
A Role for Set1/MLL-Related Components in Epigenetic Regulation of the Germ Line
The methylation of lysine 4 of Histone H3 (H3K4me) is an important component of epigenetic regulation. H3K4 methylation is a consequence of transcriptional activity, but also has been shown to contribute to “epigenetic memory”; i.e., it can provide a heritable landmark of previous transcriptional activity that may help promote or maintain such activity in subsequent cell descendants or lineages. A number of multi-protein complexes that control the addition of H3K4me have been described in several organisms. These Set1/MLL or COMPASS complexes often share a common subset of conserved proteins, with other components potentially contributing to tissue-specific or developmental regulation of the methyltransferase activity. Here we show that the normal maintenance of H3K4 di - and tri-methylation in the germ line of Caenorhabditis elegans is dependent on homologs of the Set1/MLL complex components WDR-5.1 and RBBP-5. Different methylation states that are each dependent on wdr-5.1 and rbbp-5 require different methyltransferases. In addition, different subsets of conserved Set1/MLL-like complex components appear to be required for H3K4 methylation in germ cells and somatic lineages at different developmental stages. In adult germ cells, mutations in wdr-5.1 or rbbp-5 dramatically affect both germ line stem cell (GSC) population size and proper germ cell development. RNAi knockdown of RNA Polymerase II does not significantly affect the wdr-5.1–dependent maintenance of H3K4 methylation in either early embryos or adult GSCs, suggesting that the mechanism is not obligately coupled to transcription in these cells. A separate, wdr-5.1–independent mode of H3K4 methylation correlates more directly with transcription in the adult germ line and in embryos. Our results indicate that H3K4 methylation in the germline is regulated by a combination of Set1/MLL component-dependent and -independent modes of epigenetic establishment and maintenance.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1001349
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001349
Summary
The methylation of lysine 4 of Histone H3 (H3K4me) is an important component of epigenetic regulation. H3K4 methylation is a consequence of transcriptional activity, but also has been shown to contribute to “epigenetic memory”; i.e., it can provide a heritable landmark of previous transcriptional activity that may help promote or maintain such activity in subsequent cell descendants or lineages. A number of multi-protein complexes that control the addition of H3K4me have been described in several organisms. These Set1/MLL or COMPASS complexes often share a common subset of conserved proteins, with other components potentially contributing to tissue-specific or developmental regulation of the methyltransferase activity. Here we show that the normal maintenance of H3K4 di - and tri-methylation in the germ line of Caenorhabditis elegans is dependent on homologs of the Set1/MLL complex components WDR-5.1 and RBBP-5. Different methylation states that are each dependent on wdr-5.1 and rbbp-5 require different methyltransferases. In addition, different subsets of conserved Set1/MLL-like complex components appear to be required for H3K4 methylation in germ cells and somatic lineages at different developmental stages. In adult germ cells, mutations in wdr-5.1 or rbbp-5 dramatically affect both germ line stem cell (GSC) population size and proper germ cell development. RNAi knockdown of RNA Polymerase II does not significantly affect the wdr-5.1–dependent maintenance of H3K4 methylation in either early embryos or adult GSCs, suggesting that the mechanism is not obligately coupled to transcription in these cells. A separate, wdr-5.1–independent mode of H3K4 methylation correlates more directly with transcription in the adult germ line and in embryos. Our results indicate that H3K4 methylation in the germline is regulated by a combination of Set1/MLL component-dependent and -independent modes of epigenetic establishment and maintenance.
Introduction
The modulation of chromatin architecture is a key level of regulation for potentially all eukaryotic DNA-based processes including gene expression, and DNA replication, repair, and recombination [1]. One level of chromatin modulation involves the methylation of lysines in nucleosomal histones, which can be mono-, di-, or tri-methylated. The positions of methylated lysine residues, and even the extent of methylation at any single lysine, have been associated with distinct transcriptional outcomes [2]. For example, methylation at lysine 4 of histone H3 (H3K4me) correlates with transcriptional activation, whereas methylation on lysines 9 or 27 of H3 (H3K9me/H3K27me, respectively) is most often linked to gene repression [2], [3]. Methyl groups are added to the lysine residues of histones by histone methyltransferases (HMTs) [4]. In S. cerevisae, the sole H3K4 HMT, Set1, is recruited to chromatin by the RNA polymerase II (Pol II) elongation machinery [5], [6]. The occupancy of Set1 and H3K4 methylation are highly correlated on transcribing genes, with trimethylated H3K4 (H3K4me3) enriched in the 5′-end and dimethylated H3K4 (H3K4me2) extending further into the gene body [5], [7], [8]. Set1 is the only H3K4-specific HMT identified in S. cerevisae, yet it is not essential for yeast survival under laboratory growth conditions [9].
While some HMTs have detectable in vitro activity as isolated proteins, substantial and specific in vitro activities of the known H3K4-specific SET HMTs often require the addition of other components from their in vivo complexes [10]–[12]. The COMPASS complex, within which budding yeast Set1 operates, contains seven components that are important for Set1 activity (Table 1 and [13]–[16]). Components of the COMPASS complex are highly conserved from yeast to humans, although different subsets of the subunits are distributed among different H3K4 HMT complexes in multi-cellular organisms [10], [17], [18]. At least six Set1 homologs have been identified in mammalian cells: Set1A, Set1B, and four members of the Mixed-Lineage Leukemia (MLL) family: MLL1, −2, −3, and −4 [19]–[23]. The S. cerevisiae homologs of other components in the mammalian Set1/MLL complex(es) include RbBP5 (Swd1), WDR5 (Swd3), Ash2 (Bre2), Cfp1 (Spp1), and hDPY30 (Sdc1) (Table 1 and [19]–[22], [24]). Biochemical studies suggest that WDR5/Swd3 and RbBP5/Swd1 are essential for complex stability and activity, whereas Ash2/Bre2 and Cfp1/Spp1 might play roles in the conversion from di - to tri-methylation [10], [25], [26]. Knockdown of WDR5 affects H3K4me1/2/3 to various degrees in mammalian cells [18], consistent with the idea that WDR5 plays a central role in stabilizing the complex and regulating its HMT activity [10], [26].

Important developmental roles for this complex have been identified in multiple organisms. WDR5 is required for the maintenance of H3K4me3 levels at the HOXA9 and HOXC8 loci in mammalian cells and is important for the normal expression of these two genes [18]. In X. laevis, knockdown of WDR5 leads to reduction of H3K4me1 and H3K4me3 levels and a variety of developmental phenotypes including somatic and gut defects [18]. Previous studies in C. elegans have also implicated the Set/MLL complex as playing important roles in growth and somatic gonad and vulva development, at least partly through attenuation of Ras signaling [27], [28].
Although some structural studies have suggested that WDR5 recognizes the most N-terminal residues of histone H3's “tail” [29]–[32], other studies have shown that H3 tail recognition by WDR5 may not require K4, as the interaction between WDR5 and the H3 tail can be independent of H3K4's methylation status. In contrast, asymmetric dimethylation of arginine 2 of H3 (H3R2me2a) abolishes binding of WDR5 to the H3 tail [33]–[35]. Recent studies indicate that MLL interacts with WDR5 through a WDR5-interacting (Win) motif that shares homology with the N-terminus of H3, and indeed the interaction site in WDR5 is the same for both MLL and H3 [36]–[40]. This suggests that interactions of WDR5 with H3 and MLL may be mutually exclusive. Intriguingly, in vitro-assembled complexes that lack the MLL protein still exhibit H3K4-specific HMT activity [38]. This suggests that MLL is not the sole HMT activity in the MLL complex, and that WDR5 binding to H3, to the exclusion of MLL, may not necessarily abrogate the complex's role in histone modification. In addition, histone demethylase activities are reported to copurify or otherwise interact with Set1/MLL core components, suggesting the complex may have a variety of roles in epigenetic regulation [27], [41].
These studies have provided significant insight into how H3K4 methylation may be differentially regulated by components of the HMT complex, yet the contributions of this regulation to developmental pathways are still poorly understood. Indeed, the function of the H3K4me mark itself is unclear. Although H3K4 methylation has been assigned roles in the regulation of gene activation, it is also clearly a downstream consequence of gene activity. It has been proposed that H3K4 methylation could serve as a “memory” of where transcription has occurred, which in turn may stably guide further transcriptional regulation after cell division and throughout differentiation [42]–[44]. Recent data support this and suggest that histone methylation can serve as a component of epigenetic memory that can even be transferred intact across multiple generations. For example, mutation of the H3K4 demethylase LSD1/KDM1 causes an inappropriate and continued accumulation of H3K4me2 in germ cell chromatin across many generations [45]. This H3K4me2 accumulation correlates with a progressive, generation-dependent “germline mortality” phenotype in mutant populations, a phenotype that presumably results from the heritable accumulation of H3K4me2 that is not properly removed in the absence of LSD1 [45].
Another mark normally considered a product of ongoing transcription, H3K36me, has recently been shown by our lab and others to heritably mark, in embryos, genes that had last been expressed in the germ cells of the preceding generation [46]. This heritable marking is by a metazoan-specific H3K36 HMT, MES-4, that in C. elegans can operate in a largely transcription-independent mechanism. The role of MES-4 in this mode of “maintenance histone methylation” is essential for germline viability.
Recent studies in both mammals and worms have shown that histone modifications imposed in the paternal germ cells can be transferred into subsequent generations through sperm chromatin in mammals [47]. These include chromatin regions at genes transcribed in germ cells, but also so-called bivalent domains, regions marked by both H3K4 and H3K27 methylation, which have been observed to similarly mark inactive, developmental loci in ES cells [47]–[50]. Indeed, there is significant overlap in the loci marked by bivalent domains in both sperm and ES cells, suggesting that H3K4me can be maintained at these unexpressed loci in both germ cells and in embryonic lineages that retain pluripotency. Conversely in C. elegans, transcriptional inactivity of the X chromosome during male spermatogenesis results in the X chromosome being largely depleted of H3K4me during meiosis and gametogenesis; this dearth of H3K4me persists in spermatid chromatin, and is heritably maintained through multiple cell divisions in the early embryo [51].
Although epigenetic marks, including histone modifications and DNA methylation imprints, can be inherited from parents through the gametes, there is significant erasure, or “reprogramming”, of information in the zygote, and then again during germ cell specification [52]–[54]. The purpose of this reprogramming is unclear, but it is presumably related to the removal of epigenetic content that is incompatible with developmental pluripotency. Conversely, epigenetic information that defines the pluripotent state must be recognized and protected from erasure and/or actively maintained during this process. Many regions of the genome carrying this information may not be transcriptionally active during early embryonic stages, therefore maintenance of the epigenetic content may not be obligatorily coupled to transcriptional activity. Maintenance of transcription-associated marks like H3K4me and H3K36me may therefore require mechanisms operating outside of active gene expression. Evidence is mounting that such mechanisms exist. In addition to the maintenance of bivalent silenced loci and the MES-4 system mentioned above, a recent study in zebra fish embryos revealed a class of genes in which H3K4me3 alone is present at promoters with no evidence of ongoing transcription, further uncoupling this modification from transcription [55]. In addition, unmethylated CpG-rich regions of the genome can recruit H3K4 methylation independently of transcription, and this requires the Set1/MLL complex component Cfp1/CxxC1 [56]. These data indicate that H3K4 HMT complexes may play a role in the establishment and/or maintenance of H3K4me patterns in the genome independently of transcription.
RNA Polymerase II (Pol II) is normally inactivated in the early embryonic germline of C. elegans [57]–[59]. Despite the absence of Pol II activity, and cell divisions that could dilute this mark (through replication-coupled de novo chromatin assembly), the level of H3K4me2 in the germline precursor chromatin remains relatively stable [54]. This suggests the existence of transcription-independent mechanisms capable of maintaining this mark. In these studies, we show that conserved components of Set1/MLL-like histone methyltransferase complexes are required for what appears to be a largely Pol II-independent mode of H3K4me maintenance in germ cells. Interestingly, this mode predominates in early embryonic somatic and germline precursors, and in larval and adult stages Set1/MLL complex component-dependent H3K4 methylation is most obvious in the germline stem cell (GSC) population. Mutations in some components show defects in germline stem cell maintenance, impair fertility and germ cell development, and exhibit a germline mortality phenotype [28]. Our results suggest that H3K4 methylation is an important component of the epigenetic regulation of germline establishment, maintenance, and function. We propose that a combination of transcription-dependent insertion, and transcription-independent maintenance, of this mark may be required to maintain a totipotent epigenome as it passes through the germ line across generations.
Results
Conserved Set1/MLL complex components are present in the genome of C. elegans
H3K4me2 and me3 are considered to be hallmarks of transcriptional activity, as these marks are generally correlated with active genes in genome-wide studies [5], [8], [60], [61]. However, significant levels of H3K4me2 are stably observed in the chromatin of early dividing blastomeres and germline precursors (P cells) of C. elegans embryos, which exhibit little detectable RNA Polymerase II transcription [57], [59]. This suggests that transcription-independent mechanisms of maintaining this modification exist in these cells. We wished to identify the H3K4 methyltransferase(s) involved in the maintenance of this epigenetic mark and the consequences of their absence. All known H3K4 specific methyltransferases share the conserved catalytic SET-domain [62]. C. elegans contains 34 genes that encode SET domain proteins; sequence analysis suggests that two of them, set-2 and set-16, are most closely related to yeast Set1 and human MLL H3K4 HMT's. The SET domain of worm set-2 shares 80.6% and 74.2% homology with the human Set1A and yeast Set1 proteins, respectively, whereas the SET-domains of worm set-16 and human MLL3 share 66.7% homology (not shown).
C. elegans also contains homologs of other yeast COMPASS components (see Table 1 for nomenclature). Because COMPASS is often used to refer to the specific complex found in S. cerevesiae, we hereafter refer to the C. elegans homologs of conserved components as Set1/MLL complex components to denote the metazoan complex. Note also that although homologs of most Set1/MLL complex components have been identified in C. elegans, the existence of a complex composed of these components can only be inferred at this time. Three homologues of Swd3/WDR5 (wdr-5.1, wdr-5.2, and wdr-5.3) are found in the C. elegans genome (Table 1 and [27], [28]). Other homologues include: F21H12.1 (Swd1/RbBP5), Y17G7b.2 (Bre2/Ash2), dpy-30 (Sdc1/hDPY30), cfpl-1/F52B11.1 (Spp1/Cfp1), and C33H5.6 (Swd2/Wdr82) (Table 1, and [27], [28]). No Shg1 homologs were identified in C. elegans. Previous studies have shown that a SET1/MLL complex, as in other organisms, contributes to H3K4 methylation in C. elegans [27], [28]. Inactivation of the Set1 homolog set-2 resulted in a global decrease of H4K4me3 levels in mixed-staged populations, but little detectable change in H3K4me2 levels [28]. Knockdown of each of three other worm homologs of the Set1/MLL complex components, wdr-5.1/WDR5, dpy-30/hDPY30, or cfpl-1/Spp1, resulted in decreases of both H3K4me2 and H3K4me3 to various degrees [28]. These experiments, while informative, did not assess stage - or tissue-specific effects of loss of these components, particularly in the germ line.
An essential role for Set1/MLL components in embryonic H3K4me regulation
To investigate how H3K4 methylation is regulated developmentally, we first asked if set-2 or set-16 was required for H3K4 methylation in early embryos. We dissected embryos from mutant strains and/or RNAi-treated animals and probed fixed whole mount specimens with antibodies specific to H3K4me2 or H3K4me3. In the RNAi protocol performed, treated animals are expected to exhibit reduction in both maternal and zygotic supplies of the targets. set-16(RNAi) caused >80% embryonic lethality, with survivors growing up to be Dumpy (Dpy phenotype) and sterile adults. In contrast, homozygous embryos from animals heterozygous for the set-16 (gk438) deletion allele (i.e., Maternal+/Zygotic-), developed into Dpy and sterile adults, indicating substantial maternal rescue. Appreciable loss of either di - or tri - methylated H3K4 (H3K4me2/me3) in any stage or tissue examined was not observed in either RNAi-treated or (maternally rescued) mutant embryos in our immunofluorescence assays (Figure S1 and data not shown). This result potentially differs from a recent report showing a decrease in H3K4me3 in set-16 (RNAi) adults detected by western blot assays [27]; the differences in the respective results may be due to differences in the assays and experimental focus.
However, and similar to a previous report, we observed that H3K4me3 was substantially depleted from nuclei of set-2 (tm1630) deletion mutant embryos (Figure 1A; [28]). We also confirmed that H3K4me2 was not significantly affected in set-2 mutant embryos (Figure 1B; [28]). Knockdown of set-2 by RNAi caused similar reduction in H3K4me3 levels in embryos, also without affecting H3K4me2 (data not shown). These results indicate that SET-2 is an HMT principally responsible for H3K4me3, and that other HMTs may regulate H3K4me2. We do not know if SET-2 acts to convert H3K4me2 to H3K4me3, but we did not notice any increase in H3K4me2 levels in set-2 mutants, as might be expected from a defect in such conversion (not shown).

In an attempt to identify the additional HMT(s) responsible for H3K4me2, we knocked down several additional SET domain containing H3K4 HMT candidates including: F15E6.1 (set-9), Y51H4a.2 (set-26), K09F5.5 (set-12), Y41D4b.12 (set-23), lin-59, set-25, and F25D7.3 by RNAi. We did not detect changes in H3K4me2/3 by immunofluorescence in any of these experiments (data not shown). These results confirm and extend the findings of others that SET-2 is the HMT activity that is largely responsible for H3K4 trimethylation in embryos ([28]). The dimethyl H3K4 HMT activity still remains to be identified.
To test whether the other conserved components of Set1/MLL complexes are similarly required for normal H3K4me regulation in embryos, we examined animals with mutations in the homologous genes and/or treated with RNAi targeting those loci. We first focused on the WDR5 homologs. Similar to set-2(tm1630) mutant embryos, H3K4me3 was strongly depleted from the nuclei of wdr-5.1(ok1417) embryos (Figure 1A). The wdr-5.1(ok1417) deletion allele is likely a null allele since an antibody against WDR-5.1 did not detect the WDR-5.1 band in western blot analyses (Figure S2), and wdr-5.1(RNAi) resulted in phenotypes similar to ok1417 (not shown). However, unlike set-2(tm1630), significant loss of H3K4me2 was also observed in wdr-5.1 embryos, albeit not to the near background levels observed for H3K4me3 (Figure 1B). These defects are specific to loss of WDR-5.1 activity in the deletion mutants since both H3K4me2 and H3K4me3 defects could be rescued by a WDR-5.1::GFP transgene (Figure 1C). In contrast, neither mutation of wdr-5.2, nor knockdown of wdr-5.3 by RNAi, the other Swd3/WDR5 homologs, noticeably affected H3K4me3 or H3K4me2 levels in embryos, nor did combinations with wdr-5.1(ok1417) mutants exhibit additive defects in H3K4me (data not shown). These data indicate that among the WDR5 homologs, WDR-5.1 plays the predominant role in H3K4 methylation in early embryonic stages. Protein blot analysis of H3K4me2 and H3K4me3 marks present in embryos confirmed the immunofluorescence results (Figure 1D). H3K4me1 levels were not detectably affected in any of the mutants tested (Figure 1D and data not shown).
We then examined other conserved Set1/MLL components. As with wdr-5.1(ok1417) mutants, both H3K4me3 and H3K4me2 were strongly reduced in the nuclei of rbbp-5(tm3463) mutant embryos (Figure 1A and 1B). Knockdown of rbbp-5 by RNAi resulted in similar defects, indicating our RNAi experimental conditions were efficient (data not shown). We then targeted homologs of other Set1/MLL complex components in an RNAi hyper-sensitive strain, eri-1(mg366). As shown in Figure 1A and 1B, ash-2, dpy-30, and cfp-1 are also required for normal levels of both H3K4me3 and H3K4me2 in early embryos. RNAi of wdr-82 showed no discernible effects on H3K4 methylation in our assays. Interestingly, H3K4 methylation was observed to become increasingly detectable in later stages of embryogenesis (>300 cells) in mutant and RNAi-treated embryos (Figure S3 and data not shown). This indicates that HMT activities independent of these components exist in the embryo, and that the H3K4 marks provided by these activities become increasingly evident as development progresses.
Taken together, these data show that conserved homologs of Set1/MLL components contribute to the predominant mode of H3K4me regulation in early C. elegans development. All of the predicted complex components, with the exception of wdr82/swd-2, are essential for this HMT activity. SET-2 appears to be the H3K4 HMT operating in the context of this putative complex that is specifically required for H3K4 trimethylation, whereas a separate as yet unidentified HMT activity is responsible for H3K4me2 regulation. In later embryos, H3K4 methylation becomes detectable in the absence of Set1/MLL component function; this H3K4 methylation activity may correlate with a transcription-dependent process (see below).
WDR-5.1 binding to H3 is dependent on H3R2 methylation
As mentioned, WDR5 is thought to play a central role in core complex assembly and HMT regulation by interacting with MLL and/or histone H3 N-terminal tails [10], [18]. Binding of WDR5 to the histone H3 N-terminus is not affected by lysine 4 methylation, but instead is strongly inhibited by asymmetric dimethylation of arginine 2 (H3R2me2a) [33], [34]. To test if worm WDR-5.1, like its mammalian WDR5 counterpart, has similar interactions with the H3 tail, we incubated wild-type nuclear extract with biotinylated H3 peptides and assayed for WDR-5.1 interactions by western blot analysis of peptide-bound material using a WDR-5.1 specific antibody. As shown in Figure S4, WDR-5.1 showed specific interactions with unmodified (H3K4me0) and H3K4me2 peptides, but not with an H3R2me2a-modified peptide. C. elegans WDR-5.1 thus has similar histone H3 binding characteristics to those of mammalian WDR5. These data, in addition to the H3K4 methylation defects observed in wdr-5.1, rbbp-5, and ash-2(RNAi) animals described above, strongly suggest that the these proteins probably exist in a complex that has similar attributes to other bona fide Set1/MLL complexes.
Set1/MLL-dependent H3K4 methylation maintenance in the early embryo is largely independent of transcription
The COMPASS complex is recruited to chromatin by the RNA polymerase II (RNA Pol II) holoenzyme complex in S. cerevisiae [5], and thus in yeast H3K4 methylation appears to be strictly dependent on transcriptional elongation. However, as discussed earlier, there is growing evidence that H3K4 methylation in metazoans may not always be coupled to ongoing transcription.
In C. elegans embryonic germline precursors (P cells) there is little or no Pol II transcription [58] yet H3K4 methylation persists through the four cell divisions in this lineage. However, some differences are observed between di - and tri-methylated forms (Figure 2 and [54]). In wild type embryos, H3K4me3 is present from P1 and P2, but is decreased by 30% and by ∼85% in P4, respectively (Figure 2C and 2F), whereas H3K4me2 is maintained at comparable levels in P1 through P4 (Figure 2E). The levels of both H3K4me2/3 are essentially uniform in all somatic blastomere chromatin in all early stages. In wdr-5.1/wdr-5 mutant embryos both H3K4me2 and H3K4me3 are initially observed in the chromatin of 1-2 cell nuclei; this is presumably inherited from the gamete chromatin, as it is also strongly retained in the polar bodies (Figure 2B, 2D; arrowheads) [51]. However, the maintenance of both marks in subsequent cell divisions is severely compromised in the P cells of wdr-5.1 embryos (Figure 2B, 2D). The loss is presumably through replication-coupled histone replacement, or other types of histone dynamics, as the levels decrease with cell number. Notably, H3K4me2/3 in the somatic cells were also strongly reduced in the absence of wdr-5.1 (Figure 1, Figure 2). Similar results were observed in the rbbp-5/swd-1 mutant, as well after RNAi of ash-2, dpy-30, and cfp-1 (data not shown). These data suggest that Set1/MLL component - dependent mechanisms are responsible for maintaining normal levels of H3K4me2/3 in the transcriptionally inactive P cells and early somatic blastomeres. Early development is largely driven, and cell viability largely maintained by maternal supplies in C. elegans, so the early somatic blastomeres are not thought to be robustly active [63]. The maintenance of H3K4me2/3 in these early somatic lineages may thus also be largely independent of ongoing transcription

To further investigate this discordance between Set1/MLL mediated H3K4 methylation and active transcription, we knocked down ama-1 (the large, catalytic subunit of RNA Pol II) by RNAi in both WT and wdr-5.1 mutants. Under the conditions employed, ama-1(RNAi) resulted in 100% embryonic lethality and AMA-1 protein dropped to undetectable levels by immunofluorescence in early embryos (Figure 3). In addition, the RNAi conditions employed were sufficient to abolish detection of the phospho-Ser2 modification on RNA Pol II (Ser2p; recognized by the H5 monoclonal antibody), which correlates with the elongating form of the RNA Pol II holoenzyme (Figure S5A). The depletion of AMA-1 in wild-type embryos did not significantly impact H3K4me3/me2 levels, and this ama-1(RNAi) “resistant” H3K4 methylation was not observed when wdr-5.1 was also defective (Figure 3). RNAi of cdk-9, the kinase component of the essential RNA Pol II transcription elongation factor pTEF-b, also knocked down Pol II Ser2p to undetectable levels and this also had no appreciable effect on H3K4me3/2 maintenance (Figure S5). We could not test if the WDR-5.1-independent H3K4 methylation we observed in later stages was also depleted by either RNAi, since the embryos arrested prior to those stages (not shown). Taken together with what we observed in the P cells above, these data strongly suggest that in all early embryonic nuclei, including germline precursors, H3K4 methylation maintenance is largely Set1/MLL component-dependent, and this activity is unaffected by significant reduction of ongoing transcription.

H3K4 methylation in adult germ cells has a unique mode of regulation
We next asked whether the Set1/MLL components are similarly required for H3K4me regulation in post-embryonic tissues. A recent report has shown that a worm MLL-like complex can attenuate Ras signaling in vulva development, but the vulva defects are only observed in a sensitized genetic background [27]. In our hands, mutants in some of the Set1/MLL component homologs, such as wdr-5.1/wdr-5 and rbbp-5/swd-1, exhibited fertility defects (below), so we focused on the adult germ line. In C. elegans, hermaphrodite germ cells first undergo spermatogenesis in larvae followed by a switch to oogenesis, which is maintained in adults. The adult germ cells are linearly and progressively arranged by developmental stage within the gonad arms, such that defects in any particular stage are easily identified. The mitotically active germ line stem cells (GSCs) reside in the most distal region of the gonads, and their entry into and progression through meiosis and gametogenesis occur sequentially as the cells move more proximally (e.g., left to right in Figure 4). The border between mitotic exit and meiotic entry, for example, is readily visualized by a characteristic condensation and crescent-shaped localization of all chromosomes within each nucleus in the “transition zone”, comprising leptotene and zygotene stages. After exiting the transition zone, germ cells progress proximally into pachytene and then further on into oogenesis. H3K4me3 and H3K4me2 are normally abundant in the chromatin of all adult germ cell stages, and both are broadly distributed on all autosomes (Figure 4A and Figure 5A). These marks, however, are generally absent from the X chromosomes in all germ cell stages, with the exception of oogenesis [51], [64].
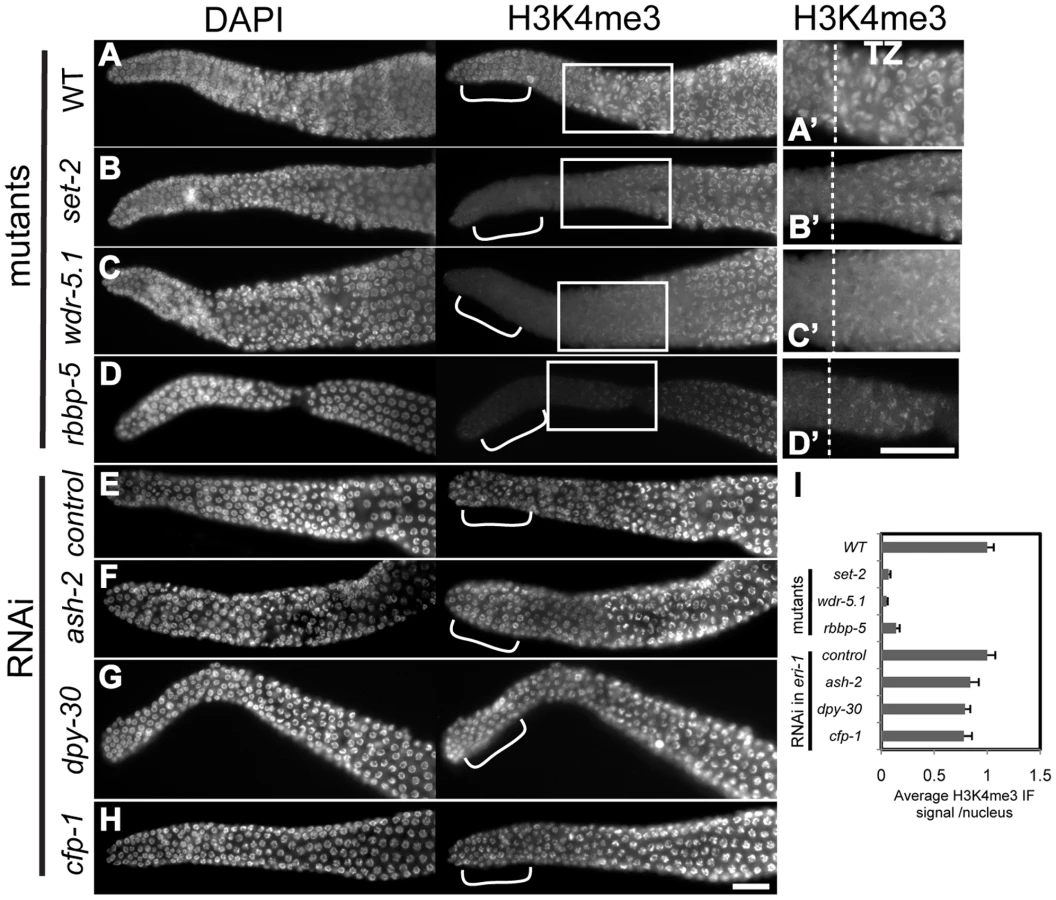

We first examined the role of the Set1/MLL component homologs in H3K4me3 regulation in adult germ cells. H3K4me3 was dramatically decreased in a specific population of nuclei in set-2(tm1630), wdr-5.1(ok1417) and rbbp-5(tm3463) deletion mutants. The decrease we observed was limited to chromatin in the most distal nuclei, the region within which the germline stem cell population (GSC) resides (Figure 4). This pattern was identical to that observed with RNAi knockdown of set-2, wdr-5.1, or rbbp-5, indicating that these cells are not specifically resistant to RNAi (not shown). Interestingly, and in contrast to what we observed in embryos, RNAi mediated knockdown of ash-2, dpy-30 or cfp-1 had minimal effects on H3K4me3 levels in adult germ cell chromatin compared to the near 90% reduction observed in set-2, wdr-5.1 and rbbp-5 mutants (Figure 4). Consistent with our results in embryos, wdr-82 knockdown also did not affect H3K4me3 in adult cells (data not shown). Significant H3K4me3 loss was also restricted to the GSCs in wdr-5.1 males, showing that the requirement for wdr-5.1 and rbbp-5 for GSC H3K4me3 is not sex-specific (Figure S6A).
The absence of redundancy and the highly overlapping H3K4me3 defect patterns in these three mutants are evidence that SET-2, WDR-5.1 and RBBP-5 work together in these cells. The absence of H3K4me defects observed with RNAi of ash-2, cfp-1, or dpy-30 indicates that, unlike in embryonic cells, these conserved components are not required for H3K4 methylation in the adult germline stem cells. The GSCs are not less susceptible to RNAi gene targeting, since RNAi of set-2, wdr-5.1, or rbbp-5 substantially knocks down methylation of H3K4 in the GSCs and ama-1(RNAi) was effective in this region of the gonad. Furthermore, RNAi mediated knockdown of ash-2, cfp-1, and dpy-30 all significantly affected H3K4 methylation in offspring from treated animals, and the lack of effect in the adult germ cells was observed in both the parents and similarly exposed adult siblings of affected embryos in these experiments (details diagrammed in Figure S7).
As the germ cells in the set-2, wdr-5.1, and rbbp-5 mutants progressed into meiosis, increasing levels of H3K4me3 was detected in the chromatin of pachytene stage and more proximal nuclei (Figure 4A-4D''). However, the levels of H3K4me3 in pachytene and diakinesis regions in wdr-5.1 were significantly reduced compared to wild type levels (Figure S8A–S8C). The wdr-5.1/rbbp-5-independent mechanism in meiotic germ cells may be additive to, or partially dependent on, what is placed in the chromatin by the WDR-5.1–dependent processes in the GSCs. Indeed it is also possible, and we cannot rule out by these experiments, that the separate mechanisms have separate targets. Transcription in adult germ cells appears to be highest in meiosis [65], so the wdr-5.1 independent H3K4 methylation may be similar to the Set1/MLL-independent H3K4 methylation observed in late-stage embryos (Figure S3). These results indicate that the regulation of H3K4me3 in embryos and adult GSCs depends on an HMT activity that requires different subsets of the “panel” of Set1/MLL components. The results also indicate that an additional, Set1/MLL component independent mode of H3K4 trimethylation is predominantly active in meiosis.
We also tested for H3K4me2 changes in mutant and/or RNAi-treated adult germ cells. We found that, like H3K4me3, H3K4me2 in the GSC region was also only slightly affected by knockdown of ash-2, dpy-30, or cfp-1 (Figure 5E–5F). As in embryos, set-2, which is required for H3K4me3 maintenance, also was dispensable for H3K4me2 in adult germ cells (Figure 5B). Similar to H3K4me3, H3K4me2 levels were decreased 90% and 85% in the GSC region of wdr-5.1(ok1417) and rbbp-5(tm3463) mutant gonads, respectively (Figure 5I). RNAi of these two genes again showed similar defects in adult germ cells (data not shown). However, as shown in Figure S9, H3K4me2 was reduced but not completely absent in the distal region of wdr-5.1(ok1417) gonads. Low levels of H3K4me2 are apparent, with longer exposure times, as discreet foci on the chromosomes, rather than the more diffuse chromatin pattern observed in wild-type GSC nuclei (Figure S9C). As with H3K4me3, H3K4me2 appeared in pachytene and diakinesis regions, and was also 50% below that of WT chromatin (Figure S9A and S9D). These defects and patterns were also observed in wdr-5.1 males (Figure S6B).
Notably, we observed similar defects in H3K4me2 staining (in both adults and embryos) in the wdr-5.1 strain using two different monoclonal antibodies from mouse [66] and rabbit (Millipore cat# 04-790). A significant decrease was also observed using a polyclonal antibody, but to a lesser extent (Millipore cat#, 07-030). Both the mouse monoclonal and polyclonal antibody signals were efficiently reduced when competed with H3K4me2 peptides, but not with H3K4me0, H3K4me1, or H3K4me3 peptides (not shown). The reason for the increased residual signal detected by the polyclonal antibody in the mutants is not known.
These results show that WDR-5.1 and RBBP-5 are essential for normal H3K4me2 and H3K4me3 in adult GSCs, and to a lesser extent, meiotic germ cells and (as in embryos) appear to rely on different HMT activities for the differently methylated products. Furthermore, some Set1/MLL components that are essential for H3K4 methylation in embryos do not appear to be required in adult germ cells. Regulation of H3K4 methylation in C. elegans, as in mammals, may therefore involve distinct HMT complexes to yield a variety of different outcomes in different tissues and developmental stages. There also appears to be H3K4-specific HMT activities in C. elegans that can be roughly divided into those that share a requirement for at least some homologs of the Set1/MLL components, and those that do not. The H3K4 methylation activities we observe in meiotic chromatin and in later embryonic stages appear to fall into the latter class.
H3K4me maintenance by WDR-5.1/RBBP-5–dependent mechanisms in adult GSCs is largely independent of ongoing transcription
As described earlier, H3K4me2/3 maintenance in the embryonic blastomeres and germline precursors was not appreciably affected by the significant disruption of RNA Pol II activity. We investigated whether this was also the case in adult germ cells by targeting ama-1 with RNAi. AMA-1 protein is detectable in all adult germ cell chromatin until diakinesis stage in oogenesis, which largely lack RNA Pol II associated with chromatin (Figure 6A-a' and c'; data not shown and [67]). In both ama-1(RNAi) and wdr-5.1;ama-1(RNAi) animals, AMA-1 protein was depleted to undetected levels in the distal GSCs and in the more distal early - to mid-pachytene meiotic nuclei, but was still faintly detectable in more proximal nuclei (Figure 6A-b' and d'). This is consistent with the more proximal (older) nuclei containing AMA-1 protein that had been translated prior to mRNA depletion by RNAi treatment. These results indicate that the RNAi had efficiently knocked down AMA-1 to undetected levels in distal - to mid-proximal germ cells in both genotypes in these experiments. Furthermore, the progression of germ cell development appeared to be crippled in these animals, as oocyte production had ceased, and 100% of all embryos produced up to the time point of analysis failed to hatch (not shown).

In parallel experiments, H3K4me3 and H3K4me2 patterns in ama-1(RNAi) gonads showed an interesting defect. In wild-type gonads treated with ama-1 RNAi, H3K4me2/3 levels in the GSCs (where H3K4me is dependent on wdr-5.1) were not significantly affected (Figure 6). However, a swath of nuclei that had recently entered into and engaged in meiosis showed a 60–70% decrease in the levels of H3K4me3 and H3K4me2 (Figure 6B-b', f', brackets; and 6C and 6D, red bars). Note that both the GSCs and this “swath” of distal meiotic nuclei showed comparable depletion of AMA-1 protein in parallel animals, but H3K4me levels were only decreased in the meiotic nuclei (Figure 6A-b' and d'). Late pachytene nuclei that exhibited AMA-1 antibody staining in parallel animals still showed significant H3K4me2/3 levels (Figure 6C, green bars). In wdr-5.1;ama-1(RNAi) animals, H3K4me2/3 was dramatically decreased in all nuclei from the distal GSC region to the mid-meiotic nuclei; i.e., the ama-1(RNAi) resistant H3K4 methylation in the GSC region was not observed (Figure 6B; d',h', 6C and 6D). We also analyzed ama-1(RNAi) wild-type and wdr-5.1(ok1417) animals using the H5 (Pol II phospho-Ser2) antibody, and confirmed that the elongating form of RNA Pol II was depleted in the treated germ cells (Figure S10A and S10B). H3K4me3 in GSC chromatin was again largely unaffected in wild-type ama-1 (RNAi), whereas that in meiotic chromatin was significantly depleted (Figure S10C). In wdr-5.1; ama-1(RNAi) germ cells, both GSC and meiotic cell again showed reduced H3K4me3 in these experiments (Figure S10D).
Taken together, our data suggest that the H3K4 methylation in GSCs, that is largely dependent on wdr-5.1/rbbp-5 mediated mechanisms, is also largely impervious to substantial depletion of RNA Pol II. We cannot conclude that all Pol II activity was completely eliminated in these experiments, but the lack of anti-AMA-1 and anti-Pol II phospho-Ser2 signals, and the fully penetrant embryonic lethality preceding the cessation of oogenesis, indicates substantial ablation of ongoing transcription in the treated germ cells. Importantly, a significant component of H3K4 methylation that occurs in meiotic cells is largely independent of WDR-5.1, and this methylation was dramatically affected by the ama-1(RNAi) conditions employed. The meiosis-coupled H3K4 HMT activity is thus more closely linked to ongoing transcription. Thus WDR-5.1/RBBP-5-dependent mechanisms can maintain H3K4 methylation in both the embryonic germline and their post-embryonic germline stem cell descendants in a largely RNA Pol II independent manner. We next investigated the role of WDR-5.1 in the larval transitions that bridge these germ cell stages.
Transitional regulation of H3K4me3/2 in proliferating larval germ cells
The H3K4 methylation that is maintained by Set1/MLL components in the transcriptionally inert P-blastomere chromatin is dramatically erased in the primordial germ cells (PGCs), named Z2 and Z3 [54]. The reduction in H3K4me is maintained throughout embryogenesis, and normally does not reappear until hatching (Figure 7A and [54]). The re-appearance of H3K4me2/3 at hatching usually coincides with reappearance of the phospho-Ser2 modification of RNA Pol II, and thus presumably the re-initiation of productive transcription in these cells [68]. PGC proliferation in early stage larvae produces the germline stem cells that ultimately contribute to and maintain the adult germ cell population. In wild-type animals, H3K4me2/3 are present at robust levels in all germ cell nuclei at all post-embryonic stages. In wdr-5.1(ok1417) mutants, H3K4me3 reappearance was initially normal in the early PGCs and their progeny in L1 larvae, and remained detectable for a few cell divisions (Figure 7B). However, as the number of germ cells increased, H3K4me3 levels substantially decreased in the proliferating (now established) GSCs in wdr-5.1 L2 and early L3 larvae.

In later larval stages, as the wdr-5.1(ok1417) germ cells began to enter meiosis, H3K4me3 was observed to again accumulate in their chromatin, as observed in wdr-5.1 adult gonads (e.g., Figure 4). The H3K4me2 pattern was similarly defective in wdr-5.1 mutants (data not shown). These results indicate that concomitant with PGC reactivation, there is a period of wdr-5.1-independent H3K4 methylation as the GSCs first become established. Shortly thereafter, and during all subsequent larval and adult stages, H3K4 methylation in the proliferating GSCs becomes largely dependent on the WDR-5.1 mechanism.
WDR-5.1 and RBBP-5 are required for normal GSC maintenance
The GSCs are the proliferative cells that support the large numbers of gametes ultimately produced by C. elegans adults. Mutations that affect the maintenance of this stem cell population affect the number of nuclei and thus the length of the pre-meiotic region of the gonad arms [69]-[71]. We examined the effect of wdr-5.1 and rbbp-5 mutations on the length of the GSC zone in young adult gonads. The established procedure is to measure the number of cell diameters from the most distal nucleus (the distal tip cell) to the first nuclei exhibiting the crescent-shaped chromosome morphology characteristic of cells entering early prophase I of meiosis (“transition zone” nuclei; e.g.; [70]). We also assayed for SYP-1 appearance, a synaptonemal complex protein [72], to help identify the boundaries between the mitotic region and transition zone (Figure 8B, 8D and 8F).

A temperature-dependent defect was observed for GSC maintenance in both wdr-5.1 and rbbp-5 mutants: at 25°C the mutants exhibited shorter GSC zones (average 11 cell diameters) compared to identically staged wild-type animals (average 18 cell diameters; Figure 8G). Indeed, some wdr-5.1 animals had GSC zones as short as 6 cell diameters (Figure 8G). The brood size of wdr-5.1 mutants was also substantially lower than wild-type animals, even at normal temperatures (Figure S11A). These data suggest that the H3K4me2/3 marks in the GSC chromatin that are maintained by the WDR-5.1/RBBP-5 dependent mechanisms, or the proteins themselves, may be important for normal maintenance of this stem cell population.
wdr-5.1 and rbbp-5 mutants display only partially overlapping somatic and germline developmental phenotypes
We further characterized the phenotypes of set-2, wdr-5.1 and rbbp-5 mutants. set-2(tm1630) mutants do not display obvious phenotypes when grown at either 20°C or 25°C (Table 2), indicating that neither SET-2 or its H3K4me3 mark are overtly essential for normal development. At 20°C, wdr-5.1 animals were fertile, but showed 12% embryo lethality (Emb) and a moderate egg laying defect (Egl) (Table 2 and Figure S12A). In contrast, at 20°C, rbbp-5(tm3465) mutants exhibited multiple phenotypes including a Dumpy phenotype (Dpy), egg-laying defect (Egl), and significant sterility (24.4%; Table 2 and Figure S12A and S12B). The Dpy phenotype was not observed in wdr-5.1(ok1417) animals at any temperature, indicating that these factors may have non-overlapping roles in some somatic developmental pathways. The basis of the Dpy phenotype in rbbp-5(tm3465) animals, for which many candidate pathways exist, is unknown.

At elevated temperatures, wdr-5.1(ok1417) embryonic lethality rose dramatically to 29% for the 1st generation at 25°C, and a previously reported germline mortality phenotype in subsequent generations was also confirmed (Figure S11B and [28]). When grown at 25°C, the mutant exhibited an increased frequency of sterile progeny with each generation. By the F4 generation, 67% of the embryos developed into sterile adults. Interestingly, sterility reached a maximum by the F4 generation, although the number of offspring produced by the few fertile animals at subsequent generations was low (Figure S11B and not shown). When rbbp-5 L4 larvae were shifted to 25°C, 12.1% of their F1 progeny arrested as embryos; however, most (82.9%) of the survivors developed into sterile adults (Table 2). The remainder appeared to be capable of producing sperm and oocytes, since some embryos were observed in these animals in utero, yet these embryos were not laid onto the plates nor did they hatch (Table 2).
In vertebrates, mutations in WDR5 and MLL cause developmental defects, presumably because of their reported roles in regulation of Hox gene expression [18], [73]. Because of the embryonic lethality we observed in wdr-5.1 and rbbp-5 mutants, we tested for defects in Hox gene expression. We examined the expression of the Hox loci lin-39, ceh-13, mab-5 and egl-5 in wdr-5.1 mutant embryos grown at 20°C by qRT-PCR. We found no significant alteration in the expression levels of these four genes in wdr-5.1(ok1417) compared to wild type embryos, suggesting that WDR-5.1 is not required for Hox gene expression at normal temperatures in C. elegans (data not shown).
wdr-5.1/WDR5 and RBBP-5/RbBP5 mutants exhibit defects in germ cell development
To investigate the mechanism underlying the sterility of wdr-5.1(ok1417) and rbbp-5(tm3463) mutants, we dissected the gonads from sterile worms and stained with DAPI. We found that 75.9% of the gonads in sterile wdr-5.1(ok1417) mutants grown at 25°C had endomitotic oocytes (Emo phenotype); i.e., unfertilized eggs had prematurely entered the cell cycle and engaged multiple rounds of replication in the absence of cytokinesis (Figure 9B and 9D; Class I). In addition, a small percentage of hermaphrodite gonads (5.1%) exhibited a Mog (masculinization of germline) phenotype in which only sperm were produced; i.e., the normal switch from spermatogenesis to oogenesis in the germline had failed to occur (Figure 9B and 9D; Class II). An additional 19% of sterile wdr-5.1 mutant animals had germ cells with severely abnormal DNA morphology and many apparently polyploid nuclei. The cells within the gonads of these animals were also disorganized; we observed what appeared to be spermatids in the mid-pachytene zone of some gonads (Figure 9B; Class III). Interestingly, 100% of the rbbp-5(tm3463) animals raised at 25°C were Emo and the other phenotypes were not observed (Figure 9C and D).

The Emo phenotype in hermaphrodites can be caused by ovulation defects, which can result from either oocyte defects or defective sperm signaling defects, leading to misregulated re-entry into the cell cycle [74]. To test if the Emo phenotype in wdr-5.1 and rbbp-5 mutants was due to defective sperm in these animals, sterile wdr-5.1 and rbbp-5 animals grown at elevated temperature were mated with wild type males. We found that fertility could be at least partially rescued by WT sperm, suggesting that sperm defects contribute to the Emo phenotype in these animals (not shown).
Discussion
Our data provide evidence that conserved Set1/MLL components regulate global H3K4 methylation in early embryos. We also show evidence that only a subset of the canonical Set1/MLL components modulates H3K4 methylation in the post-embryonic germline stem cells of C. elegans. Assuming that, as in mammals and yeast, these components function in complexes, then there may be multiple complexes that share WDR-5.1 and RBBP-5, with SET-2 providing H3K4me3-specific HMT activity in one or a subset of these. Interestingly, the Set1/MLL-dependent activities do not seem to be completely dependent on ongoing transcription, in contrast to the related COMPASS complex in yeast. Instead a separate, and as yet unidentified mechanism appears to provide transcription-coupled H3K4 methylation in C. elegans. The loss of Set1/MLL activities that require WDR-5 and RBBP-5 is detrimental to normal development, function, and generational maintenance of germ cells in C. elegans, illustrating the importance of this core complex in maintaining and regulating the germline cycle as it transits across generations. Furthermore, individual mutants in the different complex homologs exhibit different spectra of somatic and germline phenotypes, indicating that these proteins may also function in unique contexts in different tissues.
H3K4 methylation is regulated by different complexes in different tissues and developmental stages
Set1 is the only identified H3K4 HMT in S. cerevisiae and is solely responsible for H3K4 methylation in this organism. The yeast Set1-associated complex (COMPASS) is composed of seven subunits: Swd1-3, Swd2, Swd1, Bre1, Sdc1, Spp1 and Shg1 [12]–[15]. The Swd1 and Swd3 subunits are considered to be essential for complex stability and hence HMT activity. Sdc1 and Bre2 are required to stimulate transition from di - to trimethylation of H3K4, while Spp1 plays a role in H3K4 trimethylation efficiency [5], [25], [26].
The organization of the Set1/MLL complexes in metazoans seems to be different from that of COMPASS. There are multiple HMTs with non-redundant functions, and the different HMTs function in the context of slightly different complexes [10], [17], [21], [75]. Most, if not all, appear to share a common platform composed of WDR5, RbBP5 and ASH2L [10]. This metazoan “core platform” is shared among different Set1/MLL complexes that differentially regulate H3K4 methylation in different tissues or developmental stages [10], [75], [76].
All of the COMPASS and Set1/MLL components except Shg1 have orthologs in worms. We demonstrated that WDR-5.1, RBBP-5, ASH-2, CFP-1, and DPY-30 are all non-redundantly required for normal, global H3K4me2/3 maintenance in early embryos, strongly indicating that a complex that includes all of these factors is involved, although any individual complex could interact with either SET-2 and/or a different HMT (Figure 1). Interestingly, in the post-embryonic germ line only WDR-5.1/Swd3 and RBBP-5/Swd1 are essential for the normal maintenance of H3K4me2/3 in germline stem cells, while the other components appear to be dispensable.
Strikingly, we observed no significant requirement for ASH-2 for H3K4 methylation in adult germline stem cells, although ASH-2 is required in embryos and is the homolog of Ash2L, an essential member of the core complex in mammalian cells. This is inconsistent with a recent report that concluded that ASH-2 affects lifespan through regulation of H3K4 methylation in germ cells; although a germ cell-specific defect in H3K4me after ash-2(RNAi) was not directly shown in that study [77]. Our results suggest that H3K4 methylation activity in the GSCs, if indeed it is operating within a complex, involves a complex with a unique, and potentially novel, subunit composition. Importantly, whereas H3K4me3 levels are nearly undetectable in set-2, wdr-5.1, and rbbp-5 mutant GSCs, H3K4me2 loss was not as complete in the wdr-5.1 and rbbp-5 mutants, and unaffected in set-2 mutants in early embryos and the adult GSCs. It is possible this represents a partial redundancy for a separate WDR5 isoform (e.g., WDR-5.2 and WDR-5.3), a more complex dynamics of the two levels of H3K4 methylation, or a role for another mechanism that is not dependent on WDR5 or RBBP-5.
Interestingly, in both embryos and post-embryonic germ cells, SET-2 is only required for H3K4me3 but not H3K4me2, while two core complex proteins, WDR-5.1 and RBBP-5, affect both modifications. This finding indicates that H3K4me2 and H3K4me3 may require distinct HMTs that rely on the same core complex containing at least WDR-5.1 and RBBP-5. Although we have not done an exhaustive combinatorial deletion/RNAi analysis of all SET protein candidates, it is interesting to note the reports showing that the core MLL complex in other species can exhibit H3K4 HMT activity in the absence of the MLL subunit [38]. H3K4 dimethylation in early embryo and GSC chromatin could involve a complex that includes WDR-5.1 and RBBP-5, but may not include a SET domain protein.
Notably, WDR-5.1/RBBP-5-independent, and presumably transcription-coupled, H3K4 HMT activities are predominant in cells of mid-to-late stage embryos and meiotic germ cells, and at very low levels in early embryos and the GSCs. To attempt to identify the HMT involved, we also tested numerous SET domain candidates by RNAi in the wdr-5.1(ok1417) mutant background, but did not observe any significant changes in H3K4 methylation (data not shown). A recent report concluded that SET-16 is an H3K4-specific HMT required for efficient attenuation of Ras signals in some somatic lineages, and reported a decrease in total H3K4me using Western blot analyses [27]. We did not observe significant H3K4me2/3 decreases by immunofluorescence in either early embryos or adult germ cells in set-16(gk438) mutants or set-16(RNAi).
Notably, the transcription-dependent process and H3K4me1 levels in chromatin are both largely independent of Set1/MLL components. This could indicate that this activity is only capable of single methyl group transfers, and that the transcription-dependent H3K4me2 and H3K4me3 levels that we observe result from reiterative RNA Pol II initiation events (and subsequent additional methylation) at active loci. It will be interesting to determine what HMTs and/or complexes are responsible for WDR5/RBBP-5-independent H3K4 methylation.
The Set1/MLL activities can operate independently of transcription in C. elegans
Yeast Set1 is recruited to chromatin through the RNA polymerase II elongation machinery. As a result, yeast Set1 HMT activity is dependent upon transcription activation, and H3K4 methylation is thus associated with transcribing genes [8], [9]. In contrast, our data indicate that Set1/MLL and WDR-5.1/RBBP-5 mediated H3K4 methylation may not strictly rely on active transcription in C. elegans. First, H3K4me2/3 is maintained by Set1/MLL components in early germline precursors, cells that have been shown to lack significant levels of active RNA Pol II transcription [58]. The H3K4me we observe in the P cell chromatin (and indeed chromatin in all blastomeres) is densely distributed throughout all chromosomes (an exception being the paternal X [51]), and this distribution seems inconsistent with low levels of active transcription of a small number of genes, or confined to Pol I or Pol III transcription. Second, although zygotic transcription of some genes can be detected in early somatic blastomeres [78], the genome appears to be also largely quiescent in these cells, and bulk zygotic genome activation is not thought to occur until later embryonic stages [59]. Indeed, H3K4 methylation in the early somatic blastomeres is also unaffected by ama-1 and cdk-9 RNAi indicating that the bulk of H3K4me is also maintained in a transcription-independent process in these cells. Third, whereas ama-1 RNAi is able to deplete SET-2/WDR-5.1/RBBP-5 independent H3K4 methylation in meiotic nuclei, the H3K4 methylation in GSCs that depends on these proteins is unaffected by substantial loss of AMA-1 activity. Fourth, the detection of H3K4me in chromatin is not a reliable indicator of productive transcription in C. elegans germ line, in which disrupted correlations between RNA Pol II activity, or signs thereof, and H3K4me regulation are apparent at multiple developmental stages. For example, in the embryonic germline there is a transient appearance of phosphoSer2-modified RNA Pol II in the PGCs, which antithetically coincides with a dramatic genome-wide erasure of H3K4me2 [53].
We cannot rule out that the Set1/MLL components may also be capable of participating in transcription-coupled H3K4 methylation; indeed, there are reduced H3K4me2 and H3K4me3 levels observed in meiotic germ cell chromatin in the wdr-5.1 and rbbp-5 mutants. This may indicate either an additional role for these proteins in transcription-dependent accumulation of these marks, or a requirement for WDR-5.1/RBBP-5 dependent marks to achieve normal levels of H3K4 methylation during transcription (or normal transcription itself). Regardless, our evidence suggests these processes can operate independently in germ cells. A summary of the disconnected dynamics between RNA Pol II CTD phosphorylation and H3K4me3 in wild type and wdr-5.1/rbbp-5 mutants through the germline cycle, summarizing from this study and published work, is illustrated in Figure 10.

In the L1 larva, there is an initial coincidence of active Pol II transcription activation and the WDR-5.1/RBBP-5/SET-2 independent mode of methylation in the expanding population of germ cells. Interestingly, once the GSC population becomes established from the founder PGCs, the mode of H3K4 methylation switches to what appears to be a largely transcription-independent or maintenance mode in the GSCs. A hypothetical reason for this switching of modes could be that the initial reactivation of transcription, and its accompanying transcription-dependent H3K4 methylation, could be used to re-establish, or reinforce, a germ cell-specific “epigenome” that is then maintained in the GSCs by the WDR-5.1/RBBP-5 dependent mechanisms. This could be guided, in part, by other epigenetic marks, such as H3K27 and H3K36 methylation imposed by the C. elegans germ-cell enriched, PRC2-related MES-2/3/6 complex and the metazoan-specific H3K36 HMT MES-4, respectively [79]. We have also found that H3K4me2 incorporated during adult germ cell development appears to contribute to the distribution of this mark in both gametes and subsequently in early embryos (J. Arico and W. Kelly, manuscript submitted). The overall pattern inherited by the embryo from the gametes may be maintained in a Set1/MLL component-dependent manner in early cell divisions (this study), thus completing the germline epigenome cycle.
The loss of WDR-5.1 or RBBP-5 leads to defects in germ cells that include a defect in sperm development, which contributes to the Emo phenotype. Mutations in these genes also lead to progressive germ cell defects in later generations (e.g., the observed temperature sensitive mortal germline defect). This indicates that successive passage of the genome through the compromised epigenetic environment of these mutants leads to progressive defects from successive failure, during multiple rounds of the germ cell cycle, to properly maintain the epigenome. The temperature dependence of these phenotypes is not unusual for germline processes; indeed, we have previously described a null mutation in the gene emb-4 that yields a temperature-sensitive maternal effect embryonic lethality and that also shows defective H3K4 methylation dynamics in the PGCs [80]. Importantly, we cannot rule out the functions for wdr-5.1 and rbbp-5 that might be independent of their role in the maintenance of H3K4 methylation, since the H3K4me defects in these mutants were present at both 20°C and 25°C while the phenotypes are far more severe at 25°C. This is an important question that we are currently addressing.
A recent report showed that mutations in wdr-5.1, ash-2, or set-2 result in an increase in lifespan in C. elegans [77]. In that study it was shown that the enhanced lifespan increase was not observed in animals that lacked GLP-1 function, a Notch receptor required to maintain the post-embryonic proliferating germline stem cell population. It was concluded that the germline function of the Set1/MLL complex contributes to its role in lifespan. Our results could imply that it is the GSC-specific role for this complex that may play a more direct role in lifespan. However, we did not observe a role for ASH-2 in either stem cell maintenance or H3K4 methylation in our studies, whereas ash-2 mutants were observed to have an extended lifespan. This may further indicate that these Set1/MLL components play roles that may not be tied to their function in H3K4 methylation.
It is becoming increasingly clear that epigenetic processes guide germline establishment and the trans-generational maintenance of this totipotent lineage in many, if not all, metazoans. Although there has been much focus on the role of erasure mechanisms during epigenetic reprogramming, there is also a requirement to establish and/or maintain epigenetic information that contributes to pluripotency. This is especially true in the germ line, the lineage that transports both the DNA and its epigenetic content across generations. Any changes or defects in establishment, maintenance, or selective erasure of the epigenetic content required during any stage of the germ cell cycle encounters the strong selective filter of fertility. Our results suggest that this filter may act through alternating cycles of transcription-dependent establishment and transcription-independent maintenance of histone methylation. We propose that these mechanisms help provide and maintain a germline-specific epigenome that contributes to the underlying basis of the totipotency of this lineage.
Materials and Methods
Worm strains
C. elegans strains were maintained using standard conditions at 20°C unless otherwise noted. N2 (Bristol) was used as the wild-type C. elegans strain. The following mutant strains were used in this study: set-2(tm1630)III, wdr-5.1(ok1417)III, wdr-5.2(ok1444)X, rbbp-5(tm3463)II, set-16(gk438)III and eri-1(mg366)IV. The wdr-5.1::GFP transgenic strain was a generous gift from Dr. F. Palladino, Ecole Normale Superieure de lyon, France.
RNAi analysis
Double stranded RNA (dsRNA) corresponding to set-2, wdr-5.1, rbbp-5, ash-2, dpy-30, cfp-1, wdr-82, or ama-1 were generated using the Ribomax Large Scale RNA production kit (Promega). L4 staged eri-1(mg366) or WT animals were soaked in 1ug/ul of dsRNA for 24 hours at 20°C. The worms were then transferred to feeding plates containing bacteria expressing dsRNA targeting the corresponding gene, or carrying the empty L4440 vector for control experiments. After the first 24 hours, the worms (P0) were transferred to a new set of feeding plates. The worms (P0) were dissected after 24-48 hours and stained as described below. In some experiments F1 animals and their F2 progeny were also analyzed in immunofluorescence experiments, and/or assessed for embryonic lethality and sterility. For ama-1, wild-type or mutant L4 larvae were picked and soaked in either 0.5ug/ul dsRNA in 1x soaking buffer, or buffer alone for 24 hours at 20°C. The worms were then transferred to feeding plates with bacteria expressing the same dsRNA for an additional 24-30 hrs. Worms were dissected, fixed and prepared for immunofluorescence analyses 50 hours post soaking.
Immunofluorescence
Worms were dissected and fixed in paraformaldehyde/methanol [68] for H3K4me2/3, AMA-1, GFP, 8WG16 and SYP-1 staining. A methanol/acetone fixation procedure [81] was used for L1-L4 larvae staining. Samples were fixed in methanol/formaldehyde for H5 staining as described [68]. Rabbit anti-H3K4me3 (1 : 1000) and anti-H3K4me1 polyclonal antibodies were purchased from Abcam (ab8580 and ab8895, respectively). Mouse monoclonal antibodies against H3K4me3 (CMA 304; 1 : 1000) and H3K4me2 (CMA303; 1 : 20) were gifts from Dr. Hiroshi Kimura (Osaka University, Japan [66]). Mouse monoclonal antibody against GFP (MAB3580 1 : 500), Rabbit polyclonal (07-030 1 : 1000) and monoclonal (05790 1 : 1000) antibodies against H3K4me2 were purchased from Millipore. Monoclonal antibody OIC1D4 (1 : 5) and rabbit anti-PGL (1 : 10000) were gifts from Dr. S. Strome, UC Santa Cruz. The RNA polymerase II monoclonal antibodies used to detect AMA-1 protein (clone 8WG16 1 : 100) and phosphoserine 2 isotope of RNA Pol II (clone H5 1 : 50) were purchased from Covance (MMS-126R and MMS-129R, respectively). Rabbit anti-SYP-1 (1 : 100) was a gift of Dr. A. Villeneuve, Stanford University. All secondary antibodies were purchased from Molecular Probes and were used at 1 : 500 dilutions: goat anti-mouse IgG (Alexafluor 488); goat anti-rabbit IgG (Alexafluor 594), donkey anti-rabbit IgG (Alexafluor 488); donkey anti-mouse IgG (Alexafluor 594). DAPI (Sigma, 2 ug/ul) was used to counter-stain DNA. Worms were mounted in anti-fade reagent (Prolong Gold, Molecular Probes). Images were collected using a Leica DMRXA fluorescence microscope and analyzed with Simple PCI software (Hamamatsu Photonics). To quantify the immunofluorescence signals, the mean fluorescence intensity was measured for middle focal plane images of each nucleus from Z-stack images collected for each sample and normalized to the fluorescence signal obtained for DAPI. For all compared samples, the exposure time for each probe was set below image saturation for the brightest nucleus among the samples being compared.
Western blot analysis
Worms were washed from OP50 plates and bleached to collect embryos. To remove egg shells, the embryos were subsequently treated with 1 U/ml chitinase (Sigma, C6137) in egg buffer (25 mM HEPES pH 7.4, 118 mM NaCl, 48 mM KCl, 2 mM MgSO4, 2 mM CaCl2) at 20°C for 40 min, and were boiled in 1X SDS-PAGE sample buffer. The samples were loaded and run on a 15% SDS-PAGE gel and transferred to PVDF membrane. The proteins were probed with the following antibodies: rabbit polyclonal anti-H3 at 1∶10,000 (Abcam ab1791), rabbit polyclonal anti-H3K4me3 at 1∶3,000 (ab8580), mouse monoclonal anti-H3K4me2 at 1∶100 (gift from Dr. Hiroshi Kimura, Osaka University, Japan), rabbit polyclonal anti-H3K4me1 at 1∶1000 (ab8895).
Peptide pulldown assays
Biotinylated peptides corresponding to the N-terminus of histone H3 were used for peptide affinity analyses of WDR-5.1 in worm nuclear extracts. Biotinylated H3 (unmodified) and H3 (dimethyl-Lys4) peptides were purchased from Millipore (Cat#12-403 and 12-460, respectively). Biotin conjugated H3 peptides asymmetrically dimethylated at Arg2 (H3R2me2a) were synthesized by the Keck Biotechnology Resource at Yale University. For the binding assay, mixed-stage WT worms were washed off plates with PBS, washed 3X in PBS with protease inhibitor cocktail (Roche), and the worms were frozen at -80°C. To isolate the nuclei, the worm pellet was thawed and ground by mortar and pestle in 2x volume of Nuclear Isolation Buffer (NIB; 25 mM HEPES (pH 7.5), 25 mM KCl, 0.1 mM EDTA, 0.1 mM DTT, 10 mM MgCl2 and 0.5 M sucrose) in liquid nitrogen. The samples were then transferred to a homogenizer in NIB buffer and stroked 20 times on ice. Nuclei were collected by centrifugation. Nuclear proteins were extracted in Nuclear Extract Buffer (NEB 20 mM HEPES (pH 7.5), 2 mM EDTA, 25% glycerol and 0.1% NP-40) with 350 mM KCl. Prior to pulldown, the nuclear extracts were diluted with NEB buffer without KCl to obtain a solution with 150 mM KCl. The peptide pull down assay was performed according to Wysocka, et al [82]. Briefly, the extracts were incubated with peptides pre-bound to avidin beads overnight at 4°C. After washing with HEPES buffer containing 150 mM KCl for 3 times, the beads were then boiled in 1X SDS buffer and subjected to western blot analysis for WDR-5.1 detection. The WDR-5.1 rabbit antiserum was generated via immunization with a peptide comprising the last 15 C-terminal amino acids of WDR-5.1 (Pickcell Laboratories, The Netherlands). Anti-WDR-5.1 antibodies were affinity purified on Affi-Gel 15 columns (Bio-Rad Laboratories).
Temperature-sensitive (ts) phenotypic analysis
WT and mutant L4 worms (P0) were picked and shifted to 25°C. F1 embryos were counted to score embryonic lethality. To score sterility, 100 L1 larvae of animals grown at 25°C were picked onto separate plates pre-warmed to 25°C. Sterile adult offspring were counted 24 hours post L4 based on an absence of embryos in the uterus. The sterile animals were dissected to extrude gonads, and the whole-mount fixed samples were stained with anti-SYP-1 antibody and counter stained with DAPI. The images were collected on a Leica DMRA microscope. The length of mitotic regions (MR) were measured by counting the number of aligned nuclei from the distal tip of the gonads to the either the appearance of the characteristic crescent-shaped transition zone nuclei and/or the appearance of anti-SYP-1 immunofluorescence. The number of nuclei on one focus plane of DAPI images was used to reflect the length of mitotic zone.
Brood size and mortal germline assay
10 L4 worms were picked and incubate at 20°C or 25°C. F1 embryos were counted for brood size. To score sterility, 100 L1 larvae of F1 from 20°C or 25°C plates were picked onto a new set of plates and kept at the corresponding temperatures. Sterile adult F1 animals were identified and counted based on the lack of embryos in utero.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LiB
CareyM
WorkmanJL
2007 The role of chromatin during transcription. Cell 128 707 719
2. FischleW
WangY
AllisCD
2003 Histone and chromatin cross-talk. Curr Opin Cell Biol 15 172 183
3. SimsRJ3rd
ReinbergD
2006 Histone H3 Lys 4 methylation: caught in a bind? Genes Dev 20 2779 2786
4. DillonSC
ZhangX
TrievelRC
ChengX
2005 The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol 6 227
5. NgHH
RobertF
YoungRA
StruhlK
2003 Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11 709 719
6. KroganNJ
DoverJ
WoodA
SchneiderJ
HeidtJ
2003 The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 11 721 729
7. PokholokDK
HarbisonCT
LevineS
ColeM
HannettNM
2005 Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122 517 527
8. Santos-RosaH
SchneiderR
BannisterAJ
SherriffJ
BernsteinBE
2002 Active genes are tri-methylated at K4 of histone H3. Nature 419 407 411
9. BriggsSD
BrykM
StrahlBD
CheungWL
DavieJK
2001 Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev 15 3286 3295
10. DouY
MilneTA
RuthenburgAJ
LeeS
LeeJW
2006 Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 13 713 719
11. KroganNJ
DoverJ
KhorramiS
GreenblattJF
SchneiderJ
2002 COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277 10753 10755
12. ShilatifardA
2006 Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem 75 243 269
13. NagyPL
GriesenbeckJ
KornbergRD
ClearyML
2002 A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci U S A 99 90 94
14. RoguevA
SchaftD
ShevchenkoA
AaslandR
ShevchenkoA
2003 High conservation of the Set1/Rad6 axis of histone 3 lysine 4 methylation in budding and fission yeasts. J Biol Chem 278 8487 8493
15. DehePM
GeliV
2006 The multiple faces of Set1. Biochem Cell Biol 84 536 548
16. MillerT
KroganNJ
DoverJ
Erdjument-BromageH
TempstP
2001 COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci U S A 98 12902 12907
17. DouY
MilneTA
TackettAJ
SmithER
FukudaA
2005 Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121 873 885
18. WysockaJ
SwigutT
MilneTA
DouY
ZhangX
2005 WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121 859 872
19. MilneTA
DouY
MartinME
BrockHW
RoederRG
2005 MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci U S A 102 14765 14770
20. WysockaJ
MyersMP
LahertyCD
EisenmanRN
HerrW
2003 Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev 17 896 911
21. HughesCM
Rozenblatt-RosenO
MilneTA
CopelandTD
LevineSS
2004 Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 13 587 597
22. YokoyamaA
WangZ
WysockaJ
SanyalM
AufieroDJ
2004 Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 24 5639 5649
23. GlaserS
SchaftJ
LubitzS
VinterstenK
van der HoevenF
2006 Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development 133 1423 1432
24. LeeJH
SkalnikDG
2005 CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem 280 41725 41731
25. SchneiderJ
WoodA
LeeJS
SchusterR
DuekerJ
2005 Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol Cell 19 849 856
26. StewardMM
LeeJS
O'DonovanA
WyattM
BernsteinBE
2006 Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol 13 852 854
27. FisherK
SouthallSM
WilsonJR
PoulinGB
2010 Methylation and demethylation activities of a C. elegans MLL-like complex attenuate RAS signalling. Dev Biol 341 142 153
28. SimonetT
DulermoR
SchottS
PalladinoF
2007 Antagonistic functions of SET-2/SET1 and HPL/HP1 proteins in C. elegans development. Dev Biol
29. RuthenburgAJ
WangW
GrayboschDM
LiH
AllisCD
2006 Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat Struct Mol Biol 13 704 712
30. SchuetzA
Allali-HassaniA
MartinF
LoppnauP
VedadiM
2006 Structural basis for molecular recognition and presentation of histone H3 by WDR5. Embo J 25 4245 4252
31. CoutureJF
CollazoE
TrievelRC
2006 Molecular recognition of histone H3 by the WD40 protein WDR5. Nat Struct Mol Biol 13 698 703
32. HanZ
GuoL
WangH
ShenY
DengXW
2006 Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Mol Cell 22 137 144
33. KirmizisA
Santos-RosaH
PenkettCJ
SingerMA
VermeulenM
2007 Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature 449 928 932
34. GuccioneE
BassiC
CasadioF
MartinatoF
CesaroniM
2007 Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 449 933 937
35. HyllusD
SteinC
SchnabelK
SchiltzE
ImhofA
2007 PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev 21 3369 3380
36. CosgroveMS
PatelA
2010 Mixed lineage leukemia: a structure-function perspective of the MLL1 protein. Febs J 277 1832 1842
37. PatelA
DharmarajanV
CosgroveMS
2008 Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J Biol Chem 283 32158 32161
38. PatelA
DharmarajanV
VoughtVE
CosgroveMS
2009 On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem 284 24242 24256
39. PatelA
VoughtVE
DharmarajanV
CosgroveMS
2008 A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J Biol Chem 283 32162 32175
40. SongJJ
KingstonRE
2008 WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J Biol Chem 283 35258 35264
41. ChoYW
HongT
HongS
GuoH
YuH
2007 PTIP associates with MLL3 - and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282 20395 20406
42. MuramotoT
MullerI
ThomasG
MelvinA
ChubbJR
2010 Methylation of H3K4 Is required for inheritance of active transcriptional states. Curr Biol 20 397 406
43. RingroseL
ParoR
2004 Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet 38 413 443
44. PetrukS
SedkovY
SmithS
TillibS
KraevskiV
2001 Trithorax and dCBP acting in a complex to maintain expression of a homeotic gene. Science 294 1331 1334
45. KatzDJ
EdwardsTM
ReinkeV
KellyWG
2009 A C. elegans LSD1 demethylase contributes to germline immortality by reprogramming epigenetic memory. Cell 137 308 320
46. RechtsteinerA
ErcanS
TakasakiT
PhippenTM
EgelhoferTA
2010 The histone H3K36 methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny. PLoS Genet 6 e1001091 doi:10.1371/journal.pgen.1001091
47. HammoudSS
NixDA
ZhangH
PurwarJ
CarrellDT
2009 Distinctive chromatin in human sperm packages genes for embryo development. Nature 460 473 478
48. BernsteinBE
MikkelsenTS
XieX
KamalM
HuebertDJ
2006 A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125 315 326
49. BrykczynskaU
HisanoM
ErkekS
RamosL
OakeleyEJ
2010 Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nat Struct Mol Biol 17 679 687
50. AzuaraV
PerryP
SauerS
SpivakovM
JorgensenHF
2006 Chromatin signatures of pluripotent cell lines. Nat Cell Biol 8 532 538
51. BeanCJ
SchanerCE
KellyWG
2004 Meiotic pairing and imprinted X chromatin assembly in Caenorhabditis elegans. Nat Genet 36 100 105
52. SekiY
YamajiM
YabutaY
SanoM
ShigetaM
2007 Cellular dynamics associated with the genome-wide epigenetic reprogramming in migrating primordial germ cells in mice. Development 134 2627 2638
53. HajkovaP
AncelinK
WaldmannT
LacosteN
LangeUC
2008 Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452 877 881
54. SchanerC
DeshpandeG
SchedlP
KellyW
2003 A conserved chromatin architecture marks and maintains the restricted germ cell lineage in worms and flies. Dev Cell 5 747 757
55. VastenhouwNL
ZhangY
WoodsIG
ImamF
RegevA
2010 Chromatin signature of embryonic pluripotency is established during genome activation. Nature 464 922 926
56. ThomsonJP
SkenePJ
SelfridgeJ
ClouaireT
GuyJ
CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 464 1082 1086
57. EdgarLG
WolfN
WoodWB
1994 Early transcription in Caenorhabditis elegans embryos. Development 120 443 451
58. SeydouxG
DunnMA
1997 Transcriptionally repressed germ cells lack a subpopulation of phosphorylated RNA polymerase II in early embryos of Caenorhabditis elegans and Drosophila melanogaster. Development 124 2191 2201
59. BaughLR
HillAA
SlonimDK
BrownEL
HunterCP
2003 Composition and dynamics of the Caenorhabditis elegans early embryonic transcriptome. Development 130 889 900
60. BernsteinBE
KamalM
Lindblad-TohK
BekiranovS
BaileyDK
2005 Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120 169 181
61. SchneiderR
BannisterAJ
MyersFA
ThorneAW
Crane-RobinsonC
2004 Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol 6 73 77
62. MartinC
ZhangY
2007 Mechanisms of epigenetic inheritance. Curr Opin Cell Biol 19 266 272
63. AllegrucciC
ThurstonA
LucasE
YoungL
2005 Epigenetics and the germline. Reproduction 129 137 149
64. SchanerC
KellyW
2006 Germline Chromatin. Wormbook, ed. The Celegnas Research Community 1 14
65. StarckJ
BrunJ
1977 Autoradiographic localization of RNA synthesis in vitro during oogenesis in Parascaris equorum. C R Acad Sci Hebd Seances Acad Sci D 284 1341 1344
66. KimuraH
Hayashi-TakanakaY
GotoY
TakizawaN
NozakiN
2008 The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct 33 61 73
67. KellyWG
SchanerCE
DernburgAF
LeeMH
KimSK
2002 X-chromosome silencing in the germline of C. elegans. Development 129 479 492
68. FuruhashiH
TakasakiT
RechtsteinerA
LiT
KimuraH
2010 Trans-generational epigenetic regulation of C. elegans primordial germ cells. Epigenetics Chromatin 3 1 21
69. CrittendenSL
BernsteinDS
BachorikJL
ThompsonBE
GallegosM
2002 A conserved RNA-binding protein controls germline stem cells in Caenorhabditis elegans. Nature 417 660 663
70. LamontLB
CrittendenSL
BernsteinD
WickensM
KimbleJ
2004 FBF-1 and FBF-2 regulate the size of the mitotic region in the C. elegans germline. Dev Cell 7 697 707
71. AustinJ
KimbleJ
1987 glp-1 is required in the germ line for regulation of the decision between mitosis and meiosis in C. elegans. Cell 51 589 599
72. MacQueenAJ
ColaiacovoMP
McDonaldK
VilleneuveAM
2002 Synapsis-dependent and -independent mechanisms stabilize homolog pairing during meiotic prophase in C. elegans. Genes Dev 16 2428 2442
73. ByrdKN
ShearnA
2003 ASH1, a Drosophila trithorax group protein, is required for methylation of lysine 4 residues on histone H3. Proc Natl Acad Sci U S A 100 11535 11540
74. HarrisJE
GovindanJA
YamamotoI
SchwartzJ
KaverinaI
2006 Major sperm protein signaling promotes oocyte microtubule reorganization prior to fertilization in Caenorhabditis elegans. Dev Biol 299 105 121
75. CrawfordBD
HessJL
2006 MLL core components give the green light to histone methylation. ACS Chem Biol 1 495 498
76. RuthenburgAJ
AllisCD
WysockaJ
2007 Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 25 15 30
77. GreerEL
MauresTJ
HauswirthAG
GreenEM
LeemanDS
2010 Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466 383 387
78. SeydouxG
FireA
1994 Soma-germline asymmetry in the distributions of embryonic RNAs in Caenorhabditis elegans. Development 120 2823 2834
79. BenderLB
SuhJ
CarrollCR
FongY
FingermanIM
2006 MES-4: an autosome-associated histone methyltransferase that participates in silencing the X chromosomes in the C. elegans germ line. Development 133 3907 3917
80. ChecchiPM
KellyWG
2006 emb-4 is a conserved gene required for efficient germline-specific chromatin remodeling during Caenorhabditis elegans embryogenesis. Genetics 174 1895 1906
81. StromeS
WoodWB
1983 Generation of asymmetry and segregation of germ-line granules in early C. elegans embryos. Cell 35 15 25
82. WysockaJ
2006 Identifying novel proteins recognizing histone modifications using peptide pull-down assay. Methods 40 339 343
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy