
Polycomb Repressive Complex 2 Controls the Embryo-to-Seedling Phase Transition
Polycomb repressive complex 2 (PRC2) is a key regulator of epigenetic states catalyzing histone H3 lysine 27 trimethylation (H3K27me3), a repressive chromatin mark. PRC2 composition is conserved from humans to plants, but the function of PRC2 during the early stage of plant life is unclear beyond the fact that it is required for the development of endosperm, a nutritive tissue that supports embryo growth. Circumventing the requirement of PRC2 in endosperm allowed us to generate viable homozygous null mutants for FERTILIZATION INDEPENDENT ENDOSPERM (FIE), which is the single Arabidopsis homolog of Extra Sex Combs, an indispensable component of Drosophila and mammalian PRC2. Here we show that H3K27me3 deposition is abolished genome-wide in fie mutants demonstrating the essential function of PRC2 in placing this mark in plants as in animals. In contrast to animals, we find that PRC2 function is not required for initial body plan formation in Arabidopsis. Rather, our results show that fie mutant seeds exhibit enhanced dormancy and germination defects, indicating a deficiency in terminating the embryonic phase. After germination, fie mutant seedlings switch to generative development that is not sustained, giving rise to neoplastic, callus-like structures. Further genome-wide studies showed that only a fraction of PRC2 targets are transcriptionally activated in fie seedlings and that this activation is accompanied in only a few cases with deposition of H3K4me3, a mark associated with gene activity and considered to act antagonistically to H3K27me3. Up-regulated PRC2 target genes were found to act at different hierarchical levels from transcriptional master regulators to a wide range of downstream targets. Collectively, our findings demonstrate that PRC2-mediated regulation represents a robust system controlling developmental phase transitions, not only from vegetative phase to flowering but also especially from embryonic phase to the seedling stage.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1002014
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002014
Summary
Polycomb repressive complex 2 (PRC2) is a key regulator of epigenetic states catalyzing histone H3 lysine 27 trimethylation (H3K27me3), a repressive chromatin mark. PRC2 composition is conserved from humans to plants, but the function of PRC2 during the early stage of plant life is unclear beyond the fact that it is required for the development of endosperm, a nutritive tissue that supports embryo growth. Circumventing the requirement of PRC2 in endosperm allowed us to generate viable homozygous null mutants for FERTILIZATION INDEPENDENT ENDOSPERM (FIE), which is the single Arabidopsis homolog of Extra Sex Combs, an indispensable component of Drosophila and mammalian PRC2. Here we show that H3K27me3 deposition is abolished genome-wide in fie mutants demonstrating the essential function of PRC2 in placing this mark in plants as in animals. In contrast to animals, we find that PRC2 function is not required for initial body plan formation in Arabidopsis. Rather, our results show that fie mutant seeds exhibit enhanced dormancy and germination defects, indicating a deficiency in terminating the embryonic phase. After germination, fie mutant seedlings switch to generative development that is not sustained, giving rise to neoplastic, callus-like structures. Further genome-wide studies showed that only a fraction of PRC2 targets are transcriptionally activated in fie seedlings and that this activation is accompanied in only a few cases with deposition of H3K4me3, a mark associated with gene activity and considered to act antagonistically to H3K27me3. Up-regulated PRC2 target genes were found to act at different hierarchical levels from transcriptional master regulators to a wide range of downstream targets. Collectively, our findings demonstrate that PRC2-mediated regulation represents a robust system controlling developmental phase transitions, not only from vegetative phase to flowering but also especially from embryonic phase to the seedling stage.
Introduction
One common principle of flowering plants and probably one of the main reasons for their evolutionary success is the alternation of a dormant seed stage with a growing plant that will eventually reproduce and again generate seeds. Seeds harbor not only the plant embryo, i.e. the next plant generation, but typically contain a nourishing tissue, called the endosperm that supports embryo growth and often provides the nutrients for the germinating seedling. Moreover, the embryo and the endosperm are protected by a hard shall, the seed coat, that also facilitates the distribution of seeds. Remarkably, seeds often will stay dormant after ripening and require for germination a defined order of environmental conditions reflecting the progression of the seasons in moderate climates, i.e. they will germinate only after exposure to warmth after a period of cold temperatures. Many factors have been identified to influence this transition from a dormant embryonic phase to a germinating seedling (for review see [1]). However, a unifying molecular framework has not been established so far.
For the other major phase transition in plants, e.g. from vegetative growth to flowering, it has been found that Polycomb repressive complex 2 (PRC2) regulation is crucial [2]–[4]. PRC2 activity was also found to be required for repression of flower formation in young seedlings indicating a function in maintaining and/or establishing vegetative growth [5], [6]. Moreover, severely compromising PRC2 function revealed its function in maintaining overall cell and tissue organization, e.g. the distinction between root and shoot fates [5], [6].
PRC2 catalyzes the deposition of trimethylation of Lysine 27 on histone H3 (H3K27me3), a repressive chromatin mark [7], [8]. The core PRC2 complex is conserved between animals and plants and contains at least four components, which were first identified in Drosophila: the HMTase Enhancer of Zeste (E(Z)), the WD40 domain protein Extra sex combs (ESC), the Zn-finger protein Suppressor of zeste-12 (SU(Z)12) and the nucleosome-remodeling factor 55 (NURF-55) [9]–[12]. Arabidopsis contains three presumptive H3K27me3 HMTases, CURLY LEAF (CLF), SWINGER (SWN) and MEDEA (MEA) that have been found to at least partially compensate for each other. Similarly, Drosophila Su(Z)12 function is represented by three partially redundantly acting genes, EMBRYONIC FLOWER 2 (EMF2), FERTILIZATION INDEPENDENT SEED DEVELOPMENT 2 (FIS2), and VERNALIZATION 2 (VRN2). The homolog of Drosophila ESC, FIE, is the only PRC2 component that is represented by a single member in Arabidopsis.
In the past few years, much progress has been made in the understanding of the modus operandi of PRC2. However, a major obstacle in studying the function of chromatin regulators is their essential role in early development as for instance mutants in ESC in Drosophila and its murine ortholog EED are embryonic lethal [13]–[15]. Similarly, PRC2 function is crucial already for endosperm formation in flowering plants by controlling the parent-of-origin dependent activity of a number of genes in the endosperm (imprinting). PRC2 function is maternal gametophytically required and loss of the maternal PRC2 function releases targets genes from their repression leading to endosperm overproliferation and ultimately to seed abortion [16]–[19]. This requirement for endosperm formation has also precluded so far an analysis of PRC2 action during later stages of seed development and it also remained an open question whether PRC2 function is required for initial body plan formation in flowering plants during which an embryo with shoot, root, and one (Monocotyledons) or two (Dicotyledons) cotyledons is formed. In contrast to animals, the two stem cell populations established in embryogenesis, i.e. the root and shoot meristem, will produce the body of the adult plant and it has been shown previously that PRC2 is involved in postembryonic shoot meristem function [20].
We and others have previously identified a mutant in the cell cycle regulator CDKA;1 in which the second mitosis during pollen development is missing or substantially delayed [21]–[23]. However, mutant pollen can successfully fertilize the egg cell giving rise to an embryo while triggering the onset of endosperm development without a paternal contribution. This type of fertilization was found to bypass the maternal requirement of PRC2-dependent repression during endosperm development resulting in a mutual rescue of the paternal effect of cdka;1 mutant pollen and the maternal effect caused by mutations in MEA, FIS2 or FIE [24].
Here we have used cdka;1 mutant fertilization to generate homozygous fie mutant plants allowing us to functionally address the requirement of PRC2 action during embryogenesis and subsequent plant growth and development. Our results show that PRC2 is required neither for the generation nor maintenance of embryonic organization in striking contrast to animal PRC2 function. However, PRC2 in plants is vital for the reprogramming of developmental fates mediating the switch from embryonic states to growing seedlings. Furthermore, our genome-wide ChIP - and transcriptional profiling experiments gave insights into the circuitry of PRC2 action indicating that developmental phase transitions are robustly controlled by PRC2 through simultaneously targeting genes at different hierarchical levels and triggering positive feed back loops. This network design allows the transduction of environmental cues into stable and self-maintaining developmental fates likely underlying the enormously adaptable yet enduring growth of plants.
Results
Generation of homozygous fie mutant plants
Since the female gametophytic defect of mutants in FIS class genes can be bypassed by fertilization with cdka;1 mutant pollen [24], we asked whether this would allow the generation of homozygous fie mutant plants in crosses of heterozygous fie mutant mother plants with pollen of cdka;1-fie double heterozygous plants. Indeed, in the progeny of this cross and amid the descendents of a self-pollinated double heterozygous cdka;1-fie mutant a morphologically distinguishable class of plants was identified that was never found among the progeny of heterozygous fie or cdka;1 mutants. Subsequent genotyping confirmed that these plants were homozygous mutant for fie (Figure 1). Reciprocal crosses corroborated that the appearance of fie resulted solely from fertilization with paternal cdka;1 whereas maternal cdka;1 did not contribute to the generation of viable fie mutants (Table 1).
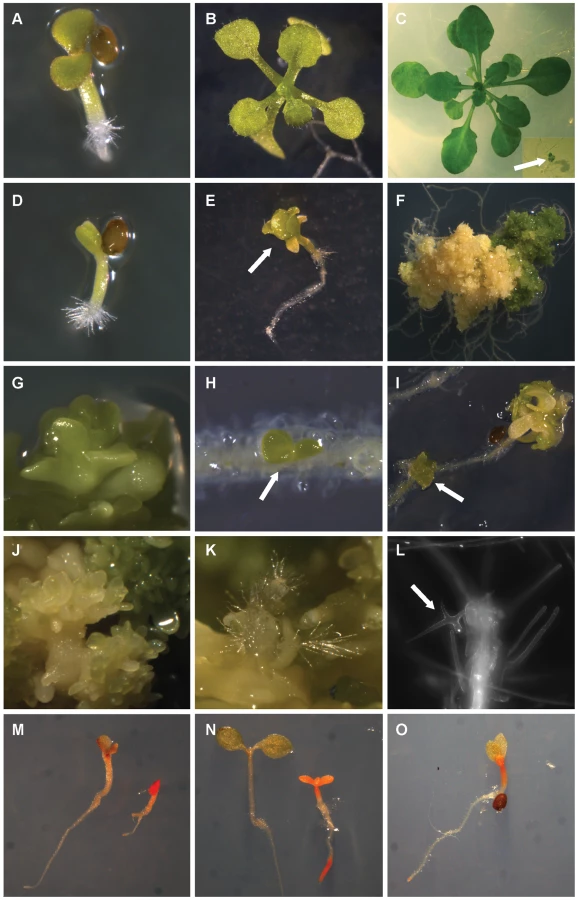

The fie mutant used as reference allele in this study is a T-DNA insertion line in a central exon and represents a transcriptional null mutant (Figure S1). In the same way generated homozygous seedlings for three additional fie alleles resulted in the same mutant phenotype (Figure S1 and data not shown). Thus, circumventing the requirement of FIS action in the endosperm is sufficient to generate homozygous null mutants for the PRC2 core gene FIE.
Homozygous fie mutants display a progressive mutant phenotype
Loss of ESC function in flies or mammals causes embryo lethality and is essential for the patterning of the body plan [15], [25]. In contrast, macro - and microscopical analyses revealed that fie mutant seedlings initially showed a wild-type-like body plan with a root and a shoot, two cotyledons, and newly forming rosette leaves that were at this stage morphologically indistinguishable from wild-type sister plants (Figure 1A, 1D). However, fie mutants grew more slowly than the wild type and around 10 days after stratification (10 DAS) already initiated flower buds (Figure 1E shows a flower bud at 15 DAS) whereas the wild type started to flower only after more than 30 DAS. During the next 10 days, homozygous fie mutants developed an increasing number of ectopic cells (Figure 1K, 1L) and organs (Figure 1H, 1I), showed signs of organ transformations (Figure 1G) and generated somatic embryos (Figure 1J). The loss of spatial and temporal organization continued and homozygous fie seedlings transformed into callus-like structures that could be maintained for several months displaying an increasing number of small cells (Figure 1F). This neoplastic behavior was confirmed by flow cytometrical analyses showing that shortly after germination fie cells started to endoreplicate as a sign of differentiation, a cellular behavior typically found in maturing wild-type plants (Figure S2A, S2B, S2D) [26], whereas at three months after germination the peaks corresponding to 8C and 16C were very much reduced and the remaining cell population gave rise to a DNA profile with cells being mostly in a G1 and a G2 phase, suggesting a massively dividing cell population (Figure S2C, S2D).
Thus, PRC2 in plants does not appear to be required for initial body plan organization, indicating a major difference with animal PRC2 function. After germination homozygous fie mutants displayed a progressive loss of cell differentiation states resembling the previously characterized clf-swn double mutant or a special fie mutant allele that results from the incomplete rescue of a fie mutant with a FIE-expressing transgene [5], [6].
H3K27me3 is lost in homozygous fie mutants
As H3K27me3 is essential in animal embryogenesis we asked if this mark is in fact missing in the viable fie mutants. Therefore we first analyzed by immuno-cytology the distribution of H3K27me3 in the nuclei of wild-type control plants and homozygous fie mutants (Figure S3). In two-week old wild-type plants, a clear nuclear signal that is widely dispersed along the entire chromatin was found (Figure S3A–S3C) consistent with previous studies [27], [28]. The spotted antibody signal is excluded from compacted heterochromatic regions, as visualized by DAPI staining. In contrast, no signal was observed in nuclei of two-week old fie plants (Figure S3D–S3F).
To obtain a high-resolution molecular map of the genome-wide distribution of H3K27me3 in wild type versus fie seedlings, chromatin immunoprecipitation (ChIP) was performed, followed by hybridization to a whole genome tiling array. A total of 5634 genes were identified as putative PRC2 targets in wild type seedlings, in good agreement (68% overlap) with a previous analysis (Table S1, Figure S4) [29].
In fie seedlings, the H3K27me3 signal was absent or extremely reduced throughout the genome (Figure 2). However, out of 5634 H3K27me3 positive genes in wild type, 1384 (24.6%) still passed the detection threshold in fie seedlings (Figure S4). Furthermore, 2014 genes appeared to be marked de novo by H3K27me3 in fie. Yet, in addition to being much weaker, the H3K27me3 signal in fie showed an atypical distribution pattern over genes and the marked genes were on average larger and slightly closer to transposable elements (Figure S5, Table S2). Notably, the most prominent signal in the mutant was found over heterochromatic regions, i.e. repeat-regions and transposable elements although H3K27me3 is typically excluded from these locations (Figure 2A, Figure S5B) [29]. Such an apparent re-localization of H3K27me3 signal to heterochromatic regions has also been seen on immuno-localization level in other mutants in PRC2 components [27].

To test the H3K27me3 signal found in wild type and fie tiling arrays, we performed locus-specific qPCR on our ChIP-derived DNA-material. We analyzed seven gene loci and corroborated a H3K27me3 signal in wild type and its absence in fie (Figure S6A, S6D). Moreover, we could detect in qPCR experiments only a slight increase in H3K27me3 over heterochromatic regions in fie in contrast to the array signal (Figure S6A, S6E, S6F). In any case, the signal over heterochromatic regions was much below the level of H3K27me3-positive genic regions in wild type. These findings suggest that the antibody recognizes additional epitopes besides the H3K27me3 mark, preferentially in the absence of the proper antigen. A weak signal may get artificially enhanced in ChIP-on-chip experiments due to the global amplification procedure that is not applied in gene-specific ChIP-qPCR experiments.
To investigate the specificity of the antibody, we performed peptide competition assays. Nuclear protein extracts isolated from wild type showed a strong signal in Western blots while no band corresponding to the H3K27me3 mark could be detected in homozygous fie mutants under standard conditions (Figure 3A). However, when over-exposed or under less stringent conditions a faint signal also became visible in fie (Figure 3A, 3B). Using increasing peptide concentrations of up to 10 µg of H3K27me2 and H3K27me1 peptides, a gradual decrease in signal strength was observed in the case of H3K27me2 and H3K27me1 in the fie mutant, with the mono-methylated peptide being the most effective (Figure 3C). As the signal was strongly reduced by the peptides the cross-reactivity of the antibody might account to a large extent for the remaining H3K27me3 signal in our Western and ChIP-experiments. Moreover the H3K27me3-peptide could not deplete the signal further in fie as would be expected when the mutant is already largely devoid of any H3K27me3 (Figure 3B). In contrast, the trimethylated peptide effectively reduced the signal in wild type to a level comparable to the detection level in fie whereas the H3K27me2 - and H3K27me1-peptides did not show any obvious effect in wild type samples up to concentrations of 10 µg (Figure 3B, 3D). Thus, we conclude that the remaining signal in fie is not H3K27me3 but to some extent H3K27me2 and more pronounced H3K27me1 demonstrating a slight cross-reactivity of the antibody. Given that H3K27me1 is found mainly over heterochromatic regions in Arabidopsis wild-type plants [30], we conclude that a cross-reactivity of the H3K27me3 antibody with H3K27me1 accounts for the gain in signal over repeat-regions and transposable elements in the fie mutant.

Genes involved in plant reproduction are particularly up-regulated in homozygous fie mutant seedlings
To unravel the transcriptional consequences upon the loss of PRC2 function we compared genome-wide expression levels of homozygous fie mutants with wild type at two different time points. At 7 DAS, no major transformations were observed, yet the plants could be unambiguously and reproducibly identified as homozygous fie mutant plants due to their aberrant root growth and subsequent genotyping (Figure 1). At the second time point at 20 DAS, substantial morphological transformations were clearly visible.
Within our reference set (Table S3), a total of 1115 genes were significantly up-regulated at 7 DAS and 1735 genes at 20 DAS in fie versus wild-type plants (Bonferroni P-value ≤0.05, see Material and Methods). Conversely, we also found genes to be significantly weaker expressed in fie versus wild type: 1308 and 1843 genes at 7 DAS and 20 DAS, respectively (Bonferroni P-value ≤0.05; Figure S7'). Next, we compared the expression data with our PRC2 target gene set. Only a fraction of all identified PRC2 target genes became up-regulated in fie mutants, indicating that PRC2 is not the only repressive system and/or besides the revelation of the repression activators are required for gene expression (Figure S7). Still, our data are consistent with the concept that H3K27me3 mark is associated with inhibition of gene expression since the overlap of the group of up-regulated genes at 7 DAS and 20 DAS with the group of H3K27me3 marked genes is larger than expected by random (7 DAS and 20 DAS: representation factor (rf) = 7.1, p<1.0e−99 *; 7 DAS and H3K27me3: rf = 1.6, p<1.8e−21; 20 DAS and H3K27me3: rf = 1.1, p<0.009; Figure S7). Conversely, for down-regulated genes at 7 DAS we see the opposite effect, i.e. the overlap of both gene sets is smaller than expected at random (7 DAS and 20 DAS: rf = 7.5, p<1.0e−99; 7 DAS and H3K27me3: rf = 0.6, p<1.3e−13; 20 DAS and H3K27me3: rf = 0.9, p<0.122; Figure S7).
To evaluate the PRC2 targets that are up-regulated, potentially in direct response to the loss of H3K27me3, we examined which gene ontology (GO) categories are overrepresented among the up-regulated genes that lost H3K27me3 in fie mutants using the BINGO analysis software [31]. Most overrepresented GO categories in the classification system biological function relate to reproduction with two distinct subcategories: Flower - and seed development (Figure 4, Figure S8). Besides reproduction, a few additional small categories were overrepresented such as abscisic acid (ABA) signaling and lipid-transport and –sequestering. However, a closer analysis of the corresponding genes revealed that they are also linked with reproduction, in particular seed development (see below).

PRC2 function in flower and seed development
H3K27me3 appears to be a key repressive mechanism for the expression of many genes controlling different aspects of flower development and consistent with this, homozygous fie mutants are very early flowering, i.e. as early as 10 DAS and produce ectopical flowers, e.g. on roots. A similar early flowering phenotype has been found in mutants with compromised PRC2 activity [5], [6], [32], and has been related to the early deregulation of LEAFY (LFY), AGAMOUS (AG) and PISTILLATA (PI), which starts as early as the embryonic stage. Our analysis identifies several additional genes controlling flower development as PRC2 targets that are significantly up-regulated in fie mutants, including genes involved in the establishment of a floral meristem (e.g. FLC, AGL24, LFY, FUL and CAL), genes involved in promoting a determinate floral meristem (e.g. ULT1, PAN, LFY) and genes involved in organ identity specification (e.g. AP3, SEP3, LFY, PI) (Figure S9). The results of our microarray experiments could be validated by qRT PCRs confirming the significant up-regulation of 5 genes (AP3, CRC, FLC, PI, SEP3). In addition, 2 genes that were only slightly (but not significantly) up-regulated in our microarray experiment were also found to have significantly elevated expression levels in the qRT PCR on fie mutant material (AG, AP1), whereas the flowering regulator FWA shows neither upregulation in the array nor in qRT-PCR experiments (Table S4).
The second principal category of PRC2 target genes that became up-regulated in fie mutants are genes functioning in late seed development (Figure 4, Figure S8, Figure S10). Among the PRC2 targets that are up-regulated in fie we find genes acting at different hierarchical levels in late seed development, from master regulators (e.g. AGL15, LEC2, ABI3, FUS3) and more specific modulators (e.g. WRI, FLC) over genes promoting ABA and/or inhibiting GA signaling (e.g. ABI4, DOG1, CHO1, SOM, SPL8) down to target genes such as storage compounds (e.g. CRU3, CRA1, LEAs, oleosins) (Figure 5).

The up-regulation of many important seed regulatory genes raised the hypothesis that fie seedlings, albeit macroscopically resembling wild type seedlings, display seed phase characteristics. To test this, we first analyzed the lipid content using the dye Fat Red that stains for triacylglycerol-lipids in red color. Whereas wild-type seedlings displayed a sharply decreasing lipid content from 5 to 8 DAS, fie mutants showed an intense red color indicating a high lipid content that is typical for late seed maturation stages in wild type (Figure 1M, 1N).
To test whether the failure to repress late seed genes during the seed maturation process interferes with germination, we performed seed germination assays of clf-swn and fie mutants in comparison with wild-type plants. Whereas all wild-type seeds germinated within 2 DAS, both clf-swn and fie mutants show delayed germination (Figure 6A). Eventually, over 90% of clf-swn mutants germinated around 4 DAS. In contrast, approximately 40 percent of the homozygous fie mutants stayed dormant for the course of the entire experiment (20 days), as revealed by dissecting dormant seeds and genotyping of the embryos.

Dissected dormant embryos were then allowed to develop on agar plates. As a reference wild-type embryos were isolated from seeds 24h after imbibition. Initially, dormant fie embryos are indistinguishable from wild type embryos (Figure 6C, 6F) and around 1/4 of these fie mutants started to develop in a similar manner as wild type, showing root - and root hair formation, unfolding and greening of the cotyledons and the accumulation of anthocyanin (Figure 6C–6H). However the remaining 3/4 of fie embryos stayed dormant for a period of several days. Some of these finally could break dormancy and started to develop although proliferation was extremely delayed (Figure 6L–6N). Notably, heterozygous cdka;1 mutants behaved like wild type seeds consistent with the previous finding that cdka;1 mutants are sporophytically recessive. Similarly, double heterozygous cdka;1-fie mutants also did not show any germination defects demonstrating that the observed dormancy phenotypes are due to the loss of PRC2 function.
Germination is associated with a low ABA to gibberellic acid (GA) ratio [1]. Intriguingly, the development of some of the dissected, initially dormant fie seedlings resembled the development of wild–type plants that germinated on high concentration of ABA, lacking proper greening of aerial tissue, root formation and expansion of true leaves (Figure 7I–7J, 7L–7M). However, applying high dosage of GA did not lead to higher germination rates of fie mutants (Figure 7B), suggesting that the primary defect in the class of non-germinating fie seeds is dormancy release and not the germination itself.

Overrepresentation of PRC2 target genes in particular gene families
Among the up-regulated genes in fie controlling seed and flower development certain gene families appeared to be overrepresented, e.g. transcription factors, consistent with previous studies showing that those are in particular marked by H3K27me3 (Table 2) [7], [29], [33]. To get a more detailed picture, we tested whether all transcription factor families are equally subject to regulation by PRC2 (Figure S11A, S11B). Approximately 2/3 of all transcription factor families have members that are marked by H3K27me3 at 20 DAS. Notably, one of the largest transcription factor families within our reference set in which none of the member was found to carry H3K27me3 was the group of AUXIN RESPONSIVE FACTORS (ARFs) that mediate auxin signaling (Table S3). However, at a more general level, we found that other genes involved in the auxin signal transduction network are targets of PRC2 regulation, for instance several IAAs and PIN auxin transport facilitators (Table S1).

Among transcription factor families that are marked with H3K27me3, the fraction of PRC2 targets varies substantially. A particular high proportion of PRC2 targets (≥60%) were found in MIKC subclass of MADS transcription factors, in the WOX-class, the HD-Zip-IV Homeobox class and in the C2C2-Dof and C2C2-YABBY zinc finger classes, for the latter even all 6 members were found to be PRC2 targets. The transcription factor subfamily with the most (in absolute numbers as well as in percentages) PRC2 targets that also showed transcriptional up-regulation in fie is the MIKC class, among which we find central regulators of seed and flower development (Figure S11, Table S1, Table S5) [34].
In addition to transcription factors, a few other gene families were overrepresented among the PRC2 targets that are up-regulated in fie; the most prominent are oleosins and LATE EMBRYONIC ABUNDANT PROTEIN genes (LEAs). Oleosins are structural components of oil bodies and were found to be expressed preferentially in seeds or the tapetum layer of developing anthers [35] (Figure 5, Table S5). 11 of the 17 oleosin genes in our reference set are H3K27me3-marked and 8 are in addition up-regulated in fie (Table 2), which matches the observation of storage lipid accumulation in fie (Figure 1M, 1N). This is a strong overrepresentation since from all genes in our reference set, we find not more than 21% marked by H3K27me3 and only 2% being up-regulated in fie as well.
Another gene family that is highly overrepresented in the class of up-regulated PRC2-targets are LEAs, most of which are expressed in seeds. Of 54 LEAs covered by our reference set, we find 27 (50%) H3K27me3-marked and 16 (30%) being in addition up-regulated in fie. Interestingly, we found 7 (13%) of the LEAs down-regulated in fie and with a single exception these are not H3K27me3 targets and show a non-seed specific expression (Table S5, Figure S10) [36], [37].
Crosstalk in chromatin regulation
In Drosophila the function of the Polycomb complex Group (PcG) is counteracted by the action of the trithorax Group (trxG) [7]. In Arabidopsis, the role of TRX has been assigned to ATX1, ATXR7/SET DOMAIN GROUP25 (SDG25), PICKLE (PKL)/PICKLE-RELATED 2 (PKR2) and ULTRAPETALA 1 (ULT1) [38]–[42]. Our data showed that ULT1, ULT2 and PKR2 are PRC2 targets and at least ULT1 and ULT2 were substantially up-regulated at 7 and 20 DAS (Table S3). ULT1 has been shown to act as an anti-repressor (i.e. limiting H3K27me3 deposition) and as an activator of the flower regulator AG by mediating Lysine 4 H3 trimethylation [42]. To test whether the loss of H3K27me3 is accompanied with a gain in H3K4me3, as suggested by our finding of a possible negative feed-back of PRC2 on ULT1/ULT2 activation, we analyzed the genome-wide distribution of H3K4me3 in wild type and fie.
Consistent with previous studies [43], we found that in wild-type plants a large number of genes (approximately 1/3 of the genome) are marked with H3K4me3 at 20 DAS (Figure 2, Figure 7). However, the number of genes that are marked by both H3K27me3 and H3K4me3 is significantly smaller than expected for an independent distribution, as was observed previously [43] (Figure 7). This indicates repulsion of these two marks in accordance with the model that H3K27me3 and H3K4me3 signifies repressed and activated genes, respectively. None-the-less, a small set of 501 genes was identified as marked by both histone modifications.
In our ChIP-chip experiments we found only a slight increase in number of genes that carry the H3K4me3 mark in fie in comparison to wild-type plants (13945 vs. 13211 genes) and we do not see an elevated level of H3K4me3 in Western blot analyses (Figure S6C). Thus, genes that loose H3K27me3 do not in general gain H3K4me3 in fie (Figure 7, Table S3). On the other hand, gene up-regulation in fie is positively correlated with loss of H3K27me3 and gain in H3K4me3. The global proportion of genes with elevated expression in fie at 7 DAS is 4.5% while the percentage of up-regulated genes reaches 28% amongst those that loose H3K27me3 and concomitantly gain H3K4me3 (Table 3). In addition, for certain gene families, such as the MIKC group of MADS transcription factors, the WOX group of Homeobox genes and the oleosins, we find those genes that gain H3K4me3 in addition to the loss of H3K27me3 to be among the most highly up-regulated for these specific classes (Table 3, Table S5).

Thus, our data supports the view that H3K4me3 and H3K27me3 are mutually exclusive marks though in general a loss in H3K27me3 is not sufficient to gain H3K4me3. However, a tightly linked interdependency between both antagonistic marks is operating for a relatively small group of genes including members of the MIKC class of major developmental regulators [42], [44], [45]. Our finding that the PRC2-antagonizing TRX-function genes ULT1/ULT2 are themselves targets of PRC2 and consequently up-regulated in fie, provides a possibility for the molecular implementation of such an interconnected control mechanism.
Discussion
Here we have generated homozygous fie mutant plants overcoming a block in the analysis of PRC2 activity in the flowering plant Arabidopsis. Our approach relies on bypassing of double fertilization and circumventing the requirement for FIE during endosperm development. This has allowed us to study the genomic and developmental consequences of the complete loss of PRC2 activity during embryo and subsequent sporophyte development.
Genomic perspective
Several lines of evidence indicate that PRC2 in plants is indeed essential for depositing H3K27me3 marks similar to its function in animals. First, our ChIP-on-chip experiments showed that there is no or only a very weak H3K27me3 signal in fie and that the remaining signal shows properties that differ from the typical H3K27me3 mark. Second, at least 3 heterochromatic regions that showed H3K27me3 signal in fie in ChIP-on-chip experiments did not show a substantial level of enrichment in gene specific ChIP-qPCR assays. Third, the little residual signal of H3K27me3 in fie mutants can be further reduced in peptide competition assays with peptides that harbor H3K27me2 or H3K27me1 epitopes. Finally, H3K27me3 peptide is not effective in reducing the signal in fie further, as would be expected when the remaining signal were H3K27me3. Conversely, the H3K27me3 peptide could reduce the antibody signal in wild type to the signal strength found in fie.
Based on the by large mutually exclusive distribution of H3K27me3 and H3K4me3, we asked if genes which lost H3K27me3 in fie would in turn acquire H3K4me3. Such a reciprocal regulation has been found for AG and FLC in Arabidopsis [42], [44], [45]. Indeed, we could confirm that AG and FLC, both members of the MIKC transcription factor class, gain H3K4me3 in the absence of FIE. Moreover, 7 other MIKC transcription factors that represent important regulators during development showed a similar response. It was recently shown that the SAND domain protein ULTRAPETALA1 (ULT1) mediates the switch from H3K27me3 to H3K4me3 at the AG locus [42]. Interestingly, we found that ULT1 itself is under the control of PRC2, as it is marked by H3K27me3 in wild type and shows strong up-regulation in fie. This might explain the switch from H3K27me3 to H3K4me3 as seen for a remarkable number of the MIKC transcription factors. In animals the maintenance of trimethylated H3K4 was shown to require permanent TRX activity to counteract PRC2, as the repressive H3K27me3 state seems to be the default state for genes that are regulated by both antagonizing HMTase machineries [9]. However, global changes in H3K4me3 levels were not observed in fie, and the change from H3K27me3 to H3K4me3 was restricted to about 5.5% of the genes marked by H3K27me3.
We also identified a small group of potentially bivalently labeled genes (1.8% of reference set), i.e. harboring the activating H3K4me3 and the repressive H3K27me3 mark. The concomitance of both tags is found in more than 10% of all genes in human and mouse embryonic stem cells and Xenopus tropicalis embryos, and is thought to maintain the target genes in a “poised state”, resulting in transcriptional silencing but allowing for fast reactivation upon commitment to differentiation [46]–[50]. Bivalency has to our knowledge only been found for the AG and FT loci in Arabidopsis and its existence is also unclear for Drosophila [44], [51], [52]. However, we showed here that in contrast to Drosophila and mouse, Arabidopsis does not require the PRC2 to establish a normal body organization (see below). This renders it unlikely that animals and plants are using the same epigenetic mechanisms to set up the body plan, at least during embryogenesis.
Developmental perspective
The observation that the plant body plan can be established without PRC2 function is an unexpected result not only because PRC2 function is essential in animal embryogenesis but also regarding the strong postembryonic phenotype of clf-swn double mutants or fie mutants with a partially complementing FIE transgene [5], [6]. The overall correct body plan of fie embryos and early seedlings suggests that there is a tight network of intercellular communication presumably maintaining positional cues in the plant embryo. Indeed, research in the last decade has unraveled several patterning mechanisms in the plant embryo, for instance polar auxin distribution and non-cell-autonomously acting transcription factors [53], [54].
However, after body-plan formation, PRC2 function is key for the correct phase transition from embryonic to vegetative growth. Much progress has been made in the understanding of chromatin regulation and in particular the function of PRC2 during the phase transition from vegetative growth to flowering (for review see [55]–[57]). In contrast, the view that chromatin regulation is important for controlling the switch from mature seed to seedling is only now emerging (for review see [58], [59]), and the involvement of PRC2 in late seed development has been unclear beyond the finding that many genes implicated in seed maturation are labeled by H3K27me3 (for review see [58], [60], [61]). Defining the role of PRC2 during seed maturation has been obscured due to a prominent function of PRC2 earlier in seed development, i.e. for endosperm growth and differentiation. The combination of our genetics and genomics studies demonstrate that PRC2 is one of the major control systems of this phase change by shutting down the entire cascade of maturation genes from master regulators to a wide range of downstream targets before or at germination (see Figure 5). Moreover, our data suggest that the PRC2 mediated phase transition from seed - to seedling stage takes place primarily at the level of the embryo, as seeds with homozygous fie mutant endosperm but heterozygous mutant embryo germinate like wild type.
A wealth of genetic and physiological experiments has demonstrated that seed development is under the tight control of plant hormones and that GA triggers while ABA inhibits seed germination (for review see [1]). High ABA and low GA levels are characteristic for maturing seeds allowing the establishment of seed dormancy, while this relationship is inverted at germination. PRC2 action in the maturing seed seems to sustain the antagonistic action of the two plant hormones ABA and GA by inhibiting positive regulators in ABA and negative players in GA signaling. For example, the PRC2 target SOMNUS (SOM), a CCCH-type zinc finger protein, down-regulates GA and up-regulates ABA levels. SOM expression is seed specific and our finding that it is a PRC2 target and up-regulated in fie suggests that in wild type down-regulation of SOM is maintained by H3K27me3 to allow for high GA and low ABA levels in the germinating seed.
Besides the regulation of the ABA-GA signaling pathway, we also found that DELAY OF GERMINATION 1 (DOG1), a major regulator of seed dormancy [62], is a H3K27me3 target and significantly up-regulated in fie seedlings. Interestingly, it was recently shown that DOG1 is also regulated by HISTONE MONOUBIQUITINATION1 (HUB1), a C3HC4 RING finger protein required for histone H2B monoubiquitination [63].
In this context it will be interesting to examine if dormancy control is generally regulated at the level of chromatin, as for example different Arabidopsis accessions from diverse environmental origins show dramatic differences in seed dormancy [64]. Since PRC2 function in the perception of cold via the repression of the flowering inhibitor FLC is well established for the transition to the generative phase [57], it is tempting to speculate that a similar mechanism functions to perceive this environmental stimulus in the seed. The need of cold stratification in order to break seed dormancy in many plant species [65] and the observation that FLC plays a role in this process as well [66], might reveal a common regulatory mechanism operating in the transition from vegetative to generative phase as well as from embryonic to vegetative phase.
Interestingly another phase transition, the switch from gametophytic to sporophytic development was recently shown to be regulated by PRC2 in moss, where PRC2 represses the differentiation of the sporophyte [67], [68]. The authors correlate their observations with the transition from gametophytic to sporophytic development in flowering plants that is as well controlled by PRC2, as Arabidopsis fie mutants for example show untimely development of the gametophytic endosperm without fertilization [22], [67], [69], [70].
The reprogramming of gene activity is a mandatory step to allow for cellular differentiation processes and the stable inheritance of these differentiation states needs to be maintained for the integrity of the organism. Plants in particular have to adapt to environmental conditions and therefore need to change their developmental phase accordingly, and the phase transition from embryo to seedling stage can be considered as the earliest adaptive phase in plants. The origin of seed dormancy in land plant evolution is regarded as a major step in the successful establishment of flowering plants to sustain in non-favorable conditions and its control by PRC2 is an exciting example for the recruitment of an evolutionary conserved molecular machinery to fulfill new functions.
Material and Methods
Plant material and growth conditions
Unless indicated otherwise, Col-0 was used as wild type for all experiments. The cdka;1-1 (AT3G48750) allele has been previously described (SALK_106809 [22]). The SALK_042962 line was used as the standard allele for fie. As additional fie alleles the T-DNA lines GK-362D08-016994 and GK-534F01-020364 were used that both displayed the previously described typical fie mutant phenotype. One previously described fie allele in WS-0 [71] was sequenced and shown to carry a base-pair exchange mutation 5′ of the fourth splice acceptor site. The curly leaf (clf-28, SALK_139371, At2g23380) and swinger (swn-4, SALK_109121, At4g02020) alleles have been previously described [72], [73]. All seeds were sterilized using chloride gas and sown on 0.8% Phyto agar plates (½ Murashige & Skoog (MS) salts and 1% sucrose) and grown under day neutral conditions (12h light 21°C, 12 h dark 17°C). After germination, plants were transferred to either new plates for long-term observation or to soil and grown in long day conditions (16 h, 22°C light; 8 h, 18 C°dark).
Germination assays and hormone treatment
To examine germination, seeds from plants that were grown under the same growth conditions and stored for at least 3 months were sterilized with Chloride-gas and stratified for at least 4 days at 4°C. Upon germination induction (light, 21°C), germination rate was monitored for the following 6 days. After approximately 14 days the plants were analyzed with respect to their phenotype to distinguish between mutants and phenotypically wild-type plants and correlated with the day of germination. Dormant seeds were dissected under a stereomicroscope using a fine needle and fine forceps and subsequently genotyped by PCR. Gibberellic acid (GA, gibberellin A3, Sigma) was dissolved in Ethanol (10 mM stock solution) and applied to the MS-plates in concentrations from 0.01 µM to 10 µM. The germination rate of fie mutants on GA-plates was analyzed 10–14 days after stratification (DAS). Abscisic acid (ABA, Sigma) was dissolved in Methanol (stock concentration 10 mM) and used in final concentration of 1 µM. Wild type germination on ABA-plates was monitored over time.
Lipid staining
Plants were first partially dehydrated in three steps (20%, 40%, 60% isopropanole solution), then incubated for 1 hour with Fat Red solution (filtered 0.5% Sudan III in 60% isopropanole) and re-hydrated again using the same dilution series in reversed direction. Subsequently samples were additionally washed twice with water and analyzed under a dissecting microscope.
RNA–extraction and qRT–PCR
RNA extraction was performed using Qiagen-RNAesy mini-kit, following the manufacturers instruction. RNA-concentration and purity was tested using nanodrop-photometric quantification (Thermo Scientific). RNA-integrity was verified by running 1 ug of total RNA on 1.5% agarose TBE-gels to detect the 28S and 16S rRNA bands. 2 µg RNA was treated with DNAseI (MBI Fermentas) according to the manual to avoid contamination of genomic DNA and subsequently processed to obtain cDNA using polyT-primer and reverse transcriptase (Superscript III, Invitrogen) following the manufacturers instruction. After reverse transcription RNA was removed by RNAseH digest. For negative control, all steps were followed in the same manner, except for adding the reverse transcriptase. The resulting cDNA was used for Reverse Transcription(RT)-PCR or quantitative Real Time-PCR (qRT-PCR) using the Roche LightCycler 480 system. Oligonucleotides were designed using either Primer3Plus-design tool (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) or QuantPrime (qPCR primer design tool: http://www.quantprime.de/main, [74] and used in final concentration of 0.25 µM each. Primers for qPCR have been tested for efficiency of >90% and are listed in Table S6. For qPCR at least two biological and three technical replicates were processed and expression was calculated relative to ACT7 (AT5G09810). Several reference genes were tested in comparison between mutant and wild type samples to confirm equal loading (see Table S6).
Transcriptome assay
Microarray analysis was carried out at the Unité de Recherche en Génomique Végétale (Evry, France), using the CATMA arrays [75], [76]. Two independent biological replicates were produced. For each biological repetition and each time point, RNA samples were obtained by pooling RNA from 100 wild-type and 100 fie seedlings at stage 7 DAS and 10 wild type and 50 fie plants at 20 DAS, respectively. 7 DAS stage plants were cultivated on plates, 20 DAS material was grown on plates for 10 days, then transferred to liquid media for another 10 days in day neutral conditions (12 h light, 21°C; 12 h dark. 17°C). Total RNA was extracted using Qiagen RNeasy plant mini kit according to the supplier's instructions. The hybridization to the slides, and the scanning were performed as described in Lurin et al. (2004) [77].
Normalization and statistical analysis were based on two dye swaps (i.e. four arrays, each containing 24,576 GSTs and 384 controls) as described in Gagnot et al. (2007) [78]. The raw P-values were adjusted by the Bonferroni method, which controls the Family Wise Error Rate, (with a type I error equal to 5%) in order to keep a strong control of the false positives in a multiple-comparison context [79]. We considered as being differentially expressed the genes with a Bonferroni P-value ≤0.05, as described in Gagnot et al (2007) [78].
Microarray data from this article were deposited at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), accession no. GSE19851, direct link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=djcjpeggkgsuirw&acc=GSE19851) and at CATdb (http://urgv.evry.inra.fr/CATdb/; Project: RS08-09_FIE) according to the “Minimum Information About a Microarray Experiment” standards.
Western blot and peptide competition assay
Nuclear enriched protein extracts were prepared after thoroughly grinding in liquid nitrogen of around 1 g of plant material. All subsequent steps are carried out in the cold. The powder was dissolved in 10 ml of Lysis buffer (45 ml Low Salt Wash buffer [see below] + 0.5 ml TritonX-100 + 5 ml glycerol + 50 µl 100 mM PMSF + 20 µl β-mercaptoethanol freshly prepared on ice), vortexed and placed on a rotation wheel for 20 min at 4C. The solution was filtered using 100 µm nylon mesh and centrifuged for 20 min at 4000 rpm at 4°C, following resuspension of the pellet in 2 ml lysis buffer. The solution was transferred to a new 2 ml tube and centrifuged for 20 min, 4000 rpm, 4°C. The resulting pellet was resuspended in 200 µl 1XSDS loading buffer. Low Salt Wash buffer: 20 ml 0.5 M HEPES pH 7.5 + 6 ml 5 M NaCl + 400 µl 500 mM EDTA + H2O up to 200 ml. 15% SDS-gel page was performed according to standard protocols. After SDS page proteins were blotted on Hybond-P PVDF membrane (Amersham Biosciences) for 75 min, 140 mA in temperature controlled condition. All membrane manipulation experiments where carried out at room temperature (RT) when not stated otherwise. Membrane was blocked using incubation with 4XBlockAce (ABD Serotec) for 3 h. Throughout all experiments we used Anti-H3 1∶20,000 (Millipore, reference nr: 06-755) as loading control, Anti-trimethyl-Histone H3 (Lys27) antibody (Millipore, reference-nr: 07-449) in final concentration between 1∶10,000 and 1∶50,000, dissolved in 5%BSA in 1xTBST (1x TBS with 0.1% TritonX-100) and Anti-trimethyl-Histone H3 (Lys4) antibody (Millipore, reference nr: 07-473) at 1∶5,000–1∶10,000. The primary antibody was incubated at 4°C over night. After washing 3 times 15 min with 1xTBST the secondary antibody (Anti-Rabbit antibody, GE-Healthcare, reference-nr: NA934-100UL) was applied at 1∶50,000 in 5%BSA-1xTBST solution for 2 h. Washing was either performed stringently with 3x 30 min or less stringently 3x10 min. For detection the two-component reagent Immobilion Western Chemiluminiscent HRP substrate (Millipore) was used. For peptide competition, first the sub-saturating antibody concentration was determined. For anti-H3K27me3 this was at a concentration of 1∶50,000. Then increasing concentrations from 0.1–10 ug of H3K27me3, H3K27me2 and H3K27me1 peptide (Millipore 12-565, 12-566, 12-567) were added to a 10 ml antibody-solution and incubated under slight agitation for 4 h at RT and an additional 1 h at 4°C before hybridizing on the membrane. Subsequent hybridization and detection were performed as described above.
ChIP-on-chip analysis
Chromatin immunoprecipitation (ChIP) experiments were done as described previously [74], in two biological replicates, using the following antibodies: H3K4me3, Millipore 07-473; H3K27me3, Millipore 07-449. DNA recovered after ChIP and directly from input chromatin was amplified using the Sigma GenomePlex Complete Whole Genome Amplification (WGA) Kit as directed, differentially labeled and hybridized in dye-swap experiments to a custom-made Roche-NimbleGen whole-genome tiling microarray. This microarray covers the entire forward strand of the Arabidopsis genome sequence (TAIR8) at 175 nt resolution with approx. 720K isothermal tiles (50–75 oligonucleotides). Following ANOVA normalization, raw data were analyzed using a linear regression mixture model (ChIPmix, [80]), which was adapted to handle multiple replicates simultaneously (script available on request). Lists of tiles reporting significant enrichment were converted in sets of chromatin domains by combining adjacent enriched tiles, allowing a maximal gap of 165 nucleotides. Only domains of at least 400 nucleotides were considered for further analysis. TAIR8 release was used for annotation of genes and transposable elements.
Several loci were additionally tested for H3K27me3 and H3K4me3 enrichment as compared to input. Input was diluted 1∶100 prior to qPCR application. From the diluted input material and from the ChIP-material 1 µl was applied for each triple replicate reaction in the Roche Lightcycler 480 Real Time System using Roche SYBR green reagent according to the supplier's instruction (Roche). Primers used for this assay are given in Table S6.
ChIP on chip data from this article were deposited at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), accession no. GSE24163, direct link: (GSE24163, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24163).
Immunocytology
10–14 DAS wild type and fie seedlings were fixed and processed as described previously [28], washed 3×5 min in 1× PBS before pre-incubation with BSA. Diluted rabbit polyclonal α - trimethyl H3K27 primary antibody (1∶100, Millipore 07-449) was incubated for 1 h at 37°C, washed 3×5 min in 1× PBS and incubated with diluted Alexa Fluor 488 conjugated goat anti-rabbit polyclonal secondary antibody (1∶200, Invitrogen (Molecular Probes) A-11008) for 1 h at 37°C, washed 3×5 min in 1× PBS and mounted in 1× PBS containing 1 µg/µl DAPI. Images were acquired using an Axioplan 2 Carl Zeiss Microscope with a cooled AxioCam HRc camera using a bandpass 515–565 nm emission filter (Carl Zeiss # 488010-9901-000) and a longpass 397 nm emission filter (Carl Zeiss # 488001-9901-000) for visualization of AF488 and DAPI, respectively. Fixed exposure settings for both florochromes were: AF488, 196 ms, 402 ms and 1002 ms (overexposure); DAPI, 50 ms, 100 ms and 305 ms (overexposure).
Data analysis
For all analyses comparing array expression and ChIP chip data a reference gene set of 24901 genes was defined that included those genes for which data from both type of experiments were available (Table S2). The Transcription factor classification was taken form the Arabidopsis transcription factor database (AtTFDB) hosted on the Arabidopsis Gene Regulatory Information Server (AGRIS, http://arabidopsis.med.ohio-state.edu/AtTFDB). Venn diagrams were generated using the VENN diagram generator designed by Tim Hulsen at http://www.venndiagram.tk/ and http://www.cmbi.ru.nl/cdd/biovenn/ (BioVenn [81]). The test for statistical significance of the overlap between two groups of genes was calculated by using software provided by Jim Lund accessible at http://elegans.uky.edu/MA/progs/overlap_stats.html. A representation factor (rf) is given and the probability (p) of finding an overlap of x genes is calculated using a hypergeometric probability formula. When p was below the calculation limits of the software (highly significant) we noted p<1.0e−99*. The representation factor is the number of overlapping genes divided by the expected number of overlapping genes drawn from two independent groups. A representation factor >1 indicates more overlap than expected of two independent groups, a representation factor <1 indicates less overlap than expected, and a representation factor of 1 indicates that the two groups by the number of genes expected for independent groups of genes. To determine which Gene Ontology (GO) categories are statistically overrepresented among the H3K27me3 targets that are up-regulated in fie, we used the BINGO 2.3 plugin for Cytoscape (http://www.psb.ugent.be/cbd/papers/BiNGO/Home.html). A custom annotation file was created using the build in annotation file for GO biological process and our reference set of 24901 genes. Other than that default parameters were used.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FinkelsteinRReevesWAriizumiTSteberC 2008 Molecular aspects of seed dormancy. Annu Rev Plant Biol 59 387 415
2. HeY 2009 Control of the transition to flowering by chromatin modifications. Molecular Plant 2 554 564
3. FarronaSCouplandGTurckF 2008 The impact of chromatin regulation on the floral transition. Seminars in Cell & Developmental Biology 19 560 573
4. KimDHDoyleMRSungSAmasinoRM 2009 Vernalization: winter and the timing of flowering in plants. Annu Rev Cell Dev Biol 25 277 299
5. KinoshitaTHaradaJJGoldbergRB 2001 Polycomb repression of flowering during early plant development. Proceedings of the National Academy of Sciences
6. ChanvivattanaYBishoppASchubertDStockCMoonYH 2004 Interaction of Polycomb-group proteins controlling flowering in Arabidopsis. Development 131 5263 5276
7. SchuettengruberBChourroutDVervoortMLeblancBCavalliG 2007 Genome regulation by polycomb and trithorax proteins. Cell 128 735 745
8. SchwartzYBPirrottaV 2008 Polycomb complexes and epigenetic states. Current Opinion in Cell Biology 20 266 273
9. PappBMüllerJ 2006 Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes & Development 20 2041 2054
10. BantigniesFCavalliG 2006 Cellular memory and dynamic regulation of polycomb group proteins. Current Opinion in Cell Biology 18 275 283
11. KohlerCVillarCB 2008 Programming of gene expression by Polycomb group proteins. Trends Cell Biol 18 236 243
12. PienSGrossniklausU 2007 Polycomb group and trithorax group proteins in Arabidopsis. BBA-Gene Structure and Expression 1769 375 382
13. SimonJChiangABenderW 1992 Ten different Polycomb group genes are required for spatial control of the abdA and AbdB homeotic products. Development 114 493 505
14. StruhlGAkamM 1985 Altered distributions of Ultrabithorax transcripts in extra sex combs mutant embryos of Drosophila. EMBO J 4 3259 3264
15. FaustCSchumacherAHoldenerBMagnusonT 1995 The eed mutation disrupts anterior mesoderm production in mice. Development 121 273 285
16. HuhJHBauerMJHsiehTFischerR 2007 Endosperm gene imprinting and seed development. Current opinion in genetics & development 17 480 485
17. BergerFChaudhuryA 2009 Parental memories shape seeds. Trends in Plant Science 14 550 556
18. KinoshitaTIkedaYIshikawaR 2008 Genomic imprinting: A balance between antagonistic roles of parental chromosomes. Seminars in Cell and Developmental Biology 19 574 579
19. BarouxCPienSGrossniklausU 2007 Chromatin modification and remodeling during early seed development. Current opinion in genetics & development 17 473 479
20. KatzAOlivaMMosqunaAHakimOOhadN 2004 FIE and CURLY LEAF polycomb proteins interact in the regulation of homeobox gene expression during sporophyte development. Plant J 37 707 719
21. AwSJHamamuraYChenZSchnittgerABergerF 2010 Sperm entry is sufficient to trigger division of the central cell but the paternal genome is required for endosperm development in Arabidopsis. Development
22. NowackMKGriniPEJakobyMJLafosMKonczC 2006 A positive signal from the fertilization of the egg cell sets off endosperm proliferation in angiosperm embryogenesis. Nat Genet 38 63 67
23. IwakawaHShinmyoASekineM 2006 Arabidopsis CDKA;1, a cdc2 homologue, controls proliferation of generative cells in male gametogenesis. Plant J 45 819 831
24. NowackMKShirzadiRDissmeyerNDolfAEndlE 2007 Bypassing genomic imprinting allows seed development. Nature 447 312 315
25. KurzhalsRLTieFStrattonCAHartePJ 2008 Drosophila ESC-like can substitute for ESC and becomes required for Polycomb silencing if ESC is absent. Developmental Biology 313 293 306
26. BramsiepeJSchnittgerA submitted Endoreplication and development. Plant Signaling & Behavior
27. LindrothAMShultisDJasencakovaZFuchsJJohnsonL 2004 Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO J 23 4286 4296
28. NaumannKFischerAHofmannIKraussVPhalkeS 2005 Pivotal role of AtSUVH2 in heterochromatic histone methylation and gene silencing in Arabidopsis. EMBO J 24 1418 1429
29. ZhangXClarenzOCokusSBernatavichuteYPellegriniM 2007 Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol 5 e129 doi:10.1371/journal.pbio.0050129
30. JacobYStroudHLeblancCFengSZhouL 2010 Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature 2010 Jul 14
31. MaereSHeymansKKuiperM 2005 BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21 3448 3449
32. YoshidaNYanaiYChenLKatoYHiratsukaJ 2001 EMBRYONIC FLOWER2, a novel polycomb group protein homolog, mediates shoot development and flowering in Arabidopsis. The Plant Cell 13 2471 2481
33. TurckFRoudierFFarronaSMartin-MagnietteML 2007 Arabidopsis TFL2/LHP1 specifically associates with genes marked by trimethylation of histone H3 lysine 27. PLoS Genet 3 e86 doi:10.1371/journal.pgen.0030086
34. de FolterSImminkRGKiefferMParenicováLHenzSR 2005 Comprehensive interaction map of the Arabidopsis MADS Box transcription factors. The Plant Cell 17 1424 1433
35. KimHUHsiehKRatnayakeCHuangAH 2002 A novel group of oleosins is present inside the pollen of Arabidopsis. J Biol Chem 277 22677 22684
36. HundertmarkMHinchaDK 2008 LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genomics 9 118
37. Bies-EthèveNGaubier-ComellaPDeburesALasserreEJobetE 2008 Inventory, evolution and expression profiling diversity of the LEA (late embryogenesis abundant) protein gene family in Arabidopsis thaliana. Plant Mol Biol 67 107 124
38. Alvarez-VenegasRPienSSadderMWitmerXGrossniklausU 2003 ATX-1, an Arabidopsis homolog of trithorax, activates flower homeotic genes. Curr Biol 13 627 637
39. TamadaYYunJYWooSCAmasinoRM 2009 ARABIDOPSIS TRITHORAX-RELATED7 is required for methylation of lysine 4 of histone H3 and for transcriptional activation of FLOWERING LOCUS C. The Plant Cell 21 3257 3269
40. BerrAXuLGaoJCognatVSteinmetzA 2009 SET DOMAIN GROUP25 encodes a histone methyltransferase and is involved in FLOWERING LOCUS C activation and repression of flowering. PLANT PHYSIOLOGY 151 1476 1485
41. AichingerEVillarCBFarronaSReyesJCHennigL 2009 CHD3 proteins and polycomb group proteins antagonistically determine cell identity in Arabidopsis. PLoS Genet 5 e1000605 doi:10.1371/journal.pgen.1000605
42. CarlesCCFletcherJ 2009 The SAND domain protein ULTRAPETALA1 acts as a trithorax group factor to regulate cell fate in plants. Genes & Development 23 2723 2728
43. ZhangXBernatavichuteYVCokusSPellegriniMJacobsenSE 2009 Genome-wide analysis of mono-, di - and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol 10 R62
44. SalehAAl-AbdallatANdamukongIAlvarez-VenegasRAvramovaZ 2007 The Arabidopsis homologs of trithorax (ATX1) and enhancer of zeste (CLF) establish 'bivalent chromatin marks' at the silent AGAMOUS locus. Nucleic Acids Research 35 6290 6296
45. PienSFleuryDMylneJSCrevillenPInzéD 2008 ARABIDOPSIS TRITHORAX1 dynamically regulates FLOWERING LOCUS C activation via histone 3 lysine 4 trimethylation. The Plant Cell 20 580 588
46. AkkersRCvan HeeringenSJJacobiUGJanssen-MegensEMFrançoijsKJ 2009 A hierarchy of H3K4me3 and H3K27me3 acquisition in spatial gene regulation in Xenopus embryos. Developmental Cell 17 425 434
47. KuMKocheRPRheinbayEMendenhallEMEndohM 2008 Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4 e1000242 doi:10.1371/journal.pgen.1000242
48. MikkelsenTSKuMJaffeDBIssacBLiebermanE 2007 Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448 553 560
49. PanGTianSNieJYangCRuottiV 2007 Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1 299 312
50. ZhaoXDHanXChewJLLiuJChiuKP 2007 Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1 286 298
51. GanQSchonesDEHo EunSWeiGCuiK 2010 Monovalent and unpoised status of most genes in undifferentiated cell-enriched Drosophila testis. Genome Biol 11 R42
52. JeongJHSongHRKoJHJeongYMKwonYESeolJH 2009 Repression of FLOWERING LOCUS T chromatin by functionally redundant histone H3 lysine 4 demethylases in Arabidopsis. PLoS ONE 4 e8033 doi:10.1371/journal.pone.0008033
53. SchlerethAMöllerBLiuWKientzMFlipseJ 2010 MONOPTEROS controls embryonic root initiation by regulating a mobile transcription factor. Nature 464 913 916
54. LauSEhrismannJSSchlerethATakadaSMayerU 2010 Cell-cell communication in Arabidopsis early embryogenesis. Eur J Cell Biol 89 225 230
55. AmasinoR 2010 Seasonal and developmental timing of flowering. Plant J 61 1001 1013
56. DennisESPeacockWJ 2007 Epigenetic regulation of flowering. Current Opinion in Plant Biology 10 520 527
57. HendersonIRDeanC 2004 Control of Arabidopsis flowering: the chill before the bloom. Development 131 3829 3838
58. ZhangHOgasJ 2009 An Epigenetic Perspective on Developmental Regulation of Seed Genes. Molecular Plant 2 610 627
59. NorthHBaudSDebeaujonIDubosCDubreucqB 2010 Arabidopsis seed secrets unravelled after a decade of genetic and omics-driven research. Plant J 61 971 981
60. JunkerAHartmannASchreiberFBäumleinH 2010 An engineer's view on regulation of seed development. Trends in plant science
61. HoldsworthMJBentsinkLSoppeWJ 2008 Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination. New Phytol 179 33 54
62. BentsinkLJowettJHanhartCJKoornneefM 2006 Cloning of DOG1, a quantitative trait locus controlling seed dormancy in Arabidopsis. Proc Natl Acad Sci USA 103 17042 17047
63. LiuYKoornneefMSoppeWJ 2007 The absence of histone H2B monoubiquitination in the Arabidopsis hub1 (rdo4) mutant reveals a role for chromatin remodeling in seed dormancy. The Plant Cell 19 433 444
64. Alonso-BlancoCBentsinkLHanhartCJBlankestijn-de VriesHKoornneefM 2003 Analysis of natural allelic variation at seed dormancy loci of Arabidopsis thaliana. Genetics 164 711 729
65. Finch-SavageWELeubner-MetzgerG 2006 Seed dormancy and the control of germination. New Phytol 171 501 523
66. ChiangGCBaruaDKramerEMAmasinoRMDonohueK 2009 Major flowering time gene, flowering locus C, regulates seed germination in Arabidopsis thaliana. Proc Natl Acad Sci USA 106 11661 11666
67. MosqunaAKatzADeckerELRensingSAReskiR 2009 Regulation of stem cell maintenance by the Polycomb protein FIE has been conserved during land plant evolution. Development 136 2433 2444
68. OkanoYAonoNHiwatashiYMurataTNishiyamaT 2009 A polycomb repressive complex 2 gene regulates apogamy and gives evolutionary insights into early land plant evolution. Proc Natl Acad Sci USA 106 16321 16326
69. OhadNMargossianLHsuYCWilliamsCRepettiP 1996 A mutation that allows endosperm development without fertilization. Proc Natl Acad Sci U S A 93 5319 5324
70. IngouffMHaseloffJBergerF 2005 Polycomb group genes control developmental timing of endosperm. Plant J 42 663 674
71. LuoMBilodeauPDennisESPeacockWJChaudhuryA 2000 Expression and parent-of-origin effects for FIS2, MEA, and FIE in the endosperm and embryo of developing Arabidopsis seeds. Proc Natl Acad Sci U S A 97 10637 10642
72. DoyleMRAmasinoRM 2009 A single amino acid change in the enhancer of zeste ortholog CURLY LEAF results in vernalization-independent, rapid flowering in Arabidopsis. PLANT PHYSIOLOGY 151 1688 1697
73. WangDTysonMDJacksonSSYadegariR 2006 Partially redundant functions of two SET-domain polycomb-group proteins in controlling initiation of seed development in Arabidopsis. Proc Natl Acad Sci USA 103 13244 13249
74. ArvidssonSKwasniewskiMRiaño-PachónDMMueller-RoeberB 2008 QuantPrime—a flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinformatics 9 465
75. CroweMLSerizetCThareauVAubourgSRouzéP 2003 CATMA: a complete Arabidopsis GST database. Nucleic Acids Research 31 156 158
76. HilsonPAllemeerschJAltmannTAubourgSAvonA 2004 Versatile gene-specific sequence tags for Arabidopsis functional genomics: transcript profiling and reverse genetics applications. Genome Research 14 2176 2189
77. LurinCAndrésCAubourgSBellaouiMBittonF 2004 Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. The Plant Cell 16 2089 2103
78. GagnotSTambyJPMartin-MagnietteMLBittonFTaconnatL 2008 CATdb: a public access to Arabidopsis transcriptome data from the URGV-CATMA platform. Nucleic Acids Research 36 D986 90
79. GeXTsutsumiSAburataniHIwataS 2003 Reducing false positives in molecular pattern recognition. Genome informatics International Conference on Genome Informatics 14 34 43
80. Martin-MagnietteMLMary-HuardTBérardCRobinS 2008 ChIPmix: mixture model of regressions for two-color ChIP-chip analysis. Bioinformatics 24 i181 6
81. HulsenTde VliegJAlkemaW 2008 BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9 488
82. van der GraaffELauxTRensingSA 2009 The WUS homeobox-containing (WOX) protein family. Genome Biol 10 248
83. OhadNYadegariRMargossianLHannonMMichaeliD 1999 Mutations in FIE, a WD polycomb group gene, allow endosperm development without fertilization. The Plant Cell 11 407 416
84. IrishVF 2010 The flowering of Arabidopsis flower development. Plant J 61 1014 1028
85. LiuCThongZYuH 2009 Coming into bloom: the specification of floral meristems. Development 136 3379 3391
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy