
Testing for an Unusual Distribution of Rare Variants
Technological advances make it possible to use high-throughput sequencing as a primary discovery tool of medical genetics, specifically for assaying rare variation. Still this approach faces the analytic challenge that the influence of very rare variants can only be evaluated effectively as a group. A further complication is that any given rare variant could have no effect, could increase risk, or could be protective. We propose here the C-alpha test statistic as a novel approach for testing for the presence of this mixture of effects across a set of rare variants. Unlike existing burden tests, C-alpha, by testing the variance rather than the mean, maintains consistent power when the target set contains both risk and protective variants. Through simulations and analysis of case/control data, we demonstrate good power relative to existing methods that assess the burden of rare variants in individuals.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1001322
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001322
Summary
Technological advances make it possible to use high-throughput sequencing as a primary discovery tool of medical genetics, specifically for assaying rare variation. Still this approach faces the analytic challenge that the influence of very rare variants can only be evaluated effectively as a group. A further complication is that any given rare variant could have no effect, could increase risk, or could be protective. We propose here the C-alpha test statistic as a novel approach for testing for the presence of this mixture of effects across a set of rare variants. Unlike existing burden tests, C-alpha, by testing the variance rather than the mean, maintains consistent power when the target set contains both risk and protective variants. Through simulations and analysis of case/control data, we demonstrate good power relative to existing methods that assess the burden of rare variants in individuals.
Introduction
High throughput sequencing of the human genome is now a reality: recent advances in sequencing technology now permit near complete ascertainment of genetic variation, including rare variants (<1% population frequency), across large portions of the genome in thousands of individuals. While this in principle can reveal the role of each gene in every medical phenotype, the analytic challenges are profound. Of particular concern are genetically complex common diseases for which the role of any gene is expected to be quite modest and an individual rare variant would have relatively small impact on the common endpoint. Under this scenario there would be little power when testing one variant at a time, as in traditional association testing. For example, while in African Americans low-frequency variants in PCSK9 can have a substantial effect on serum low-density lipoprotein cholesterol (LDL-C) [1], these variants influence risk or protection to myocardial infarction by only a factor of 2 [2], [3]. In such instances, rare variants will almost never stand out as associated individually, particularly when all variation in the genome is measured and tested in an effort to discover novel loci, rather than simply evaluate existing candidate genes.
Recently published methods show that power to detect rare risk variation can be greatly enhanced by combining information across variants in a target region, such as a gene or exon, when multiple variants influence phenotype. The “cohort allelic sums test” (CAST) [4], [5] and “Combined Multivariate and Collapsing (CMC) method” [6] use this approach. CAST contrasts the number of individuals with one or more mutations between cases and controls. Like CAST, in CMC all rare variants are collapsed and treated as a single count for analysis with common variants in a multivariate test. CMC permits a coherent test for common and rare variants (rare being defined arbitrarily, but usually at 1%). Madsen and Browning [7] introduced a non-parametric weighted sum test in which rare variants “are grouped according to function (e.g. gene), and each individual is scored by a weighted sum of the mutation counts.” The incorporation of weights improves the power of the test, and would be especially powerful when most of the rare variation is functionally relevant. While each of these rare variant tests differs in form, each seeks to assess the overall genetic burden due to rare variants, hence we call them “burden tests”. By design, they implicitly assume that all variation affecting phenotype acts in the same direction.
Even in a gene harboring phenotypically relevant variation, however, many variants will be phenotypically neutral. Indeed, the target region could include a handful of rare Mendelian mutations that cause disease, some variants that moderately increase or decrease risk, along with numerous variants of no effect. To gain insight into a new model for analysis, it is helpful to think of a coin toss associated with each variant. If the variant is phenotypically neutral, the coin is fair and the variant is as likely to appear in a case as it is in a control. In contrast, risk variants correspond to biased coins and are more likely to be observed in cases. Similarly, protective variants correspond to coins biased in the opposite direction and are more likely to be observed in controls, particularly if they are selected. See Figure 1, which illustrates this scenario and motivates a new testing procedure. Burden tests seek to determine if the coin is biased, on average, across all variants. Invariably they include phenotypically neutral variants in the burden score, which diminishes the power of the test. In addition, there are many examples in which gain or loss of function in the same gene have opposite effects on phenotype [8]-[10].

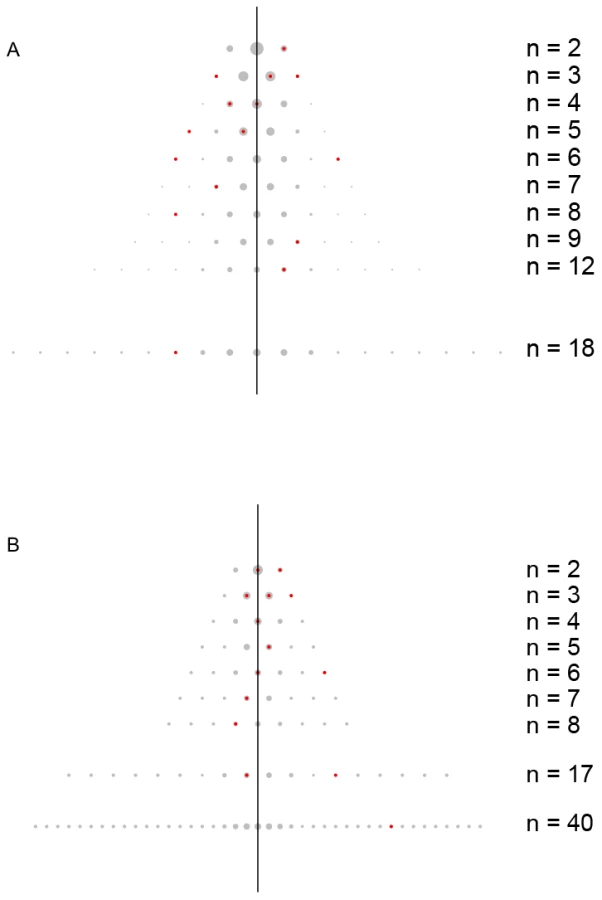
Table 1 illustrates the challenges by presenting novel sequencing of the gene APOB (Apolipoprotein-B) in 96 individuals who have high triglycerides and 96 individuals who have low triglycerides. Do rare variants in APOB influence lipid levels? Figure 2A illustrates the distribution of APOB variants amongst high/low individuals. Notice the unlikely occurrence of variants with 6 : 0 and 0 : 6 counts out of fewer than 20 variants discovered. A test that incorporates this increase in variance, or overdispersion, could in principle give better power. This observation suggests a novel approach to the problem: tally the number of copies of each variant in cases, relative to the number copies in controls, and evaluate overdispersion in the set of counts. Overdispersion in this setting measures an increase from the expected binomial variance, driven by a subset of variants seen preferentially in cases or controls (the biased coins in the example in Figure 1).

A well-established and powerful test for the presence of a mixture of biased and neutral coins is the C-alpha score-test [11], [12]. We describe here the adaptation of this test for the analysis of sequence level case-control data and demonstrate its performance in a variety of simulated and actual data examples. We then move to a few key assumptions of C-alpha and how to accommodate realistic scenarios that violate those assumptions. Finally, we propose extensions of the method.
Methods
C-alpha test
To illustrate where the information for C-alpha originates, consider a standard balanced case-control study. If the target region has no alleles associated with the phenotype, then the distribution of counts should follow a binomial distribution, indexed by n, the total number of copies of an observed variant. In Figure 2 we contrast the expected distribution of binomial counts (background) with observed counts (foreground) for three phenotypes. Each row of the triangle corresponds with a different value of n. The number of distinct variants observed for panel (b) is m = 14 with n ranging from 2 to 40. The number of variants with n copies, m(n), varies from row to row: m(2) = 2, m(3) = 3, …, and m(40) = 1. C-alpha detects unusual numbers of counts falling toward the outer edges of the triangle; In Figure 2A, one variant is observed exclusively in cases (6 : 0) and another in controls (0 : 6). Both configurations generate overdispersion. Any mixture of binomials leads to overdispersion, which can be detected by a one-sided test. This is the fundamental basis of C-alpha. See Figure S1 for further illustration.
We have tailored the C-alpha score test so that it is suitable for testing a set of rare variants for association. The binomial (n,p) distribution evaluates the probability of observing a particular variant y times in the cases out of n total, assuming the rare variants are distributed at random across the subjects. For variants seen twice (doubletons) in a balanced sample of cases and controls (p = 0.5), we expect y to be 0, 1 and 2 with probability ¼, ½ and ¼, respectively. We typically will observe a higher proportion of doubletons with y = 2 and/or y = 0 than expected, if some variants are detrimental or protective. For each variant, there will be insufficient information from which to draw firm conclusions about association. C-alpha can be used to detect a pattern across the full set of rare variants in the target region. For the i'th variant, observed ni times, we assume yi is binomial (ni,pi). Under the null hypothesis, pi = p0 (say ½ if cases and controls are equal in number and we expect rare variants to fall in either sample at random). The alternative hypothesis is that pi follows a mixture distribution across the m variants, I = 1,…,m, with some variants detrimental (pi>p0), some neutral, and some protective (pi<p0).
The C-alpha test statistic T contrasts the variance of each observed count with the expected variance, assuming the binomial distribution
To standardize this quantity we require c, the variance of T:in which m(n) is the number of variants with n copies, and denotes the probability of observing u copies of the i'th variant assuming the binomial model.
The resulting test statistic is defined as . We reject the null hypothesis when Z is larger than expected using a one-tailed standard normal distribution for reference. See the Text S1 for a derivation of the C-alpha test.
Let's examine data from two genes, PCSK9 and APOB, in which rare variation is known to affect lipid levels (LDL and Triglycerides), and treat the phenotypic extremes as binary traits (i.e. high lipid levels are cases and low lipid levels are controls). The previously reported PCSK9 data result from sequencing its coding regions [1] in 128 individuals with extremely high and 128 with extremely low plasma LDL-C levels. Applying C-alpha produces a significant association (p-value = 0.0023.) The loss of functional variants (C679X, Y142X) in PCSK9 are associated with a 28 percent reduction in mean LDL cholesterol and an 88 percent reduction in the risk for coronary heart disease [2]. The gain of function variant (H553R) in PCSK9 is associated with increased plasma levels of LDL-C [13]. The pattern of overdispersion is also apparent when examining the genotype counts derived from a pooled sequencing experiment of coding regions in APOB (Figure 2A). We do not report a P-value for the APOB example, as these data are pooled and so we cannot assess significance empirically. In this case DNA from 96 individuals with extremely high triglyceride levels was pooled, as was DNA from 96 individuals with extremely low triglyceride levels [3], and these two pools were sequenced. Another example of rare variation predisposing to disease is clearly evident from a study of Crohn's disease (Figure 2B). Association of low frequency variation at NOD2 (excluding two common coding variants R702W and insCfs1003) is demonstrated in 350 Crohn's disease cases and 350 GWAS matched controls with a P-value of less than 10−6 from individual level data.
Estimation of mixtures
It is additionally possible to estimate the underlying mixture model from the distribution of variants used for C-alpha testing. For instance, if the target region includes risk variants, phenotypically neutral variants, and variants that engender a modest protective effect, then a 3-component mixture will fit the data corresponding to the 3 genetic components. Whenever C-alpha shows significance, we can estimate the number of mixture components and the posterior probability that a particular variant is detrimental or protective using the EM algorithm (see Text S1 and Figure S4).
Singleton counts
A variant observed only once provides no direct information about over-dispersion; however the distribution of singletons as a group reflects on the question of association between the target region and phenotype. Singletons can be pooled into a single binomial count that can be included in the C-alpha test. This treatment of singletons, which is essentially identical to a burden test (see Text S1 on mixtures of biased and unbiased coins and Figure S1), allows robust integration of singletons. However, it can only be informative if the majority of the singleton variants have effects in the same direction. Other approaches to addressing singleton variants in parallel with C-alpha are considered in the discussion.
Simulation experiments
We conduct two main sets of simulations to compare C-alpha with Li and Leal burden tests and Madsen and Browning's test. Tests proposed by Li and Leal are all built around a regression model, predicting phenotype based on recoding of rare variation. For Li and Leal's approach, we sum the number of rare variants in the region for each individual as the predictor. For Madsen and Browning, the coding of variation is similar to the sum of rare variants, but a weighting scheme based on the inverse of the control allele frequency is included. The test statistic is evaluated as a nonparametric rank sum test in which each individual is scored by a weighted sum of the mutation counts. We also include a variable threshold model, which is an implementation of a burden test that selects the threshold for inclusion of variants by optimizing the test statistic. Specifically, this burden test is calculated at all allele frequency cutoffs. The test statistic is then defined by the maximum of the test statistics for all cutoffs. The distribution of the test statistic is obtained empirically by random permutation of case/control status and recalculation of the test statistic. This approach is described in Price et al [14]. In addition to these simulations under the alternative hypothesis, we present a series of simulations under the null hypothesis to investigate the small sample properties of the test statistic. For each kind of simulation and for all test statistics, we assess significance empirically, by using permutation, as described previously. For each test, we permute 50,000 times to ensure accurate assessment at P-value threshold of 0.001. Singletons are treated in accordance with the original specification for each method. For C-alpha we pooled the singletons into a single observation.
We first present a set of simulations for a population genetics model that incorporates selection [15], and generates data that matches the empirically observed allele frequency distribution derived from sequence data. This approach implements a demographic model by which variants in the region are in linkage equilibrium (simulated, not forced) and mutations have selection coefficients above 10−3. Two genes were simulated, and the mean liability value for an individual differed as a function of the number of rare alleles occurring at these genes [14], [15]. For individuals who have no rare variants, liability was simulated using a N(0,1). Each functional variant increased the mean of the liability distribution by 0.25, while maintaining the variance of 1. Each simulated gene was 1.5 kb long, with a mean and variance of number of variants at ∼38.5 and 40 respectively. Of the variants observed, 52% were functional. Note that this constitutes a favorable scenario for frequency weighted burden testing as the burden continues to increase with additional variants and the frequency of the functional variants is generally kept lower. To expand these simulations, we then randomly assigned a direction of effect for these variants, according to a range of mixing proportions (0 to 50% probability of assignment of protective in 10% increments).
For the second set of risk simulations, we assume a model with a disease prevalence of 1%, 50 sites in the region of analysis with allele frequency between 0.025% and 0.5% (this distribution is similar to that observed from the Crohn's data). These sites are variable in the population, but may be invariant in any given simulation, as they are probabilistically assigned for each member of the sample. Of these 50 variants, 6 are chosen at random for each simulation to affect the phenotype (regardless of whether they are present in the dataset), and each variant explains 0.1% of the variance of the disease under a liability threshold model (i.e. 2p [1-p]a2 = 0.001, where p is the risk allele frequency and a is the effect on mean liability [16]). The model assigns higher penetrance to rarer alleles, yet no alleles are so highly associated that they would be readily detectable by single variant analyses (see Text S1 and Figure S3 for the relationship between odds ratio and frequency). Furthermore, the functional variants may be singletons in the final dataset, especially if they have a very rare population allele frequency.
Three scenarios are explored: all 6 variants confer risk; 3 variants confer risk and 3 variants confer protection; and all 6 variants confer protection. We also consider two different study designs: 1,000 cases (individuals who exceed the threshold on the liability distribution) and 1,000 controls (individuals selected for absence of disease); and 1,000 cases and 1,000 selected controls, such that the controls are selected to be in the bottom 1% of the latent liability distribution. The latter strategy mimics experimental designs used for quantitative phenotypes.
Small sample properties
A third set of simulations explores behavior of test statistics under the null hypothesis to determine whether the type I error rate is well calibrated for C-alpha. Like many tests, C-alpha relies on asymptotic properties consistent with the central limit theorem (CLT). Specifically, the test statistic converges to a normal distribution under the null hypothesis as the number of variants tends toward infinity, with convergence being potentially faster if the frequency of all variants is similar. Thus, we varied both the number of variants and the distribution of allele frequencies to explore type I error. For these simulations, we drew N variants from the empirically observed allele frequency distribution used in the second set of simulations above and performed 25,000 replicates for each value of N.
Results
Results of power comparisons
From simulation results evaluating power (Figure 3), C-alpha shows comparable or slightly better power than burden testing in the situation where all effects are in the same direction, and much greater power when protective and risk variants exist in the test set. Thus the mixture approach shows good power for a much broader set of scenarios without sacrificing power when unidirectional burden testing is also effective.

Results assessing asymptotic properties
Simulation results (Table 2) demonstrate that the tails of the distribution are somewhat heavier than the assumed normal distribution when the number of variants is small, but as expected, they approach that of the assumed distribution when the number of variants increases. Additionally, in simulations performed under a single allele frequency, the test achieves asymptotic behavior considerably more quickly. To estimate significance accurately when small numbers of variants are under consideration, we therefore recommend a standard permutation procedure whereby we randomly reassign cases and controls and recalculate the test statistic (just as would be applied in any association scenario involving small sample numbers and/or frequencies). Regardless of the slightly heavy tails for small numbers of variants, the test statistic itself is still a fast, effective screen for identifying potential regions of interest in the genome. We note that our power calculations and any examples involving sequencing of individual samples use p-values obtained by permutation of case-control status. Additionally, C-alpha assumes independence of each observed variant. Permutation yields appropriate p-value distributions even in the presence of LD between variants because case-control permutation maintains the LD relationship between all SNPs [17].

Discussion
We have demonstrated here the adaptation of the C-alpha test statistic and its broad applicability to medical sequence data on a gene or pathway level. The approach, distinct from more traditional burden testing, has several advantages over the previously proposed test statistics. Its primary advantage is sensitivity to risk and protective variants in the same gene or pathway. Yet, even if the effects of rare alleles are uniformly in one direction, such as increasing risk, C-alpha maintains comparable power to burden tests. Grouping genes together into pathways and testing rare variants falling into these groups of genes could provide greater statistical power and biological insight into the functionally relevant processes affecting the phenotype of interest. In such groups the presence of both risk and protective variants is even more likely. As demonstrated here, the C-alpha test is well calibrated to incorporate such divergent effects on risk. Moreover because it is a single degree of freedom test with normal asymptotic properties, C-alpha enhances power and allows for rapid and straightforward calculation.
As with other burden-style tests, C-alpha is designed for situations in which numerous rare variants are observed in the target region. We recommend permutation testing for accurate significance estimation in scenarios where the asymptotic behavior is not assured - in particular, for small numbers of variants, when LD is present, or when the test is driven by a single more common variant. In this latter scenario, as in the example of NOD2, it will likely be desirable to analyze the more common variant with Fisher's exact test and to reanalyze the remaining rarer variation to search for additional signal.
While singleton variants can be combined in C-alpha, it seems reasonable to consider a distinct analysis of singletons in a more biologically motivated fashion. For example, when a truly rare and fully penetrant mutational origin is suspected, one might focus on variants seen only in cases and not seen in external reference data such as from 1000 Genomes Project. One might then further filter the identified variants to leave only putatively deleterious non-synonymous, splice and obvious loss of function variants and then compare the rate of these to the parallel set found in controls only. Such a Mendelian-style analysis (already successfully applied in several cases such as Miller syndrome [18]), focuses predominantly or exclusively on singleton variants and is almost completely independent of the complementary C-alpha analysis applied to variants seen twice or more in a study. For a polygenic disease, it is reasonable to predict that each of these models could be relevant for different genes (and in some cases, such as APOB, the same gene). Thus complementary and independent analyses may be a desirable approach to surveying large, exome-scale datasets.
As with any statistical test, the presence of confounders can seriously bias the results. For instance, we have demonstrated that population stratification has a significant impact on rare variation tests. C-alpha assumes cases and controls have the same balance of ancestry, because the distribution of rare variants likely depends on ancestry. Otherwise unequal representation could increase the rate of false positives for any rare variant test. It is important to note that the balanced sampling assumption is not equivalent to requiring that samples be homogenous. Balanced sampling requires solid experimental design. While statistical methods for controlling for bias can be effective, proper study design is far preferable. If a large set of genotypes were available, such as from a genome-wide association (GWA) study, principal component analyses and related procedures can be used to match or control for ancestry [19]–[21]. For well-matched case-control samples, under the null hypothesis, it is reasonable to assume the distribution of rare variants is independent of case/control status; i.e., a variant is as likely to be identified in a case as a control. One approach to determining whether the assumption of balanced ancestry is violated is to calculate the number of rare synonymous variants observed in cases versus controls across the sequenced regions. If there is a substantive difference between cases and controls in the number of synonymous variation, one explanation is that the matching of ancestry between cases and controls was not successful. If the deviation is modest, that implies that the level of ancestral mismatching is in all likelihood modest. One other major source of bias is differential genotyping bias. For sequencing experiments, variability in coverage and variant discovery between cases and controls can behave similarly to population stratification if such errors are not balanced between cases and controls. As with population structure, appropriate balance of subjects to control for technical variability before analysis should insulate sequencing experiments from major bias, yet subtle excess variability might persist. For well designed studies we propose that Genomic Control [22] will help to correct for the effects of minor stratification and technical variability (for details, see Text S1 and Figure S2).
More generally, the choice of target region and set of variation affects power. A test based on all potentially functional variants (non-synonymous, splice, nonsense, etc.) within a single gene will often be effective as it strikes a balance between sufficient numbers of variants and the enrichment of functional variation. In contrast, a single exon is not likely to have enough variants for the test to be powered adequately or to achieve asymptotic properties and, while including introns and synonymous variants would dramatically increase the number of variants, the expectation is that the vast majority of these variants will be phenotypically neutral and thus will mute the signal. Alternatively, a group of exons from related genes (e.g., a biological pathway) could be analyzed jointly. The test would then determine if some unspecified variants in the pathway are associated with the phenotype via a deviation from the expected distribution of variation. Using this strategy, if several of the genes have an effect, then the power will be enhanced; however, if only one gene in the pathway has an effect, including the other genes in the test will reduce power to detect the effect. For target regions that do show evidence of association, using a nonparametric mixture model we can estimate the distribution of pi across variants and estimate the posterior probability the i'th variant is detrimental or protective [23]. In the application to a pathway of genes, one is more likely to uncover both risk and protective variants, making the proposed test even more desirable.
Selecting a subset of variants, as suggested above, is just one form of weighting variants in the analysis. C-alpha allows for valid weights to be incorporated into the calculation of the test statistic (see Text S1). Tests that up-weight variants based on allele frequencies find that power can be improved, if there is a relationship between allele frequency and effect size [7]. However, we found that using the allele frequencies derived from the experiment bias the test. The 1000 Genomes Project provides an independent source for weights for allele frequency weighting. Alternatively, if weights are obtained from the data at hand, a permutation procedure can be used to obtain P-values. Aside from allele frequency, bioinformatic tools that predict the functionality of a variant offer another source for weights. Many computational tools classifying coding variants regarding impact on protein function are already published In Silico Funcitonal Profiling [24] POLYPHEN [25], SIFT [26], SNPs3D [27], and Pmut [28]. For example, for POLYPHEN the larger the position-specific score the more likely the substitution is deleterious and applying such weights to each variant in C-alpha might further enhance power. Similar tools are available to score functionality for consensus transcription factor binding sites (TFSEARCH, MATINSPECTOR) and 3’-UTR (ASTRA) [29].
Sequencing technology will continue to develop and reduce in cost for the foreseeable future. Large medically-focused sequencing efforts involving thousands of exomes or whole genomes are now underway and are introducing a host of novel computational challenges not encountered in GWAS and previous large-scale medical genetics studies. By enabling powerful analyses of genes and pathways, without concern for effect direction, C-alpha promises to be a flexible and powerful approach for the identification of functionally relevant regions from experiments involving deep sequencing.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. CohenJ
PertsemlidisA
KotowskiIK
GrahamR
GarciaCK
2005 Low ldl cholesterol in individuals of african descent resulting from frequent nonsense mutations in pcsk9. Nat Genet 37 2 161 165
2. CohenJC
BoerwinkleE
MosleyTH
HobbsHH
2006 Sequence variations in PCSK9, low ldl, and protection against coronary heart disease. N Engl J Med 354 12 1264 1272
3. KathiresanS
MelanderO
AnevskiD
GuiducciC
BurttNP
2008 Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med 358 12 1240 9, PMID 18354102
4. CohenJC
KissRS
PertsemlidisA
MarcelYL
McPhersonR
2005 Multiple rare alleles contribute to low plasma levels of hdl cholesterol. Science 305 5685 869 872
5. MorgenthalerS
ThillyWG
2007 A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (cast). Mutat Res 615 1-2 28 56
6. LiB
LealSM
2008 Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 83 3 311 321
7. MadsenBE
BrowningSR
2009 A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet 5 e1000384 doi:10.1371/journal.pgen.1000384
8. AbifadelM
RabesJP
DevillersM
MunnichA
ErlichD
2009 Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum Mutat 30 4 520 529
9. BennM
2009 Apolipoprotein b levels, apob alleles, and risk of ischemic cardiovascular disease in the general population, a review. Atherosclerosis 206 1 17 30
10. Newton-ChehC
ShahR
2007 Genetic determinants of qt interval variation and sudden cardiac death. Curr Opin Genet Dev 17 3 213 221
11. NeymanJ
ScottE
1966 On the use of c(α) optimal tests of composite hypotheses. Bulletin of the International Statistical Institute 41 477 497
12. ZeltermanD
ChenC
1988 Homogeneity tests against central-mixture alternatives. Journal of the American Statistical Association 83 401 179 182
13. KotowskiIK
PertsemlidisA
LukeA
CooperRS
VegaGL
2006 A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet 78 3 410 422
14. PriceAL
KryukovGV
deBakkerPIW
PurcellSM
StaplesJ
2010 Pooled Association Tests for Rare Variants in Exon-Resequencing Studies. Am J Hum Genet 86 6 832 838
15. KryukovGV
ShpuntA
StamatoyannopoulosJA
SunyaevSR
2009 Power of deep, all-exon resequencing for discovery of human trait genes. Proc Natl Acad Sci U S A 106 10 3871 3876
16. FisherRA
1918 The correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinburgh 52 399 433
17. CheverudJM
2001 A simple correction for multiple comparisons in interval mapping genome scans. Heredity 87 52 58
18. NgSB
BuckinghamKJ
LeeC
BinghamAW
TaborHK
2010 Exome sequencing identifies the cause of a mendelian disorder Nature Genetics 42 30 35
19. PriceAL
PattersonNJ
PlengeR
WeinblattM
ShadickN
2006 Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38 8 904 909
20. LucaD
RingquistS
KleiL
LeeAB
GiegerC
2008 On the use of general control samples for genome-wide association studies: genetic matching highlights causal variants. Am J Hum Genet 82 2 453 63
21. DevlinB
RoederK
1999 Genomic control for association studies. Biometrics 55 4 997 1004
22. LeeAB
LucaD
KleiL
DevlinB
RoederK
2010 Discovering genetic ancestry using spectral graph theory. Genet Epidemiol 34 1 51 59
23. LindsayBG
RoederK
1992 Residual diagnostics for mixture models. Journal of the American Statistical Association 87 419 785 794, 1992
24. MortM
EvaniUS
KrishnanVG
KamatiKK
BaenzigerPH
2010 In Silico Functional Profiling of Human Disease-Associated and Polymorphic Amino Acid Substitutions. Human Mutation 31 3 335 46
25. SunyaevS
RamenskyV
KochI
LatheW
KondrashovAS
2001 Prediction of deleterious human alleles. Hum Mol Genet 10 6 591 597
26. NgPC
HenikoffS
2001 Predicting deleterious amino acid substitutions. Genome Res 11 5 863 874
27. YueP
MelamudE
MoultJ
2006 Snps3d: candidate gene and snp selection for association studies. BMC Bioinformatics 7 166
28. Ferrer-CostaC
GelpiJL
ZamakolaL
ParragaI
de la CruzX
2005 Pmut: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 21 14 3176 3178
29. MignoneF
GrilloG
LicciulliF
IaconoM
LiuniS
2005 Utrdb and utrsite: a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mrnas. Nucleic Acids Res 33 Database issue D141 6
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy