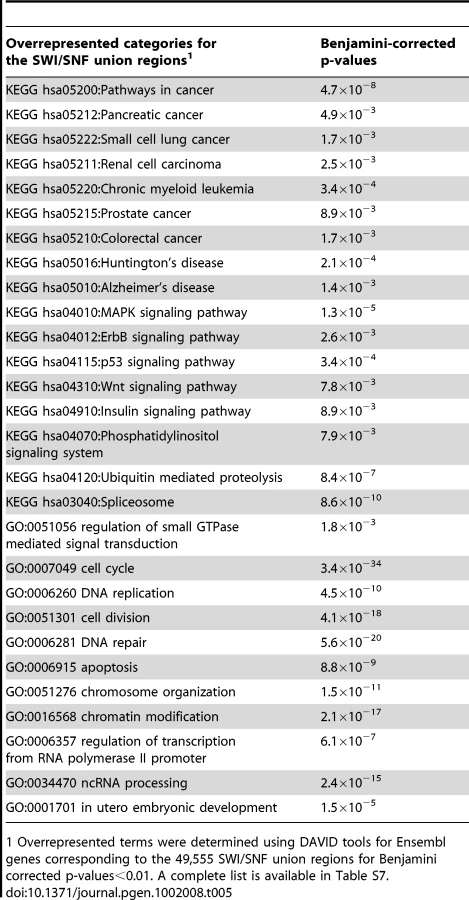
The SWI/SNF-enriched pathways are highly interconnected. Using the 49,555 SWI/SNF targets we identified a total of 24 KEGG signaling or biochemical pathways (Figure 6, yellow nodes). Interestingly, these pathways partition into three groups (Figure 6A–6C). Two of the groups (Figure 6A and 6B) comprise sets of pathways exhibiting at most one degree of separation, e.g. ‘inositol phosphate metabolism’ and ‘amino sugar and nucleotide sugar metabolism’. The third group (Figure 6C) consists of three pathways that are unrelated to any other pathways in the KEGG database. As displayed in Figure S2 directly related pathways such as ‘p53 signaling’ and the ‘cell cycle’ have shared components and many of the genes encoding these components are occupied by SWI/SNF factors. Thus, our results demonstrate that SWI/SNF is involved in many closely related signaling pathways and cellular processes and may help serve to coordinate expression of genes involved in these processes.

SWI/SNF components associate with proteins involved in multiple aspects of gene regulation and are nodes in a highly integrated network
The genomic binding data demonstrates that SWI/SNF localization is coupled with a broad range of functional elements, suggesting that SWI/SNF may also be found with a broad range of associated proteins. To further examine the scope of SWI/SNF's roles in the nucleus we analyzed proteins associated with SWI/SNF subunits using co-immunoprecipitation followed by mass spectrometry. The SWI/SNF components Ini1, BAF155, BAF170, Brg1, Brm and ARID1A were immunoprecipitated from HeLa S3 nuclei, the resulting proteins were gel-separated and peptides were generated for analysis by mass spectrometry (See Materials and Methods; Table S9). In addition to the factor-specific antibodies, parallel immunoprecipitations were performed using non-specific IgG antibodies. Proteins identified in these “control IgG” immunoprecipitations were excluded as potential SWI/SNF co-purifying factors.
We identified a total of 101 proteins that were specifically associated with at least one of the SWI/SNF components assayed (Figure 7, turquoise edges; Table S10). Of the non-SWI/SNF subunits detected, 5 of these interactions were found previously in HeLa cells (e.g. estrogen receptor alpha [42], and 96 were new to this study. Interestingly one of the novel interactions we observed in HeLa cells, BAF155 with NUF2, has been previously observed in yeast between the yeast homolog of BAF155 (SWI3) and NUF2 [15]. Using the 101 nodes that we identified as proteins co-purifying with SWI/SNF in our undirected approach we ascertained overrepresented GO categories (Table 6). Several of these designations such as ‘cell cycle’ and ‘chromosome organization’ coincide with the categories obtained from GO and pathway analyses of SWI/SNF ChIP-Seq targets, suggesting the possibility of highly-interactive network structures.

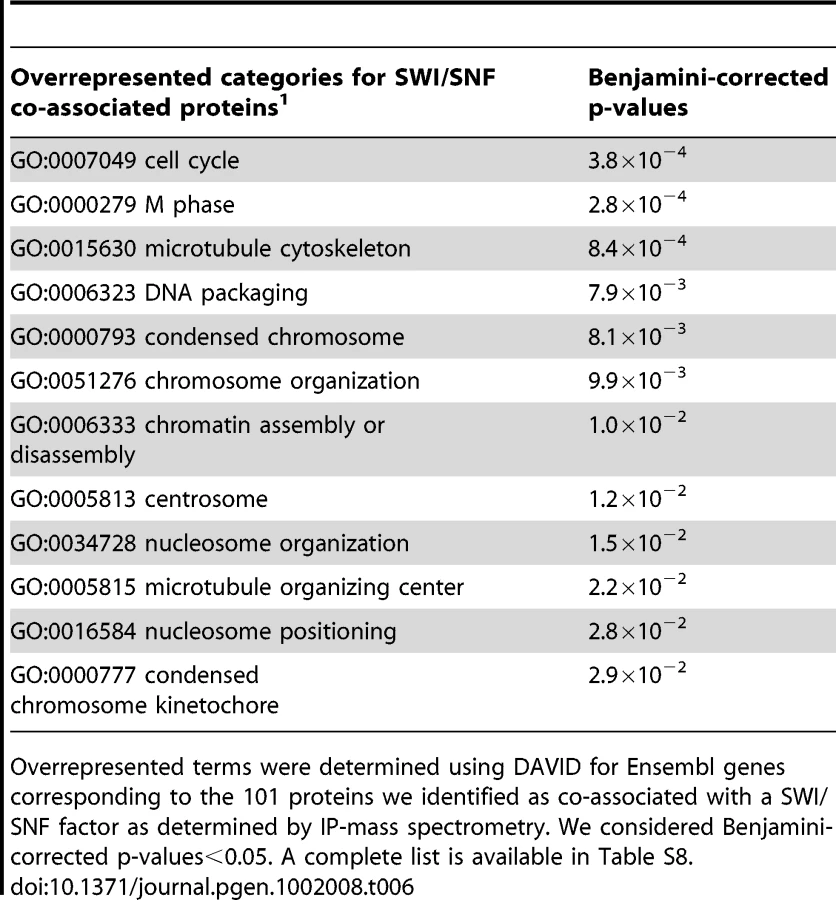
Many of the proteins that were novel to this study reinforce and expand upon other published reports of SWI/SNF characterizations. For example SWI/SNF components have been localized by immunofluorescence to mitotic kinetochores and spindle poles [43], and Brg1-deficient mice show dissolution of pericentromeric heterochromatin domains [44]. From our immunoprecipitations BAF155 and BAF170 were associated with a number of kinetochore and centrosomal proteins (e.g. BUB1B, CENPE and NUF2, Figure 8, green circles). The role of SWI/SNF in the maintenance of kinetochore and spindle function is unknown. We detected a variety of transcription factor activators and repressors (e.g. NFκB1, NFκB2, RelA, PML and NFX1) as well as DNA repair (ERCC5 and RAD50) and cell cycle (e.g. CCNB3 and CDCA2) proteins (Figure S3). Some of the SWI/SNF interacting proteins themselves interact with one another. For example we detected several different proteins integral to estrogen and insulin signaling (Figure 7; Table S10). We also identified proteins associated with only one SWI/SNF factor; these may either be interactions with a specific SWI/SNF component or an inability to detect the protein in the immunoprecipitations.

We developed an expanded network of SWI/SNF associations by including proteins that were found by others to co-purify with SWI/SNF subunits (Figure 7, black edges). Only those factors that showed a one-degree separation with a SWI/SNF component in HeLa cells are displayed and all interactions are annotated in Table S10. SWI/SNF interacting proteins are associated with numerous UniProt keywords (Figure 8; [45]). Overall these results suggest a role for SWI/SNF components in a wide array of nuclear processes and diseases. Some of these processes may take place in nuclear substructures. Higher order chromatin structure is facilitated by the nuclear lamina and tethering of genes to the nuclear periphery is one epigenetic mechanism of gene regulation [46], [47]. Intriguingly we and others have detected SWI/SNF components with various nuclear envelope-associated proteins (Figure 7 and Table S10) including lamin A, EMD (emerin) and BAF/BANF1 (Barrier to Autointegration Factor, which although similar in name is not a SWI/SNF subunit). Two of the nuclear membrane proteins, SYNE1 and C14orf49, that we isolated in association with BAF155 are part of LINC complexes that link the nucleoskeleton and cytoskeleton [48], [49].
A fraction of SWI/SNF regions are associated with the nuclear lamina
Numerous studies point to a high degree of functional organization in cell nuclei [46]. Emerging nuclear organization models would benefit greatly from a catalogue of processes and chromatin characteristics mapped to particular genomic elements. For example, the nuclear lamins are thought to influence chromatin organization, DNA replication and transcription [47], [50]. Our immunoprecipitation results demonstrating that SWI/SNF components are associated with lamin A/C (Figure 7 and Table S10) along with immunoprecipitation, immunolocalization and cell fractionation experiments from others demonstrating an association between SWI/SNF and nuclear lamina (e.g. emerin Figure 7; [51]) prompted us to investigate whether SWI/SNF and the lamins can be located to the same genomic sequences.
We isolated lamin A/C and lamin B ChIP DNA from HeLa S3 nuclei and performed ChIP-chip on tiling arrays covering the ENCODE pilot regions (see Materials and Methods and Table S1). Most of the 1,770 lamin A/C regions mapped to H3K27me3 domains (76%; 1,337/1,770) whereas the 1,270 lamin B regions were less commonly associated with H3K27me3 (29%; 372/1,270). Comparing regions where signal was detectable for the SWI/SNF, lamin A/C and lamin B experiments revealed that SWI/SNF has a much higher overlap with lamin B than lamin A/C. We found that 38% (297/784) of SWI/SNF sites are within 100 bp of a lamin B region whereas only 5% (41/784) of SWI/SNF sites are within 100 bp of a lamin A/C region (Table 7). For both lamin types the colocalization with SWI/SNF regions is significant relative to random target lists (lamin B, p<1×10−16; lamin A/C, p<1×10−15; GSC tests). SWI/SNF-lamin B intersecting regions contained approximately the same proportion of CTCF sites in the ENCODE regions as did all SWI/SNF sites in the ENCODE regions (p>0.05 hypergeometric test; Table 7). Enhancers are underrepresented in the SWI/SNF-lamin B regions relative to all SWI/SNF locations in the ENCODE regions (p<1.9×10−36; hypergeometric test). The SWI/SNF-lamin B regions are overrepresented for Pol II (p<2.9×10−39; hypergeometric test) and 5′ ends (p<7.3×10−37; hypergeometric test) relative to all SWI/SNF locations in the ENCODE regions.

In crosslinked chromatin SWI/SNF is detected primarily with lamin B, but as noted from the above mass spectrometry experiments, in solubilized, non-cross-linked cells SWI/SNF is detected with lamin A/C and not lamin B (Figure 1 and Figure 7). We interpret these results to indicate that SWI/SNF, lamin A/C and lamin B co-associate in different nuclear contexts but are all part of a broader interacting network with specific sub-associations.
Association of SWI/SNF with DNA replication origins
SWI/SNF and the lamins have each been implicated in DNA replication (see above; [52], [53]). One of the proteins we detected as associated with SWI/SNF is the replication protein RepA and another regulator of DNA replication, geminin, has been found to co-purify with SWI/SNF in HeLa cells (Figure 7, red circles; [54]). We investigated whether there might be a relationship among SWI/SNF, lamins and DNA replication origins. We obtained a set of 282 DNA replication origins identified in HeLa cells for the ENCODE regions ([55]; Table S1). Of these 282 replication origins, 90 (32%) occur within 100 bp of a SWI/SNF region (p<1×10−16, GSC test), 86 (31%) occur at the 5′ ends of protein-coding genes and 151 (54%) occur within 100 bp of a lamin B region. In contrast to lamin B, only 17% (48/282) of the replication origins were near a lamin A/C region. These results are consistent with nuclear staining patterns observed in mouse 3T3 cells showing colocalization between lamin B and sites of DNA replication whereas the same colocalization patterns were not observed for replication foci and lamin A [52].
Of the 86 replication origins in promoter regions, 88% (76/86) intersected a lamin B region and most (78% or 67/86) were within a 100 bp of a SWI/SNF region. These data indicate that SWI/SNF components are located near many DNA replication origins, particularly those located in promoter regions. The coincidence of chromatin remodeling factors, promoters, lamins and replication origins at the same subset of genomic regions suggests that these loci may be particularly favorable for the formation of both DNA and RNA polymerase assembly and chromatin tethering. As shown in Figure 1 the interplay among these elements as well as with Pol II, CTCF and heterochromatin regions is complex and interwoven, such that each may share many different supporting and counteracting roles.
Discussion
SWI/SNF performs a crucial function in gene regulation and chromosome organization by directly altering contacts between nucleosomes and DNA. In the work presented here we undertook a two-pronged approach (ChIP-Seq and IP-mass spectrometry) to move towards a more thorough understanding of these functions. Our ChIP-Seq analyses demonstrate that SWI/SNF components overlap extensively with important regions that require tight control of the dynamics of nucleosome occupancy such as promoters, enhancers and CTCF sites. Not only does the SWI/SNF complex change the accessibility of DNA but it also acts in concert with an extensive host of cooperating factors, thereby facilitating combinatorial control among various genomic elements. In addition to our ChIP-Seq results, the diversity and number of proteins that co-purify with SWI/SNF as identified in our mass spectrometry experiments further supports SWI/SNF's involvement with a variety of functionally distinct complexes.
RNA polymerases II and III are extensively colocalized with SWI/SNF components. Studies of transcription in HeLa cells have estimated that the number of active RNA II polymerases exceeds the number of transcriptionally active sites by at least one order of magnitude, leading to the proposal of “transcription factories” [1]–[3]. The number of RNA Pol II transcription factories in HeLa cells has been estimated between 5,000 and 8,000 where each factory can be typified by several looped loci, their resulting transcripts and distal elements such as enhancers. We infer that SWI/SNF regions are prevalent in transcriptional assemblages and their associated regulatory loops, given that >90% of our high-confidence union targets are associated with genic or regulatory regions and that 65% of Pol III and 84% of Pol II regions colocalize with at least one SWI/SNF factor (Table 4, Tables S5 and S6).
Interestingly we observed that SWI/SNF components often occur independently of each other and in various configurations across the genome, and similarly our mass spectrometry data point to heterogeneity of SWI/SNF complexes. We speculate that several mechanisms may underlie these various configurations and their associated genomic features, including 1) synergism or antagonism of the individual SWI/SNF factors in influencing expression (e.g. Figure 5); 2) failure to detect individual subunits due to epitope masking as a consequence of variation with local environments; 3) the capture of incomplete complexes that may in fact be completed upon superposition of genome-wide 3C data once such data become available (e.g. Figure 4); 4) the existence of SWI/SNF sub-complexes that deviate from the conventional composition of SWI/SNF assemblies (e.g. [56]) or 5) the capture of intermediates in a multistep assembly or remodeling process. This last view is consistent with a model of stochastic assembly that may occur through intermediate interactions and that has been described for several other large, multifactor complexes such as RNA polymerases and associated transcription factors [57], spliceosomes [58], and DNA repair complexes [59].
As shown in Figure 6 SWI/SNF occurs throughout many interconnected pathways. The assembly of functional SWI/SNF complexes at many locations in the genome may require the activation of one or more of these related pathways. Consequently some of the SWI/SNF associated regions we observed may reflect constitutive binding of partially assembled complexes that may be poised to receive additional signal inputs for subsequent regulatory activity. Indeed it has been shown that SWI/SNF components are present at regulatory regions even in the absence of stimulatory conditions or tissue-specific cofactors. For example Brg1 is present constitutively at the interferon-inducible genes IFITM3 [60] and CIITA [30] in unstimulated HeLa cells, which is consistent with our own finding of Brg1 and Ini1 at IFITM3 and various combinations of BAF155, BAF170, Ini1 and Brg1 at different elements in CIITA. In solution SWI/SNF factors are associated constitutively with RelB (HEK293 cells, [61]), RelA, NFkB1 and NFkB2 (HeLa cells, this study), the glucocorticoid receptor (T4D7 cells, [62]) and estrogen receptor alpha (HeLa cells, this study and [42]; SW13 cell extracts, [63]). The prevalence of SWI/SNF and the high degree of connectivity of its overrepresented pathways implies that SWI/SNF may assist in many related processes and may even facilitate crosstalk across many constituents of the transcriptional machinery. Notably SWI/SNF binds in the genes of its own subunits (Table S19) suggesting that SWI/SNF may contribute to auto - and cross-regulation of its subunit levels. Loss-of-function of a particular subunit, as may occur in certain cancers, could initiate oscillations and alter the relative abundance of the levels of the other SWI/SNF subunits through a variety of feedback and feed-forward loops. Aberrant SWI/SNF expression has been proposed to result in new combinatorial assemblies of SWI/SNF, some of which may deleterious [64].
The gene attributes revealed by our ChIP-Seq data substantiate that SWI/SNF is proximal to targets that comprise sets of fundamental biological processes. Many of the functional categories we found to be significantly overrepresented have disease implications, especially as related to cancer (Figure S2). For example failures in DNA repair and unchecked cell cycle activity are common characteristics of pre-cancerous cells, and our SWI/SNF analyses identified the p53 and MAPK signaling pathways, which are well known for maintaining checkpoint functions. Growth dysregulation particularly in the context of hormone signaling is another common cancer phenotype. Extracellular growth signals are transduced from the cell membrane to the nucleus by the ErbB, insulin and phosphtidylinositol signaling pathways, all of which we recovered as overrepresented (Table 5). The existence of phosphoinositide signaling in the nucleus and the ability of Brg1 to act as an effector for phosphatidylinositol 4,5-bisphosphate (PIP2) raises the prospect of several levels of control of this signaling pathway with respect to SWI/SNF [65], a hypothesis that can be examined in future studies.
Several of the overrepresented pathways we identified through our ChIP-Seq analyses share proteins detected in SWI/SNF co-purification experiments, thereby providing a resource to explore potential, highly-interactive network structures. For example we found that genes with products critical for ‘nucleotide excision repair’ were enriched using our SWI/SNF union list (Figure 6). Within this pathway the excision repair protein ERCC5 co-purified with both BAF155 and BAF170 in our IP (immunoprecipitation)-mass spectrometry experiments. The excision repair protein, XPC, associates with SWI/SNF in response to UV irradiation in HeLa cells, and BRCA1 and ATR also cooperate with SWI/SNF in DNA repair (Figure 7; Table S10; [66]). Thus we speculate SWI/SNF may participate in DNA repair through both transcriptional regulation as well as recruitment to regions undergoing repair.
Our study uses two strategies to attempt to comprehensively collect a SWI/SNF interaction network. We limited our network to a single model system, HeLa cells, because many attributes of SWI/SNF have been documented in these cells and it has been noted that SWI/SNF associations vary by cell type [67]. We extensively collated SWI/SNF protein interactions described in the literature. This undertaking was necessary because many of the proteins described in the literature as co-associated with SWI/SNF factors are not represented in interaction databases such as BioGRID, Molecular Interactions Database (MINT), IntAct, Human Protein Reference Database (HPRD), Nuclear Protein Database (NPD) and Interologous Interaction Database (I2D). Therefore we attempted to comprehensively collect such information to overcome these limitations. In total 158 SWI/SNF interacting proteins have been described in HeLa cells (Figure 8 and Table S10), which is similar to the number of SWI/SNF interacting proteins that have been described in other cell types [67]. Published molecular associations that were not discerned here might be due to interactions that are: 1) transient or of low affinity, 2) dependent on a specific set of biochemical conditions or 3) undetectable due to masking by the presence of more abundant protein(s) of similar size. In working with protein interaction data, similar degrees of overlap have been noted when comparisons are made across data sets [68], [69] and even in a well-studied model such as yeast, mass spectrometry analyses have found a plasticity of complexes and many previously undetected interactions [70]–[72]. From the ChIP-Seq and ChIP-chip results we expected that CTCF and lamin B may be among the proteins that co-associate with SWI/SNF, however neither of these factors was recovered in any of the non-directed experiments (Table S10), including a CTCF immunoprecipitation-mass spectrometry experiment performed in HeLa cells. In addition to the above considerations one possibility is that CTCF or lamin B may associate more strongly with one of the SWI/SNF factors not studied, e.g. BAF53A or one of the BAF60 subunits.
SWI/SNF is most often described in a chromatin remodeling context however data derived from a variety of sources suggests that SWI/SNF has other facets. It is possible that not all of SWI/SNF's functions involve DNA localization and therefore other types of global experiments, such as the IP-mass spectrometry, are valuable as first steps towards recognizing previously unknown roles. Unlike cytoplasmic compartments, nuclear compartments are not separated by a physical barrier but rather are functional assemblies that are typically organized around sets of molecules engaged in common functions. Data from both ChIP-Seq and IP-mass spectrometry illuminate the sectors in which SWI/SNF operates and the integration of these two methods is better than each alone for furnishing a broad comprehension of SWI/SNF action. For example ChIP-Seq enables the global identification of SWI/SNF chromosomal elements except for those regions with highly repetitive sequence such as human centromeres (Figure 2A). In this respect IP-mass spectrometry is complementary to ChIP-Seq because it strongly suggests that SWI/SNF occurs at kinetochores as evidenced by its co-purification with CENPE, NUF2, BUB1B and CLASP2 (Figure 7 and Figure 9). In addition to kinetochore proteins the SWI/SNF co-purification experiments also uncovered proteins from other substructures including centrosomes, microtubules, the nuclear periphery and PML nuclear bodies, the latter of which is characterized by cryptic foci of PML (promyelocytic leukemia protein) and has been implicated in a variety of diseases [73]. The ChIP-Seq and IP-mass spectrometry data are synergistic as well. Notably both methods found an overrepresentation of regions or proteins enriched for ‘cell cycle’ and ‘chromosome organization’. One possible inference from these studies is that SWI/SNF is well positioned to integrate signals across multiple signaling pathways both by its presence in a variety of cellular structures and its role in gene regulation through chromatin remodeling.

A fraction of SWI/SNF complexes co-associate with elements of the nuclear periphery where they are well situated to contribute to the nuclear organization and position-dependent gene expression (Figure 7; [74]). We found that in crosslinked cells SWI/SNF localizes more widely with lamin B than lamin A whereas in non-crosslinked cells SWI/SNF co-purifies with lamin A. As mentioned above lamin B may have escaped detection in SWI/SNF protein interaction studies. A related possibility is that SWI/SNF may exist in different nuclear pools that have varying solubilities and associations, such that recovery of particular SWI/SNF complexes depends upon the proteins with which SWI/SNF is associated. For example lamins A and B are known to have different nucleoplasmic mobilities and localization patterns [50], [52]. Immunolocalization experiments in HeLa nuclei have revealed that the A/C - and B-type lamins form distinct meshworks with occasional points of intersection [50], which is consistent with the interspersed patterns of lamin A/C and B that we detected (Figure 1). Hence it is reasonable to expect that SWI/SNF associated with lamin A would behave differently than when associated with lamin B. We surmise that in a chromatin context the dominant association of SWI/SNF with the nuclear lamins occurs in regions where lamin B is present. The purification of SWI/SNF with lamin A may indicate other biological roles, such as cell cycle progression or nuclear assembly [75], [76].
Gaining a more detailed understanding of SWI/SNF's activities in or near various heterochromatin environments will be central to comprehending nuclear events over the cell cycle as well as during development. Among the numerous molecular and epigenetic factors that have been found to affect heterochromatin formation or maintenance, the heterochromatin protein 1 alpha (HP1α, also known as CBX5; Figure 7) and Polycomb complexes (PcG) are of particular relevance to SWI/SNF [77]–[79]. Polycomb complexes promote gene silencing by catalyzing the trimethylation of H3K27 in its target regions, and SWI/SNF antagonizes this epigenetic silencing [80]. It is tempting to speculate that SWI/SNF found near the edges of H3K27me3 domains (Figure 1A and 1C) may be contributing to the establishment or maintenance of boundary elements. SWI/SNF may also engage in heterochromatin dynamics through its interaction with HP1α, which is often located in the centromeric regions (reviewed in [81]). Curiously HP1α interacts with the lamin B receptor [82] thus providing a potential bridge between heterochromatin and the inner nuclear membrane. Both H3K27me3 and lamin B are associated with spatially regulated genes whose conversion between active and inactive states depends on access to their regulatory regions, as may be conferred by SWI/SNF.
The work presented here provides new insights into the scope of SWI/SNF's influence in gene regulation and nuclear organization. The integration of numerous studies is beginning to reveal the complexities contributing to the regulation of any given locus. Contemporary models of transcriptional control propose that a series of factors transiently associate with a regulatory region before a decisive event tilts these intermediate reactions towards a productive outcome [57], [83]. SWI/SNF may contribute to such intermediate reactions or trigger switches between inactive and active states. The capacity for SWI/SNF to preserve many aspects of homeostasis also makes it vulnerable to being ensnared for aggressive cell proliferation. Our work demonstrates that SWI/SNF in particular and perhaps chromatin remodeling proteins in general will contribute unique insights to our understanding of gene regulation and disease mechanisms through the integration of target regions, spatial positioning and functional annotations. For example the co-occurrence of SWI/SNF with centrosomes, microtubules, kinetochores and the nuclear periphery may suggest that a pool of SWI/SNF is sequestered by these structures during mitosis to assist in the post-mitotic reformation of chromosomal territories. Our collective findings help inform a comprehensive view of SWI/SNF function as well as form a valuable compendium for future studies of nuclear functions as related to chromatin remodeling.
Materials and Methods
Chromatin immunoprecipitations
Suspension HeLa S3 cells were cultured by the National Cell Culture Center (Biovest International Inc., Minneapolis, MN) in modified minimal essential medium (MEM), supplemented with 10% FBS at 37°C in 5% CO2, to a density of 6×105 cells/mL. Cells were fixed with 1% formaldehyde at room temperature for 10 min. Fixation was terminated with 125 mM glycine (2 M stock made in 1x PBS). Formaldehyde-fixed cells were washed in cold Dulbecco's PBS (Invitrogen) and swelled on ice in a 10-mL hypotonic lysis buffer [20 mM Hepes (pH 7.9), 10 mM KCl, 1 mM EDTA (pH 8.0), 10% glycerol, 1 mM DTT, 0.5 mM PMSF, and Roche Complete protease inhibitors, Cat#1697498]. To isolate nuclei, whole cell lysates were homogenized with 30 strokes in a 7 mL Dounce homogenizer (Kontes, pestle B). Nuclear pellets were collected by centrifugation and lysed in 10 mL of RIPA buffer per 3×108 cells [RIPA buffer: 10 mM Tris-Cl (pH 8.0), 140 mM NaCl, 1% Triton X-100, 0.1% SDS, 1% deoxycholic acid, 0.5 mM PMSF, 1 mM DTT, and protease inhibitors]. Chromatin was sheared with an analog Branson 250 Sonifier (power setting 2, 100% duty cycle for 7×30-s intervals) to an average size of less than 500 bp, as verified on a 2% agarose gel. Lysates were clarified by centrifugation at 20,000× g for 15 min at 4°C.
Clarified nuclear lysates from 1×108 cells were agitated overnight at 4°C with 20 µg of one of the following antibodies: 1) anti-Ini1 (C-20), Santa Cruz Biotechnology, sc-16189; 2) anti-BAF155 (H-76), Santa Cruz Biotechnology, sc-10756; 3) anti-BAF170 (H-116), Santa Cruz Biotechnology, sc-10757; 4) anti-Brg1 (G-7), Santa Cruz Biotechnology, sc-17796; 5) anti-lamin A/C (H-110), Santa Cruz Biotechnology, sc-20681; 6) anti-lamin B antibody, EMD Biosciences, NA12; or 7) normal IgG, Santa Cruz Biotechnology, sc-2025. Antibody incubations were followed by addition of either protein A (Millipore #16-156) or protein G agarose beads (Millipore #16-266). Beads were permitted to bind to protein complexes for 60 min at 4°C. Immunoprecipitates were washed three times in 1x RIPA, once in 1x PBS, and then eluted in 1xTE/1%SDS. Crosslinks were reversed overnight at 65°C. ChIP DNA was purified by incubation with 200 µg/ml RNase A (Qiagen #19101) for 1 h at 37°C, with 200 µg/ml proteinase K (Ambion AM2548) for 2 h at 45°C, phenol:chloroform:isoamyl alcohol extraction, and precipitation with 0.1 volumes of 3 M sodium acetate, 2 volumes of 100% ethanol and 1.5 µL of pellet paint (Novagen #69049-3). ChIP DNA prepared from 1×108 cells was resuspended in 50 µL of Qiagen Elution Buffer (EB). Three biological replicates were prepared per antibody.
Construction and sequencing of Illumina libraries
ChIP-Seq libraries were prepared and sequenced as previously described [26], [84]. Biological replicates for each factor were converted into separate and distinct libraries. To summarize, ChIP DNA samples were loaded onto Qiagen MinElute PCR columns, eluted with 15 µL of Qiagen buffer EB, size-selected in the 100–350 bp range on 2% agarose E-gels (Invitrogen) and gel-purified using a Qiagen gel extraction kit. DNA was end-repaired and phosphorylated with the End-It kit from Epicentre (Cat# ER0720). The blunt, phosphorylated ends were treated with Klenow fragment (3′ to 5′ exo minus; NEB, Cat# M0212s) and dATP to yield a protruding 3′-‘A’ base for ligation of Illumina adapters (100 RXN Genomic DNA Sample Prep Oligo Only Kit, Part# FC-102-1003), which have a single ‘T’ base overhang at the 3′ end. After adapter ligation (LigaFast, Promega Cat# M8221) DNA was PCR-amplified with Illumina genomic DNA primers 1.1 and 2.1 for 15 cycles by using a program of (i) 30 s at 98°C, (ii) 15 cycles of 10 s at 98°C, 30 s at 65°C, 30 s at 72°C, and (iii) a 5 min extension at 72°C. The final libraries were band-isolated from an agarose gel to remove residual primers and adapters. Library concentrations and A260/A280 ratios were determined by UV-Vis spectrometry on a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). Purified and denatured library DNA was capture on an Illumina flowcell for cluster generation and sequenced on an Illumina Genome Analyzer II following the manufacturer's protocols [85].
Identification of proteins by mass spectrometry
Immunoprecipations were performed using the same conditions as for ChIP experiments except the HeLa S3 cells were not crosslinked. In addition to the ChIP antibodies described above we also used anti-Brm, Abcam Cat# ab15597 and anti-BAF250a (PSG3), Santa Cruz Biotechnology, sc-32761. Complexes were resolved on BioRad 4–20% precast Tris-HCl gels (Cat#161-1159) such that a single gel was used for each specific antibody and normal IgG immunoprecipitation pair. Gels were silver stained using Pierce SilverSNAP stain for mass spectrometry (Cat#24600) and each lane was excised into 10–12 molecular weight regions. Gel slices were destained, dried in a Savant speed-vac and digested overnight at 42°C with Sigma's Trypsin Profile IGD kit for in-gel digests (Cat# PP0100). Following the overnight incubation the liquid was removed from each gel piece and volume reduced by drying to approximately 10 µL. The individual gel slices were analyzed separately.
Mass spectrometry
The samples were subjected to nanoflow chromatography using nanoAcquity UPLC system (Waters Inc.) prior to introduction into the mass spectrometer for further analysis. Mass spectrometry was performed on a hybrid ion trap LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific) in positive electrospray ionization (ESI) mode. The spectra was acquired in a data dependent fashion consisting of full mass spectrum scan (300–2000 m/z) followed by MS/MS scan of the 3 most abundant parent ions. For the full scan in the orbitrap the automatic gain control (AGC) was set to 1×106 and the resolving power for 400 m/z of 30,000. The MS/MS scans were done using the ion trap part of the mass spectrometer at a normalized collision energy of 24 V. Dynamic exclusion time was set to 100 s to avoid loss of MS/MS spectral information due to repeated sampling of the most abundant peaks.
Sequence data from MS/MS spectra was processed using the SEQUEST database search algorithm (Thermo Fisher Scientific). The resulting protein identifications were brought into the Scaffold visualization software (Proteome Software) where the information was further refined resulting in improved protein id conformation. Scaffold search criteria were set at 98% probability and required at least 2 unique peptides per id.
Determination of enriched regions in SWI/SNF ChIP-Seq data
All ChIP-Seq data sets (Ini1, Brg1, BAF155, BAF170, and Pol II) were scored against a normal IgG control using PeakSeq [26] with default parameters (q-value<0.05) to determine an initial set of enriched regions. These lists were then filtered by removing those regions that did not meet all of the following requirements: 1) the q-value from PeakSeq was further restricted to a q-value of<0.01; 2) a minimum of 20 sequencing reads per peak from the specific antibody ChIP; 3) an enrichment of 1.5-fold of the specific antibody relative to the normal IgG control; and 4) an excess of at least 10 of the specific antibody reads relative to the normal IgG control reads. Enriched regions satisfying these criteria comprised our initial list of enrichment sites for each factor (Table 1 and Tables S11, S12, S13, S14, S15, and S16). Among these data sources, Pol II and the normal IgG control have been published as part of prior studies and are available in GEO (accession numbers GSE14022 and GSE12781, respectively) [26], [84]. Data for Ini1, Brg1, BAF155 and BAF170 can be accessed through GEO series GSE24397.
Generation of a SWI/SNF union list from ChIP-Seq results
After obtaining our initial list of enriched regions for each factor subjected to chromatin immunoprecipitation, we generated a union list of SWI/SNF component targets. Using the method described in Euskirchen et al. [86], we formed the union of Ini1, BAF155, BAF170, and Brg1 enriched regions as identified by ChIP-Seq and merged any unioned regions that were separated by ≤100 bp. Each union region was then classified by whether it intersected with one or more of BAF155, BAF170, Ini1, and Brg1. The resulting list consists of 69,658 SWI/SNF union regions (Table S2).
Determination of the “high-confidence” and “core” SWI/SNF regions from the ChIP-Seq union regions
We compared our ChIP-Seq target lists for the 69,658 SWI/SNF union regions against genomic features at which chromatin remodeling is expected to play a prominent role: RNA polymerase II sites [26], 5′ ends of Ensembl protein-coding genes, CTCF sites [28], and regions predicted to be enhancers in HeLa cells [29]. We also compared individual SWI/SNF component lists against each other. Only those SWI/SNF regions which intersect another SWI/SNF component or which intersect at least one of the above genomic features were retained for the ‘high-confidence’ union list. For gene promoter regions, we define overlap as a target region with at least 1 shared bp within ±2.5 kb of the annotated transcription start site (TSS). SWI/SNF region intersections were calculated both for all genes in the Ensembl 52 database build using annotations from NCBI36 (human genome build hg18) as well as for a subset of genes that Ensembl identifies as protein-coding. The resulting target list consists of 49,555 ‘high-confidence’ SWI/SNF union regions (Table S3). Union regions containing all three of the BAF155, BAF170, and Ini1 subunits are designated as the 9,760 ‘core’ SWI/SNF regions (Table 3).
Generating co-occurrence tables
To determine the co-occurrences of features of interest we used a similar intersection strategy as was used for determining the high-confidence SWI/SNF regions. For all pairwise comparisons, one of the two data sets was extended by 100 bp on each side of the region and then intersected against the other, non-extended dataset. We required an overlap of at least 1 bp to deem two regions as associated. Using a Perl script, the intersection results for all comparisons were combined to form the co-occurrence table. The same procedure was followed to generate SWI/SNF-centric (Tables S2 and S3), Pol II-centric (Table S5) and Pol III-centric (Table S6) co-occurrence tables.
Determination of expressed regions
Using the HeLa RNA-Seq data of Morin et al. [37], we subdivided each list by the expression status of the corresponding gene targets. Expressed genes were defined as any Ensembl gene with an associated Ensembl transcript having an adjusted depth of ≥1, representing an average coverage of 1x across all bases in the transcript. A total of 9,711 expressed protein-coding genes satisfied these criteria.
Comparison of expression levels associated with different SWI/SNF sub-complexes
We created a series of lists based upon the combinations of SWI/SNF components that could co-occur using the 49,555 high-confidence SWI/SNF regions derived from Table S3. Using the RNA-Seq data of Morin et al. [37], we intersected each list against the 5′ ends of transcripts queried by that study and recorded the corresponding adjusted depth for any transcript with a 5′ end within ±2.5 kb of a SWI/SNF region. Morin et al. treats adjusted depth as a measurement of transcription level for the corresponding transcript. For each list, these measurements were used to build a series of violin plots showing the probability distribution of transcription levels associated with different compositions of SWI/SNF subunits. Note that each SWI/SNF region from Table S2 can only be assigned to one list (e.g. a region containing BAF155, BAF170, and Ini1 is not also assigned to the list of regions containing BAF155 and BAF170).
Pathway analyses of SWI/SNF factors
Overrepresented GO categories [87] and KEGG pathways [88] were determined using DAVID tools [16]. Figures S2 and S3 were drawn using KGML-ED [89].
ChIP-chip experimental procedures and array scoring
The ENCODE tiling arrays (NimbleGen Systems Inc., Madison, WI) interrogate the regions from the pilot phase of the ENCODE project [90] and tile the non-repetitive forward strand DNA sequence with 50-mer oligonucleotides spaced every 38 bp (overlapping by 12 bp) for a total of approximately 390,000 features. For array hybridizations ChIP DNA samples from 1×108 cells were labeled according to the manufacturer's protocol by Klenow random priming with Cy5 nonamers (lamin A/C or lamin B ChIP DNA) or Cy3 nonamers (normal IgG ChIP DNA). Biological replicates, defined as ChIP DNA isolations prepared from distinct cell cultures, were each hybridized to separate microarrays. Each lamin data set consists of three biological replicates. ChIP DNA labeling and array hybridizations were conducted by the NimbleGen service facility (Reykjavik, Iceland). Briefly, arrays were hybridized in Maui hybridization stations for 16–18 h at 42°C, and then washed in 42°C 0.2% SDS/0.2x SSC, room temperature 0.2x SSC, and 0.05x SSC. Arrays were scanned on an Axon 4000B scanner.
For each pair of arrays the files (in GFF file format) corresponding to the two channels for ChIP DNA (635 nm) and reference DNA (532 nm), were uploaded to the TileScope pipeline for normalization and scoring [91]. Data were scored with the following TileScope program parameters: quantile normalization of replicates, iterative peak identification, window size = 500, oligo length = 50, pseudomedian threshold = 1.0, p-value threshold = 4.0, peak interval = 1000, and feature length = 1000. Regions called by Tilescope were then filtered and corrected for multiple hypothesis testing by false discovery rate (FDR). To generate our set of background regions for FDR analysis, we randomly shuffle the probe values within each replicate, ensuring that the same probes are swapped for each replicate. This shuffled data set is then used as input to Tilescope and the scores compared against the lamin A/C and the lamin B data sets. The final lists of enriched regions for lamin A/C and lamin B have a final FDR of 0.1. Target coordinates were converted to hg18 using the UCSC ‘liftOver’ utility (http://genome.ucsc.edu/cgi-bin/hgLiftOver). Lamin A/C and lamin B data are available through GEO series GSE24382 and Tables S17 and S18.
Comparison of features across the ENCODE regions
To facilitate comparisons between sequencing and array data we retained only those regions that could be queried by both platforms. To this end, we first identified sequences represented on the ENCODE tiling array that possess less than 25% mappability in ChIP-Seq experiments using 30 bp reads. Any enriched regions in the lamin A/C and the lamin B data sets that were entirely contained within these regions of low mappability were removed from our lists, as corresponding signal levels are unlikely to be detected accurately via ChIP-Seq. Mappability was determined using a 30 bp read length and reported in 100 bp windows according to [26]. The end result is a list of lamin A/C and lamin B enriched regions identified by ChIP-chip in areas of the genome that can be queried by ChIP-Seq. Accordingly, regions that are not represented on the ENCODE tiling arrays were also removed from our SWI/SNF ChIP-Seq experiments for this comparison. Because our ChIP-Seq data covers the entire genome, we began by restricting our enriched SWI/SNF regions only to those that occur in the ENCODE pilot regions. We further refined our ChIP-Seq data set by discarding any SWI/SNF regions that occur in a region of the tiling array for which a signal level of 0 was observed via ChIP-chip. Once our SWI/SNF, lamin A/C, and lamin B lists were limited to those regions that could be queried by both platforms, we intersected the remaining lamin regions and the SWI/SNF regions using the same method that generated the all features table for enhancers, Pol II, and other elements, as described above. Similar procedures were followed for intersections with DNA replication origins identified in the ENCODE regions using tiling arrays [55].
Evaluating enrichment of SWI/SNF components with respect to other genomic features
To determine whether SWI/SNF components, core regions, and union regions are enriched for factors such as enhancers, small RNAs, lamin A/C and B, CTCF sites, Pol II regions, Pol III sites, 5′ ends and DNA replication origins, we used the genome structure correction test (GSC). This test determines the significance of observations where there “exists a complex dependency structure between observations” and was specifically designed for large-scale genomic studies [27]. Given two lists of genomic regions to compare and a list of coordinates defining the overall sample space (i.e. the length of each chromosome), a p-value for the significance of the overlap of the two lists is calculated and we report this value where noted.
Data deposition
All data produced for this study can be accessed through GEO and accession numbers for individual series are provided in the relevant sections. Alternatively, data from the lamin ChIP-chip experiments and the Ini1, Brg1, BAF155, and BAF170 ChIP-Seq experiments can be accessed through GEO using the SuperSeries accession number GSE24398.
Supporting Information
Zdroje
1. JacksonDAIborraFJMandersEMCookPR 1998 Numbers and organization of RNA polymerases, nascent transcripts, and transcription units in HeLa nuclei. Mol Biol Cell 9 1523 1536
2. CookPR 1999 The organization of replication and transcription. Science 284 1790 1795
3. CookPR 2010 A model for all genomes: the role of transcription factories. J Mol Biol 395 1 10 doi:10.1016/j.jmb.2009.10.031
4. ClapierCRCairnsBR 2009 The biology of chromatin remodeling complexes. Annu Rev Biochem 78 273 304 doi:10.1146/annurev.biochem.77.062706.153223
5. de la SernaILOhkawaYImbalzanoAN 2006 Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nat Rev Genet 7 461 473 doi:10.1038/nrg1882
6. WuJILessardJCrabtreeGR 2009 Understanding the words of chromatin regulation. Cell 136 200 206 doi:10.1016/j.cell.2009.01.009
7. PhelanMLSifSNarlikarGJKingstonRE 1999 Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell 3 247 253
8. ChenJArcherTK 2005 Regulating SWI/SNF subunit levels via protein-protein interactions and proteasomal degradation: BAF155 and BAF170 limit expression of BAF57. Mol Cell Biol 25 9016 9027 doi:10.1128/MCB.25.20.9016-9027.2005
9. SohnDHLeeKYLeeCOhJChungH 2007 SRG3 interacts directly with the major components of the SWI/SNF chromatin remodeling complex and protects them from proteasomal degradation. J Biol Chem 282 10614 10624 doi:10.1074/jbc.M610563200
10. PercipallePVisaN 2006 Molecular functions of nuclear actin in transcription. J Cell Biol 172 967 971 doi:10.1083/jcb.200512083
11. CastanoEPhilimonenkoVVKahleMFukalováJKalendováA 2010 Actin complexes in the cell nucleus: new stones in an old field. Histochem Cell Biol 133 607 626 doi:10.1007/s00418-010-0701-2
12. RandoOJZhaoKJanmeyPCrabtreeGR 2002 Phosphatidylinositol-dependent actin filament binding by the SWI/SNF-like BAF chromatin remodeling complex. Proc Natl Acad Sci U.S.A 99 2824 2829 doi:10.1073/pnas.032662899
13. VersteegeISévenetNLangeJRousseau-MerckMFAmbrosP 1998 Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394 203 206 doi:10.1038/28212
14. SévenetNLellouch-TubianaASchofieldDHoang-XuanKGesslerM 1999 Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum Mol Genet 8 2359 2368
15. WongJNakajimaYWestermannSShangCKangJ 2007 A protein interaction map of the mitotic spindle. Mol Biol Cell 18 3800 3809 doi:10.1091/mbc.E07-06-0536
16. HuangDWShermanBTLempickiRA 2009 Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 44 57 doi:10.1038/nprot.2008.211
17. WiegandKShahSAl-AghaOZhaoYTseK 2010 ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med
18. JonesSWangTShihIMaoTNakayamaK 2010 Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330 228 231 doi:10.1126/science.1196333
19. Van MaeleBBusschotsKVandekerckhoveLChristFDebyserZ 2006 Cellular co-factors of HIV-1 integration. Trends Biochem Sci 31 98 105 doi:10.1016/j.tibs.2005.12.002
20. TurelliPDoucasVCraigEMangeatBKlagesN 2001 Cytoplasmic recruitment of INI1 and PML on incoming HIV preintegration complexes: interference with early steps of viral replication. Mol Cell 7 1245 1254
21. DasSCanoJKalpanaGV 2009 Multimerization and DNA binding properties of INI1/hSNF5 and its functional significance. J Biol Chem 284 19903 19914 doi:10.1074/jbc.M808141200
22. IsakoffMSSansamCGTamayoPSubramanianAEvansJA 2005 Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci U.S.A 102 17745 17750 doi:10.1073/pnas.0509014102
23. LeeYSSohnDHHanDLeeHSeongRH 2007 Chromatin remodeling complex interacts with ADD1/SREBP1c to mediate insulin-dependent regulation of gene expression. Mol Cell Biol 27 438 452 doi:10.1128/MCB.00490-06
24. XiQHeWZhangXHLeHMassaguéJ 2008 Genome-wide impact of the BRG1 SWI/SNF chromatin remodeler on the transforming growth factor beta transcriptional program. J Biol Chem 283 1146 1155 doi:10.1074/jbc.M707479200
25. SimoneC 2006 SWI/SNF: the crossroads where extracellular signaling pathways meet chromatin. J Cell Physiol 207 309 314 doi:10.1002/jcp.20514
26. RozowskyJEuskirchenGAuerbachRKZhangZDGibsonT 2009 PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol 27 66 75 doi:10.1038/nbt.1518
27. BickelPBoleyNBrownJHuangHZhangN 2010 Subsampling methods for genomic inference. Annals of Applied Statistics 4 1660 1697 doi:10.1214/10-AOAS363
28. CuddapahSJothiRSchonesDERohTCuiK 2009 Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res 19 24 32 doi:10.1101/gr.082800.108
29. HeintzmanNDHonGCHawkinsRDKheradpourPStarkA 2009 Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459 108 112 doi:10.1038/nature07829
30. NiZAbou El HassanMXuZYuTBremnerR 2008 The chromatin-remodeling enzyme BRG1 coordinates CIITA induction through many interdependent distal enhancers. Nat Immunol 9 785 793 doi:10.1038/ni.1619
31. Bazett-JonesDPCôtéJLandelCCPetersonCLWorkmanJL 1999 The SWI/SNF complex creates loop domains in DNA and polynucleosome arrays and can disrupt DNA-histone contacts within these domains. Mol Cell Biol 19 1470 1478
32. NomaKCamHPMaraiaRJGrewalSIS 2006 A role for TFIIIC transcription factor complex in genome organization. Cell 125 859 872 doi:10.1016/j.cell.2006.04.028
33. RaabJRKamakakaRT 2010 Insulators and promoters: closer than we think. Nat Rev Genet 11 439 446 doi:10.1038/nrg2765
34. OlerAJAllaRKRobertsDNWongAHollenhorstPC 2010 Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat Struct Mol Biol 17 620 628 doi:10.1038/nsmb.1801
35. BarskiAChepelevILikoDCuddapahSFlemingAB 2010 Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat Struct Mol Biol 17 629 634 doi:10.1038/nsmb.1806
36. UrnovFDWolffeAP 2001 Chromatin remodeling and transcriptional activation: the cast (in order of appearance). Oncogene 20 2991 3006 doi:10.1038/sj.onc.1204323
37. MorinRBainbridgeMFejesAHirstMKrzywinskiM 2008 Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. BioTechniques 45 81 94 doi:10.2144/000112900
38. SaundersACoreLJLisJT 2006 Breaking barriers to transcription elongation. Nat Rev Mol Cell Biol 7 557 567 doi:10.1038/nrm1981
39. FudaNJArdehaliMBLisJT 2009 Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461 186 192 doi:10.1038/nature08449
40. Affymetrix and Cold Spring Harbor Laboratory ENCODE Transcriptome Projects 2009 Post-transcriptional processing generates a diversity of 5'-modified long and short RNAs. Nature 457 1028 32 doi:10.1038/nature07759
41. HanahanDWeinbergRA 2000 The hallmarks of cancer. Cell 100 57 70
42. MalovannayaALiYBulynkoYJungSYWangY 2010 Streamlined analysis schema for high-throughput identification of endogenous protein complexes. Proc Natl Acad Sci U.S.A 107 2431 2436 doi:10.1073/pnas.0912599106
43. XueYCanmanJCLeeCSNieZYangD 2000 The human SWI/SNF-B chromatin-remodeling complex is related to yeast rsc and localizes at kinetochores of mitotic chromosomes. Proc Natl Acad Sci U.S.A 97 13015 13020 doi:10.1073/pnas.240208597
44. BourgoRJSiddiquiHFoxSSolomonDSansamCG 2009 SWI/SNF deficiency results in aberrant chromatin organization, mitotic failure, and diminished proliferative capacity. Mol Biol Cell 20 3192 3199 doi:10.1091/mbc.E08-12-1224
45. UniProt Consortium 2010 The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res 38 D142 148 doi:10.1093/nar/gkp846
46. MisteliT 2007 Beyond the sequence: cellular organization of genome function. Cell 128 787 800 doi:10.1016/j.cell.2007.01.028
47. DechatTPfleghaarKSenguptaKShimiTShumakerDK 2008 Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 22 832 853 doi:10.1101/gad.1652708
48. CrispMLiuQRouxKRattnerJBShanahanC 2006 Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 172 41 53 doi:10.1083/jcb.200509124
49. HaqueFLloydDJSmallwoodDTDentCLShanahanCM 2006 SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 26 3738 3751 doi:10.1128/MCB.26.10.3738-3751.2006
50. ShimiTPfleghaarKKojimaSPackCSoloveiI 2008 The A - and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev 22 3409 3421 doi:10.1101/gad.1735208
51. ReyesJCMuchardtCYanivM 1997 Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J Cell Biol 137 263 274
52. MoirRDMontag-LowyMGoldmanRD 1994 Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J Cell Biol 125 1201 1212
53. CohenSMChastainPDRossonGBGrohBSWeissmanBE 2010 BRG1 co-localizes with DNA replication factors and is required for efficient replication fork progression. Nucleic Acids Res 38 6906 6919 doi:10.1093/nar/gkq559
54. SeoSHerrALimJRichardsonGARichardsonH 2005 Geminin regulates neuronal differentiation by antagonizing Brg1 activity. Genes Dev 19 1723 1734 doi:10.1101/gad.1319105
55. CadoretJMeischFHassan-ZadehVLuytenIGuilletC 2008 Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc Natl Acad Sci U.S.A 105 15837 15842 doi:10.1073/pnas.0805208105
56. RymeJAspPBöhmSCavellánEFarrantsAO 2009 Variations in the composition of mammalian SWI/SNF chromatin remodelling complexes. J Cell Biochem 108 565 576 doi:10.1002/jcb.22288
57. DinantCLuijsterburgMSHöferTvon BornstaedtGVermeulenW 2009 Assembly of multiprotein complexes that control genome function. J Cell Biol 185 21 26 doi:10.1083/jcb.200811080
58. RinoJCarvalhoTBragaJDesterroJMPLührmannR 2007 A stochastic view of spliceosome assembly and recycling in the nucleus. PLoS Comput Biol 3 2019 2031 doi:10.1371/journal.pcbi.0030201
59. LuijsterburgMSvon BornstaedtGGourdinAMPolitiAZMonéMJ 2010 Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J Cell Biol 189 445 463 doi:10.1083/jcb.200909175
60. LiuHKangHLiuRChenXZhaoK 2002 Maximal induction of a subset of interferon target genes requires the chromatin-remodeling activity of the BAF complex. Mol Cell Biol 22 6471 6479
61. BouwmeesterTBauchARuffnerHAngrandPBergaminiG 2004 A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol 6 97 105 doi:10.1038/ncb1086
62. HsiaoPFryerCJTrotterKWWangWArcherTK 2003 BAF60a mediates critical interactions between nuclear receptors and the BRG1 chromatin-remodeling complex for transactivation. Mol Cell Biol 23 6210 6220
63. BelandiaBOrfordRLHurstHCParkerMG 2002 Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J 21 4094 4103
64. WeissmanBKnudsenKE 2009 Hijacking the chromatin remodeling machinery: impact of SWI/SNF perturbations in cancer. Cancer Res 69 8223 8230 doi:10.1158/0008-5472.CAN-09-2166
65. BarlowCALaishramRSAndersonRA 2010 Nuclear phosphoinositides: a signaling enigma wrapped in a compartmental conundrum. Trends Cell Biol 20 25 35 doi:10.1016/j.tcb.2009.09.009
66. RayAMirSNWaniGZhaoQBattuA 2009 Human SNF5/INI1, a component of the human SWI/SNF chromatin remodeling complex, promotes nucleotide excision repair by influencing ATM recruitment and downstream H2AX phosphorylation. Mol Cell Biol 29 6206 6219 doi:10.1128/MCB.00503-09
67. HoLRonanJLWuJStaahlBTChenL 2009 An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc Natl Acad Sci U.S.A 106 5181 5186 doi:10.1073/pnas.0812889106
68. EwingRMChuPElismaFLiHTaylorP 2007 Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol 3 89 doi:10.1038/msb4100134
69. LambertJMitchellLRudnerABaetzKFigeysD 2009 A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol Cell Proteomics 8 870 882 doi:10.1074/mcp.M800447-MCP200
70. GavinABöscheMKrauseRGrandiPMarziochM 2002 Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415 141 147 doi:10.1038/415141a
71. GavinAAloyPGrandiPKrauseRBoescheM 2006 Proteome survey reveals modularity of the yeast cell machinery. Nature 440 631 636 doi:10.1038/nature04532
72. BreitkreutzAChoiHSharomJRBoucherLNeduvaV 2010 A global protein kinase and phosphatase interaction network in yeast. Science 328 1043 1046 doi:10.1126/science.1176495
73. Lallemand-BreitenbachVde ThéH 2010 PML nuclear bodies. Cold Spring Harb Perspect Biol 2 a000661 doi:10.1101/cshperspect.a000661
74. ReyesJCMuchardtCYanivM 1997 Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J Cell Biol 137 263 274
75. MuchardtCReyesJCBourachotBLeguoyEYanivM 1996 The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J 15 3394 3402
76. GüttingerSLaurellEKutayU 2009 Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol 10 178 191 doi:10.1038/nrm2641
77. NielsenALSanchezCIchinoseHCerviñoMLerougeT 2002 Selective interaction between the chromatin-remodeling factor BRG1 and the heterochromatin-associated protein HP1alpha. EMBO J 21 5797 5806
78. LavigneMEskelandRAzebiSSaint-AndréVJangSM 2009 Interaction of HP1 and Brg1/Brm with the globular domain of histone H3 is required for HP1-mediated repression. PLoS Genet 5 e1000769 doi:10.1371/journal.pgen.1000769
79. WilsonBGWangXShenXMcKennaESLemieuxME 2010 Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18 316 328 doi:10.1016/j.ccr.2010.09.006
80. SchuettengruberBChourroutDVervoortMLeblancBCavalliG 2007 Genome regulation by polycomb and trithorax proteins. Cell 128 735 45 doi:10.1016/j.cell.2007.02.009
81. KwonSHWorkmanJL 2008 The heterochromatin protein 1 (HP1) family: put away a bias toward HP1. Mol Cells 26 217 227
82. YeQCallebautIPezhmanACourvalinJCWormanHJ 1997 Domain-specific interactions of human HP1-type chromodomain proteins and inner nuclear membrane protein LBR. J Biol Chem 272 14983 14989
83. MétivierRGallaisRTiffocheCLe PéronCJurkowskaRZ 2008 Cyclical DNA methylation of a transcriptionally active promoter. Nature 452 45 50 doi:10.1038/nature06544
84. AuerbachRKEuskirchenGRozowskyJLamarre-VincentNMoqtaderiZ 2009 Mapping accessible chromatin regions using Sono-Seq. Proc Natl Acad Sci U.S.A 106 14926 14931 doi:10.1073/pnas.0905443106
85. BentleyDRBalasubramanianSSwerdlowHPSmithGPMiltonJ 2008 Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456 53 59 doi:10.1038/nature07517
86. EuskirchenGMRozowskyJSWeiCLeeWHZhangZD 2007 Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array - and sequencing-based technologies. Genome Res 17 898 909 doi:10.1101/gr.5583007
87. Gene Ontology Consortium 2010 The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res 38 D331 335 doi:10.1093/nar/gkp1018
88. KanehisaMGotoSFurumichiMTanabeMHirakawaM 2010 KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38 D355 360 doi:10.1093/nar/gkp896
89. KlukasCSchreiberF 2007 Dynamic exploration and editing of KEGG pathway diagrams. Bioinformatics 23 344 350 doi:10.1093/bioinformatics/btl611
90. ENCODE Project Consortium 2004 The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306 636 640 doi:10.1126/science.1105136
91. ZhangZDRozowskyJLamHYKDuJSnyderM 2007 Tilescope: online analysis pipeline for high-density tiling microarray data. Genome Biol 8 R81 doi:10.1186/gb-2007-8-5-r81
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy