
Triplet Repeat–Derived siRNAs Enhance RNA–Mediated Toxicity in a Drosophila Model for Myotonic Dystrophy
More than 20 human neurological and neurodegenerative diseases are caused by simple DNA repeat expansions; among these, non-coding CTG repeat expansions are the basis of myotonic dystrophy (DM1). Recent work, however, has also revealed that many human genes have anti-sense transcripts, raising the possibility that human trinucleotide expansion diseases may be comprised of pathogenic activities due both to a sense expanded-repeat transcript and to an anti-sense expanded-repeat transcript. We established a Drosophila model for DM1 and tested the role of interactions between expanded CTG transcripts and expanded CAG repeat transcripts. These studies revealed dramatically enhanced toxicity in flies co-expressing CTG with CAG expanded repeats. Expression of the two transcripts led to novel pathogenesis with the generation of dcr-2 and ago2-dependent 21-nt triplet repeat-derived siRNAs. These small RNAs targeted the expression of CAG-containing genes, such as Ataxin-2 and TATA binding protein (TBP), which bear long CAG repeats in both fly and man. These findings indicate that the generation of triplet repeat-derived siRNAs may dramatically enhance toxicity in human repeat expansion diseases in which anti-sense transcription occurs.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1001340
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001340
Summary
More than 20 human neurological and neurodegenerative diseases are caused by simple DNA repeat expansions; among these, non-coding CTG repeat expansions are the basis of myotonic dystrophy (DM1). Recent work, however, has also revealed that many human genes have anti-sense transcripts, raising the possibility that human trinucleotide expansion diseases may be comprised of pathogenic activities due both to a sense expanded-repeat transcript and to an anti-sense expanded-repeat transcript. We established a Drosophila model for DM1 and tested the role of interactions between expanded CTG transcripts and expanded CAG repeat transcripts. These studies revealed dramatically enhanced toxicity in flies co-expressing CTG with CAG expanded repeats. Expression of the two transcripts led to novel pathogenesis with the generation of dcr-2 and ago2-dependent 21-nt triplet repeat-derived siRNAs. These small RNAs targeted the expression of CAG-containing genes, such as Ataxin-2 and TATA binding protein (TBP), which bear long CAG repeats in both fly and man. These findings indicate that the generation of triplet repeat-derived siRNAs may dramatically enhance toxicity in human repeat expansion diseases in which anti-sense transcription occurs.
Introduction
Trinucleotide repeat expansions within non-coding regions of RNA cause pathogenesis in a number of human diseases, including myotonic dystrophy type 1 (DM1), fragile X-associate tremor and ataxia syndrome (FXTAS), spinocerebellar ataxia type 8 (SCA8), and Huntington's disease-like 2 (HDL2) [1]-[3]. The causative mutations of DM1, SCA8 and HDL2 are CTG repeat expansions. In DM1, the CTG expansion is located within the 3′ untranslated region (3′UTR) of the dystrophia myotonica-protein kinase (DMPK) gene [4], [5]. The expanded CUG repeat RNA forms ribonuclear foci, and mislocalizes and misregulates RNA binding proteins such as CUG-BP1 and MBNL1 that influence alternative splicing [6]-[8]. Similarly, expanded CUG RNA also contributes to pathophysiology of SCA8 and HDL2 [9], [10]. These findings indicate that CTG-based RNA expansion diseases may have the accumulation of RNA foci, sequestration of MBNL1, and disruption of alternative splicing as common components.
It is now recognized, however, that more than 70% of genomic loci show evidence of transcription from both sense and anti-sense strands in the mammalian genome [11], thus many of the trinucleotide repeat disease loci may display bidirectional transcription. Indeed, anti-sense transcripts have been detected for most trinucleotide repeat disease loci, including DM1 and SCA8 [12]-[16]. In DM1 cells, the sense and antisense transcripts can cause regional chromatin modification [15]. In SCA8, an antisense CAG transcript can be translated into a polyglutamine-encoding protein [14]. Given that anti-sense transcription occurs widely in the human genome [11], [17], [18], defining the range of potential roles and impact of anti-sense repeat transcripts on trinucleotide repeat diseases may provide novel insight into disease pathogenesis.
Drosophila has proven a powerful system to reveal insight into neurological and neurodegenerative disease with relevance to the human situation [19]-[22]. Thus, to gain insight into CUG RNA toxicity in the DM1 disease situation, we established transgenic flies that express pure, uninterrupted CTG repeat expansions in the 3′UTR of a control protein DsRed. These flies recapitulate major features of human CUG RNA expansion diseases. Given the finding that ∼70% of genes show anti-sense transcription, we then tested the effect of co-expressing CTG and CAG disease transcripts. These data revealed dramatically enhanced toxicity upon co-expression of these transcripts; studies indicate this is due to the generation of triplet repeat-derived siRNAs which can target other repeat containing transcripts. These findings suggest that sense and anti-sense expanded repeat transcripts may interact in vivo to generate small RNAs that may dramatically enhance pathology in disease situations.
Results
Expression of expanded CUG RNA causes repeat-length dependent toxicity
To study CUG repeat RNA toxicity in flies, we generated UAS constructs with a pure CTG repeat expansion of 250 in length, (CTG)250, within the 3′UTR of DsRed (Figure 1A). Due to instability of the repeat in E. coli, we obtained clones with various repeat lengths and generated a series of transgenic lines that together encompassed a range of CTG repeat lengths (Figure 1B). Among these, we selected six lines bearing different repeat lengths that expressed the transcript at comparable levels (Figure 1C and 1D).

With expression ubiquitously using daughterless-gal4 (da-gal4), in the nervous system with elav-gal4, or in muscle with 24B-gal4, repeat-length dependent lethality was observed (Table 1). When expression was targeted selectively to the eye, the animals showed abnormal eye pigmentation and disruption of retinal integrity, the severity of which was dependent on repeat length (Figure 1E). Flies bearing the longest repeats also showed variability in severity which may be a feature of a pure repeat sequence (Figure 1E and Figure S1, see Discussion). Taken together, these studies indicate that non-coding, uninterrupted CTG repeats confer length-dependent toxicity when expressed in brain and muscle in flies. As in mammals, the expanded CTG repeat transcripts formed RNA accumulations in muscle nuclei in flies (Figure S2; also [23], [24]), and affected alternative splicing (Figure S2B and S2C). These data indicate that the fly recapitulates fundamental key features of the CTG expansion disease DM1 (also [23]-[25]).

Enhancement of CTG-repeat toxicity by CAG-repeat transcripts
Bi-directional transcription is prevalent in the mammalian genome and is thought to occur in DM1 [11], [12], [15], [16]. We therefore asked whether expression of a non-coding CAG repeat transcript together with a CTG repeat transcript would have an effect distinct from that of the CTG repeat transcript alone.
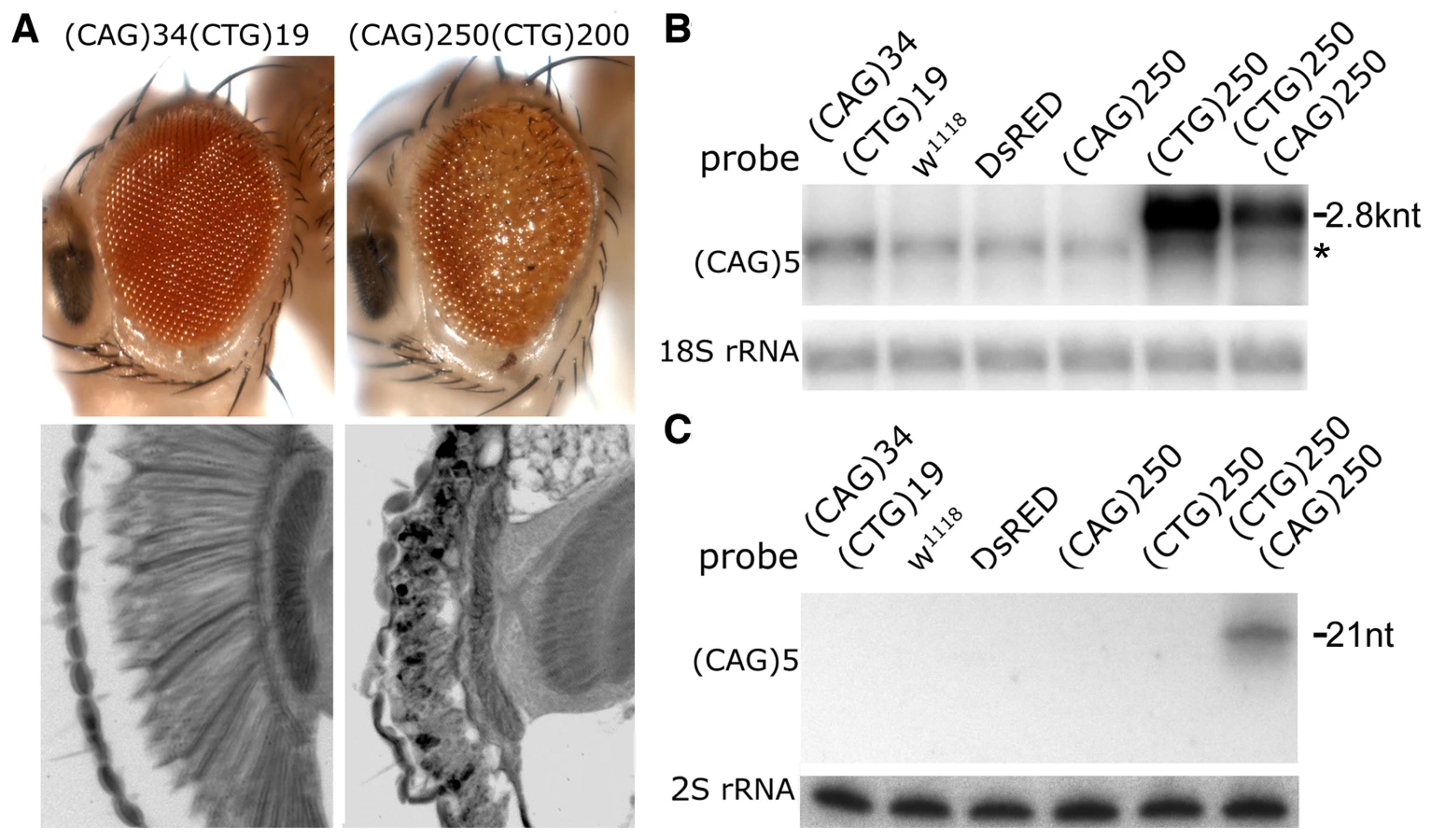
Toxicity of non-coding (CAG)250 repeat transcripts has been previously characterized, as adult-stage late-onset neurological dysfunction and loss [26]. Expression of either (CAG)250 or (CTG)200 in the eye with gmr-gal4, however, causes minimal effects (Figure 1E) [26]. Co-expression of (CTG)200 together with a (CAG)250 transcript, however, resulted in dramatic toxicity: the eye was now severely rough with abnormal pigmentation, demonstrating severe loss of retinal integrity (Figure 2A). The effect was synergistic, as expression of either two copies of a (CAG)250 or two copies of (CTG)200 repeat alone had limited or no effects (Figure S3). Additional combinations of repeat lengths with gmr-gal4 indicated that co-expression of (CTG)250 or (CTG)270 with (CAG)250 repeats caused lethality at pre-adult pupal stages and/or generated adults with severely disrupted eyes, depending upon the precise combination of transgenes (Figure 3A and 3D, and data not shown). The interaction was dependent on disease-length repeat expansions, since flies co-expressing small (CTG)19 and (CAG)34 repeats did not show toxicity (Figure 2A).

A toxic interaction between CTG/CAG transcripts was also seen using a heat shock driver in adults, and a muscle driver 24B. Adult flies with a 30 min heat shock induction of (CAG)250 and (CTG)250 transcripts started to die at ∼24 h, with 90% flies dead at 50 h (Figure 3B). In contrast, flies expressing (CAG)250 alone, (CTG)250 alone or non-pathogenic (CTG)19/(CAG)34 transcripts were not affected. In muscle, co-expression of (CAG)100 with (CTG)130 caused developmental lethality, while (CTG)19/(CAG)34, (CAG)100, and (CTG)130 flies were viable (Figure 3C). Taken together, these data indicate that co-expression of disease-length CTG repeat transcripts together with comparable CAG repeat transcripts causes synergistic pathogenesis.
CTG/CAG transcripts are processed into small RNAs
To define the basis of the enhanced toxicity upon co-expressed CTG and CAG transcripts, we reasoned that the stability of each transcript may become greater, such that each transcript then displays greater toxicity. However, northern analysis indicated that the levels of the full-length repeat mRNAs were reduced (Figure 2B and Figure S4A), arguing against increased transcript stability accounting for the enhanced toxicity.
A second possibility was that novel interactions between the two transcripts were taking place, causing an effect distinct from either transcript on its own. Small RNA northern analysis revealed that small RNAs of ∼21 nt, detected with either (CAG)5 or (CUG)5 probes, were generated in the co-expression situation (Figure 2C and Figure S4B). These data indicated that expanded CAG and CTG transcripts are processed into triplet repeat-derived small RNAs when co-expressed.
Toxicity of co-expressed CTG/CAG transcripts is dependent on Dcr2 and Ago2
Given that the CTG/CAG transcripts produced small RNAs, we determined whether the enhanced toxicity as well as generation of the small RNAs were dependent on dcr2, the enzyme in flies that cleaves double-stranded RNA for siRNA biogenesis[27]. Flies expressing (CTG)250/(CAG)250 with gmr-gal4 were lethal at late developmental stages; dissection of animals from the pupal case revealed severely disrupted eye morphology (Figure 3A). Flies expressing the repeat transcripts, but homozygous mutant for dcr2 gene function, were now viable and displayed significantly restored eye structure (Figure 3A). Homozygous loss of dcr2 activity also rescued organismal lethality with heatshock induction of (CTG)250/(CAG)250 transcripts, and rescued developmental lethality upon expression of (CTG)130/(CAG)100 transcripts in muscle (Figure 3B and 3C). Loss of dcr2 had a minimal or no effect on toxicity of (CTG)250 alone, (CAG)250 alone or a mutant tau protein, the latter being associated with frontotemporal dementia (Figure S5A and S5B and data not shown). This indicates that dcr2 function is required for the toxicity associated with CTG/CAG co-expression. The dcr2 null background also blocked the biogenesis of the triplet repeat-derived small RNAs generated upon co-expression of the two transcripts, restoring the full length repeat RNA levels, concomitant with mitigation of toxicity (Figure 3E and 3F).
To characterize the triplet repeat-derived small RNAs, we asked whether they were methylated at the 3′ end, a modification specific to siRNAs loaded to Ago2-RISC [28]. They were resistant to oxidation/ß-elimination normally, but sensitive to ß-elimination in the hen1 mutant background which prevents methylation (Figure 3G and Figure S4C), arguing that the repeat-derived small RNAs are siRNAs and are assembled into the Ago2-RISC complex. Further, loss of ago2 also dramatically mitigated (CTG)200/(CAG)250 toxicity (Figure 3D and Figure S6). To address specificity, we determined whether genes that modulate the miRNA pathway contributed to the CTG/CAG interaction. Reduction in gene dosage of dcr1 or ago1 showed no effect (data not shown). In addition, whereas upregulation of dcr2 dramatically enhanced CTG/CAG toxicity with concomitant increase of the triplet repeat-derived small RNAs, there was no effect of dcr1 upregulation (Figure S7). These data suggest that the triplet repeat-derived small RNAs are siRNAs in nature and that their toxic effects are dependent on Dcr2 and Ago2 activity.
Triplet repeat derived siRNAs compromise the expression of genes containing short CAG stretches
We tested whether the siRNAs may be competing with endogenous small RNAs for the biogenesis machinery, and thus by a titration mechanism causing toxicity. However, up-regulation of dcr2, which should suppress according to a titration mechanism, instead enhanced toxicity of expanded CTG/CAG (see Figure S7). Moreover, generation of miRNAs, with analysis of miR-277 and miR-8, and generation of endogenous small RNAs, with analysis of hp-CG4068B and esiRNA-sl-1 (dependent on dcr2 activity [29]), were not affected upon co-expression of the CTG/CAG transcripts (Figure 4A). Transcription of retrotransposon 412 was also not affected in flies expressing expanded CTG/CAG (Figure 4B), suggesting that the RNA interference pathways in these flies were largely intact. Together, these data argue that overwhelming endogenous RNA interference pathways cannot account for the enhanced toxicity.

In light of the requirement for ago2 and dcr2, we then asked whether the triplet repeat-derived siRNAs targeted other transcripts with small CAG or CUG stretches, such that disruption or loss of the activity of those genes may subsequently caused the deleterious effects. We selected two endogenous fly genes which, like their human counterparts, contain CAG repeat stretches, atx2 (containing (CAG)9) and tbp (containing (CAG)5CAA(CAG)2), and analyzed their expression levels by realtime PCR. In flies expressing expanded CTG/CAG transcripts, the levels of these CAG-containing mRNAs were downregulated ∼60–70%; transcripts without such repeats and control transcripts such as tubulin and appl were unaffected (Figure 4B). Further analysis revealed that the atx2 and tbp transcripts were being cleaved within their CAG repeat stretches in flies expressing expanded CTG/CAG repeats in a dcr-2 dependent manner (Figure 4C and 4D). In contrast, we did not observe down-regulation of CUG containing transcripts by realtime PCR nor did we detect cleavage of these transcripts by RLM-RACE in flies expressing expanded CTG/CAG (data not shown; see Discussion). These data indicate that co-expression of CTG/CAG repeat transcripts generates triplet repeat-derived siRNAs that target other CAG-containing transcripts within the genome; deleterious effects on the levels and activity of these genes may contribute to the pathogenic effects of genes with expanded repeats that are bi-directionally transcribed.
Discussion
Like many genes within the mammalian genome [11], the DM1 gene displays bi-directional transcription, generating an anti-sense CAG repeat transcript in addition to the disease-associated CTG transcript [15]. These transcripts have been shown to interact in human cells to generate small RNAs, with one effect being local gene silencing [15]; however additional ways in which this may contribute to pathogenicity in disease is largely unknown. In order to provide new insight into DM1, we generated a Drosophila model by expressing pure, uninterrupted CTG repeat expansions; fly models for various disorders have revealed critical insight into a number of human disease situations (Clark et al., 2006; Fernandez-Funez et al., 2000; Jin et al., 2003; Meulener et al., 2005; Warrick et al., 1998). Interestingly, targeted expression of the long CTG repeats in the fly eye caused a variable toxic effect (see Figure S1). This was also observed in a fly model of SCA8, which carries an uninterrupted CTG repeat expansion [30]. In contrast, fly models generated using interrupted CTG repeats were not reported to show variable phenotype [24], [25]. It is thus possible that phenotypic variability may be a feature of pure repeat sequences, which is in line with the fact that DM1 is among the most variable human disorders. To define potential effects of bi-directional transcription, we then co-expressed expanded CAG repeat transcripts with the DM1 CTG repeats. This resulted in dramatically enhanced toxicity concomitant with the generation of triplet repeat-derived siRNAs. Our results are in striking contrast with previous findings that co-expression of CGG and CCG expansions in flies leads to mitigated toxicity in a ago2-dependent manner [31], suggesting that toxicity derived from interactions between sense and anti-sense repeat transcripts may be specific to CTG/CAG situations. Both CAG and CUG strands can be processed into ∼21 nt small RNAs when coexpressed and small RNAs derived from both strands are methylated in a Hen1-dependent manner (see Figure 2C, Figure 3G, Figure S4B and S4C). These results suggest that both CAG and CUG small RNAs can be loaded into mature, holo-RISCs presumably due to the symmetrical thermodynamic properties of the repeat small RNA duplex [28], [32]-[34]. In our studies, we detected direct cleavage of CAG containing transcripts, and we were unable to detect cleavage of CUG containing transcripts mediated by CAG small RNAs (see Figure 4B–4D). Although underlying reasons for this differential effect remain unclear, CUG and CAG transcripts may have differential expression levels or translation efficiencies, and/or CUG-containing and CAG-containing transcripts may be associated with different RNA binding proteins of various affinities, making CUG-transcripts less accessible to the RISC complex than CAG-containing transcripts [35]. A number of CUG-binding proteins have been defined, such as MBNL1, CUGBP1 and PKR [36]-[38]. Interestingly, in-vitro gel retardation analysis indicated that MBNL1 has a much lower affinity for CAG repeat RNA than CUG repeat RNA[36]. Moreover, expanded CAG transcripts, although co-localizing with MBNL1 in ribonuclear foci similarly to expanded CUG transcripts, do not appear to cause mis-regulation of alternative splicing in cells[39], further highlighting differential properties of these repeats in interacting with RNA binding proteins.
The toxicity caused by co-expression of expanded CAG and CTG was associated with deleterious effects on transcripts of other CAG containing genes within the genome; additional mechanisms that contribute to toxicity may also exist. A large number of genes contain CAG stretches in fly and human genomes (Table S1 and Table S2). The enhanced toxicity we observed in flies expressing expanded CAG and CTG may therefore be reflecting an additive effect of knockdown of multiple CAG-containing genes, with each individual gene contributing only partially to the overall outcome. Although further reducing atx2 dosage did not enhance toxicity of co-expressed CTG/CAG expansions (ZY and NB, unpublished observations), the compromised activities of many target genes may be involved and further compromising any single one has minimal impact. The toxic effects seen of the CAG/CTG situation may also be complicated by the later-onset and progressive nature of the toxicity. Further study will clarify the contribution of this mechanism, and key targets among all possible transcripts, to the overall phenotype of the disease. Moreover, the deleterious effects caused by triplet repeat derived small RNAs may be further exacerbated by the wide prevalence of CAG stretches in the human transcriptome (Table S2) and the relative low specificity of RNA interference when siRNAs and/or RNA targets contain simple repeats like CAG [40], [41]. Such interactions may represent a novel activity of endo-siRNAs that characterize disease situations where bi-directional transcription spanning the repeat region occurs (Figure 4E).
We confirmed that two of CAG containing genes, atx2 and tbp, are targets of the triplet repeat-derived siRNAs. Interestingly, CAG repeat expansions in ATXN2 (the human Ataxin-2 gene) and TBP define two of the CAG-repeat expansion diseases (SCA2 and SCA17, respectively). In such diseases, the expanded polyglutamine domain is thought to confer toxicity [1], [2]; however, increasing evidence suggests that the loss-of-function of gene activity, and not just dominant activities of the protein with an expanded polyglutamine region, occur in disease [42], [43]. Our findings raise the possibility that bi-directional transcription of the repeat region in diseases like DM1 may confer additional components of pathogenicity due to deleterious interactions between the two overlapping repeat-containing transcripts through the generation and activity of triplet repeat-derived siRNAs.
Studies indicate that bi-directionally transcribed RNAs, and presumably resultant endogenous double-stranded RNAs, are processed into ∼21–23 nt small RNAs in human cells [44], [45]. This is despite the fact that in most mammalian cells, long exogenous double-stranded RNAs can elicit the interferon response [46], [47]. That response presumably occurs in a threshold-dependent manner; cells may also respond differentially to long exogenous double-stranded RNAs versus endogenous double-stranded RNAs. Thus, these findings suggest that the biogenesis pathway of small RNAs from endogenous double-stranded RNAs is conserved in mammalian cells. Many loci are bi-directionally transcribed throughout the mammalian genome [11], [17], [18], and among these are a number of human trinucleotide disease genes, including SCA8 and DM1 [12], [16]. In SCA8, an anti-sense transcript is proposed to encode a polyglutamine protein, which itself may have deleterious actions [14]. In DM1, the two transcripts interact to produce small RNAs that can have local effects on gene silencing [15]. Our findings raise another possibility, that processing of co-expressed transcripts containing CUG/CAG expansions into triplet repeat-derived siRNAs in vivo, may contribute to toxicity with widespread deleterious effects. These effects may include downregulating the expression of other genes containing CAG repeats. Among the genes that could be targets are the polyglutamine disease genes themselves, one of which is TBP. Expansion of the TBP polyglutamine repeat underlies SCA17 [48]; intriguingly, general transcriptional compromise has been shown to be a component of repeat expansion diseases [49], [50]. Our studies raise the possibility that perhaps another reason why these diseases share transcriptional compromise may be that they share bi-directional transcript interactions that compromise common elements like TBP. This possibility underscores the idea of shared therapeutic targets and mechanisms in repeat expansion diseases.
It has been proposed that siCAG and siCUG may be used for therapy of triplet repeat expansion diseases based on findings in cell culture that these siRNAs seem to specifically target mutant transcripts with expanded repeats [51]. Our data suggest caution in designing such siRNA-based therapy, as in the intact organismal situation, pathogenic activities may be noted. Although previous findings suggest that expanded CUG alone can be processed into small RNAs, [51], our data suggest that both expanded CAG and CTG are required for triplet repeat-derived siRNA generation and toxicity in vivo (see Figure 2C and Figure S5). Thus, co-expressed CAG and CTG expansions may contribute to DM1 pathogenesis through a fundamentally different mechanism from that of CTG expansions alone. Although our studies were conducted in fly models, the findings may apply to human trinucleotide expansion diseases. Targeting these diseases at the transcriptional level may therefore be a promising therapeutic approach that would minimize not only the effects of single expanded repeat transcripts, but deleterious interactions between sense and anti-sense repeat transcript domains.
Materials and Methods
Fly lines
General fly lines were ordered from public stock centers and maintained at 25 °C on standard medium unless otherwise indicated. CTG repeats of various length (DNA templates kindly provided by C. Thornton, University of Rochester) were inserted into the 3′UTR of DsRed2 gene (Clontech) in pUAST. All transgenic constructs were confirmed by sequencing. Fly lines were generated by P-element mediated transformation. Repeat lengths were determined by Southern blot and confirmed by Genescan for select lines showing variability. dcr2L811fsX, ago2414, Hen1 mutant Pimetf00810, UAS-dcr1, UAS-dcr2, UAS-DsRed-(CAG)100 and UAS-DsRed-(CAG)250 lines are described [26], [27], [52]-[54].
Western, Southern, and Northern blots
Standard techniques were used. Primary antibodies for Westerns were anti-DsRed (1∶400, anti-rabbit, Clontech), anti-actin (1∶4,000, anti-mouse, Abcam). HRP conjugated secondary anti-mouse (1∶4000, Chemicon) and anti-rabbit antibodies (1∶4000, Zymed) were used with ECL+ reagents (Amersham). For Southern blots, genomic DNA was extracted from ∼50 flies using the Gentra Puregene Cell Kit (Qiagen) and 5 µg of genomic DNA was fully digested with 200 units of EcoRI and XbaI. DsRed DNA was PCR amplified using primers: forward 5′-GGCCCCCTGCCCTTCGCC-3′ and reverse 5′-CTACAGGAACAGGTGGTGGCGG-3′, purified using QIAquick Gel Extraction Kit (Qiagen) and labeled using the High Prime DNA Labeling Kit (Roche Applied Science). For Northern blots, flies were heatshocked at 37 °C for 30 min and allowed to recover at 25 °C for 20 h. Total RNA was extracted using Trizol Reagent (Invitrogen) from either whole flies (for comparing transgene levels among various lines) or heads. 2–10 µg of total RNA was loaded on 1% denaturing formaldehyde/MOPS agarose gels for regular Northern blots. For small RNA Northerns, total RNA was further purified using the mirVana miRNA Isolation Kit (Ambion) to enrich small RNA. 100 ng small RNA was loaded on 15% TBE-Urea polyacrylamide gel (Invitrogen). The SV40 probe for Northern blots was PCR amplified using primers: forward 5′-TGTGGTGTGACATAATTGGACA-3′ and reverse 5′-AGATGGCATTTCTTCTGAGCA-3′, purified using QIAquick Gel Extraction Kit (Qiagen) and labeled using the High Prime DNA Labeling Kit (Roche Applied Science). Other probes for Northern blot were made using the MAXIscript Kit (Ambion) from the annealed double stranded DNA template containing T7 promoter. Oligo sequences were:
T7 promoter forward oligo: 5′-GATAATACGACTCACTATAGGGAGA-3′
r(CAG)5 probe: 5′-GGGGGCTGCTGCTGCTGCTGTCTCCCTATAGTGAGTCGTATTATC-3′
r(CUG)5probe: 5′-GGGGGCAGCAGCAGCAGCAGTCTCCCTATAGTGAGTCGTATTATC-3′
2S rRNA: 5′-TGCTTGGACTACATATGGTTGAGGGTTGTATCTCCCTATAGTGAGTCGTATTATC-3′
18S rRNA: 5′-AGGGAGCCTGAGAAACGGCTACCACATCTAAGGAATCTCCCTATAGTGAGTCGTATTATC-3′
hp4068B: 5′-TTGACTCCAACAAGTTCGCTCCTCTCCCTATAGTGAGTCGTATTATC-3′
mir277 : 5′-TAAATGCACTATCTGGTACGACATCTCCCTATAGTGAGTCGTATTATC-3′.
In situ hybridization
In-situ hybridization was performed as described [26].
Radioactive PCR
Radioactive PCR for the alternative splicing assay was performed as described [55]. Total RNA was extracted from fly heads using RNeasy Mini Kit (Qiagen) and cDNA was synthesized using the SuperScript First-Strand Synthesis for RT-PCR (Invitrogen). Primers used for radioactive PCR reaction were: forward 5′-GCCATTTGACCATTCACCACATTGGTGTG-3′, reverse 5′-TTGCTGGAGCATAGCACTCTTCAGGTG-3′. The forward primer was labeled using the T4 polynucleotide kinase (New England Biolabs). PCR reactions were run 21–23 cycles and separated on 5% non-denaturing TBE polyacrylamide gels. The gel was then dried and exposed to Storage Phosphor Screens (GE Healthcare). Band densitometry was quantified using Image J (NIH).
Real-time PCR
0-3d flies were heatshocked at 37 °C for 30 min and recovered at 25 °C for 9 h. Total RNA was extracted from fly heads using Trizol Reagent (Invitrogen), treated with Turbo DNase (Ambion) and further purified using the mirVana miRNA Isolation Kit (Ambion). cDNA was synthesized in a 20 µl reaction volume from 0.2 µg of total RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). 0.2 µl of cDNA was used as the template in a 20 µl reaction volume diluted from the Power SYBR Green PCR Master Mix Kit (Applied Biosystems). Real-time PCR was performed in triplet or quadruplicate using a 7500 Fast Real-Time PCR System (Applied Biosystems). Data were analyzed using the ΔΔCt method. Endogenous control was rp49. Each experiments was repeated at least three times on independent RNA preparations.
Real-time PCR primers were designed using the Primer Express software (Applied Biosystems) and the sequences were:
atx2 forward: 5′-CGCACGCGCGATAACC-3′
atx2 reverse: 5′-AGTTGGAAGTCCTGGCCAAA-3′
tbp forward: 5′-AAGCTCGGTTTCCCTGCAA-3′
tbp reverse: 5′-GCAGGAGCCGACCATGTTT-3′
412 forward: 5′-CACCGGTTTGGTCGAAAG-3′
412 reverse: 5′-GGACATGCCTGGTATTTTGG-3′
appl forward: AGGTCACGCGCGTTATGAA
appl reverse: GGCGCATGTCCTGGTACTTC
ß-tubulin forward 5′-CATCCAAGCTGGTCAGTGC-3′
ß-tubulin reverse 5′-GCCATGCTCATCGGAGAT-3′
rp49 forward: 5′-CAACATCGGTTACGGATCGA-3′
rp49 reverse: 5′-AATCCGGTGGGCAGCAT-3′
RNA ligase mediated amplification of cDNA ends (RLM-RACE)
To detect cleavage of atx2 and tbp transcripts, RLM-RACE and cloning of RLM-RACE products were carried out using the GeneRacer Kit (Invitrogen, Carlsbad, CA). Briefly, 3-4d flies were heatshocked at 37 °C for 30 min and then maintained at 25 °C for 14 hr. Total RNA was extracted from fly heads using the RNAeazy kit (Qiagen, Valencia, CA). The 5′ GeneRacer RNA adaptor molecule was ligated onto the total RNA population. Ligated products were reverse transcribed using random primers and nested PCR was performed using primers derived from the 5′ GeneRace adaptor and gene specific primers, respectively, to detect RISC cleavage products. PCR products were analyzed on 1% agarose TAE gel. To analyze cleavage sites, PCR products were gel purified using the gel purification kit (Qiagen, Valencia, CA), cloned using TOPO TA cloning kit (Invitrogen, Carlsbad, CA) and sequenced. Primers used for nested PCR were:
dTBPclv5RACEnest1 : 5′-GGGCCCATCGTCTGGTGGATGTT-3′
dTBPclv5RACEnest2 : 5′-TGGTGGATGTTGCTCAGGGCATCT-3′
dAtx2clv5RACEnest1 : 5′-TGTGGCGGCGGCATTGTATGGTAAA-3′
dAtx2clv5RACEnest2 : 5′-TGTGGCGGCGGCTGCTGCACTT-3′
GeneRacer5′ Primer: 5′-CGACTGGAGCACGAGGACACTGA-3′
GeneRacer5′ nested Primer: 5′-GGACACTGACATGGACTGAAGGAGTA-3′
5′ GeneRacer RNA adaptor: 5′-CGACUGGAGCACGAGGACACUGACAUGGACUGAAGGAGUAGAAA-3′
Heatshock survival assay
0-3d flies were heat shocked at 37 °C for 30 min and then maintained at 25 °C for 50 hr. Numbers of dead/living flies were recorded. At least ∼100 flies were scored for each genotype and the experiments were repeated three times.
Viability assay
24B-gal4 driver flies were outcrossed to flies of appropriate genotype and progeny flies were scored for viability. At least 100 progeny flies were scored for each cross and experiments were repeated three times.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. RanumLP
CooperTA
2006 RNA-mediated neuromuscular disorders. Annu Rev Neurosci 29 259 277
2. OrrHT
ZoghbiHY
2007 Trinucleotide Repeat Disorders. Annu Rev Neurosci
3. LiLB
BoniniNM
2010 Roles of trinucleotide-repeat RNA in neurological disease and degeneration. Trends Neurosci 33 292 298
4. BrookJD
McCurrachME
HarleyHG
BucklerAJ
ChurchD
1992 Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68 799 808
5. FuYH
PizzutiA
FenwickRG
KingJ
RajnarayanS
1992 An unstable triplet repeat in a gene related to myotonic muscular-dystrophy. Science 255 1256 1258
6. PhilipsAV
TimchenkoLT
CooperTA
1998 Disruption of Splicing Regulated by a CUG-Binding Protein in Myotonic Dystrophy. Science 280 737 741
7. SavkurRS
PhilipsAV
CooperTA
2001 Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 29 40 47
8. MankodiA
TakahashiMP
JiangH
BeckCL
BowersWJ
2002 Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 10 35 44
9. RudnickiDD
HolmesSE
LinMW
ThorntonCA
RossCA
2007 Huntington's disease–like 2 is associated with CUG repeat-containing RNA foci. Ann Neurol 61 272 282
10. DaughtersRS
TuttleDL
GaoW
IkedaY
MoseleyML
2009 RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet 5 e1000600 doi:10.1371/journal.pgen.1000600
11. KatayamaS
TomaruY
KasukawaT
WakiK
NakanishiM
2005 Antisense transcription in the mammalian transcriptome. Science 309 1564 1566
12. BatraR
CharizanisK
SwansonMS
2010 Partners in crime: bidirectional transcription in unstable microsatellite disease. Hum Mol Genet 19 R77 82
13. LaddPD
SmithLE
RabaiaNA
MooreJM
GeorgesSA
2007 An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet 16 3174 3187
14. MoseleyML
ZuT
IkedaY
GaoW
MosemillerAK
2006 Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 38 758 769
15. ChoDH
ThienesCP
MahoneySE
AnalauE
FilippovaGN
2005 Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol Cell 20 483 489
16. DionV
WilsonJH
2009 Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet 25 288 297
17. GeX
WuQ
JungY-C
ChenJ
WangSM
2006 A large quantity of novel human antisense transcripts detected by LongSAGE. Bioinformatics 22 2475 2479
18. HeY
VogelsteinB
VelculescuVE
PapadopoulosN
KinzlerKW
2008 The antisense transcriptomes of human cells. Science 322 1855 1857
19. ZoghbiHY
BotasJ
2002 Mouse and fly models of neurodegeneration. Trends Genet 18 463 471
20. MarshJL
ThompsonLM
2006 Drosophila in the study of neurodegenerative disease. Neuron 52 169 178
21. CauchiRJ
van den HeuvelM
2006 The fly as a model for neurodegenerative diseases: is it worth the jump? Neurodegener Dis 3 338 356
22. BilenJ
BoniniNM
2005 Drosophila as a model for human neurodegenerative disease. Annu Rev Genet 39 153 171
23. HouseleyJM
WangZ
BrockGJR
SolowayJ
ArteroR
2005 Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Human Molecular Genetics 14 873 883
24. de HaroM
Al-RamahiI
De GouyonB
UkaniL
RosaA
2006 MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Human Molecular Genetics 15 2138 2145
25. Garcia-LopezA
MonferrerL
Garcia-AlcoverI
Vicente-CrespoM
Alvarez-AbrilMC
2008 Genetic and chemical modifiers of a CUG toxicity model in Drosophila. PLoS ONE 3 e1595 doi:10.1371/journal.pone.0001595
26. LiLB
YuZ
TengX
BoniniNM
2008 RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453 1107 1111
27. LeeYS
NakaharaK
PhamJW
KimK
HeZ
2004 Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell 117 69 81
28. HorwichMD
LiC
MatrangaC
VaginV
FarleyG
2007 The Drosophila RNA methyltransferase, DmHen1, modifies germline piRNAs and single-stranded siRNAs in RISC. Curr Biol 17 1265 1272
29. CzechB
MaloneCD
ZhouR
StarkA
SchlingeheydeC
2008 An endogenous small interfering RNA pathway in Drosophila. Nature 453 798 802
30. MutsuddiM
MarshallCM
BenzowKA
KoobMD
RebayI
2004 The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr Biol 14 302 308
31. SofolaOA
JinP
BotasJ
NelsonDL
2007 Argonaute-2-dependent rescue of a Drosophila model of FXTAS by FRAXE premutation repeat. Hum Mol Genet 16 2326 2332
32. SchwarzDS
HutvagnerG
DuT
XuZ
AroninN
2003 Asymmetry in the assembly of the RNAi enzyme complex. Cell 115 199 208
33. KhvorovaA
ReynoldsA
JayasenaSD
2003 Functional siRNAs and miRNAs exhibit strand bias. Cell 115 209 216
34. HutvagnerG
2005 Small RNA asymmetry in RNAi: function in RISC assembly and gene regulation. FEBS Lett 579 5850 5857
35. PeiY
TuschlT
2006 On the art of identifying effective and specific siRNAs. Nat Methods 3 670 676
36. MillerJW
UrbinatiCR
Teng-UmnuayP
StenbergMG
ByrneBJ
2000 Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J 19 4439 4448
37. TimchenkoLT
MillerJW
TimchenkoNA
DeVoreDR
DatarKV
1996 Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res 24 4407 4414
38. TianB
WhiteRJ
XiaT
WelleS
TurnerDH
2000 Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. Rna 6 79 87
39. HoTH
SavkurRS
PoulosMG
ManciniMA
SwansonMS
2005 Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci 118 2923 2933
40. JasinskaA
MichlewskiG
de MezerM
SobczakK
KozlowskiP
2003 Structures of trinucleotide repeats in human transcripts and their functional implications. Nucleic Acids Res 31 5463 5468
41. MaY
CreangaA
LumL
BeachyPA
2006 Prevalence of off-target effects in Drosophila RNA interference screens. Nature 443 359 363
42. LimJ
Crespo-BarretoJ
Jafar-NejadP
BowmanAB
RichmanR
2008 Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452 713 718
43. ThomasPSJr
FraleyGS
DamianV
WoodkeLB
ZapataF
2006 Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy. Hum Mol Genet 15 2225 2238
44. KawajiH
NakamuraM
TakahashiY
SandelinA
KatayamaS
2008 Hidden layers of human small RNAs. BMC Genomics 9 157
45. YangN
KazazianHHJr
2006 L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol 13 763 771
46. Ui-TeiK
ZennoS
MiyataY
SaigoK
2000 Sensitive assay of RNA interference in Drosophila and Chinese hamster cultured cells using firefly luciferase gene as target. FEBS Lett 479 79 82
47. CaplenNJ
FleenorJ
FireA
MorganRA
2000 dsRNA-mediated gene silencing in cultured Drosophila cells: a tissue culture model for the analysis of RNA interference. Gene 252 95 105
48. NakamuraK
JeongSY
UchiharaT
AnnoM
NagashimaK
2001 SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10 1441 1448
49. FreimanRN
TjianR
2002 Neurodegeneration. A glutamine-rich trail leads to transcription factors. Science 296 2149 2150
50. RileyBE
OrrHT
2006 Polyglutamine neurodegenerative diseases and regulation of transcription: assembling the puzzle. Genes Dev 20 2183 2192
51. KrolJ
FiszerA
MykowskaA
SobczakK
de MezerM
2007 Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell 25 575 586
52. OkamuraK
IshizukaA
SiomiH
SiomiMC
2004 Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev 18 1655 1666
53. SaitoK
SakaguchiY
SuzukiT
SuzukiT
SiomiH
2007 Pimet, the Drosophila homolog of HEN1, mediates 2′-O-methylation of Piwi - interacting RNAs at their 3′ ends. Genes Dev 21 1603 1608
54. DietzlG
ChenD
SchnorrerF
SuKC
BarinovaY
2007 A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448 151 156
55. CooperTA
2005 Use of minigene systems to dissect alternative splicing elements. Methods 37 331 340
56. KoobMD
MoseleyML
SchutLJ
BenzowKA
BirdTD
1999 An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nature Genetics 21 379 384
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy