
Roles of () in Oocyte Nuclear Architecture, Gametogenesis, Gonad Tumors, and Genome Stability in Zebrafish
Mild mutations in BRCA2 (FANCD1) cause Fanconi anemia (FA) when homozygous, while severe mutations cause common cancers including breast, ovarian, and prostate cancers when heterozygous. Here we report a zebrafish brca2 insertional mutant that shares phenotypes with human patients and identifies a novel brca2 function in oogenesis. Experiments showed that mutant embryos and mutant cells in culture experienced genome instability, as do cells in FA patients. In wild-type zebrafish, meiotic cells expressed brca2; and, unexpectedly, transcripts in oocytes localized asymmetrically to the animal pole. In juvenile brca2 mutants, oocytes failed to progress through meiosis, leading to female-to-male sex reversal. Adult mutants became sterile males due to the meiotic arrest of spermatocytes, which then died by apoptosis, followed by neoplastic proliferation of gonad somatic cells that was similar to neoplasia observed in ageing dead end (dnd)-knockdown males, which lack germ cells. The construction of animals doubly mutant for brca2 and the apoptotic gene tp53 (p53) rescued brca2-dependent sex reversal. Double mutants developed oocytes and became sterile females that produced only aberrant embryos and showed elevated risk for invasive ovarian tumors. Oocytes in double-mutant females showed normal localization of brca2 and pou5f1 transcripts to the animal pole and vasa transcripts to the vegetal pole, but had a polarized rather than symmetrical nucleus with the distribution of nucleoli and chromosomes to opposite nuclear poles; this result revealed a novel role for Brca2 in establishing or maintaining oocyte nuclear architecture. Mutating tp53 did not rescue the infertility phenotype in brca2 mutant males, suggesting that brca2 plays an essential role in zebrafish spermatogenesis. Overall, this work verified zebrafish as a model for the role of Brca2 in human disease and uncovered a novel function of Brca2 in vertebrate oocyte nuclear architecture.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1001357
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001357
Summary
Mild mutations in BRCA2 (FANCD1) cause Fanconi anemia (FA) when homozygous, while severe mutations cause common cancers including breast, ovarian, and prostate cancers when heterozygous. Here we report a zebrafish brca2 insertional mutant that shares phenotypes with human patients and identifies a novel brca2 function in oogenesis. Experiments showed that mutant embryos and mutant cells in culture experienced genome instability, as do cells in FA patients. In wild-type zebrafish, meiotic cells expressed brca2; and, unexpectedly, transcripts in oocytes localized asymmetrically to the animal pole. In juvenile brca2 mutants, oocytes failed to progress through meiosis, leading to female-to-male sex reversal. Adult mutants became sterile males due to the meiotic arrest of spermatocytes, which then died by apoptosis, followed by neoplastic proliferation of gonad somatic cells that was similar to neoplasia observed in ageing dead end (dnd)-knockdown males, which lack germ cells. The construction of animals doubly mutant for brca2 and the apoptotic gene tp53 (p53) rescued brca2-dependent sex reversal. Double mutants developed oocytes and became sterile females that produced only aberrant embryos and showed elevated risk for invasive ovarian tumors. Oocytes in double-mutant females showed normal localization of brca2 and pou5f1 transcripts to the animal pole and vasa transcripts to the vegetal pole, but had a polarized rather than symmetrical nucleus with the distribution of nucleoli and chromosomes to opposite nuclear poles; this result revealed a novel role for Brca2 in establishing or maintaining oocyte nuclear architecture. Mutating tp53 did not rescue the infertility phenotype in brca2 mutant males, suggesting that brca2 plays an essential role in zebrafish spermatogenesis. Overall, this work verified zebrafish as a model for the role of Brca2 in human disease and uncovered a novel function of Brca2 in vertebrate oocyte nuclear architecture.
Introduction
People who are heterozygous for strong mutations in the tumor suppressor gene BRCA2(FANCD1) have increased susceptibility to breast, ovarian, prostate, and pancreatic cancers [1]-[3]. Breast cancer risk for females heterozygous for germline mutations in BRCA2 is nearly 60% by age 50 [4] and for ovarian cancer is 11% [5]. BRCA2 is expressed in a broad range of mammalian tissues [6], [7] and null activity alleles are embryonic lethal in mouse and humans but are viable in rats [8]-[11]. Biallelic inheritance of hypomorphic BRCA2 mutations in the germline results in Fanconi anemia (FA), a disease characterized by catastrophic anemia, genome instability, characteristic morphological defects, and enormously elevated risk for leukemia (800 fold) and squamous cell carcinomas (2000 fold) [12]-[14]. The BRCA2 subtype of Fanconi anemia represents complementation group D1 [15] and results in a severe form of the disease with nearly 100% incidence of leukemia and/or solid tumors by 5 years of age [16], [17]. The role of BRCA2 in tumor suppression and maintenance of genomic integrity is associated with its function in error-free, homology-directed recombination (HDR) [18]. HDR helps repair DNA breaks associated with meiosis, and mouse mutants in FA genes have defects in meiotic cells [19]. Zebrafish fancl mutants experience female-to-male sex reversal due to the apoptotic loss of meiotic oocytes at the time of sex determination [20], consistent with the abnormal activation of the apoptotic pathway in the absence of Fanconi gene activity [21].
The involvement of BRCA2 in HDR, ovarian cancer, hypogonadal phenotypes, and the expression of Brca2 in mouse spermatocytes [22] converge to suggest a role for Brca2 in gonadogenesis. Homozygous Brca2 knockout mice die as embryos [23], but transgenic mice carrying a BRCA2-containing human BAC that expresses the human gene at high levels everywhere except the gonads survive as sterile males and females [24]. In contrast, rats bearing a premature stop codon survive, but show slow growth and sterility [10], reflecting conserved and lineage-specific roles of brca2.
To help understand the roles of Brca2 in vertebrates, we characterized zebrafish bearing an insertional mutation in brca2. We show here that comparative analysis of zebrafish brca2 [25] identifies a few conserved, and hence putatively functional, coding regions and is expressed in proliferating somatic cells and in meiotic oocytes and spermatocytes. Surprisingly, brca2 transcript is asymmetrically localized to the animal pole of the cytoplasm in developing wild-type oocytes. The insertional brca2 null activity allele causes genome instability, slow growth of tissue culture cells, male sterility, testicular neoplasias, and female-to-male sex reversal that is rescued by mutation of the tumor suppressor gene tp53(p53). Male and female double mutants are sterile and develop testicular neoplasias and invasive ovarian tumors. Nuclear symmetries are strikingly altered in oocytes of double mutant females, revealing a novel role of Brca2 in establishing or maintaining the architecture of the vertebrate oocyte nucleus. This work reveals that this zebrafish brca2 mutant is a model for unraveling gene functions as well as a valuable tool for small-molecule screens to help discover therapeutic compounds for human patients.
Results
Zebrafish brca2 Shares with Human BRCA2 Features of the Genome, Gene, and Protein
We isolated, cloned, and sequenced a zebrafish brca2 cDNA (NM_001110394) and a BAC clone (AC149226). Because the zebrafish Brca2 protein shares only 21% identity with human BRCA2, we confirmed orthology by conserved syntenies [26]. Our meiotic mapping on the HS panel [27] showed that brca2 lies on zebrafish chromosome 15 (Dre15, Figure 1A top), and sequence data at Ensembl (http://www.ensembl.org/Danio_rerio/Info/Index) showed that its genomic neighborhood contains 14 genes with conserved synteny to the orthologous region on human chromosome 13 (Hsa13, Figure 1A middle), as would be expected if zebrafish brca2 and human BRCA2 are orthologs. The absence of a second copy of brca2 in the co-orthologous region in Dre10 (Figure 1A bottom) provides evidence that, like all 13 other zebrafish fanc genes [25], [28], brca2 evolved to single copy in the zebrafish lineage after the teleost genome duplication [27], [29]-[31].

A comparison of our cDNA and BAC sequences revealed that zebrafish brca2 has 26 exons (numbered 2–27 to follow human nomenclature; Figure 1B) like its tetrapod ortholog [7], [32]-[34]. Despite low sequence identity (21%), zebrafish Brca2 conserves an N-terminal acidic transcriptional activation domain and a C-terminal DNA binding domain (DBD) [35]-[37] (Figure 1C). Exon-11, with 1,397 amino acid residues in zebrafish and 1,643 residues in human, is one of the longest vertebrate exons, 28 times larger than average [38]. Use of the stickleback brca2 sequence (Figure S1) to help inform alignments showed that exon-11 of zebrafish brca2 contains a central array of BRC repeats conserved in approximate number, relative position, and sequence identity to those in tetrapods [34] (Figure S2A). Phylogenetic analysis of BRC repeats revealed orthology between chicken and human repeats 1, 5, 7, and 8 (Figure 1D) but not a one-to-one orthology between zebrafish and tetrapod repeats, suggesting that some individual repeats may have evolved independently by tandem duplication and/or gene conversion. Despite differences in BRC repeat sequences, the correlation of hydrophobicity indexes among repeats revealed great structural similarity (Figure 1E). The DBDs of zebrafish and human Brca2 contain three oligonucleotide binding folds (OB1-3) and a helical domain (HD) [37] (Figure 1C and Figure S3). The mapping of human tumor-derived mutations to these conserved features [37] supports the hypothesis that they are critical for functionally similar molecular interactions across vertebrates.
brca2 Is Expressed in Meiotic Germ Cells
In zebrafish embryos, brca2 has been shown to be expressed maternally and zygotically [25], and this is confirmed here by RT-PCR (Figure S4A) and histological sections that show broad expression that is elevated in rapidly proliferating cells in the embryonic and larval central nervous system, in the proliferating ventricle margins of adult brains, in the blood-forming kidney marrow, and in the proliferative intervillus region of the intestine [39] (Figure S4B-S4N). Germ line cells expressed brca2 in both male and female gonads in transitional stages (Figure 2A, 2B), in immature gonads (Figure 2C, 2D), and in the mature gonads of adults (Figure 2E, 2F). Somatic cells of the zebrafish gonad either do not express brca2, or do so at a low level like mouse Sertoli cells [40].

Unexpectedly, in stage III and IV oocytes, brca2 transcripts became asymmetrically distributed to a small peripheral patch of the ooplasm (Figure 2E). Comparison with the distribution of pou5f1(oct4) [41], [42] revealed that brca2 mRNA transcripts accumulated asymmetrically at the animal pole of the oocyte (Figure 2G, 2H). Expression of brca2 was obvious in spermatocytes (sc, Figure 2F), but not in spermatids and sperm (sp, Figure 2F), as in mouse [40]. Expression of brca2 in meiotic cells is consistent with a role in repairing DNA breaks associated with meiotic HDR. Furthermore, the accumulation of brca2 transcript at the animal pole suggests a role of maternal message in provisioning embryos with Brca2 protein that could help effect DNA repair during the rapid cleavage divisions that occur before the initiation of zygotic transcription at the mid-blastula transition.
A Zebrafish brca2 Null Allele Shows That brca2 Is Required for Genome Stability
To understand brca2 function, we studied a zebrafish line with the insertional mutation ZM_00057434, which disrupts brca2 exon-11 in BRC repeat-z (Figure 1C, Figure 3A and 3B, Figure S2). Reverse transcriptase-PCR and sequence analysis detected no normal transcript in mutants, but instead identified two aberrant transcripts, one lacking the DBD and the other lacking all BRC repeats (Figure 3C–3G). Because Brca2 protein cannot function without either of these features [37], we conclude that ZM_00057434 is a null allele.
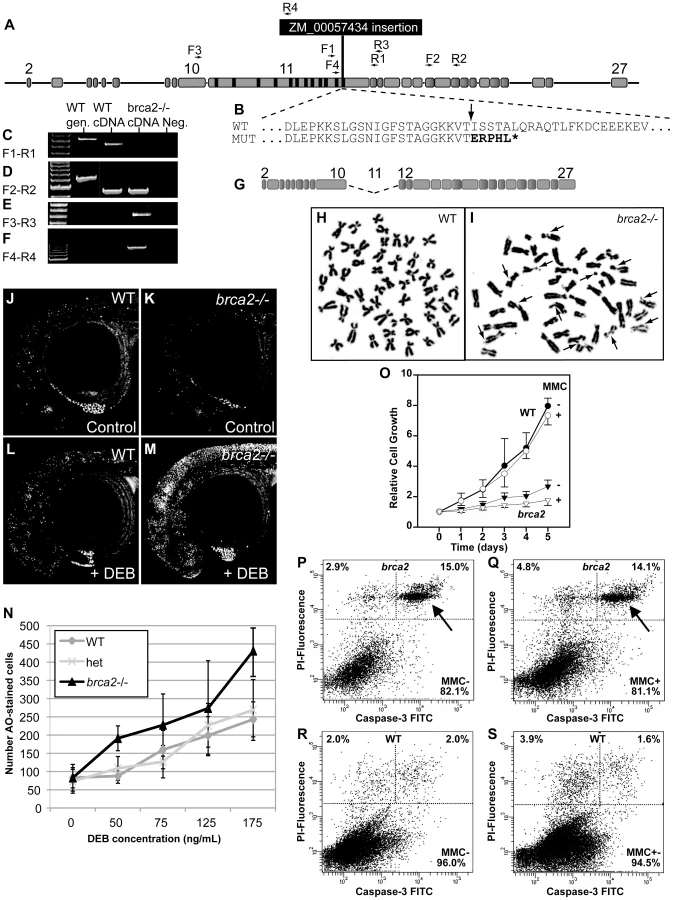
Genome instability is a cardinal characteristic of Fanconi anemia [43], [44]. Cell cultures from fins of homozygous brca2 zebrafish mutants and wild-type controls revealed normal karyotypes (2n = 50) with low levels of spontaneous breakage (Figure 3H). After treatment with 10 ng/ml of the DNA-damage agent MMC, however, mutant cells showed many chromosome aberrations, including chromatid and chromosome breaks, radial reunion figures, and acentric chromosome fragments. Of 100 metaphases counted in mutant cells, 66 showed chromosome aberrations, including 32 that showed one or two anomalies, 23 with 3 or 4 abnormal chromosomes, and 11 with more than 5 aberrations (Figure 3I). In contrast, all 24 metaphases from wild-type cells treated with MMC were normal (Figure 3H). These results show that zebrafish cells require brca2 activity to prevent chromosome aberrations.
To test genome stability in living animals, we crossed brca2 heterozygotes, stained resulting embryos at 28hpf with acridine orange (AO, which fluoresces strongly when it intercalates into DNA with double-strand breaks [45]), scored the amount of AO staining in individual embryos, and genotyped embryos by PCR. Untreated mutants and wild-type controls had about the same amount of AO staining (Figure 3J, 3K). In contrast, after treatment with the DNA damage agent diepoxybutane (DEB) at 4hpf, mutants accumulated substantially more AO-positive cells than wild-type siblings (Figure 3L-3N). Thus, we conclude that Brca2 helps protect zebrafish embryos from DNA damage.
To learn the role of brca2(fancd1) in zebrafish somatic cells, we established tissue cultures from fin biopsies of brca2 mutants and wild types and studied their growth rates. Mutant cultures showed significantly slower growth compared to wild-type cultures (Figure 3O, p<0.001 at day 5). Addition of MMC further delayed culture growth both for brca2 mutants (p<0.5 vs. untreated) and for wild types, although delay in wild types was not statistically significant (Figure 3O). The poor growth of brca2 mutant cultures was due to high rates of spontaneous apoptosis (15%, Figure 3P-3S), as evidenced by propidium iodide exclusion and anti-active Caspase-3 staining. In brca2 mutants, addition of MMC increased the non-apoptotic cell death rate from 2.9% to 4.8% while the proportion of apoptotic cells remained essentially the same (15.0 and 14.1%, respectively, Figure 3P, 3Q). Untreated wild-type cultures revealed much less spontaneous apoptosis than mutant cultures (2% vs. 15%; Figure 3R) and showed just a small effect of MMC on the non-apoptotic cell death rate (2.0% untreated, 3.9% treated, Figure 3S). We conclude that mutant cultures grow more slowly than wild-type cultures due to high rates of spontaneous apoptosis.
Lack of brca2 Activity Causes Female-to-Male Sex Reversal
To test the viability of zebrafish brca2 mutants, we mated heterozygotes, and among 414 adult offspring, 24.9% were homozygous wild types, 44.9% were heterozygotes, and 30.2% were homozygous mutants, a ratio indistinguishable from the expected 1 : 2:1 ratio (X2 test, p = 0.37, df = 2). We conclude that zebrafish brca2 mutants survive about as well as wild types, as in Drosophila [46]. In addition, zebrafish brca2(fancd1) mutants expressed genes for primitive and definitive hematopoiesis normally (Figure S5), providing no evidence for the early hematopoietic defects found in human FA patients.
Remarkably, however, all homozygous brca2 mutants developed exclusively as males. A series of heterozygote in-crosses gave 199 wild-type homozygotes and heterozygotes, about half of which were females (50.2%±0.1% (sd)). In contrast, of the 61 homozygous mutants, none were female (X2 test, p = 0.00003, df = 2). Genotypic ratios following Mendelian principles ruled out female-specific lethality; thus, we conclude that individuals that would otherwise have become females experienced female-to-male sex reversal. Homozygous brca2 mutant males were sterile (the 35 males tested fertilized no eggs, with an average clutch size of 197 eggs tested per male), but wild-type sibling males were all fertile (the 28 males tested fertilized an average of 80% of the eggs per male with an average clutch size of 210 eggs tested per male). These data show that brca2 plays a role in male fertility and is necessary for female development in otherwise wild-type fish.
Juvenile Gonads of brca2 Mutants Lack Perinucleolar Oocytes and Sperm
To investigate the developmental basis of sex reversal in brca2 mutants, we analyzed transitional and immature (but differentiated) gonads. All juvenile zebrafish, regardless their definitive sex, initially develop oocytes; in females, these oocytes continue to develop but in males, they disappear [47], [48]. In our experiments, some wild types at 21dpf contained perinucleolar oocytes (early stage IB) and other wild types contained a few pyknotic cells and oocytes at earlier stages of development (early oocytes at stage IA (leptotene to pachytene) [49]) (Figure 4A, 4B). In contrast, all eight homozygous brca2 mutants examined at 21dpf lacked perinucleolar oocytes and contained earlier stage oocytes and large numbers of pyknotic cells (Figure 4C). By 27dpf, wild types contained either ovaries or testes (Figure 4D, 4E). All eight brca2 mutants analyzed, however, showed testis-like gonads that lacked perinucleolar oocytes but retained a few early oocytes and pyknotic cells (Figure 4F). At 32dpf, perinucleolar oocytes in wild-type animals reached late stage IB and entered diplotene, as indicated by the presence of lampbrush chromosomes (Figure 4G), while testes of wild-type males showed all stages of spermatogenesis including sperm (Figure 4H). In contrast, all eight brca2 mutants analyzed at 32dpf had only testes that possessed spermatogonia (sg) and spermatocytes (sc) but lacked later developmental stages (spermatids and sperm) (Figure 4I). In addition, 32dpf mutant gonads showed abnormal clusters of cells with pyknotic nuclei (pc, outlined by dashed lines in Figure 4I) and contained tubules abnormally depleted of germ cells (asterisk, Figure 4I).

The lack of perinucleolar oocytes in brca2 mutant gonads during the critical period for sex determination is consistent with the finding that gonads lacking oocytes during this period assume a male fate [20]. In addition, results showed that brca2 mutant germ cells became pyknotic, disappeared, and left empty spermatogenic tubules.
brca2 Spermatocytes Undergo Apoptosis
The presence of pyknotic spermatocytes, lack of spermatids and sperm, and the existence of empty tubules in brca2 mutant testes suggested the hypothesis that spermatocytes did not progress through meiosis and died. To test if the activation of apoptotic pathways is involved in spermatocyte death, we used immunoassays to detect active-Caspase-3, a marker of apoptosis [50]. In contrast to wild-type gonads, brca2 mutant testes showed clusters of cells with active-Caspase-3 that were clearly pyknotic after hematoxylin and eosin staining (Figure 4J–4M), confirming that brca2 spermatocytes undergo apoptosis.
Immature brca2 Gonads Have a Male-Expression Profile
At 47dpf, brca2(fancd1) mutants already showed hypogonadism (Figure S6I), a characteristic shared by many FA patients. Germ cell distribution as revealed by vasa expression [51] was similar in mutants and wild-type gonads (Figure S6A, S6E, S6I). In mutants, somatic cells expressing the Sertoli cell marker amh (anti-Müllerian hormone) [52], [53] failed to form neat borders surrounding tubules as in wild-type males (Figure S6F,S6J) and lacked expression of the female marker cyp19a1a (aromatase) [54] (Figure S6C, S6G, S6K). The early meiotic marker sycp3 (synaptonemal complex protein 3, [55]) was expressed by groups of spermatocytes in wild-type and mutant males (Figure S6H, S6L). We conclude that mutant gonads develop a molecular profile similar to wild-type testis accompanied by disorganization of amh-expressing somatic cells that surround testis tubules.
Neoplasia and Impaired Spermatogenesis in Adult Mutant Testes
To understand the cellular basis of male infertility in brca2 mutants, we compared adult testis histology in wild types (n = 3) and brca2 mutants (n = 7). Comparison of the anterior part of the testes (anterior testes) of wild types and brca2 mutants revealed persistent hypogonadism (smaller diameter gonads) in the mutants (Figure 5A, 5B). Strikingly, brca2 mutant testes lacked sperm and showed tubules with central empty cavities (asterisks). Wild-type and mutant testes both contained spermatocytes at the bouquet stage (late-zygotene/early-pachytene [56], [57]) (sc-b, Figure 5C, 5D), but mutants lacked later stages (spermatids and sperm). Mutant testes also contained abnormal clusters of pyknotic cells (pc, Figure 5D). Occasionally, bouquet stage spermatocytes and pyknotic cells occupied the same tubule (Figure 5E), suggesting that spermatocytes blocked in meiosis became pyknotic. Eosinophils (eos, Figure 5F) invaded some cavities containing pyknotic cells, suggesting an inflammation-like response in mutant gonads. In the posterior part of the testes (posterior testes) of wild types, tubules demarcated by interstitial cells were filled with sperm (sp, Figure 5G), but in the posterior testes of brca2 mutants, tubules were devoid of sperm (Figure 5H). This observation can account for the infertility phenotype of adult brca2 mutant males.

In addition to truncated spermatogenesis, brca2 mutant testes displayed abnormal regions of accumulating cells (Figure 5I-5L). Some of these neoplasias contained both spermatogonia (sg) and interstitial cells (ic, Figure 5I, 5K) and others contained only spermatogonia (sg, Figure 5J, 5L). We conclude that brca2 provides some function that regulates proliferation of spermatogonia and interstitial cells.
Sertoli Cells Are Altered in Adult Mutant Testes
To help understand the altered morphologies of adult mutant testes, we studied gene expression patterns. In the anterior testis, brca2 mutants contained more clusters of vasa-expressing cells than did wild types (Figure 6A, 6A′, 6D, 6D′), reflecting the accumulation of vasa-expressing early germ cell stages (spermatogonia and spermatocytes, Figure 6A′, 6D′)) and the depletion of non-vasa expressing late stages (spermatids and sperm; purple circle, Figure 6A′). In brca2 mutants, amh-expressing cells were less frequent and did not surround tubules normally (Figure 6B, 6E). In addition, sycp3-expressing pachytene spermatocytes accumulated abnormally in mutant testes (Figure 6C, 6F), as expected from the histological data that showed the lack of post-meiotic cells.

The posterior testes of adult wild types did not express vasa and amh, consistent with the presence of interstitial cells and late germ cells (sperm), which no longer express vasa, and the absence of Sertoli cells (Figure 6G–6I). In contrast, posterior testes of adult mutants contained empty cavities and abnormally proliferating cells (Figure 6J–6O) similar to those observed in histological analyses (Figure 5I–5L). Neoplasias contained either mixtures of vasa-expressing and non-vasa-expressing cells (Figure 6J–6L) or possessed only vasa-expressing, early spermatogenic cells (Figure 6M–6O). These results revealed the abnormal presence of early spermatogenic cells in the posterior part of the testes in mutants. Moreover, the presence of scattered amh-expressing cells revealed the abnormal presence of Sertoli cells in mutant posterior testes (Figure 6K, 6N).
Mutation of tp53(p53) Rescues brca2 Sex Reversal
All zebrafish gonads initially form oocytes but these die in wild-type juvenile males [47], [48], and increased germ cell apoptosis leads to oocyte loss and female-to-male sex reversal in fancl mutant zebrafish [20]. Tp53 (alias p53) is an important activator of apoptosis and zebrafish with hypomorphic mutations in tp53 are viable and fertile despite reduced apoptosis [58], [59]. To learn the role of apoptosis in sex reversal of brca2 mutants, we made double mutants for brca2 and the hypomorphic allele tp53M214K [58], [59]. Results showed that no brca2−/− mutants with at least one wild-type tp53 allele developed into females (Figure 7A). In contrast, brca2−/− mutants that lacked a normal tp53 allele became males and females with about equal frequency (Figure 7A). This result shows that a tp53 mutation can rescue the female-to-male sex reversal caused by the lack of brca2 activity. Because double mutant females develop ovaries containing oocytes (Figure 7E), we conclude that Tp53-mediated apoptotic cell death is important for sex reversal in brca2 mutants and interpret these results to mean that the survival of oocytes in brca2 mutant gonads can allow individuals to become females. The rescue of sex reversal by Tp53 mutation reveals that brca2 function is required for oocyte survival, which secondarily leads to female gonad fate and ovarian development.

Brca2 Function Is Not Required to Localize Animal or Vegetal Transcripts in Developing Oocytes
In wild-type late stage II to early stage III oocytes, brca2 and pou5f1 transcripts localize to the animal pole and vasa transcripts gradually spread out cortically from the vegetal pole (Figure 2G, 2H, Figure S7A–S7C, and [41], [42], [60]-[62]). To test the hypothesis that brca2 function is important for the localization of these transcripts, we examined mRNA distribution in oocytes of brca2;tp53 double mutants. In situ hybridization on adjacent serial sections showed that double mutant females produced oocytes with brca2 and pou5f1 transcripts localized to one pole and vasa transcripts positioned at the opposite pole of the same individual oocytes (Figure S7D–S7F). We conclude that brca2 activity is not necessary to localize the messages tested to their proper location in developing zebrafish oocytes.
Mutation of tp53 Does Not Rescue Infertility in brca2 Mutants
To learn if tp53 mutation can rescue the infertility phenotype observed in brca2 single mutants, we mated brca2 homozygous mutants that were either wild type, heterozygous, or homozygous for the tp53 mutation to wild-type animals and scored fertility. Results showed that all brca2 mutant males were sterile regardless of their tp53 genotype (for brca2−/−;tp53+/+, brca2−/−;tp53+/−, and brca2−/−;tp53−/−: we found 0/200 offspring (8 males tested), 0/349 offspring (13 males tested), and 0/148 offspring (6 males tested), respectively). Female brca2;tp53 double mutants were also sterile when mated to wild-type males (of 549 eggs produced by 9 females, 79 initiated cleavage (the average double mutant female had 20%±18% fertility compared to doubly heterozygous siblings with 85%±27% fertility). Non-developing eggs laid by double mutant females were milky and were of highly variable size. Some double mutant females mated to wild-type males produced doubly heterozygous eggs that completed cleavage and gastrulation, but failed to develop to later stages (Figure 7B). Because doubly heterozygous individuals from doubly heterozygous mothers develop normally but doubly heterozygous embryos from homozygous brca2 mutants are lethal, and because homozygous tp53 mutant females have normal fertility [58], we conclude that maternal brca2 function is important for proper embryo development.
Oocytes in brca2;tp53 Double Mutants Have Altered Nuclear Architecture
Histological sections of 6mpf (months post-fertilization) adult ovaries from wild types (brca2+/+;tp53+/+; n = 3), tp53 homozygous mutants (brca2+/+;tp53−/−; n = 3) and double mutants (brca2−/−;tp53−/−; n = 4) revealed oocytes at a variety of developmental stages in all three genotypes (Figure 7C–7E). In tp53 mutants and in wild types, late stage IB oocytes (L-IB) contained nucleoli distributed uniformly along the nuclear periphery (Figure 7C, 7D, 7D′), consistent with the normal fertility of homozygous tp53M214K mutants [58]. In contrast, brca2;tp53 double mutants contained degenerating late stage oocytes (d, Figure 7E) with a granulosa cell layer (gc) that was poorly organized and sometimes separated from the vitelline envelope (ve, Figure 7E′). Wild-type nuclei of late stage IB to early stage II, stage II, and stage III oocytes were radially symmetrical, containing peripheral nucleoli and central chromosomes (Figure 7F–7H). In contrast, brca2−/−;tp53−/− double mutant oocytes were polarized, showing abnormally enlarged and variably shaped nucleoli that accumulated asymmetrically towards one pole of the nucleus while chromosomes concentrated towards the opposite pole (Figure 7I–7K). We conclude that brca2 activity is essential to establish or to maintain a normal architecture of the oocyte nucleus.
In addition to their abnormal location, oocyte chromosomes in double mutants had altered morphology. In wild-type oocytes, chromosomes were distributed independently in the center of the nucleus (arrows in Figure 7L), but chromosomes in oocytes of double mutants were interconnected and formed abnormal loops (arrows in Figure 7M). These chromosome phenotypes were not observed in oocytes of tp53 single mutant females. Aberrant chromosome structure would be expected if recombination-induced chromosome breaks are left unrepaired or are repaired by an error-prone pathway. Our experiments showed that MMC-induced DNA breaks caused chromatid and chromosome damage that led to radial reunion formation in somatic cells (Figure 3I). Likewise, inappropriate repair of recombination-induced DNA breaks could prevent dispersal of oocyte chromosomes. These results suggest a role of brca2 in repairing DNA breaks originating either artificially by MMC or naturally in meiotic recombination.
Ovarian Tumors in brca2;tp53 Double Mutants
Because humans heterozygous for BRCA2 mutations have elevated risk of tumors, we investigated older brca2:tp53 mutants for abnormal growths. By 6mpf, tumors formed that invaded the ovarian cavity and intercalated between oocytes in two of four brca2−/−;tp53−/− double mutants examined (asterisks, Figure 7N–7Q). Tumors were not detected in wild types (n = 3) or in tp53 homozygous mutants (n = 3). One tumor appeared to originate at the ovarian cavity membrane (arrow, Figure 7N). Tumor origin in the other female was unclear because it was metastatic and contacted both the ovarian cavity membrane and the swim bladder (Figure 7P). Both tumors involved spindle-shaped cells that invaded the ovary and surrounded the oocytes (Figure 7N′, 7O, 7Q). The high incidence of ovarian cancer we observed in brca2−/−;tp53−/− double mutants contrasts with the prior finding that animals homozygous for either of two mutant tp53 alleles (tp53M214K, the mutation used here, or tp53I166T) are viable and fertile, but at 8.5 or 8.8mpf (ten weeks later than our double mutants), 1 of 144 and 1 of 417 fish, respectively, began to show tumors and by 16.5 or 22mpf, 28% or 100%, respectively, of the tp53 mutants had developed tumors [58], [59]. Of several hundred tumors previously described in tp53 mutants, none were reported to involve the ovarian cavity membrane [58], [59]. The early appearance and unique ovarian location of tumors in brca2−/−;tp53−/− double mutants suggest a specific association with brca2 activity, potentiated by impaired tp53 function and Tp53 deficiency is cooperative with Brca2 in tumorigenesis in humans [8]. Future long-term investigations are required that focus on tumor development in a large cohort of brca2−/−;tp53−/− double mutants (1/16th of the progeny of double heterozygotes) compared to their single mutant tp53−/− siblings to more clearly define the role of brca2 in the development of ovarian tumors.
Neoplasia and Megalospermatogonia in brca2;tp53 Double Mutants
Testis development was abnormal in double mutants. Analyses of wild-type (n = 4) and tp53 single mutant (n = 3) testes in 6mpf adults revealed germ cells at all stages of spermatogenesis in all the animals analyzed (Figure 8A, 8B). In contrast, all testes analyzed of brca2−/− single mutants (brca2−/−;tp53+/+; n = 4) and double mutants (brca2−/−;tp53−/−, n = 6) abnormally lacked spermatids and sperm (Figure 8C, 8D). Furthermore, in all brca2−/−;tp53+/+ single mutants and all brca2−/−;tp53−/− double mutants, posterior tubules contained empty cavities lacking germ cells (asterisks in Figure 8C, 8D) consistent with our finding that adult double mutants failed to recover fertility. Unexpectedly, some testes in brca2−/−;tp53−/− double mutants (n = 2), but not in the other genotypes, contained germ cells enlarged up to ten times normal diameter (Figure 8E). Some of these enormous cells we call megalospermatogonia (ms) because, like normal spermatogonia [63], they contained an enlarged central nucleolus (nc). Testes of some brca2−/−;tp53−/− double mutants (n = 3) contained other large cells that showed the peripheral distribution of numerous nucleoli, which constitutes an oocyte-like morphology [63], so we interpret these as large early oocytes (eo), and additionally we observed the presence of enlarged pyknotic cells (pc) (ms, pc, and eo in Figure 8E, 8F, 8F′, compare to normal spermatogonia (sg) outlined by dashed lines in Figure 8E, 8F). The lack of sex-specific markers for early gonial cells precludes a more precise definition of cell type. Somatic cell neoplasias appeared in the posterior testis of all double mutants (but showing variability on the neoplastic tissue size) (Figure 8G, 8G′) suggesting the hypothesis that late stage spermatogenic cells negatively regulate the proliferation of somatic cells in testes. To test this hypothesis and to investigate whether the absence of brca2 activity or merely the absence of germ cells allows over-proliferation of the somatic component of the testis, we examined animals depleted of germ cells by dead end-morpholino (dnd−/−; n = 10; see also [64]). Dnd is an RNA binding protein that is essential for germ cell survival in mice and zebrafish [64], [65]. Our results revealed that testes in 18mpf dnd-morpholino treated animals developed neoplastic somatic proliferation (Figure 8H, 8H′) similar to those observed in brca2 single and brca2;tp53 double mutants (Figure 5I, 5K, Figure 8G). In five of ten dnd-injected animals, neoplasias invaded the intestine and body wall musculature (Figure 8H). Although we did not detect invasive somatic proliferation in brca2 mutants, these dnd-knockdown animals were 12 months older than our brca2 mutants, suggesting that invasive proliferation might arise in brca2 mutants as they age. Overall, these results would be expected if somatic cell neoplasias in brca2 and brca2;tp53 mutants were not due to a direct effect of the lack of brca2 activity, but arose as a secondary effect of germ cell loss in brca2 mutants. A possible mechanism to explain these results is that late stage germ cells in the wild-type spermatogenic pathway exert a negative control over the proliferation of somatic cells in zebrafish testes. Loss of Dead end function in mouse results in germ cell tumors in a strain-specific manner [65]. Because dnd-knockdown zebrafish have no germ cells, the origin of gonad tumors after dnd-knockdown differs between mouse and zebrafish. Long-term investigations are required to examine whether the somatic testicular tumors we observed in zebrafish lacking dnd function are subject to effects of the genetic background.

Discussion
The biological mechanisms that underlie Fanconi anemia and hereditary breast, ovarian, and prostate cancer intersect at Brca2(Fancd1). Null alleles of Brca2 are embryonic lethal in mouse and human [11], [23], [66], which precludes study of the gene's full function in homozygous null-allele adults in these species. Rats lacking Brca2 activity are viable [10], but mammals are not favorable for a whole-animal small molecule screen for therapeutic substances related to Brca2 disease. Here we exploit a viable zebrafish brca2 null-allele. We show that zebrafish brca2 is the ortholog of human BRCA2, it shares important coding features with the human gene, and it is expressed in rapidly dividing and meiotic cells. Importantly, we show that brca2 maintains genome stability in response to DNA damaging agents and it is essential for the survival of post-recombinant spermatocytes. Loss of brca2 function causes female-to-male sex reversal that is rescued by mutating tp53, indicating that brca2 subverts female development by apoptosis and is required for normal oogenesis. Unexpectedly, we found that brca2 transcript localizes asymmetrically to the animal pole of wild-type oocytes and that brca2 activity is essential for establishing or maintaining the architecture of the oocyte nucleus. Moreover, results showed that brca2 activity is necessary in zebrafish as it is in humans to prevent ovarian tumors in the absence of tp53 function. Therefore, this work validates the zebrafish brca2 mutant as a useful tool for small-molecule screens to help discover potential therapeutic compounds for human patients.
A Zebrafish brca2-Null Allele Disrupts Genome Stability
Genomic and genetic evidence shows that zebrafish has a single-copy of brca2 that is orthologous to its mammalian counterpart. We found that the zebrafish genome has duplicate copies of the human chromosome segment that contains BRCA2, and that these duplicates arose from the teleost genome duplication (TGD). In one of these duplicated segments, the BRCA2 ortholog disappeared, leaving zebrafish with a single copy of brca2. About 75% of genes from the TGD event reverted to singletons [27], but all of the 13 zebrafish orthologs of FA pathway genes are present in single copy [25], which would happen rarely (2.4%) solely by chance. We conclude that evolutionary forces probably acted to reduce brca2 and other FA pathway genes to single copy after the TGD. This finding is predicted by the duplication-degeneration-complementation hypothesis [67], which suggests that genes with simple tissue - and time-specific regulatory elements would be more likely to revert to singletons than those with complex regulation. In addition, many Fanc proteins join to form molecular machines in a 1 : 1 stoichiometry, so that if one gene in the network evolves to single copy, the others might follow by natural selection or neutral evolutionary forces.
Expression analyses showed that maternal brca2 message accumulated in zebrafish embryos. This message would be available to provide embryos with Brca2 protein that could function to help resolve stalled replication forks [68] during the rapid cleavage divisions that precede the mid-blastula transition, the stage at which zygotic transcription initiates [69]. Our finding that the heterozygous offspring of homozygous brca2 mutant mothers fail to develop much past gastrulation supports this conclusion. Expression of brca2 in meiotic cells of zebrafish, as in mammals [24], suggests a role in the repair of DNA breaks incurred during meiotic homologous recombination [46].
Zebrafish brca2 ZM_00075660 mutants generate only aberrant transcripts that lack domains essential for Brca2 activity and provide a vertebrate null allele model to unravel the effects of brca2 during embryonic and post-embryonic development. Mutant tissue culture cells and developing embryos show more chromosome damage and excess staining of broken DNA, respectively, than wild-type cells or embryos after exposure to DNA damaging agents. Similarly, loss of BRCA2 function in humans results in hypersensitivity to DNA crosslinking agents [44], [70], thus leading to chromosome breaks [43], [71], showing that zebrafish and human Brca2 orthologs share functions in maintaining genome stability.
Flow cytometry showed that the poor growth of zebrafish brca2 mutant cell cultures results from high rates of spontaneous apoptotic cell death. This finding parallels our finding of increased apoptosis in juvenile mutant gonads that results in oocyte loss and sex reversal and that the inhibition of cell death in brca2;tp53 double mutants rescues sex reversal. Spontaneous apoptosis leading to bone marrow failure is also a problem in hematopoietic stem cells in human FA patients, [21]. Zebrafish therefore appears to be a valid model to study the basic influence of Brca2 deficiency on apoptosis. In contrast, the effect of damage caused by MMC in zebrafish brca2 mutant cell cultures on non-apoptotic cell death rates is surprisingly low compared to humans; the cause of which remains unexplained.
Lack of Brca2 Activity Results in Female-to-Male Sex Reversal
Results showed that brca2 mutant zebrafish developed exclusively as males due to female-to-male sex reversal. Sex ratios also appear to be skewed in the offspring of human carriers of BRCA2 mutations, suggesting a possible role in sex determination or differential survival [72]. During the sex determination period, zebrafish mutant gonads contained apoptotic cells and lacked diplotene oocytes, the presence of which is essential to tip gonad fate towards the female pathway in fancl zebrafish mutants [20]. In contrast to fancl mutants, which become fertile males, fancd1(brca2) mutants become sterile males. The more severe phenotype of brca2 mutants compared to fancl mutants parallels the fact that human homozygotes for FANCD1(BRCA2) null alleles are lethal as embryos [11]. In the FA-BRCA network, FANCL should act upstream of BRCA2 (see for review [73]. Because the phenotype of brca2 mutant zebrafish is more severe than that of fancl mutant zebrafish, BRCA2 likely plays roles in addition to its function downstream of fancl. Because FANCD1(BRCA2) is the only FA complementation group that fails to form RAD51 foci after ionizing radiation and crosslink damage, FANCD1(BRCA2), but not FANCL, is required for RAD51-mediated DNA repair [74], [75].
The inhibition of apoptosis in brca2 mutants by the mutation of tp53 rescued female-to-male sex reversal and led to the development of females, consistent with the idea that brca2 mutant oocytes die by apoptosis when unable to repair DNA breaks associated with meiotic recombination. This conclusion parallels that from zebrafish fancl mutants, in which oocytes die in juveniles followed by female-to-male sex reversal [20] and supports the notion that fanc-related sex reversal acts via Tp53-mediated apoptosis.
The brca2(fancd1) and fancl results combine to support the following model for zebrafish sex determination. (1) The FA network facilitates DNA repair associated with meiosis and hence the survival of oocytes during the critical period for zebrafish sex determination. (2) Activation of Tp53-dependent germ cell apoptosis, at least in fanc mutants, alters the total number of germ cells and thus reduces the number of surviving oocytes below the threshold necessary to maintain female fate. (3) Post-recombinant oocytes release a signal that down-regulates amh and/or maintains cyp19a1a (aromatase) expression in somatic cells of the bipotential gonad [20], [48], [52], [64], [76]-[80]; the fewer the number of post-recombinant oocytes, the less aromatase-maintenance signal. (4) Aromatase converts testosterone to estrogen, thereby reinforcing ovary development and the female fate. (5) In normally developing males at the juvenile hermaphrodite stage [47], unknown genetic factors that may be influenced by the environment stimulate the death of oocytes and hence loss of the aromatase-maintenance signal. According to this model, in the absence of either brca2(fancd1) or fancl, oocytes do not effectively repair the DNA breaks of meiosis, DNA-damaged oocytes die by apoptosis before they liberate the aromatase-maintenance signal, the gonad becomes a testis, and individuals that otherwise would have become females develop into males.
The study of zebrafish brca2 mutants verifies the importance of Brca2 for gonad development and provides a new vertebrate model for the adult roles of Brca2 that is obscured by null mutant lethality in human and mouse. Neither Brca2 knockout rats nor Brca2 knockout mice rescued by a human BRCA2 BAC showed sex reversal [10], [24], reflecting lineage-specific sex determining mechanisms. Zebrafish brca2 mutants developed gonads without diplotene oocytes, but rescued mice did develop oocytes, many of which disappeared post-natally, but some of which progressed through meiotic prophase I, became fertilized, and produced embryos [24]; in contrast, Brca2 mutant rats were sterile. A possible explanation for the difference between mouse and rat is that the transgenic mice might not be total null mutants because expression of human BRCA2 was detected in their gonads [24].
Brca2 Activity Is Essential for Normal Spermatogenesis
Zebrafish brca2 mutants contained spermatocytes arrested in meiosis, as did transgenic mice rescued by human BRCA2 [24] and rats mutant for Brca2−/− [10]. We found that zebrafish brca2 mutant testes (1) showed hypogonadism like human FA patients; (2) developed spermatogonia that entered meiosis as shown by sycp3 expression and histological data; (3) contained bouquet stage spermatocytes that arrested in late zygotene-early pachytene; (4) lacked post-meiotic spermatogenic stages, including spermatids and sperm; (5) failed to properly organize Sertoli cells, as shown by amh expression; (6) contained abnormal pyknotic cells that were positive for the apoptotic marker active-Caspase-3; and (7) formed tubules that lacked germ cells but contained eosinophils, blood cells involved in inflammation. Together, these results suggest a mechanistic model in which brca2 mutant spermatogenic cells develop rather normally until meiotic recombination (pachytene), fail to repair double strand DNA breaks associated with homologous recombination, then die, leaving empty testis tubules in hypogonadal sterile males.
Because spermatogenic cells die even in brca2;tp53 double mutants, we conclude that, after meiotic failure, brca2 spermatogenic cells die by a Tp53-independent pathway in double mutants, or alternatively, that the hypomorphic nature of the tp53M214K allele may allow cells to disappear by a Tp53-dependent pathway. Megalospermatogonia with enormous nuclei appeared in double mutants, possibly due to continued DNA replication in spermatogonia damaged by inadequate DNA repair in the absence of brca2 activity. In brca2 single mutant testes, cells may experience extra rounds of replication but tp53-mediated cell death may delete them. Alternatively, in the absence of brca2 function, tp53 activity might be required to prevent abnormal functions that lead to cell enlargement and megalospermatogonia. The presence of oocytes in testes of double mutant animals might be explained by the alteration of somatic cell-to-germ cell signaling in testes that are developing with abnormal Sertoli cell distribution and lack of spermatids and sperm. Normal Tp53 function might be necessary to induce oocyte apoptosis in brca2 single mutant testes.
brca2 Mutant Testes, Neoplasia, and Germ Cell-to-Soma Signaling
Six-month old brca2 single mutants accumulated neoplastic growths involving spermatogonia with or without disorganized clumps of interstitial cells. The posterior part of the testis, which completely lacked germ cells in brca2 single and brca2;tp53 double mutants, showed abnormal proliferation of somatic cells in the testes. The investigation of genetically wild-type animals lacking germ cells due to dnd knockdown uncovered somatic neoplasias of the testes similar to those found in brca2 mutant testes. Knockdown of dnd was not previously reported to cause neoplasias, but the oldest dnd-knockdown animals previously reported were 6 months old [64] and our dnd-knockdowns were 18 months old, suggesting that these neoplasias arise with age, although we can't rule out strain-specific effects. We conclude that post-meiotic germ cells, specifically the spermatids or sperm that are lacking from brca2 mutants, control growth of surrounding somatic cells by an as yet unknown mechanism that is secondary to brca2-impaired spermatogenesis.
The Animal Pole Distribution of brca2 Transcript Does Not Appear to Generate Oocyte Polarity
Our gene expression analyses showed unexpectedly that brca2 transcript localizes asymmetrically at the animal pole of wild-type oocytes. The asymmetrical distribution of certain mRNAs in the oocyte cytoplasm helps to promote animal or vegetal pole identity [41], [42], [60]-[62]. In brca2;tp53 double mutants, however, the localization of messages for pou5f1, vasa, and even brca2 itself were normal, suggesting that brca2 activity is not essential to polarize the oocyte cytoplasm. Furthermore, brca2 transcript begins to be localized in stage III oocytes but ccnb1 transcripts begin to be localized earlier [41], [42], a timing that is incompatible with the hypothesis that brca2 initiates ooplasm asymmetry. The germinal vesicle (the oocyte nucleus) lies in the center of stage I-III oocytes, but moves to the animal pole by stage IV [41], [49]; thus, the oocyte nucleus - - and hence the resulting zygote and cleavage nuclei - - occupy cytoplasm enriched in brca2 transcript. The translation of this transcript would be available to support the repair of DNA damage incurred in the rapid divisions of cleavage before zygotic transcription initiates at the mid-blastula transition. The observation that brca2;tp53 double mutant females formed doubly heterozygous embryos that cleaved normally, completed gastrulation, but then died by 24 hpf supports the notion that maternally transmitted brca2 transcript is important for normal early development and that zygotic brca2 transcript is too little or too late to rescue the phenotype.
Brca2 Activity Is Essential for Normal Architecture of the Oocyte Nucleus
brca2;tp53 double mutant females produced oocytes with normally organized ooplasm but aberrantly organized nuclei. Developing oocytes that lacked brca2 activity partitioned nucleoli aberrantly to one side of the nucleus rather than their usual radial location and distributed chromosomes opposite to the nucleoli rather than their normal central position. Although these ovaries were also homozygous mutant for tp53 (which was necessary to obtain homozygous brca2 mutant females), the oocyte nucleus architecture defect is due to brca2 deficiency because homozygous tp53 mutant females form normal oocytes [58], [59], as we confirmed. Oocytes in brca2;tp53 females reached the bouquet stage of meiosis in which telomeres cluster at one side of the oocyte. The asymmetric localization of chromosomes in the oocytes of brca2;tp53 females may result from a deficiency in DNA repair that inhibits exit from the bouquet stage. This could result, for example, if Brca2 is necessary for the relaxation of telomere clustering that generates the bouquet stage, a proposition supported by the high rate of recombination in subtelomeric sequences [81]: a high rate of recombination near the telomeres could create interlinked chromosomes like the radial reunion figures we observed in somatic cell chromosomes and these links might not permit normal chromosome dispersal in the nucleoplasm. It is unclear, however, how the abnormal persistence of chromosome clustering would generate the asymmetric distribution and abnormal morphologies of nucleoli that we found in oocytes of mutant females. Drosophila brca2 mutants develop oocytes with an abnormally asymmetric karyosome and dorso-ventral defects [46], phenotypes that may be functionally related to those we observe in zebrafish. Transgenic female mice rescued with a human BRCA2-containing BAC have abnormal polar bodies in meiosis [24], a phenotype that may be a consequence of nuclear symmetry problems we demonstrate here in zebrafish. We conclude that brca2 is generally important for the organization of oocyte nuclei both in protostomes and in vertebrates.
brca2;tp53 Double Mutant Females and Invasive Ovarian Tumors
Tp53 deficiency coupled with diminished Brca2 activity promotes mammalian breast tumors [8]. Likewise, zebrafish brca2;tp53 double mutants develop invasive ovarian tumors, and these appear earlier and more frequently than tumors in animals with lower Tp53 activity alone. We conclude that zebrafish shares with human and rat [10] a requirement for Brca2 activity to help suppress the formation of ovarian cancers. The genome instability we observed in brca2 mutant tissue culture cells may contribute to tumor formation because zebrafish gin mutations identified on the basis of genomic instability have elevated cancer risk [82]. Future studies are necessary to better understand the etiology of brca2-dependent ovarian tumors in zebrafish.
FANCD1 is an alias of BRCA2 because homozygous hypomorphic mutations in this gene cause Fanconi Anemia while heterozygous null mutations increase the risk of breast and ovarian tumors and homozygous null mutations cause lethality [11]. In addition to genome instability, bone marrow failure, leukemia, and squamous cell carcinomas, many FA patients experience hypogonadism, impaired gametogenesis, defective meiosis, and sterility [19], [83]. Thus, zebrafish brca2(fancd1) shares genome instability and gonad developmental phenotypes with FA patients and ovarian tumors with human heterozygotes for BRCA2 mutations. These findings indicate that zebrafish brca2 mutants provide a suitable model for human BRCA2-related disease. In a result of special significance, the embryonic sensitivity of zebrafish brca2 mutants to a DNA damage agent provides an assay for a small molecule screen to identify compounds that can rescue the DNA damage phenotype and thus has the potential to contribute to the discovery of substances that can ameliorate at least some of the phenotypes observed in human patients.
Methods
DNA Amplification and Cloning
A partial gene model for zebrafish brca2 was inferred using GenomeScan (http://genes.mit.edu/genomescan.html) to match the human BRCA2 protein to a zebrafish genomic BAC clone we identified and sequenced (Genbank accession #AC149226). Primers for RACE (Clontech) were designed using this preliminary gene model. RACE template was 5′ first-strand zebrafish cDNA synthesized from pooled mRNA from embryos at 12, 24, and 48 hours post-fertilization (hpf). A BLAST search of the zebrafish EST database (http://www.ncbi.nlm.nih.gov/genome/seq/BlastGen/BlastGen.cgi?taxid=7955) using the 3′ UTR modeled with GenomeScan recovered EST CT605096. This EST and our GenomeScan model were used to design primers for amplification of the entire brca2 cDNA (primers in Table S1) using as template second strand cDNA from 60hpf embryos. Amplified fragments were cloned using the TOPO Cloning Kit for Sequencing (Invitrogen).
Bioinformatics
We applied reciprocal best BLAST “hit” (RBH) [84] as an initial test for orthology of zebrafish and human BRCA2 genes. We queried the stickleback genome (http://www.ensembl.org/Gasterosteus_aculeatus/blastview) using zebrafish Brca2 and found stickleback brca2 on contig 2939. This contig, together with human BRCA2, was used to develop a preliminary gene model for stickleback brca2 (Figure S1). The Synteny Database identified paralogous and orthologous chromosome segments [26].
Animals
The insertional brca2 mutant (ZM_00075660) was purchased from Znomics, which randomly inserted derivatives of a Moloney murine leukemia-based retroviral vector into zebrafish [85]. To genotype ZM_00075660, we used primers F4/R4 to amplify the mutant allele, and primers F1/R1 to amplify the wild-type allele. Table S1 lists primer sequences. To reduce Tp53 activity, the hypomorphic mutation tp53M214K was obtained from ZIRC (http://zebrafish.org/zirc/home/guide.php) and was used and genotyped as described [58]. All animals were reared and collected under standard conditions [86]. The University of Oregon Institutional Animal Care and Use Committee approved all animal work (Animal Welfare Assurance Number A-3009-01, IACUC protocol #08-13). Genetic nomenclature follows guidelines from ZFIN (http://zfin.org/zf_info/nomen.html), e.g., human gene, BRCA2; mouse gene, Brca2; zebrafish gene, brca2; human protein BRCA2 and mouse and zebrafish protein, Brca2.
In Situ Hybridization and Histology
Whole mount in situ hybridizations were performed as described [87] using several individuals for each developmental stage. In situ hybridization experiments on zebrafish cryosections were performed following [52]. Probes for amh and cyp19a1a were made following [52] and probe for vasa was made from its 3′end as described [51]. A brca2 cDNA fragment of 725nt (nucleotides 2627-3351 of NM_001110394), a pou5f1 cDNA fragment of 778nt (nucleotides 705-1482 of NM_131112), and a sycp3 cDNA fragment of 620nt (nucleotides 265-884 of NM_001040350) were used to synthesize DIG-labeled riboprobe (Boehringer Mannheim). For gonad histology, paraffin embedded Bouin's fixed tissue was sectioned at 7 microns and stained with hematoxylin and eosin.
Immunohistochemistry
Animals were fixed at 60dpf in 4% PFA overnight at 4°C, embedded in paraffin, and sectioned at 7 microns. Apoptotic cells were detected by immunofluorescence using anti-active Caspase-3 (Pharmingen, # 559565 Purified Rabbit anti-active caspase-3) following published protocols [20], [39].
dead end Morpholino Injections
Animals depleted of germ cells were obtained by injecting wild-type zebrafish embryos from the AB strain at the 1–2 cell stage with dead end antisense morpholino oligonucleotide (Gene Tools) as described [88]. Injected and non-injected animals were raised to adulthood and collected at 18 months post-fertilization.
Acridine Orange Staining
Embryos were exposed to diepoxybutane (DEB) in embryo medium from 7–28hpf, or left untreated. Embryos were stained with acridine orange (AO) and mounted following [89]. The initial focal plane was the otolith, and a z-series consisting of seven 10-micron steps was captured on a Bio-Rad Radiance 2100 confocal microscope. Images were merged into a single plane with Velocity 4.4.0 and the number of AO-positive cells in the central nervous system anterior to the beginning of the yolk extension was quantified using ImageJ.
Karyotyping
Distal tips of caudal and dorsal fins from mutant and wild-type adult fish were cultured in 1∶1 (vol/vol) DMEM (Gibco) and Amniopan (PAN) media supplemented with 100 µg/ml penicillin and 0.1 mg/ml streptomycin in a 5% CO2 atmosphere at 28°C. Adherent fibroblast-like cells grew from primary explants. When culture flasks were confluent, cells were trypsinized and subcultured at split ratios of 1 : 4. Cells of the 20th or 21st passage were exposed to 5 or 10 ng/ml mitomycin C for 24 hrs and screened for chromosome morphology. Rates of chromatid and chromosome breaks, radial reunion figures, and other categories of breakage were scored by analyzing 100 cells from each cell line. To visualize mitotic chromosomes, subconfluent cultures were exposed to colcemid for 3 hrs. Accumulated metaphases were prepared following standard methods. Slides were stained with 5% Giemsa solution. Metaphases were screened under a light microscope. Line authenticity was confirmed by PCR genotyping using the primer set Brca2zmwt.F2 5′-GCAGGTTGTGATGAAGCCACC-3′ and Brca2zmwt.R1 5′-GTGGTGTGAGGCCAGAGGTT-3′ for amplification of a 888-bp fragment of the wt brca2 sequence and the primer set 5Fd1ins.F 5′-CTTGCGCACCAAGGCTTCAC-3′ and 5Fd1ins.R 5′-ACCGCATCTGGGGACCATCT-3′ for amplification of a 971-bp fragment of the insert. For cell growth studies, 1×105 cells per line were seeded into six flasks. Following trypsinization, one flask per line was counted daily until day 5. Resulting numbers were plotted as multiples of the initial cell count. Mitomycin C (MMC) was added at given concentrations at time 0 h. For flow cytometric assays, cultures were harvested and cells were washed twice with PBS and fixed in 4% paraformaldehyde at 37°C for 10 min. The reaction was stopped on ice for 2 min, then cells were pelleted and resuspended in 100% methanol at −20°C for permeabilization. For immunostaining, we used the CaspGLOW Fluorescein Active Caspase-3 Staining Kit (BioVision # K183-25) and propidium iodide (PI) counterstain at a final concentration of 8 µg/ml. Histograms were recorded on an analytical, triple-laser equipped flow cytometer (LSRII, Becton Dickinson) using Sapphire 488 nm solid state laser excitation of 5(6)-fluorescein isothiocyanate (FITC) and propidium iodide (PI) with appropriate filter sets to discern fluorescence intensity of different emission wavelengths. Quantification of cell distributions was by FACSDiva Software, version 6.1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WoosterR
WeberBL
2003 Breast and ovarian cancer. N Engl J Med 348 2339 2347
2. EdwardsSM
Kote-JaraiZ
MeitzJ
HamoudiR
HopeQ
2003 Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet 72 1 12
3. GaytherSA
de FoyKA
HarringtonP
PharoahP
DunsmuirWD
2000 The frequency of germ-line mutations in the breast cancer predisposition genes BRCA1 and BRCA2 in familial prostate cancer. The Cancer Research Campaign/British Prostate Group United Kingdom Familial Prostate Cancer Study Collaborators. Cancer Res 60 4513 4518
4. EastonD
1997 Breast cancer genes--what are the real risks? Nat Genet 16 210 211
5. AntoniouA
PharoahPD
NarodS
RischHA
EyfjordJE
2003 Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 72 1117 1130
6. TavtigianSV
SimardJ
RommensJ
CouchF
Shattuck-EidensD
1996 The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat Genet 12 333 337
7. ConnorF
SmithA
WoosterR
StrattonM
DixonA
1997 Cloning, chromosomal mapping and expression pattern of the mouse Brca2 gene. Hum Mol Genet 6 291 300
8. MoynahanME
2002 The cancer connection: BRCA1 and BRCA2 tumor suppression in mice and humans. Oncogene 21 8994 9007
9. FrappartPO
McKinnonPJ
2007 BRCA2 function and the central nervous system. Cell Cycle 6 2453 2457
10. CotroneoMS
HaagJD
ZanY
LopezCC
ThuwajitP
2007 Characterizing a rat Brca2 knockout model. Oncogene 26 1626 1635
11. NevelingK
EndtD
HoehnH
SchindlerD
2009 Genotype-phenotype correlations in Fanconi anemia. Mutat Res 668 73 91
12. D'AndreaA
2003 Fanconi anemia. Curr Biol 13 R546
13. BagbyGC
AlterBP
2006 Fanconi anemia. Semin Hematol 43 147 156
14. RosenbergPS
AlterBP
EbellW
2008 Cancer risks in Fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica 93 511 517
15. HowlettNG
TaniguchiT
OlsonS
CoxB
WaisfiszQ
2002 Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297 606 609
16. HirschB
ShimamuraA
MoreauL
BaldingerS
Hag-alshiekhM
2004 Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 103 2554 2559
17. AlterBP
RosenbergPS
BrodyLC
2007 Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet 44 1 9
18. MoynahanME
CuiTY
JasinM
2001 Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res 61 4842 4850
19. WongJC
AlonN
McKerlieC
HuangJR
MeynMS
2003 Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet 12 2063 2076
20. Rodríguez-MaríA
CañestroC
BreMillerRA
Nguyen-JohnsonA
AsakawaK
2010 Sex Reversal in Zebrafish fancl Mutants Results from Tp53-Mediated Germ Cell Apoptosis. PLoS Genet 6 e1001034
21. LiX
Le BeauMM
CicconeS
YangFC
FreieB
2005 Ex vivo culture of Fancc−/ − stem/progenitor cells predisposes cells to undergo apoptosis, and surviving stem/progenitor cells display cytogenetic abnormalities and an increased risk of malignancy. Blood 105 3465 3471
22. ChenPL
ChenCF
ChenY
XiaoJ
SharpZD
1998 The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci U S A 95 5287 5292
23. HakemR
de la PompaJL
MakTW
1998 Developmental studies of Brca1 and Brca2 knock-out mice. J Mammary Gland Biol Neoplasia 3 431 445
24. SharanSK
PyleA
CoppolaV
BabusJ
SwaminathanS
2004 BRCA2 deficiency in mice leads to meiotic impairment and infertility. Development 131 131 142
25. TitusTA
YanYL
WilsonC
StarksAM
FrohnmayerJD
2009 The Fanconi anemia/BRCA gene network in zebrafish: embryonic expression and comparative genomics. Mutat Res 668 117 132
26. CatchenJM
ConeryJS
PostlethwaitJH
2009 Automated identification of conserved synteny after whole-genome duplication. Genome Res 19 1497 1505
27. PostlethwaitJH
WoodsIG
Ngo-HazelettP
YanY-L
KellyPD
2000 Zebrafish comparative genomics and the origins of vertebrate chromosomes. Genome Res 10 1890 1902
28. TitusTA
SelvigDR
QinB
WilsonC
StarksAM
2006 The Fanconi anemia gene network is conserved from zebrafish to human. Gene 371 211 223
29. AmoresA
ForceA
YanY-L
JolyL
AmemiyaC
1998 Zebrafish hox clusters and vertebrate genome evolution. Science 282 1711 1714
30. TaylorJ
BraaschI
FrickeyT
MeyerA
Van De PeerY
2003 Genome duplication, a trait shared by 22,000 species of ray-finned fish. Genome Res 13 382 390
31. JaillonO
AuryJM
BrunetF
PetitJL
Stange-ThomannN
2004 Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431 946 957
32. TengDH
BogdenR
MitchellJ
BaumgardM
BellR
1996 Low incidence of BRCA2 mutations in breast carcinoma and other cancers. Nat Genet 13 241 244
33. TakataM
TachiiriS
FujimoriA
ThompsonLH
MikiY
2002 Conserved domains in the chicken homologue of BRCA2. Oncogene 21 1130 1134
34. WarrenM
SmithA
PartridgeN
MasabandaJ
GriffinD
2002 Structural analysis of the chicken BRCA2 gene facilitates identification of functional domains and disease causing mutations. Hum Mol Genet 11 841 851
35. MilnerJ
1997 Structures and functions of the tumor suppressor p53. Pathol Biol (Paris) 45 797 803
36. BignellG
MicklemG
StrattonMR
AshworthA
WoosterR
1997 The BRC repeats are conserved in mammalian BRCA2 proteins. Hum Mol Genet 6 53 58
37. YangH
JeffreyPD
MillerJ
KinnucanE
SunY
2002 BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 297 1837 1848
38. DeutschM
LongM
1999 Intron-exon structures of eukaryotic model organisms. Nucleic Acids Res 27 3219 3228
39. CheesmanSE
NealJT
MittgeE
SeredickBM
GuilleminK
2010 Microbes and Health Sackler Colloquium: Epithelial cell proliferation in the developing zebrafish intestine is regulated by the Wnt pathway and microbial signaling via Myd88. Proc Natl Acad Sci U S A
40. ZabludoffSD
WrightWW
HarshmanK
WoldBJ
1996 BRCA1 mRNA is expressed highly during meiosis and spermiogenesis but not during mitosis of male germ cells. Oncogene 13 649 653
41. HowleyC
HoRK
2000 mRNA localization patterns in zebrafish oocytes. Mech Dev 92 305 309
42. AbramsEW
MullinsMC
2009 Early zebrafish development: it's in the maternal genes. Curr Opin Genet Dev 19 396 403
43. SchroederTM
AnschutzF
KnoppA
1964 Spontane Chromosomenaberrationen bei familiärer Panmyelopathie. Humangenetik 1 194 196
44. AuerbachAD
1993 Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol 21 731 733
45. DarzynkiewiczZ
1990 Differential staining of DNA and RNA in intact cells and isolated cell nuclei with acridine orange. Methods Cell Biol 33 285 298
46. KlovstadM
AbduU
SchupbachT
2008 Drosophila brca2 is required for mitotic and meiotic DNA repair and efficient activation of the meiotic recombination checkpoint. PLoS Genet 4 e31
47. TakahashiH
1977 Juvenile hermaphroditism in the zebrafish, Brachydanio rerio. Bull Fac Fish Hokkaido Univ 28 57 65
48. UchidaD
YamashitaM
KitanoT
IguchiT
2002 Oocyte apoptosis during the transition from ovary-like tissue to testes during sex differentiation of juvenile zebrafish. J Exp Biol 205 711 718
49. SelmanK
WallaceRA
SarkaA
Q.X
1993 Stages of oocyte development in the zebrafish Brachydanio rerio. J Morphol 218 203 224
50. DaiC
KrantzSB
1999 Interferon gamma induces upregulation and activation of caspases 1, 3, and 8 to produce apoptosis in human erythroid progenitor cells. Blood 93 3309 3316
51. YoonC
KawakamiK
HopkinsN
1997 Zebrafish vasa homologue RNA is localized to the cleavage planes of 2 - and 4-cell-stage embryos and is expressed in the primordial germ cells. Development 124 3157 3165
52. Rodriguez-MariA
YanYL
BremillerRA
WilsonC
CanestroC
2005 Characterization and expression pattern of zebrafish Anti-Mullerian hormone (Amh) relative to sox9a, sox9b, and cyp19a1a, during gonad development. Gene Expr Patterns 5 655 667
53. von HofstenJ
LarssonA
OlssonPE
2005 Novel steroidogenic factor-1 homolog (ff1d) is coexpressed with anti-Mullerian hormone (AMH) in zebrafish. Dev Dyn 233 595 604
54. ChiangEF
YanYL
GuiguenY
PostlethwaitJ
ChungB
2001 Two Cyp19 (P450 aromatase) genes on duplicated zebrafish chromosomes are expressed in ovary or brain. Mol Biol Evol 18 542 550
55. MoensPB
2006 Zebrafish: chiasmata and interference. Genome 49 205 208
56. ZicklerD
KlecknerN
1998 The leptotene-zygotene transition of meiosis. Annu Rev Genet 32 619 697
57. ScherthanH
2001 A bouquet makes ends meet. Nat Rev Mol Cell Biol 2 621 627
58. BerghmansS
MurpheyRD
WienholdsE
NeubergD
KutokJL
2005 tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci U S A 102 407 412
59. ParantJM
GeorgeSA
HoldenJA
YostHJ
2010 Genetic modeling of Li-Fraumeni syndrome in zebrafish. Dis Model Mech 3 45 56
60. KosakaK
KawakamiK
SakamotoH
InoueK
2007 Spatiotemporal localization of germ plasm RNAs during zebrafish oogenesis. Mech Dev 124 279 289
61. MarlowFL
MullinsMC
2008 Bucky ball functions in Balbiani body assembly and animal-vegetal polarity in the oocyte and follicle cell layer in zebrafish. Dev Biol 321 40 50
62. LindemanRE
PelegriF
2010 Vertebrate maternal-effect genes: Insights into fertilization, early cleavage divisions, and germ cell determinant localization from studies in the zebrafish. Mol Reprod Dev 77 299 313
63. LealMC
CardosoER
NobregaRH
BatlouniSR
BogerdJ
2009 Histological and stereological evaluation of zebrafish (Danio rerio) spermatogenesis with an emphasis on spermatogonial generations. Biol Reprod 81 177 187
64. SlanchevK
SteblerJ
de la Cueva-MendezG
RazE
2005 Development without germ cells: the role of the germ line in zebrafish sex differentiation. Proc Natl Acad Sci U S A 102 4074 4079
65. YoungrenKK
CoveneyD
PengX
BhattacharyaC
SchmidtLS
2005 The Ter mutation in the dead end gene causes germ cell loss and testicular germ cell tumours. Nature 435 360 364
66. SharanSK
MorimatsuM
AlbrechtU
LimDS
RegelE
1997 Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 386 804 810
67. ForceA
LynchM
PickettFB
AmoresA
YanY-L
1999 Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151 1531 1545
68. HussainS
WilsonJB
MedhurstAL
HejnaJ
WittE
2004 Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet 13 1241 1248
69. KaneDA
KimmelCB
1993 The zebrafish midblastula transition. Development 119 447 456
70. AuerbachAD
WolmanSR
1976 Susceptibility of Fanconi's anaemia fibroblasts to chromosome damage by carcinogens. Nature 261 494 496
71. HoughtalingS
NewellA
AkkariY
TaniguchiT
OlsonS
2005 Fancd2 functions in a double strand break repair pathway that is distinct from non-homologous end joining. Hum Mol Genet 14 3027 3033
72. DomchekSM
MerillatSL
TiggesJ
TweedAJ
WeinarM
2005 Sex ratio skewing of offspring in families with hereditary susceptibility to breast cancer. J Med Genet 42 511 513
73. MoldovanGL
D'AndreaAD
2009 How the fanconi anemia pathway guards the genome. Annu Rev Genet 43 223 249
74. GodthelpBC
WiegantWW
WaisfiszQ
MedhurstAL
ArwertF
2006 Inducibility of nuclear Rad51 foci after DNA damage distinguishes all Fanconi anemia complementation groups from D1/BRCA2. Mutat Res 594 39 48
75. TarsounasM
DaviesD
WestSC
2003 BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene 22 1115 1123
76. MaackG
SegnerH
2003 Morphological development of the gonads in zebrafish. J Fish Biol 62 895 906
77. WangX
BartfaiR
Sleptsova-FreidrichI
OrbanL
2007 The timing and extent of ‘juvenile ovary’ phase are highly variable during zebrafish testis differentiation. Journal of Fish Biology 70 33 44
78. SiegfriedKR
Nusslein-VolhardC
2008 Germ line control of female sex determination in zebrafish. Dev Biol
79. DraperBW
McCallumCM
MoensCB
2007 nanos1 is required to maintain oocyte production in adult zebrafish. Dev Biol 305 589 598
80. HouwingS
KammingaLM
BerezikovE
CronemboldD
GirardA
2007 A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell 129 69 82
81. PaigenKea
2008 The recombinational anatomy of a mouse chromosome. PLoS Genet 4 e1000119
82. MooreJL
RushLM
BrenemanC
MohideenMA
ChengKC
2006 Zebrafish genomic instability mutants and cancer susceptibility. Genetics 174 585 600
83. AuerbachAD
2009 Fanconi anemia and its diagnosis. Mutat Res 668 4 10
84. HirshAE
FraserHB
2001 Protein dispensability and rate of evolution. Nature 411 1046 1049
85. AmsterdamA
HopkinsN
2006 Mutagenesis strategies in zebrafish for identifying genes involved in development and disease. Trends Genet 22 473 478
86. WesterfieldM
1995 The zebrafish book: a guide for the laboratory use of zebrafish (Danio rerio). Eugene, , OR University of Oregon Press
87. YanYL
MillerCT
NissenR
SingerA
LiuD
2002 A zebrafish sox9 gene required for cartilage morphogenesis. Development 129 5065 5079
88. WeidingerG
SteblerJ
SlanchevK
DumstreiK
WiseC
2003 dead end, a novel vertebrate germ plasm component, is required for zebrafish primordial germ cell migration and survival. Curr Biol 13 1429 1434
89. ReimersMJ
La DuJK
PerieraCB
GiovaniniJ
TanguayRL
2006 Ethanol-dependent toxicity in zebrafish is partially attenuated by antioxidants. Neurotoxicol Teratol 28 497 508
90. HigginsDG
SharpPM
1988 CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene 73 237 244
91. PerriereG
GouyM
1996 WWW-query: an on-line retrieval system for biological sequence banks. Biochimie 78 364 369
92. KyteJ
DoolittleRF
1982 A simple method for displaying the hydropathic character of a protein. J Mol Biol 157 105 132
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy