
Ultra-Deep Sequencing of Mouse Mitochondrial DNA: Mutational Patterns and Their Origins
Somatic mutations of mtDNA are implicated in the aging process, but there is no universally accepted method for their accurate quantification. We have used ultra-deep sequencing to study genome-wide mtDNA mutation load in the liver of normally - and prematurely-aging mice. Mice that are homozygous for an allele expressing a proof-reading–deficient mtDNA polymerase (mtDNA mutator mice) have 10-times-higher point mutation loads than their wildtype siblings. In addition, the mtDNA mutator mice have increased levels of a truncated linear mtDNA molecule, resulting in decreased sequence coverage in the deleted region. In contrast, circular mtDNA molecules with large deletions occur at extremely low frequencies in mtDNA mutator mice and can therefore not drive the premature aging phenotype. Sequence analysis shows that the main proportion of the mutation load in heterozygous mtDNA mutator mice and their wildtype siblings is inherited from their heterozygous mothers consistent with germline transmission. We found no increase in levels of point mutations or deletions in wildtype C57Bl/6N mice with increasing age, thus questioning the causative role of these changes in aging. In addition, there was no increased frequency of transversion mutations with time in any of the studied genotypes, arguing against oxidative damage as a major cause of mtDNA mutations. Our results from studies of mice thus indicate that most somatic mtDNA mutations occur as replication errors during development and do not result from damage accumulation in adult life.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1002028
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002028
Summary
Somatic mutations of mtDNA are implicated in the aging process, but there is no universally accepted method for their accurate quantification. We have used ultra-deep sequencing to study genome-wide mtDNA mutation load in the liver of normally - and prematurely-aging mice. Mice that are homozygous for an allele expressing a proof-reading–deficient mtDNA polymerase (mtDNA mutator mice) have 10-times-higher point mutation loads than their wildtype siblings. In addition, the mtDNA mutator mice have increased levels of a truncated linear mtDNA molecule, resulting in decreased sequence coverage in the deleted region. In contrast, circular mtDNA molecules with large deletions occur at extremely low frequencies in mtDNA mutator mice and can therefore not drive the premature aging phenotype. Sequence analysis shows that the main proportion of the mutation load in heterozygous mtDNA mutator mice and their wildtype siblings is inherited from their heterozygous mothers consistent with germline transmission. We found no increase in levels of point mutations or deletions in wildtype C57Bl/6N mice with increasing age, thus questioning the causative role of these changes in aging. In addition, there was no increased frequency of transversion mutations with time in any of the studied genotypes, arguing against oxidative damage as a major cause of mtDNA mutations. Our results from studies of mice thus indicate that most somatic mtDNA mutations occur as replication errors during development and do not result from damage accumulation in adult life.
Introduction
A decline of mitochondrial function has been observed in a variety of aging mammalian tissues and is implicated as a driving force behind the aging process [1]–[2]. A somatic mammalian cell carries thousands of copies of the mitochondrial DNA (mtDNA) chromosome, which encodes essential subunits of the respiratory chain protein complexes as well as rRNAs and tRNAs needed for mitochondrial translation. Expression of mtDNA is required for maintenance of oxidative phosphorylation and accumulation of somatic mtDNA mutations has been suggested as a cause of the observed decrease in respiratory chain function during aging [3]–[4]. A variety of low levels of point mutations and rearrangements of mtDNA are found in aging mammals. Rare mutational events tend to undergo clonal expansion as exemplified by human aging where clonal expansion of somatic mtDNA mutations cause a mosaic respiratory chain deficiency in tissues such as brain, heart, skeletal muscle and large intestine [4]. Point mutations as well as insertions and deletions (indels) of mtDNA are observed in tissues of aging humans [5]–[9], primates [10] and rodents [11], but the relative contribution of each of these different types of mutations to the aging process is unknown.
The mtDNA mutator mice (genotype PolgAmut/PolgAmut) express a proof-reading-deficient mtDNA polymerase (PolgAD257A) and have provided experimental support that accumulation of mtDNA mutations can lead to a premature aging syndrome [12]–[14]. These mice contain high levels of point mutations in their mtDNA and high levels of several species of large linear deleted mtDNA molecules. Although the linear deleted molecules are abundant (∼25–30% of total mtDNA in liver) the corresponding reduction in levels of full-length mtDNA molecules is on its own not sufficient to cause an impairment of respiratory chain function [13]. A detailed molecular characterization of the mtDNA mutator mice has shown that the high levels of point mutations are the likely explanation for the respiratory chain deficiency and the premature aging syndrome [15]. One report claims that the presence of a third type of mtDNA mutation, i.e. circular mtDNA molecules with large deletions, may be of critical importance in driving the premature aging phenotype of mtDNA mutator mice [16]–[17]. However, this finding has been refuted by several other reports showing that the levels of such deleted molecules are extremely low [15], [18]–[21]. In addition, the biochemical phenotype in mtDNA mutator mice can be fully explained by the finding that high levels of point mutations in mtDNA leads to the synthesis of respiratory chain subunits with abundant amino acid replacements, which, in turn, cause instability of the respiratory chain complexes [15].
Oxidative damage has for more than 50 years been proposed as a central mechanism in aging, but the supporting evidence is mainly correlative. Interestingly, the mtDNA mutator mice have no or minor increase in levels of reactive oxygen species (ROS) production and oxidative damage despite a severe decline in oxidative phosphorylation capacity. This finding refutes the popular notion of a vicious cycle whereby somatic mtDNA mutations lead to increased ROS production, which, in turn, creates additional mtDNA mutations that further increase ROS production [12], [14].
Genome-wide estimates of intra-individual mtDNA variability are needed to determine the importance of the various types of mtDNA mutations in aging. Recent studies using next-generation sequencing of the human mitochondrial genome have identified a small number of sites, in which normal individuals carry both the wildtype copy and a high-frequency mutant allele [22]–[23]. In their efforts to eliminate false positive calls, these studies limited their analyses to high-frequency mutations and excluded rare variants.
We hypothesised that a DNA sequencing technology that minimize the levels of false positive calls caused by technical errors could be applied to assess the intra-individual mtDNA variability caused by low frequency mutations. We investigated this possibility by using the ABI SOLiD technology to sequence mtDNA of normal aging mice and prematurely aging mtDNA mutator mice. To control for technical errors we sequenced a complete mtDNA genome inserted in a lambda clone.
Results
We used the SOLiD sequencing platform to sequence mtDNA purified from liver mitochondria of normal C57Bl/6N mice (henceforth denoted wtB6) at 30, 40 and 84 weeks of age. In addition, we analysed liver mtDNA from mtDNA mutator mice (PolgAmut/PolgAmut; henceforth denoted mutators), heterozygous mtDNA mutator mice (+/PolgAmut; henceforth denoted heterozygotes) and their wildtype siblings (henceforth denoted wtmut) at 30 and 40 weeks of age. An mtDNA molecule cloned in the lambda phage (henceforth referred to as λmtDNA) was also sequenced and used as a control and for correction of errors introduced by the SOLiD sequencing technique. Samples and sequence read information is shown in Table S1.
Sequence coverage
The sequence coverage for 99% of the mtDNA bases was at least 1800x in each sample and the coverage for 70% of the mtDNA bases was at least 10,000x (Figure S1). The wtB6, heterozygotes and wtmut mice showed a similar sequencing coverage between samples. The coverage profiles were quite similar to that obtained by sequencing λmtDNA (Figure 1). By contrast, in the mutator samples there was a pronounced decrease in sequence coverage for approximately one third of the genome (Figure 1). This under-represented region corresponds to the small arc between the two origins of replication for the mtDNA molecule. The mtDNA mutator mice have been reported to contain ∼25–30% of truncated, linear double-stranded mtDNA molecules, ranging from the origin of heavy strand replication (OH) to the origin of light strand replication (OL) [13], [19]. The unequal sequence coverage curve thus likely reflects the presence of these linear mtDNA molecules with a large deletion in the mutator.

Point mutation loads
The point mutation frequency was estimated as the median number of mutations per nucleotide site and varied dramatically in mice of different genotypes (Figure 2, Figure 3). The wtB6 mice showed a median mutation frequency of 1.3–1.8×10−4 per site, while the mutators had frequencies of about 12×10−4 (Table 1). The heterozygotes also had an elevated point mutation frequency both at 30 and 40 weeks of age, as compared with their wildtype siblings. The wtmut and wtB6 mice showed similar point mutation frequencies (1.3–1.8×10−4, Table 1). We also determined the number of high frequency point mutation sites, defined as the number of sites with single nucleotide variant (SNV) frequencies >0.5%. Interestingly, there are approximately the same number of such sites in heterozygotes and wtmut (Figure 3), arguing that this mutation load is inherited from their common mother which is heterozygous for the mtDNA mutator allele. In contrast, the number of high frequency point mutation sites in wtB6 mice was only half of the value of wtmut mice (Figure 3). We observed no difference in mutation frequencies (Table 1) or in the number of high frequency point mutation sites in wtB6 mice between the ages of 30 and 84 weeks (Figure 3).

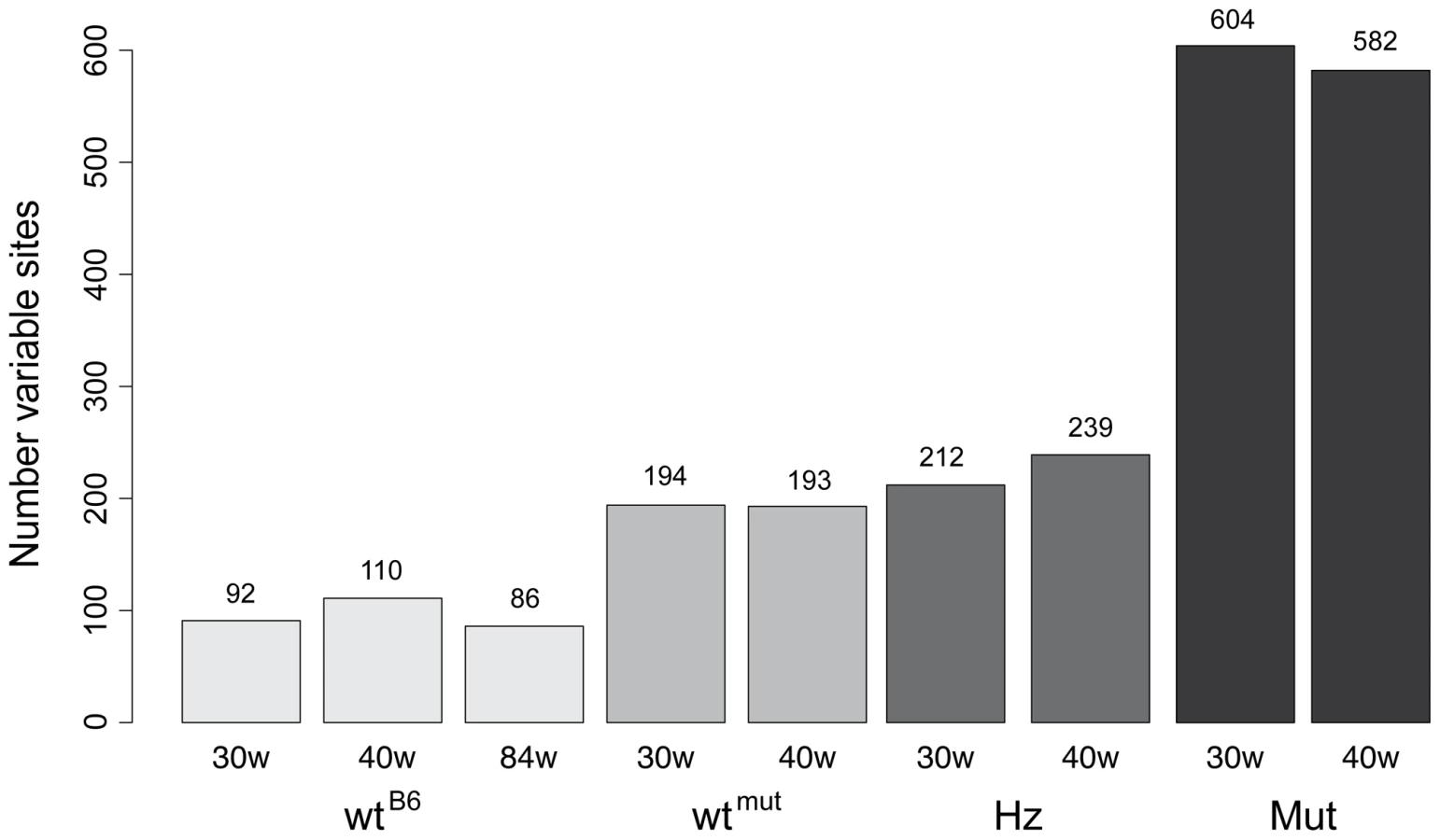

Genomic distribution of point mutations
In the mutators the protein coding genes, tRNA genes and rRNA genes showed similar mutation loads, whereas the mutation load was 59–66% lower in the control region (also referred to as the major non-coding region or D-loop region) (Table 1; Figure 4). A particularly conserved part of the control region, the conserved sequence blocks (CSB), had an almost 80% reduction in the mutation load compared with the coding regions. There was also a modest reduction (34–42% decrease) in mutation load in the control region of heterozygotes, whereas no difference was observed in wtB6 or wtmut mice (Table 1).

Absence of a signature of oxidative damage
The obtained mutational spectrum allowed us to investigate whether oxidative damage is a source of mtDNA damage during aging, since oxidative damage is expected to increase the number of observed transversion mutations, as exemplified by the G/C to T/A transversions caused by the oxidative adduct 8-oxo-guanine [4]. The number of transitions versus transversions did not change as a function of age in any of the samples, implying that mtDNA polymerase errors are responsible for the majority of the observed point mutations (Table S2). The heterozygotes and the mutators showed increased relative levels of transitions in the two samples, as expected under conditions of excess polymerase errors. These observations argue against oxidative damage as main contributor to the observed mutation pattern.
Shared mutations among litter mates
In order to identify mutations that are common to littermates, we calculated the number of high frequency point mutation sites that are shared among offspring (mutators, heterozygotes and wtmut mice) obtained by mating heterozygous mtDNA mutator mice (Figure 5). We found more than 800 high frequency point mutation sites in all siblings within litters of 30 and 40-weeks of age. Approximately 85% of the high frequency variable point mutation sites were present in single animals and most of these were mutators (56–59%). Approximately 5% of the sites (n = 44-42) were shared between the littermates. Interestingly, the wtB6 mice obtained from independent matings shared a substantial number of these high frequency point mutation sites (n = 35), suggesting the occurrence of mutational hotspots.

Mutational hotspots
The point mutation frequency varied between nucleotide sites, with some sites experiencing 100–1,000 times higher mutation frequencies than an average variable site. A number of regions also showed several neighbouring, but not necessarily adjacent, positions with high mutation frequencies (Table S3). Seven of the twelve identified hotspot regions in this study corresponded to the regions with increased levels of inherited mtDNA mutations we previously have described in wildtype mouse strains derived from female mtDNA mutator mice [24]. Similar hotspot regions have been reported in human mtDNA [22], [25], [26] and such mutational hotspots may play an important role in the generation of the common disease alleles reported for mtDNA [27].
Insertion/deletion mutations
Although we did not utilize paired-end sequencing, it was still possible to detect structural re-arrangements, such as indel mutations, by using the SplitSeek method [28]. This method splits the sequence reads and aligns the two parts independently to the reference sequence. We found that indels had a median frequency of 10−3–10−4 (Table 1). The mutators had 4–6.6 times more indels than their wtmut siblings and 2–10.5 times more indels than wtB6 mice (Table 1). Most indels were small (Table S4) and only five deletions involved more than 1 kb of DNA. Four of these large deletions were found in a single mutator and the remaining deletion was present in a heterozygote (Table S5). The number of indels did not increase with age in mice of any of the studied genotypes (Table 1).
The indels showed an uneven distribution across the mtDNA genome with a clustering of sites in two regions around genome positions 1,000–5,000 and 12,000–15,000 (Figure S2). Small indels found in the mutator mouse sibling sets were often observed in proximity to mononucleotide stretches. These small indels were present in mutators, heterozygotes and wtmut, but were very infrequent in wtB6 mice and essentially absent in λmtDNA (Table S4). These data demonstrate that these small indel events are induced by the PolgAmut allele. The presence of indels in wtmut animals, who lack the PolgAmut allele, can be explained by inheritance of these indels from their heterozygous mothers.
Functional consequence of mutations
Adult homozygous mtDNA mutator mice are predicted to encode about 7 amino acid substitutions per mtDNA molecule, compared with 2 substitutions in their wtmut siblings and 1 or 2 substitutions in wtB6 mice (Table 1). The presence of many amino acid substitutions in the mutators has been demonstrated to impair respiratory chain function due to destabilization of the respiratory chain complexes [15]. Our results provide additional support for the conclusion that the premature aging syndrome in mtDNA mutator mice is due to accumulation of point mutations in mtDNA, which, in turn, cause amino acid substitutions that impair mitochondrial function.
Discussion
Our analysis has revealed a number of novel aspects of the accumulation of mtDNA mutations in wildtype and mtDNA mutator mice. The mutators show highly elevated point mutation frequencies as compared to their wtmut siblings, consistent with previous results [12]–[14], [16], [21], [29]–[30]. The SOLiD sequencing estimates of mutation loads presented here are similar to the mutation load estimates that we and others previously have obtained by Sanger sequencing of cloned PCR fragments [12]–[14]. In a recent study using a different next-generation DNA sequencing technology, He et al. [22] reported eight sites with a mutation frequency >1.6% in a human mtDNA sample. If we apply their detection threshold to our data, we identify five such sites in the wtB6 mice. Techniques that enable identification of low frequency variants, such as those used in our study, are likely to uncover additional variability and provide a more complete understanding of the mitochondrial mutation load.
We hypothesized that our analysis criteria should also uncover gradual increase in the mutation loads with natural aging, consistent with published results obtained by other mutation detection methods (reviewed in [4]). However, we neither detected a difference in the mtDNA mutation load in liver mtDNA from wtB6 mice at different ages (30–84 weeks), nor did we see a shift in the mutational spectrum consistent with oxidative damage causing mtDNA mutations in mice of the different genotypes. Together, the data we present here suggest that most mtDNA mutations are due to mtDNA replication errors and that oxidative damage of mtDNA does not drive the aging process in liver.
Our estimates of the mutation load in the wtB6 and wtmut mice are in good agreement with estimates based on sequencing of cloned PCR fragments [12]–[14], but they are higher than estimates obtained by a restriction enzyme digestion-based assay [30] (summarized in Table S6). Also, we observed no increase in mutation load in wtB6 mice with age, as reported elsewhere [11], [30]. A possible explanation for this difference from results in the literature is that the next-generation sequencing method has an inherent limitation in detecting extremely low levels of mutations. It remains possible that there is a slight increase of mutation load with age in wtB6 mice, as reported in other studies [11], and the true mutation levels may be below the detection threshold of the SOLiD sequencing method. But in any case, the mutation levels seen in the aging wtB6 mice were very modest in comparison to those of age-matched mtDNA mutator mice. Also, we chose to only analyze mtDNA from mouse liver as this tissue made it possible for us to obtain sufficient quantities of pure mtDNA for direct sequencing, without excess DNA amplification. A continuously dividing tissue like liver may show a different pattern of accumulation of mtDNA point mutations with age in comparison with a postmitotic tissue such as brain or heart.
The different coding regions showed similar mutation frequencies, while the control region had a much lower mutation frequency. A reduction in the overall mutation load has previously been observed in the mitochondrial control region of mtDNA mutator mice [12]–[13], [29]. The control region contains crucial sequence elements controlling replication and transcription of mtDNA [31]. It is therefore likely that mutations that inhibit mtDNA maintenance or expression could undergo strong selection and be eliminated from the mtDNA pool.
Some of the mtDNA point mutations observed in siblings to mtDNA mutator mice are likely to have been inherited via the maternal gamete of their heterozygous mother. The wtmut mice from this cross carry approximately twice the number of high frequency point mutations (>0.5%) in comparison with wtB6 mice. In addition, an elevated number of small indel mutations are observed in the wtmut mice, but not in wtB6 mice (Table S4). We cannot exclude that some of the shared point mutations represent extreme mutational hotspots, however, a more likely explanation is that these shared mutations are maternally inherited. A similar phenomenon has been observed in humans, where the variability at several positions was shown to be inherited from the common maternal cytoplasm instead of representing repeated de novo mutational events [22].
In mtDNA mutator mice, approximately 30% of the mtDNA molecules are non-replicating, linear mtDNA molecules with large deletions [13], [19]. It could be speculated that these molecules contain an elevated mutation load and that most of the mutation load is sequestered in these molecules. By ultra-deep sequencing, we were able to determine that the mutation load in the region covering the linear molecule did not vary from the global mtDNA mutation load in the mutators. A recent publication suggests the PolgAmut polymerase may pause at the control and OriL regions during mtDNA replication, which may explain the generation of the linear molecules with large deletions [19]. Our results are consistent with a hypothesis that altered processivity of the mutator polymerase and not the point mutations per se, are responsible for the creation of the linear molecules. The physiological consequences of the linear molecules, and their contribution to the premature aging in the mtDNA mutator mice, remain unclear and worthy of further investigation.
Circular mtDNA molecules with deletions have been suggested to be the driving force of the aging phenotype in the mutator mice, and are reliably detected in human tissues during aging. High levels of these circular mtDNA molecules with deletions lead to mitochondrial dysfunction in human patients [32] or mice engineered to contain these mutations [33]–[34]. However, we found that the circular mtDNA molecules with deletions are exceedingly rare in mtDNA mutator mice, with only 4 breakpoints being detected in the millions of reads of two mutator samples. Very low levels of this type of deleted molecules have also been reported by studies using different PCR based analyses [15], [20]. Recently, an independent next-generation sequencing analysis detected exceedingly low levels of this type of mutation in brain and heart of mtDNA mutator mice [21]. Large deletion of mtDNA are known to impair mitochondrial translation due to lack of one or more tRNA genes [35]. However, mtDNA mutator mice do not display impaired mitochondrial translation in heart or liver [15] and the levels of deleted mtDNA are much lower than the levels observed in respiratory chain deficient mouse strains with single [33] and multiple [34] deletions of mtDNA. Together, these observations provide strong evidence that the circular deleted mtDNA molecules are not the causative factor in the aging phenotype of the mtDNA mutator mice. A recent study made a remarkable observation that similar low levels of circular deletions were accumulating in a mouse model with a tissue-specific disruption of the mitochondrial fusion process [36]. The presence of very low levels of circular mtDNA molecules with deletions in two very different models of mitochondrial dysfunction suggests these rare events are being generated as a secondary consequence of mitochondrial dysfunction. Another possibility is that these molecules are continuously generated at low frequency during normal mtDNA replication and that mitochondrial dysfunction limit their clearance.
The large numbers of point mutations in adult mtDNA mutator mice result in production of highly mutated mtDNA-encoded respiratory chain subunits, causing the experimentally observed instability of the respiratory chain complexes [15], [37]. There is likely a threshold for the tolerance of point mutations, where eventually the combined effect of the many amino acid changes in mtDNA mutator mice cause destabilization of respiratory chain complexes and lead to mitochondrial dysfunction. Our results support the assertion that the accumulation of point mutations has an adversary effect on mitochondrial function and cause the premature aging syndrome in the mtDNA mutator mice.
Materials and Methods
Preparation of purified mitochondria and mitochondrial DNA
This animal study was approved by the animal welfare ethics committee and performed in compliance with Swedish law. Three C57Bl/6N males mice were obtained from the animal unit's breeding colony. Two sibling sets of three males were also obtained, each containing the three genotypes expected from a PolgAmut/PolgAwt intercross. These two litters were not from the same heterozygous mother.
Mitochondria were isolated using standard protocols. Briefly, whole livers were homogenised under ice-cold conditions and cell debris pelleted by low speed centrifugation (600 g, 4°C for ten minutes). The supernatant was transferred and the mitochondria pelleted by centrifugation at 5000 g for 10 min. Resuspended mitochondria were isolated by centrifugation in a 1.0M/1.5M two-phase sucrose gradient. Isolated mitochondria were lysed in 1% sarkosyl and DNA purified by organic extraction, followed by salt precipitation. The DNA preparation was treated with RNase and the DNA precipitated prior to use.
Isolated mtDNA from a liver preparation from a C57Bl/6N mouse was digested in BglII and cloned into lambda using the Lambda FIX II/Xho I and Gigapack III Gold Packaging kits (Stratagene). A single clone was expanded and the DNA was extracted by standard phage DNA extraction protocols.
Sequencing library preparation and DNA sequencing using SOLiD
Sequencing libraries were prepared from 1 µg of purified mtDNA following manufacturer's instructions (ABI). Emulsion-PCR was performed according to the manufacturer's instructions (ABI), and then applied to standard slide and sequenced with 50 base pair read length, using an ABI SOLiD 3 sequencing system. The reads were aligned to the C57Bl/6J mouse mtDNA reference sequence (NC_005089.1), using the corona lite mapping algorithm (Applied Biosystems) with default settings. The first 49 bases of the mtDNA sequence were appended to the end of the reference to avoid that reads fail to align due to the circularity of the mitochondrial genome. This alignment procedure attempts to map each read at full length to the reference sequence, allowing for at most 6 mismatches for each 50 bp read.
Calculating point mutation frequencies
In the SOLiD sequencing technology, a SNV is represented by two valid adjacent mismatches in an aligned read. We used the valid adjacent mismatch calls to calculate the mutation frequencies for all samples at every position of the mtDNA molecule by the following method. At each position the number of nucleotide substitutions were calculated for each of the three alternative bases. By dividing these numbers with the total read coverage we obtained SNV frequencies for each of the three possible mutant alleles, with their sum representing the total mutation frequency at the specific position. This method may include some false positive SNV calls, which implies that our mutation frequencies will be over-estimated. The error frequencies may be dependent both on the sequence context and the sequencing technology used, and is likely to vary substantially at different positions of the mtDNA molecule. We therefore used a λmtDNA control sample as a means to correct the mutation rates.
Correcting mutation frequencies using cloned mtDNA
The mutation frequencies were calculated as described above for all samples, including the λmtDNA control. For each of the mtDNA samples, the SNV frequencies for the λmtDNA clone were subtracted to obtain corrected frequencies of all nucleotide changes at each position. In the cases where the λmtDNA showed a higher rate of some nucleotide compared to the mtDNA sample, the corrected value was set to zero. Several sites in the λmtDNA control sample showed elevated rates of mutations. The mutations found in λmtDNA can partly be explained by technical errors in the SOLiD sequencing, but there may also be a set of true polymorphisms that were incorporated during the replication of the λmtDNA clone. Since we cannot distinguish between these two sources of variability we have taken a conservative approach and subtracted the entire λmtDNA mutation signal from the mouse mtDNA samples. As a consequence there may be sites were the λmtDNA mutation rate is higher than in the native mtDNA.
Estimating number of protein coding nucleotide changes per mtDNA molecule
We used our SNV frequencies as an estimate of the average number of mutations per mtDNA molecule. All SNVs within protein coding genes were extracted and their frequencies were calculated. These SNVs were then grouped into three different categories, ‘synonymous’, ‘amino acid change’ or ‘stop codon’, depending on the effect on the protein sequence. For each of the categories the number of changes per mtDNA molecule was calculated as the sum of all SNV frequencies belonging to that group. To remove the effect of extreme mutational hotspots, all mutations with and observed per site frequency of >0.5% were excluded from this analysis. If such sites are not removed there is a risk that a few sites with extremely high frequency will have too large an effect on the estimate since their frequencies can sometimes be 1,000 times higher than at other sites. By removing the highest frequency sites we instead focus on the combined effect of the lower frequency mutations on the proteome. These estimates are therefore likely to be conservative.
Indel analysis
Reads containing insertions and deletions will not be aligned to the reference sequence with the corona lite program. The unmapped reads were analyzed for indels using the SplitSeek strategy [28]. This strategy was originally developed for junction detection in RNA-seq data, but it can also be used for detecting insertions and deletions. The unmapped reads were aligned using version 1.1 of the AB WT pipeline (http://solidsoftwaretools.com/gf/project/transcriptome/), a software that performs split read alignment of SOLiD data, using the same settings as in the RNA-seq study [28] and the alignment results were used as input to the SplitSeek program. We required each indel to be supported by at least 5 reads with unique starting points, and by reads on both strands. For each indel we calculated a frequency for its occurrence in the mtDNA samples by the ratio ri/(ri+rc). In this formula ri is the number of reads that supports an indel, while rc is the total coverage over the indel calculated from the initial full-length mapping of reads.
Identifying mutational hotspots
Mutational hotspots were defined as regions with elevated mutation frequency over at least 20 bases. The regional hotspots were identified by first calculating the median SNV frequency in a 20 bp window around each base in each sample to obtain a smoothed signal of the mutation rates. For each sample, we then selected those positions with a median window frequency above the 90th percentile of all SNV frequencies in the entire mtDNA genome. In this way we select only those positions with a substantially elevated mutation rate over a number of neighbouring bases. Furthermore, we required the same region to be identified in at least three of the nine mice. This analysis resulted in the 12 hotspot regions presented in Table S2.
Identifying shared high frequency mutation sites
We used the same cut-off to detect high frequency SNVs as was used for filtering out positions with high signal in the negative control sample (i.e. a per site frequency of 0.005 or 0.5%). For each of the samples we extracted all positions with mutation frequencies of at least 0.5%.
Mitochondrial fragments in the nuclear genome
Parts of the mitochondrial genome can exist as nuclear-inserted mitochondrial pseudogenes (NucMts) [38]. In mice, these NucMts normally differ substantially from the mtDNA, though a large insert with few variations from the C57Bl/6J mtDNA sequence is known [39]. Since the purified mitochondrial fraction will be contaminated with small amounts of nuclear DNA, sequences that appear to be mtDNA but are derived from NucMts could be present in the sequenced reads and affect the mutation rate. However, mtDNA represents about 1% of the DNA in a cell. Assuming a ratio of 90∶10 between mitochondrial and nuclear DNA in the sample, a sequencing reaction that generates a total of 1×109 bases will then include 9×108 bases of mtDNA from the mitochondria and the remaining from the nuclear genome. If there are 100 mtDNA fragments inserted in each nuclear genome, the 1×108 bases that derives from the nuclear genome represent roughly 4% of a nuclear mouse genome, or about 4 mtDNA fragments. These sequences should be compared to the 9×108 bases of mtDNA sequence from the mitochondria, which correspond to about 5×104 mtDNA genomes. Given that only a fraction of the nuclear insert sequences have a nucleotide that deviates from the consensus, nuclear inserts are likely to contribute less than one deviating read out of 5×104 reads at each position. Thus, the effect of nuclear inserts on the estimate is likely to be very small.
Data accession
The SOLiD data is available from the EMBL-EBI Sequence Read Archive. Study accession number: ERP000469 (http://www.ebi.ac.uk/ena/data/view/ERP000469).
Supporting Information
Zdroje
1. HarmanD 1972 The biologic clock: the mitochondria? J Am Geriatr Soc 20 145 147
2. MiquelJEconomosACFlemingJJohnsonJEJr 1980 Mitochondrial role in cell aging. Exp Gerontol 15 575 591
3. KrishnanKJGreavesLCReeveAKTurnbullD 2007 The ageing mitochondrial genome. Nucleic Acids Res 35 7399 7405
4. LarssonNG 2010 Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 79 683 706
5. CortopassiGAArnheimN 1990 Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res 18 6927 6933
6. IkebeSTanakaMOhnoKSatoWHattoriK 1990 Increase of deleted mitochondrial DNA in the striatum in Parkinson's disease and senescence. Biochem Biophys Res Commun 170 1044 1048
7. KatayamaMTanakaMYamamotoHOhbayashiTNimuraY 1991 Deleted mitochondrial DNA in the skeletal muscle of aged individuals. Biochem Int 25 47 56
8. YenTCSuJHKingKLWeiYH 1991 Ageing-associated 5 kb deletion in human liver mitochondrial DNA. Biochem Biophys Res Commun 178 124 131
9. HattoriKTanakaMSugiyamaSObayashiTItoT 1991 Age-dependent increase in deleted mitochondrial DNA in the human heart: possible contributory factor to presbycardia. Am Heart J 121 1735 1742
10. SchwarzeSRLeeCMChungSSRoeckerEBWeindruchR 1995 High levels of mitochondrial DNA deletions in skeletal muscle of old rhesus monkeys. Mech Ageing Dev 83 91 101
11. PikoL 1992 Accumulation of mtDNA defects and changes in mtDNA content in mouse and rat tissues with aging. Ann N Y Acad Sci 663 450 452
12. TrifunovicAHanssonAWredenbergARovioATDufourE 2005 Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A 102 17993 17998
13. TrifunovicAWredenbergAFalkenbergMSpelbrinkJNRovioAT 2004 Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429 417 423
14. KujothGCHionaAPughTDSomeyaSPanzerK 2005 Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309 481 484
15. EdgarDShabalinaICamaraYWredenbergACalvarusoMA 2009 Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab 10 131 138
16. VermulstMWanagatJKujothGCBielasJHRabinovitchPS 2008 DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet 40 392 394
17. VermulstMWanagatJLoebLA 2009 On mitochondria, mutations, and methodology. Cell Metab 10 437
18. EdgarDLarssonNGTrifunovicA 2010 Response: Point Mutations Are Causing Progeroid Phenotypes in the mtDNA Mutator Mouse. Cell Metab 11 93
19. BaileyLJCluettTJReyesAProllaTAPoultonJ 2009 Mice expressing an error-prone DNA polymerase in mitochondria display elevated replication pausing and chromosomal breakage at fragile sites of mitochondrial DNA. Nucleic Acids Res 37 2327 2335
20. KraytsbergYSimonDKTurnbullDMKhrapkoK 2009 Do mtDNA deletions drive premature aging in mtDNA mutator mice? Aging Cell 8 502 506
21. WilliamsSLHuangJEdwardsYJUlloaRHDillonLM 2010 The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab 12 675 682
22. HeYWuJDressmanDCIacobuzio-DonahueCMarkowitzSD 2010 Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 464 610 614
23. LiMSchonbergASchaeferMSchroederRNasidzeI 2010 Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am J Hum Genet 87 237 249
24. StewartJBFreyerCElsonJLWredenbergACansuZ 2008 Strong Purifying Selection in Transmission of Mammalian Mitochondrial DNA. PLoS Biol 6 e10 doi:10.1371/journal.pbio.0060010
25. KhrapkoKCollerHAAndrePCLiXCHanekampJS 1997 Mitochondrial mutational spectra in human cells and tissues. Proc Natl Acad Sci U S A 94 13798 13803
26. ZhengWKhrapkoKCollerHAThillyWGCopelandWC 2006 Origins of human mitochondrial point mutations as DNA polymerase gamma-mediated errors. Mutat Res 599 11 20
27. ElliottHRSamuelsDCEdenJAReltonCLChinneryPF 2008 Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet 83 254 260
28. AmeurAWetterbomAFeukLGyllenstenU 2010 Global and unbiased detection of splice junctions from RNA-seq data. Genome Biol 11 R34
29. RovioAT 2006 DNA Polymerase Gamma Mutaions in Male Infertility and Ageing. Tampere University of Tampere 137
30. VermulstMBielasJHKujothGCLadigesWCRabinovitchPS 2007 Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet 39 540 543
31. FalkenbergMLarssonNGGustafssonCM 2007 DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem 76 679 699
32. HoltIJHardingAEMorgan-HughesJA 1988 Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331 717 719
33. InoueKNakadaKOguraAIsobeKGotoY 2000 Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat Genet 26 176 181
34. TyynismaaHMjosundKPWanrooijSLappalainenIYlikallioE 2005 Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A 102 17687 17692
35. NakaseHMoraesCTRizzutoRLombesADiMauroS 1990 Transcription and translation of deleted mitochondrial genomes in Kearns-Sayre syndrome: implications for pathogenesis. Am J Hum Genet 46 418 427
36. ChenHVermulstMWangYEChomynAProllaTA 2010 Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141 280 289
37. KukatATrifunovicA 2009 Somatic mtDNA mutations and aging–facts and fancies. Exp Gerontol 44 101 105
38. Jensen-SeamanMIWildschutteJHSoto-CalderonIDAnthonyNM 2009 A comparative approach shows differences in patterns of numt insertion during hominoid evolution. J Mol Evol 68 688 699
39. Bayona-BafaluyMPAcin-PerezRMullikinJCParkJSMoreno-LoshuertosR 2003 Revisiting the mouse mitochondrial DNA sequence. Nucleic Acids Res 31 5349 5355
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy