
The Gyc76C Receptor Guanylyl Cyclase and the Foraging cGMP-Dependent Kinase Regulate Extracellular Matrix Organization and BMP Signaling in the Developing Wing of
Signaling between cells regulates many processes, including the choices cells make between different fates during development and regeneration, and misregulation of such signaling underlies many human pathologies. To understand how such signals control developmental decisions, it is necessary to elucidate both how cells regulate and respond to different levels of signaling, and how different types of signals combine and regulate each other. We have used genetic screening in the fruitfly Drosophila melanogaster to identify mutations that reduce or eliminate signals carried by Bone Morphogenetic Proteins (BMPs), and show that BMP signaling is sensitive Gyc76C, a peptide receptor that stimulates the production of cGMP in cells. We identify downstream intracellular effectors of this cGMP activity, but provide evidence that the effects on the BMP pathway are not mediated at the intracellular level, but rather through cGMP’s effects upon the extracellular matrix and matrix-remodeling proteinases, which in turn affects the activity of extracellular BMP-binding proteins. We discuss differences and parallels with other examples of cGMP activity in Drosophila melanogaster and mammals.
Published in the journal:
. PLoS Genet 11(10): e32767. doi:10.1371/journal.pgen.1005576
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005576
Summary
Signaling between cells regulates many processes, including the choices cells make between different fates during development and regeneration, and misregulation of such signaling underlies many human pathologies. To understand how such signals control developmental decisions, it is necessary to elucidate both how cells regulate and respond to different levels of signaling, and how different types of signals combine and regulate each other. We have used genetic screening in the fruitfly Drosophila melanogaster to identify mutations that reduce or eliminate signals carried by Bone Morphogenetic Proteins (BMPs), and show that BMP signaling is sensitive Gyc76C, a peptide receptor that stimulates the production of cGMP in cells. We identify downstream intracellular effectors of this cGMP activity, but provide evidence that the effects on the BMP pathway are not mediated at the intracellular level, but rather through cGMP’s effects upon the extracellular matrix and matrix-remodeling proteinases, which in turn affects the activity of extracellular BMP-binding proteins. We discuss differences and parallels with other examples of cGMP activity in Drosophila melanogaster and mammals.
Introduction
The vein cells that develop from the ectodermal epithelia of the Drosophila melanogaster wing are positioned, elaborated and maintained by a series of well-characterized intercellular signaling pathways. The wing is easily visualized, and specific mutant vein phenotypes have been linked to changes in specific signals, making the wing an ideal tissue for examining signaling mechanisms, for identifying intracellular and extracellular crosstalk between different pathways, and for isolating new pathway components [1–3].
We and others have been using one venation defect, the loss of the posterior crossvein (PCV), to identify and characterize participants in Bone Morphogenetic Protein (BMP) signaling. The PCV is formed during the end of the first day of pupal wing development, well after the formation of the longitudinal veins (LVs, numbered L1-L6) (Fig 1B), and requires localized BMP signaling in the PCV region between L4 and L5 [4]. Many of the homozygous viable crossveinless mutants identified in early genetic screens have now been shown to disrupt direct regulators of BMP signaling, especially those that bind BMPs and regulate BMP movement and activity in the extracellular space [5, 6]. The PCV is especially sensitive to loss of these regulators because of the long range over which signaling must take place, and the role many of these BMP regulators play in the assembly or disassembly of a BMP-carrying “shuttle”.

As summarized in Fig 1A, the BMP Decapentaplegic (Dpp) is secreted by the pupal LVs, possibly as a heterodimer with the BMP Glass bottom boat (Gbb). This stimulates autocrine and short-range BMP signaling in the LVs that is relatively insensitive to extracellular BMP regulators. However, Dpp and Gbb also signal over a long range by moving into the intervein tissues where the PCV forms [7–9]. In order for this to occur, the secreted BMPs must bind the D. melanogaster Chordin homolog Short gastrulation (Sog) and the Twisted gastrulation family member Crossveinless (Cv, termed here Cv-Tsg2 to avoid confusion with other “Cv” gene names). The Sog/Cv-Tsg2 complex facilitates the movement of BMPs from the LVs through the extracellular space, likely by protecting BMPs from binding to cell bound molecules such as their receptors [8–11]. In order to stimulate signaling in the PCV, BMPs must also be freed from the complex. The Tolloid-related protease (Tlr, also known as Tolkin) cleaves Sog, lowering its affinity for BMPs, and Tsg family proteins help stimulate this cleavage [12, 13]. Signaling is further aided in the PCV region by a positive feedback loop, as BMP signaling increases localized expression of the BMP-binding protein Crossveinless 2 (Cv-2, recently renamed BMPER in vertebrates). Cv-2 also binds Sog [14](Olsen, Halbisen, Li and Blair, in preparation), cell surface glypicans and the BMP receptor complex, and likely acts as a co-receptor and a transfer protein that frees BMPs from Sog [8, 15, 16]. The lipoprotein Crossveinless-d (Cv-d) also binds BMPs and glypicans and helps signaling by an unknown mechanism [17].
PCV development takes place in a complex and changing extracellular environment, but while there is some evidence that PCV-specific BMP signaling can be influenced by changes in tissue morphology [18] or loss of the cell-bound glypican heparan sulfate proteoglycans [17], other aspects of the environment have not been greatly investigated. During the initial stages of BMP signaling in the PCV, at 15–18 hours after pupariation (AP), the dorsal and ventral wing epithelia form a sack that retains only a few dorsal to ventral connections from earlier stages; the inner, basal side of the sack is filled with extracellular matrix (ECM) proteins, both diffusely and in laminar aggregates (Fig 1B) [19–24]. As BMP signaling in the PCV is maintained and refined, from 18–30 hours AP, increasing numbers of dorsal and ventral epithelial cells adhere, basal to basal, flattening the sack. The veins form as ECM-filled channels between the two epithelia, while in intervein regions scattered pockets of ECM are retained basolaterally between the cells within each epithelium; a small amount of ECM is also retained at the sites of basal-to-basal contact. This changing ECM environment could potentially alter BMP movement, assembly of BMP-containing complexes, and signal reception, as has been demonstrated in other developmental contexts in Drosophila [25–30].
We will here demonstrate the strong influence of the pupal ECM on PCV-specific long-range BMP signaling, through the identification of a previously unknown ECM-regulating pathway in the wing. In a screen we conducted for novel crossveinless mutations on the third chromosome, we found a mutation in the guanylyl cyclase at 76C (gyc76C) locus, which encodes one of five transmembrane, receptor class guanylyl cyclases in D. melanogaster [31–33]. Gyc76C has been previously characterized for its role in Semaphorin-mediated axon guidance; Malpighian tubule physiology, and the development of embryonic muscles and salivary glands [34–39]. Like the similar mammalian natriuretic peptide receptors NPR1 and NPR2 [40], the guanylyl cyclase activity of Gyc76C is likely regulated by secreted peptides [35], and can act via a variety of downstream cGMP sensors.
Our evidence suggests that Gyc76C influences BMP signaling in the pupal wing by changing the activity of the cGMP-dependent kinase Foraging (For; also known as Dg2 or Pkg24A) [41], also a novel role for this kinase. But rather than controlling BMP signal transduction in a cell-autonomous manner, we will provide evidence that Gyc76C and Foraging regulate BMP signaling non-autonomously by dramatically altering the wing ECM during the period of BMP signaling in the PCV. This effect is largely mediated by changing the levels and activity of matrix metalloproteinases (Mmps), especially Drosophila Mmp2. Genetic interactions suggest that the ECM alterations affect the extracellular mobility and activity of the BMP-binding protein Sog.
This provides the first demonstration of Gyc76C and For activity in the developing wing, and the first evidence these proteins can act by affecting Mmp activity. Moreover, our demonstration of in vivo link from a guanylyl cyclase to Mmps and the ECM, and from there to long-range BMP signaling, may have parallels with findings in mammalian cells and tissues. NPR and NO-mediated changes in cGMP activity can on the one hand change matrix metalloproteinase expression secretion and activity (e.g. [42–47]), and on the other change in BMP and TGFβ signaling [48–52]; we will discuss these below.
Results
Genetic screen for new crossveinless mutations
As many critical regulators of BMP signaling are likely to be required at earlier stages of D. melanogaster development, we screened for novel BMP regulators in the PCV by creating large mitotic recombinant clones homozygous for mutagenized third chromosomes in the posterior, PCV territory of the developing wing. We utilized posteriorly-expressed engrailed (en)-Gal4 and UAS - FLPase to induce mitotic recombination between mutagenized FRT-bearing chromosome [53, 54]; the non-mutagenized FRT chromosomes carried Minute (M) mutations that dominantly slow the rate of cell divisions (RpS17 for the 3R chromosome arm and RpS3 for 3L), allowing the homozygous mutagenized clones to divide more quickly and outcompete their M+/M- neighbors [55]. In en-Gal4 / UAS-FLP; FRT82B RpS3Plac92 ubi-GFP / FRT82B wing discs, homozygous wild type (M+) clones, identified by the absence of GFP, fill almost the entire posterior (Fig 1E). Inducing clones homozygous for a recessive mutation in dystrophin (dys) using en-Gal4 / UAS-FLP; FRT82B RpS3Plac92 ubi-GFP / FRT82B dysEP3397 reliably generated adults with a partial, “detached” PCV phenotype (Fig 1F), similar to that caused by loss of dys function in the entire wing [56].
We screened 14,500 F2 adults; the 9 independent mutant chromosomes we found that reliably disrupted the PCV in large posterior clones were recessive and homozygous lethal. One 3L mutant chromosome gave crossveinless phenotypes over vvlM638 and vvlsep and three 3R mutant chromosomes were lethal over dysEP3397. The remaining 5 (Fig 2) (S1 Fig) complemented these and other candidates known to be required for PCV or vein formation, and complemented each other. In addition, one viable mutant found originally in an F2 male was not caused by third chromosome recombinant clones, but mapped to the X chromosome, and is allelic to ade5 as will be discussed below.

3L043 mutates guanylyl cyclase at 76C
Large posterior 3L043 clones result in the complete loss of the PCV in adult wings (Fig 2E). Deletion mapping placed the lethality under Df(3L)Exel9061, a molecularly-defined deletion [58] that removes part of CG14101 and the coding exons of gyc76C (Fig 2A). 3L043 was lethal over gyc76CKG0373ex33, a homozygous lethal 8kb genomic deletion that leaves CG14101 intact but removes part of the 5’ UTR of gyc76C [34] (Fig 2A). Sequencing 3L043 DNA identified an A to T transversion within exon 15 of gyc76C, resulting in the missense mutation L635H (Fig 2B and 2C).
Loss of gyc76C function mimicked the 3L043 phenotype. Flies carrying small homozygous gyc76CKG0373ex33 clones, generated using heat shock promoter 70 (hs)-driven FLPase, often had wings with disrupted PCVs (Fig 2F). Flies carrying larger posterior gyc76CKG0373ex33 clones, generated using en-Gal4 UAS-FLPase and the Minute technique, did not survive to adulthood, but driving expression of a UAS-RNAi constructs directed against gyc76C (UAS-gyc76C-RNAi, VDRC stocks 3057 and 6552), with either the general disc driver A9-Gal4 or the posterior wing driver hh-Gal4, caused partial or complete loss of the PCV (Fig 2G and 2H). gyc76CKG0373ex33 clones and hh-Gal4-driven expression of UAS-gyc76C-RNAi also caused occasional morphological defects and wing blistering not seen with large posterior gyc76C3L043 clones (Fig 2G and 2H), suggesting that the gyc76C3L043 allele is hypomorphic.
Gyc76C shares the intracellular domain structure of vertebrate NPRs (Fig 2B), including an intracellular protein tyrosine-like kinase (PTKc) domain, a putative coiled-coil dimerization domain (“dimer” in Fig 2B), and a guanylyl cyclase catalytic (CYC) domain [59]. The L635H missense mutation in gyc76C3L043 alters a residue within the PTKc domain that is conserved in vertebrate NPRs (Fig 2C). However, the Gyc76C PTKc domain, like that of the NPRs, lacks a glutamate that is required to catalyze phosphate transfer and thus is likely kinase dead [60]; this domain is thought to regulate the activity of the guanylyl cyclase domain [39, 59].
Gyc76C likely acts via cGMP and the cGMP-dependent kinase For
While receptor guanylyl cyclases can increase cGMP levels, vertebrate NPR-A can also act independently of cGMP by directly binding to and altering the activity of TRPC3/C6 Ca2+ channels [61]. Two lines of evidence strongly suggest, however, that Gyc76C is acting in the wing via the production of cGMP. First, we compared the effects of overexpressing wild type and cyclase-dead versions of the Gyc76C. Overexpression of wild type gyc76C in the wing induces ectopic venation (Fig 2I and 2J). In other contexts expressing a form of Gyc76C carrying a D945A mutation within its CYC domain has a dominant negative effect on endogenous Gyc76C cyclase activity, likely through the formation of non-functional homodimers as occurs with a similar mutation in NPR [34, 62]. Overexpression of UAS-myc-gyc76CD945A using hh-Gal4 or A9-Gal not only failed to induce ectopic venation, but caused crossveinless phenotypes (Fig 2L). This is in marked contrast to the equivalent mutation in NPR-A, which retained its ability to alter TRPC3/C6 channel activity [61].
Second, cGMP can act by stimulating cGMP-dependent protein kinases (PRKGs), and loss of one of these mimicked the gyc76C mutant phenotype. D. melanogaster has three PRKGs: Pkg21D (also known as Dg1), CG4839, and For (also known as Dg2) [41, 63, 64]. RNAi-mediated knockdown of Pkg21D mimics the loss of Gyc76C in Malpighian tubule function, axon guidance and embryonic muscle and salivary gland formation [35, 37–39]. However, en-Gal4-driven or A9-Gal4-driven expression of UAS-pkg21D-RNAi (VDRC 34594 or 34595) did not disrupt PCV formation. CG4839MB10509 flies have a Minos transposable element inserted into one of the gene’s coding exons but are homozygous viable with normal wing venation.
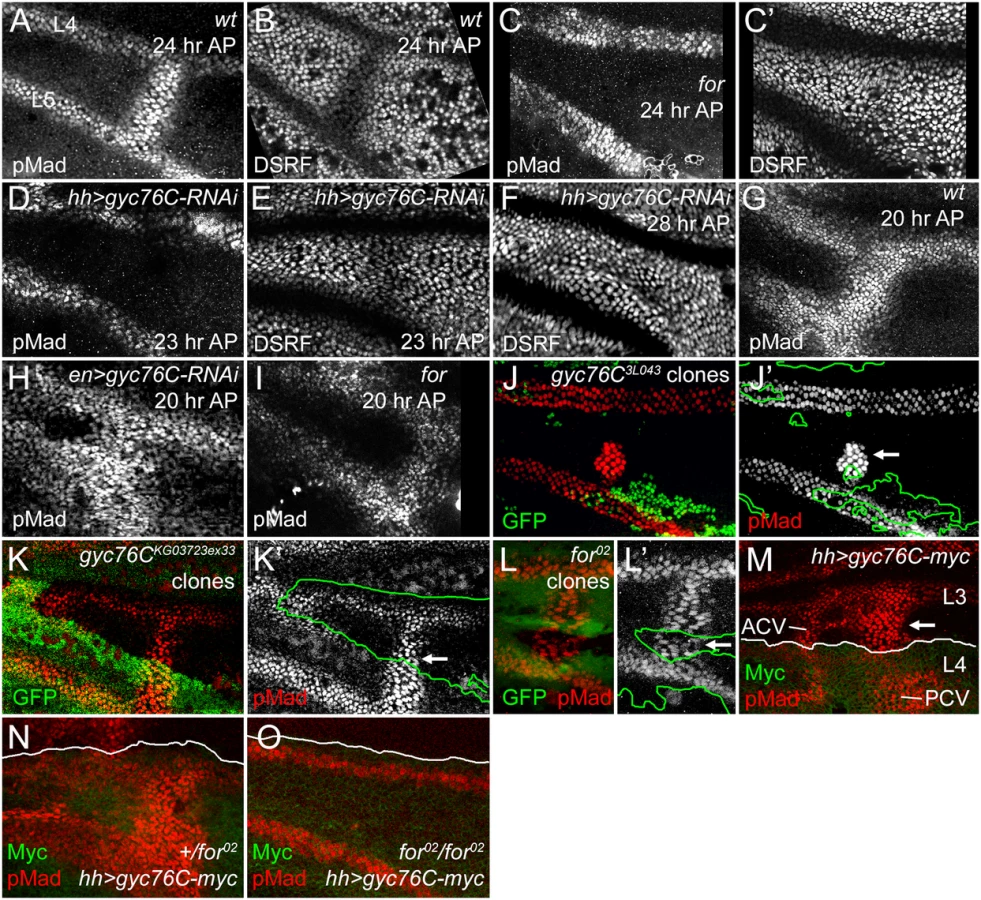
By contrast, PCVs were lost from the wings of pupae homozygous for the adult lethal fork04703 and for02 alleles. PCVs can normally be visualized from 16 to 32 hours AP using antisera specific to the C-terminal phosphorylated form of the BMP receptor-activated Smad, Mothers against Dpp (anti-pMad), and after 22–24 hours AP by reduced expression of D. melanogaster Serum Response Factor transcription factor (DSRF, also known as Blistered) (Fig 3A and 3B) [4]. 24 hour AP for homozygotes lacked anti-pMad staining and DSRF downregulation in the PCV (Fig 3C and 3C’). Removal of for also blocks the effects of Gyc76C overexpression: hh-gal4-induced overexpression of gyc76C induced ectopic anti-pMad staining in pupal wild type or for02/+ wings, especially in a central region of the wing near the normal crossveins (Fig 3M and 3N), but did not do so in for02 homozygotes (8/8 cases) (Fig 3O). We will show below that mutation of for not only mimics the effects of gyc76C knockdown on BMP signaling, but also its effects on the ECM, strongly supporting the involvement of Gyc76C and the PRKG For in a common pathway linked by cGMP.
A role for cGMP in crossvein-specific BMP signaling provides an explanation for the crossveinless wings produced by defects in purine synthesis, a part of the “purine syndrome” [65–67]. As noted above, a homozygous viable X chromosome crossveinless mutation found in our screen (X1) maps to and is allelic to the ade5 gene (S3 Fig), which encodes an enzyme with 5-aminoimidazole ribonucleotide carboxylase and 4-[(N-succinylamino)carbonyl]-5-aminoimidazole ribonucleotide synthetase activities, the sixth and seventh steps in purine synthesis [68]. The pupal crossvein defects in ade51 wings were similar to, although milder than, those caused by for mutations (S3E and S3F Fig).
We also attempted to manipulate cGMP in the wing using the D. melanogaster cGMP phosphodiesterase PDE6, which can reduce levels of cGMP (and cAMP in one assay) after extraction from S2 cells [69]; overexpression of PDE6 causes a 25% reduction of cGMP levels in Malpighian tubules [70]. However, UAS-PDE6-RNAi driven with hh-Gal4 or en-Gal4 UAS-dcr2 UAS-PDE6-RNAi only rarely produced the ectopic venation expected from increased cGMP, and overexpression of wild type UAS-PDE6 or a mutated form lacking a prenylation site that alters its subcellular localization [71], did not produce crossveinless wings with either hh-Gal4, en-Gal4, or MS1096-Gal4. While the cGMP reductions caused by PDE might be expected to block PRKG activity, even ubiquitous PDE5/6 overexpression with actin promoter-driven Gal4 failed to reproduce the lethality of for or Pkg21D mutants, and adults appeared normal. PDE activity can be regulated at several levels, and cGMP, PDE and PRKG activities can depend greatly on subcellular localization [72, 73]. Given our other experimental support for cGMP’s role in the gyc76C and for phenotypes, we think it likely that PDE6 does not cause a large enough cGMP change, in the correct subcellular compartment, to greatly affect For activity.
We next investigated the role of the only known ligand for Gyc76C, but found it plays only a weak role in the wing. The VQQ neuropeptide, one of several produced from the Nplp1 peptide precursor protein, can stimulate Gyc76C-dependent cGMP production in S2 cells and Malpighian tubules, although the effects of its removal have not been tested [35]. Nplp1EY11089 is a P element insertion that introduces stop codons into the first coding exon of Nplp1, 3’ to the signal peptide-coding region needed for secretion, but 5’ to the peptide coding region (S4A Fig). But while this mutation blocked Nplp1 peptide production in the CNS (S4B and S4C Fig), it failed to reproduce the lethality of strong gyc76C mutants, and caused only occasional ectopic branching from the PCV rather than its loss (S4D and S4E Fig). Thus, either there are redundant Gyc76C-stimulating peptides, or Gyc76C has significant activity in the absence of peptide binding. Plexin A-mediated Semaphorin signaling can affect Gyc76C activity in embryonic axons and in vitro [34, 39], but we have reduced Plexin A signaling in the wing and found no effects on PCV development (hh-Gal4 UAS-Plexin A-RNAi).
Gyc76C and For act non-autonomously to refine and maintain normal BMP signaling in the pupal wing
The LVs are specified early in wing development by localized EGF-receptor-mediated MAPK activity, well prior to the appearance of the crossveins, but begin to express the BMP Dpp during early pupal stages [1, 8, 74]. Anti-pMad provides a measure of BMP signaling immediately downstream of receptor activation; anti-pMad staining appears around both the LVs and the PCV at 15 hours AP; by 18–20 hours the PCV always forms a continuous, gap-less line of pMad staining between L4 and L5, despite the PCV not expressing Dpp or requiring EGF receptor-mediated MAPK activity until after 24 hours AP [4, 8]. As in for mutants, knockdown of gyc76C using hh-Gal4-driven or en-Gal4-driven expression of UAS-gyc76C-RNAi, always blocked or created large gaps in anti-pMad staining in the 23–24 hour AP PCV, as shown by comparing pMad levels with those in the adjacent LVs (Fig 3D and S5D Fig). This was accompanied by loss of DSRF downregulation in the PCV between 24 and 28 hours AP, slightly later than the equivalent effect in for mutants (Fig 3C’, 3E and 3F, S5D’ Fig).
However, BMP signaling was still initiated in the PCVs of for mutant or gyc76C knockdown wings, and visible at 20–22 hours AP (Fig 3H and 3I, S5C Fig). In for mutants signaling was often reduced at stages prior to formation of a vein lumen in the PCV region (S6 Fig). The early BMP signaling was more robust after gyc76C knockdown than in for mutants; in fact, en-Gal4-driven knockdown of gyc76C often led to broader anti-pMad staining than in wild type wings at 20 hours AP, in both the PCV and the LVs (Fig 3G and 3H). The broadening of the PCV and adjacent LVs was also apparent in the dorsal epithelium after driving dorsal-specific knockdown using ap-Gal4 UAS-gyc76C-RNAi (S5A Fig) and the width of L5 in adult hh-Gal4 UAS-gyc76C-RNAi wings was also significantly greater than in control hh-Gal4 wings (S7A–S7E Fig).
Thus, the effects on BMP signaling were complex: Gyc76C suppressed and refined BMP signaling around the LVs and the early PCV, but Gyc76C and For maintained BMP signaling in the older PCV. This is difficult to reconcile with an intracellular, “cell-autonomous” effect of Gyc76C and For on BMP signal transduction, which would be expected to lower pMad levels in all the vein cells. Instead, the effect is quite reminiscent of extracellular alterations in long-range BMP signaling: reducing the BMP shuttling mediated by extracellular BMP-binding proteins like Sog and Cv-Tsg2 can increase short-range signaling near the Dpp-expressing LVs, but decrease long-range signaling from the LVs into the PCV region [10].
As a more rigorous test of cell autonomy, we generated large homozygous gyc76C3L043 or gyc76CKG0373ex33 clones using the Minute technique and hs-FLPase, examining these at 28 hours AP which, because of the slowed development of M-/+ flies, corresponds to approximately 24 hours AP in wild type flies. Clones that encompassed the region of PCV formation on both the dorsal and ventral epithelia could result in the complete or near-complete loss of pMad from the PCV (S8A, S8A’, S8C and S8C’ Fig. Effects of additional gyc76C and for mutant clones on PCV development). However, individual PCV cells within smaller clones often had pMad levels identical to those in neighboring heterozygotic cells (Fig 3J and 3K’, S8B and S8B’ Fig). We observed similar non-autonomy in for02 mutant clones in 24 hour AP or older wings (Fig 3L and 3L’); for02 clones could even occasionally disrupt PCV formation in neighboring for/+ or +/+ cells (S8D and S8E Fig). These non-autonomous effects are quite similar to those caused by clones lacking the extracellular BMP-binding regulators Sog, Cv-Tsg2 and Cv-2 [10, 15].
The overexpression of Gyc76C also induced ectopic venation and anti-pMad staining in a non-autonomous fashion. hh-Gal4 expression is limited to the posterior of the wing (Fig 2K), but hh-Gal4-driven expression of UAS-myc-gyc76C resulted in ectopic venation in the anterior compartment of adult wings (Fig 2J), and ectopic pMad anterior to the region of Gyc76C overexpression in pupal wings (Fig 3M and 3N).
Gyc76 interacts genetically with Sog and other extracellular regulators of BMP signaling
The clonal analyses above strongly suggest that Gyc76C and For do not regulate BMP signaling in the pupal wing at the level of cell-autonomous signal transduction, but rather influence the extracellular regulation of BMP secretion, movement or reception. We therefore next examined the roles of BMPs and BMP-binding proteins in Gyc76C activity using genetic interactions. In the results that follow, at least 10 wings of each genotype were compared, and results were identical in all of them.
First, Gyc76C can act downstream of Dpp expression. Overexpression of UAS-dpp-GFP using an L5-specific Gal4 driver [75] expanded the width of the adult L5 (S7H Fig), but co-expression of UAS-Gyc76C-RNAi in L5 significantly reduced this expansion (S7I and S7J Fig).
Gyc76C’s vein-promoting activity also depended on the presence of the secreted BMP binding protein Cv-Tsg2. Loss of Cv-Tsg2 prevents BMP signaling in the pupal PCV and thus PCV formation in adults [9–11] (Fig 4A). The ectopic venation normally caused by en-Gal4 UAS-gyc76C was blocked in a cv hemizygous background, and the overexpression of gyc76C did not rescue crossvein formation (Fig 4B and 4C).

However, altering the levels of any single BMP, BMP regulator or effector did not rescue the defects caused by moderate gyc76C knockdown. The PCV disruption caused by A9-Gal4-driven expression of UAS-gyc76C-RNAi (Fig 4D) was not improved by individual co-expression of UAS-gbb-Flag, UAS-tlr, UAS-cv-His, cvEP1349, UAS-myc-cv-2-V5, cv-2EP1103, UAS-sog, UAS-tkv-HA, UAS-punt, UAS-mad-Flag or UAS-medea (the sole D. melanogaster co-Smad).
Nonetheless, we found that greatly increasing Sog cleavage could counteract the PCV-disrupting effects. en-Gal4-driven overexpression of the Tlr protease rescued the crossveinless disruption of ade5X1 mutants (S6G Fig). And while Tlr overexpression did not rescue PCV disruption in A9-Gal4, UAS-gyc76CRNAi wings, expressing an activated form of Tld (TldA53) did (Fig 4E). This suggests that gyc76C knockdown decreases BMP signaling by increasing the affinity of the Sog/Cv-Tsg2 complex for BMPs.
The effects of gyc76C overexpression are consistent with this hypothesis. Strong overexpression of UAS-sog with en-Gal4 always blocks PCV formation (Fig 4G); the BMPs can likely still move as part of the Sog/Cv-Tsg2 complex, but the excess Sog overwhelms the available Tlr and Cv-2 so that BMPs remain sequestered in the complex [8, 74]. Increasing Sog cleavage with Tlr overexpression can rescue the crossvein defects caused by Sog overexpression [13], as can overexpression of Cv-2 [8]. en-Gal4-driven expression of UAS-gyc76C also rescued the PCV loss normally caused by UAS-sog expression (Fig 4H).
Since Tsgs and Cv-2 can also decrease the BMP-sequestering activity of the Sog/Cv complex, we tested whether overexpression of Cv-Tsg2 or Cv-2 could rescue gyc76C knockdown in combination with each other or with Sog. The PCV disruption caused by A9-Gal4-driven expression of UAS-gyc76C-RNAi (Fig 4D) was not improved by co-expression of cvEP1349, cv-2EP1103 or UAS-sog in any single or pair-wise combination. It was rescued, however, by triple co-expression of UAS-sog, cvEP1349, and cv-2EP1103 (Fig 4F). This result cannot be explained if gyc76C knockdown simply increased the affinity of the Sog/Cv-Tsg2 complex for BMPs, since adding excess Sog should increase BMP sequestration, not reduce it. Rather, we hypothesize that gyc76C knockdown also reduces the movement of the Sog/Cv-Tsg2 complex into the PCV region. Excess Sog can overcome this defect in diffusion, but only increases BMP signaling in a genetic background (excess Cv-Tsg2 and Cv-2) that frees BMPs from the excess Sog. In summary, our results suggest that Gyc76C knockdown has complex effects on Sog function, both increasing Sog’s affinity for BMPs, but also decreasing the range of Sog movement (see Discussion).
Gyc76C and For affect the wing extracellular matrix
The non-autonomous, complex effects of Gyc76C and For on BMP signaling and Sog function are reminiscent of similar effects caused by altering the ECM in different developmental contexts (see Discussion). Moreover, the adult wing blistering caused by very strong loss of Gyc76G activity (Fig 2H detail) suggests a failure to properly adhere the two wing epithelia, an effect that can also be caused by altering the wing ECM and its receptors. Gal4-driver overexpression mediated by a UAS-containing EP insertion near the for locus was also reported to induce blistering [76]. We therefore examined the effects of cGMP activity on the levels and distribution of the ECM components Collagen IV using the 6G7 monoclonal antibody, LamininB2 (LanB2, also called Lanγ1) using anti-LanB2, and the secreted perlecan heparan sulfate proteoglycan (HSPG) Terribly reduced optic lobes (Trol, previously named l(1)zw1) using anti-Trol and a trol-GFP protein trap. In the normal pupal wing all three of these formed a diffuse ECM with scattered laminar aggregates; the aggregates were especially prominent with the 6G7 anti-Collagen IV. ECM proteins also concentrate in the hemocytes that circulate between the wing epithelia, and anti-LanB2 also stained the apical surfaces of the epithelia.
We did not detect gross histological changes in the ECM after gyc76C knockdown prior to 24 hours AP, although the more open, pocket-like architecture of younger pupal wings makes it more difficult to detect ECM organization at this stage. Profound defects appeared, however, around 24–28 hours AP and strengthened from 28–34 hours AP, appearing slightly earlier in for mutants (Fig 5 and S9G–S9J’ Fig). By 24 hours AP the ECM normally fills both the large vein channels and smaller basolateral pockets between cells in intervein regions (Figs 1B and 5A–5C). Since the ECM is prominent in the normal veins, the PCV loss and LV expansion in for mutant wings, or in the posterior hh-Gal4 UAS-gyc76C-RNAi wings, caused parallel losses or gains of vein ECM (Fig 5D–5E”‘, 5I and 5I’). However, we also observed histological changes in the organization of the ECM that were not simply reflections of altered venation.

First, the basolateral pockets of intervein ECM, although initially normal, were progressively depleted in the posterior after posterior gyc76C knockdown (Fig 5D–5E”‘), or throughout the wing in for homozygotes (Fig 5I and 5I’) (for time course in S9G–S9J’ Fig). As this occurred the vein ECM became broader and more diffuse, and was often retained in abnormal vein-like blobs near the site of the PCV. The broadened ECM accumulation near veins did not strictly correlate with altered vein specification: the extremely broad L5 ECM caused by posterior gyc76C knockdown extended into regions lacking vein markers such as heightened pMad or reduced DSRF, and the vein-like blobs near the normal PCV site were retained after molecular markers of PCV development vanished (S9A and S9A’ Fig). In hh-Gal4 UAS-gyc76C-RNAi wings diffuse Trol-GFP was especially strong in L5 and the PCV-like blobs (Fig 5D”‘). 6G7 anti-Collagen IV staining showed an abnormally high accumulation of laminar aggregates in the posterior of hh-Gal4 UAS-gyc76C-RNAi wings (Fig 5E”‘ and S9J Fig).
These gross organizational defects were preceded by a more subtle change: at 24 hours AP abnormally large intracellular vesicles appeared in the interveins (Fig 5D”‘ and 5F–5H’, S9B–S9F Fig). These containing Trol-GFP and were likely endocytotic, since many co-localized with the late endocytic marker Rab9.YFP (Fig 5H and 5H’), although Trol-GFP only rarely overlapped the late endocytic marker Rab7, and did not significantly overlap the early endocytic marker Rab5, or the recycling vesicle marker Rab11 (S9C–S9E’ Fig). Intriguingly, anti-Trol staining did not accumulate in the GFP-containing vesicles (Fig 5G and 5G’) or co-localize with Rab9.YFP (S9B and S9B’ Fig). Since the GFP tag in Trol-GFP is inserted into the N terminal domain II, while the anti-Trol was produced against the C-terminal domain V [77], it is possible that the GFP represents uptake of an abnormal cleavage product of Trol-GFP. We will present results below suggesting that the vesicles are a cellular reaction to breakdown of the ECM.
The ECM phenotypes are not a general result of changes in BMP signaling or a crossveinless condition. cv null mutations block most or all PCV BMP signaling during the initial stages of PCV formation and broaden signaling in the LVs [9–11], but did not obviously alter wing ECM outside the missing PCV (Fig 5K and S9L Fig). Nor did we detect ECM defects outside the missing PCV in crossveinless wings mutant for the Rho-Rac GAP Crossveinless c (cv-c1) or expressing en-Gal4 UAS-dys-RNAi (Fig 5J, S9M and S9N Fig).
The effects of cGMP activity on the ECM were not strictly cell autonomous. Moderate-sized gyc76C3L043 or for homozygous clones did not deplete the intervein pockets or increase accumulation of Collagen IV aggregates, even where dorsal and ventral clones overlapped (S9K and S9K’ Fig). Posterior overexpression of gyc76C with hh-Gal4 also altered wing ECM in a non-autonomous fashion. While ECM in the LVs appeared normal, ECM in the intervein pockets was fainter and more diffuse in the posterior; this effect extended up to L3, well anterior of the region of hh-Gal4 expression (Fig 5L and 5L’).
Gyc76C regulates matrix metalloproteinase levels
Since reductions in Gyc76C or For activity disrupt several components of the wing ECM, we next searched for effects on components known to organize or modify the ECM. Posterior gyc76C knockdown did not cause posterior-wide changes in the levels of ECM receptors such as the glypican Dlp, Dystroglycan, or the integrins Mys, Mew and If, nor alter expression of the mys expression regulator Delilah [78]. Nor could we detect posterior-wide changes in the BMP receptor Thickveins or the vein-width regulator Notch. Changes were limited to those caused by altered venation, and only for those proteins whose levels are normally different in vein and intervein (S10 Fig).
The depletion of ECM from the interveins and the diffuse ECM appearance the veins, next suggested the involvement of the extracellular matrix metalloproteinases (Mmps). This is also consistent with the possible appearance of a Trol-GFP cleavage product noted above, as vertebrate perlecan can be cleaved by Mmps [79]. There are two D. melanogaster Mmps: Mmp2, which is predicted to be GPI-linked to the cell surface, and Mmp1, which is diffusible [80–82]. An engineered Mmp2::GFP produced by the endogenous Mmp2 locus [83] is normally expressed in a slightly patchy pattern in intracellular structures and the cell cortex, more weakly in vein cells and more strongly near the wing hinge; the cytoplasmic GFP is stronger apically (Fig 6C and 6D’), without any strong anterior-posterior bias (11 of 11 23–25 hour AP wings). After posterior hh-Gal4-driven knockdown of Gyc76C, posterior Mmp-2 levels were higher and more uniform, an effect especially noticeable in more apical focal planes, and after a summing projection of all cross-sections along the proximo-distal (X) (Fig 6A and 6B’). Posterior apical Mmp2::GFP was increased by 25% or greater over anterior in 13 out of 16 23–25 hour AP wings, and the increase was significant in a comparison of all experimental and control wings (Fig 6E). Those wings lacking the effect may reflect variation in its timing.

Anti-Mmp1 staining in normal pupal wings was largely extracellular and concentrated in the diffuse ECM of veins and intervein pockets. hh-Gal4 UAS-gyc76C always increased the posterior levels of Mmp1 in the ECM of both veins, especially L5 and PCV-like blobs, and intervein pockets beginning at 24 hours AP (details in Fig 6F–6I’; low magnification in S9S–S9U Fig; anti-Mmp1 in control wings in S9W and S9X Fig) This increased staining was retained in the veins but was transient in the intervein pockets: as the intervein pockets became depleted of ECM beginning at 27–28 hours AP, Mmp1 levels in the pockets also decreased, although increased Mmp1 was sometime still observed in pockets partially depleted of anti-Trol (Fig 6H and 6H’). The increased Mmp1 is likely due to changes in Mmp1 secretion or accumulation rather than transcription, as an Mmp1-lacZ enhancer reporter that reproduces Mmp1 expression in other contexts [84] was not obviously altered in hh-Gal4 UAS-gyc76C-RNAi wings (S9Q and S9R Fig). Given the strong association of Mmp1 with wing ECM, Mmp1’s abnormal accumulation in the abnormal ECM of gyc76C knockdown wings could be both a result and a cause.
Reducing Mmp activity largely rescues the effects gyc76C knockdown on the ECM and BMP signaling
To test the role of Mmp activity in the ECM and BMP signaling changes caused by reduced Gyc76C activity, we first overexpressed the D. melanogaster member of the diffusible Tissue inhibitor of metalloproteases (Timp) protein family, which can inhibit both Mmp1 and Mmp2 activities [85, 86]. hh-Gal4 UAS-gyc76C-RNAi UAS-Timp pupae did not produce adults and pupal wings were foreshortened, likely due to Timp’s effects on wing disc eversion [87]. Nonetheless, Timp greatly improved PCV development in hh-Gal4 UAS-gyc76C-RNAi pupal wings, forming normal or nearly normal PCVs as assessed by heightened pMad or reduced DSRF pMad in 9/9 28 hour AP or older wings (Fig 6J). hh-Gal4-driven expression of UAS-Timp in a gyc76C knockdown background (Fig 6K) also increased the ECM, but largely in an in an abnormal proximal clump likely caused by excess inhibition of Mmp activity. Intriguingly, BMP signaling also expanded in the proximal wing adjacent to the excess ECM (Fig 6J).
We next tested the roles of the Mmps individually using RNAi lines with proven efficacy [84]. UAS-Mmp1-RNAi did not improve the defects of hh-Gal4 UAS-gyc76C-RNAi wings, but UAS-Mmp2-RNAi did. hh-Gal4 UAS-gyc76C-RNAi UAS-Mmp2-RNAi larvae and pupae were occasionally unhealthy-appearing, generating fragile wings with poor morphology and development, but the ECM in those that were healthy appeared almost normal: the width and strength of the staining around posterior veins and the strength and number of intervein ECM pockets were nearly normal (Fig 6O and 6P). The abnormally large intracellular vesicles normally found in the interveins of gyc76C knockdown wings were also greatly reduced (S9Y and S9Z Fig), suggesting that these are a cellular reaction to Mmp-induced breakdown of the ECM. While 6G7 anti-Collagen IV staining still showed the abnormally high numbers of Collagen IV aggregates observed in hh-Gal4 UAS-gyc76C-RNAi wings (Fig 6P’), it should be noted that hh-Gal4 UAS-Mmp2-RNAi in otherwise wild type wings also increases abnormal anti-Collagen IV aggregates in the posterior (S9O and S9P Fig). BMP signaling in the PCVs was also largely rescued, as assessed by heightened pMad or reduced DSRF in 15/17 24–32 hour AP wings (Fig 6L–6N). These results strongly suggest that most of the ECM and BMP signaling defects caused by reduced Gyc76C activity are caused by increased Mmp activity.
We next asked whether the ECM and BMP signaling defects were linked, or were independent effects of Mmp activity, by looking at the effects of manipulated the ECM directly. It is difficult to remove most ECM components from the wing: null mutants are lethal or cause morphological abnormalities at earlier stages, and mosaic techniques cannot be used for those ECM components, like Trol and Collagen IV, that are likely transported into the wing via hemolymph and hemocytes [88, 89]. Instead, we tested the ability of overexpressed ECM to rescue the effects of gyc76C knockdown, choosing Trol because hh-Gal4-driven expression of UAS-trol in a wild type background led to few abnormalities beyond a slight broadening of the PCV (Fig 6Q). hh-Gal4-driven expression of both UAS-trol and UAS-gyc76C-RNAi increased the blistering sometimes observed in adult wings after gyc76C knockdown, but partially or wholly rescued PCV formation in adult (Fig 6R) and pupal wings (6/7 28 hours AP or older; Fig 6S). Unlike UAS-Mmp2-RNAi, the very strong overexpression caused by UAS-trol did not obviously improve the other ECM components in the posterior of hh-Gal4 UAS-gyc76C-RNAi pupal wings, but did cause an abnormal accumulation of diffuse 6G7 anti-CgIV staining between L4 and L5 proximal to the PCV (Fig 6T and 6T’), leaving open the possibility that the rescue was mediated by reorganization of the ECM, rather than by Trol directly.
Discussion
Here we report that mutation 3L043, uncovered by a genetic screen to identify homozygous lethal mutations required for PCV development, is a novel allele of gyc76C, a transmembrane peptide receptor that, like vertebrate NPRs, acts as a guanylyl cyclase. We further show that gyc76C is likely linked by cGMP production to the activity of the cGMP-dependent kinase For, and that Gyc76C and For define a new pathway for the regulation of wing ECM (Fig 7). This pathway appears to act largely through changes in the activity of ECM-remodeling Mmp enzymes. Loss of gyc76C or For alter both the organization of the wing ECM and the levels of the two D. melanogaster Mmps, and the gyc76C knockdown phenotype can be largely reversed by knockdown of Mmp2. This is the first indication of a role for cGMP, Gyc76C and For function in the developing wing, and their effects on the ECM provides a novel molecular output for each.

We have also shown that Gyc76C and For are necessary for the normal refinement and maintenance of long-range BMP signaling in the posterior crossvein region of the pupal wing; in fact, crossvein loss is the most prominent aspect of the adult gyc76C knockdown phenotype. Our evidence suggests that this effect is also mediated by changes in Mmp activity, and most likely the Mmp-dependent reorganization of the ECM (Fig 7). In fact, our analysis using genetic mosaics finds no evidence for a reliable, cell autonomous effect of cGMP activity on BMP signal transduction in the wing. Thus, this apparent crosstalk between receptor guanylyl cyclase activity and BMP signaling in the wing is mediated by extracellular effects.
cGMP, Mmp and BMP/TGFβ in mammals
It is noteworthy that the cGMP activity mediated by NPR or nitric oxide signaling can change also Mmp gene expression, secretion or activation in many different mammalian cells and tissues (e.g. [42–47]). Both positive and negative effects have been noted, depending on the cells, the context, and the specific Mmp. Given the strong role of the ECM in cell-cell signaling, the contribution of cGMP-mediated changes in Mmp activity to extracellular signaling may be significant.
There is also precedent for cGMP activity specifically affecting BMP and TGFβ signaling in mammals. cGMP-dependent kinase activity increases BMP signaling in C2C12 cells, and this effect has been suggested to underlie some of the effects of nitric oxide-induced cGMP on BMP-dependent pulmonary arterial hypertension [50, 52]. Conversely, atrial natriuretic peptide stimulates the guanylyl cyclase activities of NPR1 and NPR2 and can inhibit TGFβ activity in myofibroblasts; this inhibition has been suggested to underlie the opposing roles of atrial natriuretic peptide and TGFβ during hypoxia-induced remodeling of the pulmonary vasculature [48, 49, 51]. However, unlike the pathway we observed in the fly wing, these mammalian effects are thought to be mediated by the intracellular modulation of signal transduction, with cGMP-dependent kinases altering BMP receptor activity or the phosphorylation and nuclear accumulation of receptor-activated Smads [50, 51]. Nonetheless, it remains possible that there are additional layers of regulation mediated through extracellular effects, underscoring the importance of testing cell autonomy.
Gyc76C and For in other contexts
Aside from its role in adult Malpighian tubule physiology, Gyc76C was previously shown to have three developmental effects: in the embryo it regulates the repulsive axon guidance mediated by Semaphorin 1A and Plexin A [34, 39], the proper formation and arrangement of somatic muscles [37], and lumen formation in the salivary gland [38]. All these may have links to the ECM. Loss of gyc76C from embryonic muscles affects the distribution and vesicular accumulation of the βintegrin Mys [37], and reduces laminins and the integrin regulator Talin in the salivary gland [38]. The axon defects likely involve a physical interaction between Gyc76C and semaphorin receptors that affects cGMP levels [39]; nonetheless, gyc76C mutant axon defects are very similar to those caused by loss of the perlecan Trol [90].
The parallels between the different contexts of Gyc76C action are not exact, however. First, only the wing phenotype has been linked to a change in Mmp activity. Second, unlike the muscle phenotype, the wing phenotype is not accompanied by any obvious changes in integrin levels or distribution, beyond those caused by altered venation (S10 Fig). Finally, most gyc76C mutant phenotypes are reproduced by loss of the Pkg21D (Dg1) cytoplasmic cGMP-dependent kinase [35, 37, 38, 91], instead of For (Dg2, Pkg24A) as found in the wing, and thus may be mediated by different kinase targets.
For has been largely analyzed for behavioral mutant phenotypes [92], and the overlap between Pkg21D and For targets is unknown. While many targets have been identified for the two mammalian cGMP-dependent kinases, PRKG1 (which exists in alpha and beta isoforms) and PRKG2, it is not clear if either of these is functionally equivalent to For. One of the protein isoforms generated by the for locus has a putative protein interaction/dimerization motif with slight similarity to the N-terminal binding/dimerization domains of alpha and beta PRKG1, but all three For isoforms have long N-terminal regions that are lacking from PRKG1 and PRKG2. In fact, a recent study suggested that For is instead functionally equivalent to PRKG2: Like PRKG2, For can stimulate phosphorylation of FOXO, and is localized to cell membranes in vitro [93]. But For apparently lacks the canonical myristoylation site that is thought to account for the membrane localization and thus much of the target specificity of PRKG2. FOXO remains the only identified For target, and foxo null mutants are viable with normal wings [94].
The regulation of long-range BMP signaling by candidate Mmp2 targets
The loss of long range BMP signaling in the PCV region caused by knockdown of gyc76C can, like the ECM, be largely rescued by knockdown of Mmp2. Two results suggest that it is the alteration to the ECM that affects long-range BMP signaling, rather than some independent effect of Mmp2. First, the BMP signaling defects caused by gyc76C knockdown were rescued by directly manipulating the ECM through the overexpression of the perlecan Trol. Second, when Mmp activity is inhibited by overexpression of the diffusible Mmp inhibitor TIMP, this not only rescued the PCV BMP signaling defects caused by gyc76C knockdown, but also led to ectopic BMP signaling, not throughout the region of TIMP expression, but only in those regions with abnormal accumulation of ECM.
The Mmp2-mediated changes in the ECM likely affect long-range BMP signaling by altering the activity of extracellular BMP-binding proteins, particularly Sog. The BMPs Dpp and Gbb produced in the LVs bind Sog and Cv-Tsg2, shuttle into the PCV region, and are released there by Tlr-mediated cleavage of Sog and transfer to Cv-2 and the receptors (see Introduction and Fig 1B). Our genetic interaction experiments suggest that knockdown of gyc76C both increases Sog’s affinity for BMPs and reduces the movement of the Sog/Cv-Tsg2/BMP complex into the crossvein region.
Collagen IV provides the best-studied example for how the ECM might affect Sog activity. The two D. melanogaster collagen IV chains regulate BMP signaling in other contexts, and they bind both Sog and the BMP Dpp [25–27]. Results suggest that collagen IV helps assemble and release a Dpp/Sog/Tsg shuttling complex, and also recruits the Tld protease that cleaves Sog cleavage and releases Dpp for signaling [25, 27, 28]. D. melanogaster Mmp1 can cleave vertebrate Collagen IV [80]. Since reduced Gyc76C and For activity increases abnormal Collagen IV aggregates throughout the wing and diffuse Collagen IV in the veins, we hypothesize that these Collagen IV changes both foster the assembly or stability of Sog/Cv-Tsg2/BMP complexes and tether them to the ECM, favoring the sequestration of BMPs in the complex and reducing thelong-range movement of the complex into the region of the PCV (Fig 7).
While few other D. melanogaster Mmp targets have been identified, it is likely that Mmp1 and Mmp2 share the broad specificity of their mammalian counterparts [80, 81], so other ECM components, known or unknown, might be involved. For instance, vertebrate Perlecan and can be cleaved by Mmps [79]. Trol regulates BMP signaling in other D. melanogaster contexts [29, 30], and Trol overexpression rescue gyc76C knockdown’s effects on BMP signaling. But while null trol alleles are lethal before pupal stages, normal PCVs were formed in viable and even adult lethal alleles like trolG0023, and actin-Gal 4-driven expression of trol-RNAi using any of four different trol-RNAi lines did not alter adult wing venation. Loss of the D. melanogaster laminin B chain shared by all laminin trimers strongly disrupts wing venation [24], and a zebrafish laminin mutation can reduce BMP signaling [95].
Finally, it was recently shown that Dlp, one of the two D. melanogaster glypicans, can be removed from the cell surface by Mmp2 [96]. While gyc76C knockdown did not detectably alter anti-Dlp staining in the pupal wing (S10G and S10H Fig), it is noteworthy that Dlp and the second glypican Dally are required non-autonomously for BMP signaling in the PCV and that they bind BMPs and other BMP-binding proteins [15, 17].
Methods
D. melanogaster stocks
The following were generated from Bloomington Drosophila Stock Center stocks, unless otherwise indicated.
A9-Gal4 w
y w; ap-Gal4 UAS-GFP/CyO
y w; en-Gal4
y w; en-Gal4 UAS-FLP
UAS-GFP; hh-Gal4/TM6,Tb
hh-Gal4 UAS-GFP/TM6,Tb (recombinant generated in lab)
L5-Gal4 (3.7KX-lacZ/UAS) kindly provided by J. de Celis [75]. L5-Gal4 UAS-dpp-GFP recombinant generated in lab.
y w; UAS-FLP
y w hs-FLP; ubi-mRFP.nls FRT40A /CyO
y w; FRT2A
y w hs-Flp; hs-GFP RpS174 [also known as M(3)i55] FRT2A/TM3, Sb (kindly provided by G. Struhl)
y w; FRT82B
y w, FRT82B, RpS3Plac92[also known as M(3)w or M(3)95A] ubi-GFP/TM6B, Tb
FRT82B dysEP3397/TM6,Tb (FRT recombinant kindly provided by D. Olson)
gyc76CKG03723ex33/TM3,Sb, UAS-myc-gyc76C and UAS-myc-gyc76D945A, kindly provided by A. Kolodkin [34]. gyc76CKG03723ex33 FRT2A, en-Gal4 UAS-myc-gyc76C and hh-Gal4 UAS-myc-gyc76C recombinants were lab-generated. Gyc76C overexpression experiments used a second chromosome UAS-myc-gyc76C, except for those of Fig 3M and 3N which used a lab-generated for02; hh-Gal4 UAS-myc-gyc76C /CyO-TM6,Tb stock.
y cv1 v ade51 fl/FM6, kindly provided by D. Clark [68]. Crossveinless males were crossed to Df(1)ED7165/FM7h; the deficiency covers ade5 but not cv.
y w; Nplp1EY11089
for02/CyO,Tb, forK04703/CyO,Tb
for02 FRT40A/CyO, recombinant generated in lab.
UAS-PDE6-RNAi, UAS-PDE6 and UAS-PDE6C1128S kindly provided by Dr. S. Davies [70, 71].
cv43 [11]
UAS-dpp-GFP, UAS-tlr-HA, UAS-tldA53, UAS-sog-HA, cv70, kindly provided by M. O’Connor [10, 13].
cv-2P(EP)1103 (Szeged Stock Center) [4]
cvP(EP)1349; cv-2P(EP)1103 UAS-sog-HA stock generated in lab.
cv-c1
cvP(EP)1349
y w; P(UASp-YFP.Rab9) [97]
CG4839MB10509
UAS-gyc76C-RNAi, UAS-dys-RNAi and UAS-Pkg24-RNAi lines were from the VDRC. UAS-gyc76C RNAi; hh-Gal4/CyO-TM6,Tb and en-Gal4 UAS-dys-RNAi stocks generated in lab.
trol-GFP (P{PTT-un1}trolG00022) [98] from Kyoto DGRC.
Mmp2::GFP/CyO, kindly provided by J. Sun [83]
UAS-TIMP, UAS-Mmp1-RNAi, UAS-Mmp2-RNAi and Mmp1-lacZ kindly provided by D. Bohmann [84].
P(GSV2)trolGS7407 (UAS-trol) kindly provided by J. Pastor-Pareja [88].
Bloomington deficiency kits and molecularly defined deletions and P element w+ insertions for mapping.
Mutagenesis
50 or more males were transferred to empty vials, allowed to dehydrate for 30 minutes, and then transferred overnight to new bottles contained filter paper soaked in a solution of 24mM EMS, 10Mm Tris pH 7.5 and 1% sucrose. The males were then transferred to dry tubes containing damp filter paper for 30 minutes before being crossed to 50 females for two days.
Mapping
Mapping was as described in the Results, except that 3L043 lethality was initially mapped using Bloomington deficiency kits DK3,3L and DK3,3R (Bloomington), while ade5X1 was initially mapped using meiotic recombination relative to molecularly mapped w+ P element insertions [99]. The 3L044 missense mutation gyc76CL635H was identified by Sanger sequencing (University of Wisconsin-Madison Biotechnology Center) using primers 5’ ATGGATTGTTTGCCACCAACAG Fwd, and 5’ TCAAACAATCGGAATGAAGCTG Rev.
Immunohistochemistry
Wing disc and pupal wing dissection, fixation and staining were as described previously [4, 8]; identical methods were used for larval CNS staining. Images were captured on BioRad and Olympus FV1000 confocal microscopes. Projections, cross-sections and quantifications were generated from Z-series images using ImageJ.
Concentrations and sources of primary antibodies were: 1 : 2000 rabbit anti-phosphoSmad3 (Epitomics); 1 : 500–1000 mouse anti-DSRF (Cold Spring Harbor Laboratory Antibody Facility); 1 : 50 mouse anti-Engrailed 4D9, mouse anti-FasIII or mouse anti-Mmp1 (Developmental Studies Hybridoma Bank); 1 : 500 rabbit anti-MTYamide [100] kindly provided by L. Schoofs; 1 : 500 rabbit anti-Trol [77] kindly provided by S. Baumgartner; 1 : 1000 rabbit anti-Drosophila Lamininγ1 (LanB2) (ABCAM); 1 : 50 mouse 6G7 anti-Collagen IV [22] kindly provided by J. Palka; 1 : 500 rabbit anti-GFP (MBL); 1 : 500 rabbit anti-Rab5 (ABCAM); 1 : 3000 rabbit anti-Rab7 [101] kindly provided by A. Nakamura; 1 : 1000 rabbit anti-Rab11 [102] kindly provided by D. Ready; 1 : 50 rabbit anti-Dei [78] kindly provided by A. Salzberg. Secondary staining used Jackson ImmunoResearch fluorescently-tagged (FITC, RITC, Cy2, Cy3, or Cy5) Min X anti-mouse IgG(H+L) or anti-rabbit IgG(H+L) antisera.
Supporting Information
Zdroje
1. Blair SS. Wing vein patterning in Drosophila and the analysis of intercellular signaling. Ann Rev Cell Dev Biol. 2007;23 : 293–319.
2. De Celis JF, Diaz-Benjumea FJ. Developmental basis for vein pattern variations in insect wings. Int J Dev Biol. 2003;47(7–8):653–63. 14756341
3. De Celis JF. Pattern formation in the Drosophila wing: The development of the veins. BioEssays: news and reviews in molecular, cellular and developmental biology. 2003;25(5):443–51.
4. Conley CA, Silburn R, Singer MA, Ralston A, Rohwer-Nutter D, Olson DJ, et al. Crossveinless 2 contains cysteine-rich domains and is required for high levels of BMP-like activity during the formation of the cross veins in Drosophila. Development. 2000;127(18):3947–59. 10952893
5. O'Connor MB, Umulis D, Othmer HG, Blair SS. Shaping BMP morphogen gradients in the Drosophila embryo and pupal wing. Development. 2006;133(2):183–93. 16368928
6. Umulis D, O'Connor MB, Blair SS. The extracellular regulation of bone morphogenetic protein signaling. Development. 2009;136(22):3715–28. doi: 10.1242/dev.031534 19855014
7. Ray RP, Wharton KA. Context-dependent relationships between the BMPs gbb and dpp during development of the Drosophila wing imaginal disk. Development. 2001;128(20):3913–25. 11641216
8. Ralston A, Blair SS. Long-range Dpp signaling is regulated to restrict BMP signaling to a crossvein competent zone. Developmental biology. 2005;280(1):187–200. 15766758
9. Matsuda S, Shimmi O. Directional transport and active retention of Dpp/BMP create wing vein patterns in Drosophila. Developmental biology. 2012;366(2):153–62. doi: 10.1016/j.ydbio.2012.04.009 22542596
10. Shimmi O, Ralston A, Blair SS, O'Connor MB. The crossveinless gene encodes a new member of the Twisted gastrulation family of BMP-binding proteins which, with Short gastrulation, promotes BMP signaling in the crossveins of the Drosophila wing. Developmental biology. 2005;282(1):70–83. 15936330
11. Vilmos P, Sousa-Neves R, Lukacsovich T, Marsh JL. crossveinless defines a new family of Twisted-gastrulation-like modulators of bone morphogenetic protein signalling. EMBO Rep. 2005;6(3):262–7. 15711536
12. Shimmi O, O'Connor MB. Physical properties of Tld, Sog, Tsg and Dpp protein interactions are predicted to help create a sharp boundary in Bmp signals during dorsoventral patterning of the Drosophila embryo. Development. 2003;130(19):4673–82. 12925593
13. Serpe M, Ralston A, Blair SS, O'Connor MB. Matching catalytic activity to developmental function: Tolloid-related processes Sog in order to help specify the posterior crossvein in the Drosophila wing. Development. 2005;132(11):2645–56. 15872004
14. Ambrosio AL, Taelman VF, Lee HX, Metzinger CA, Coffinier C, De Robertis EM. Crossveinless-2 Is a BMP feedback inhibitor that binds Chordin/BMP to regulate Xenopus embryonic patterning. Developmental cell. 2008;15(2):248–60. doi: 10.1016/j.devcel.2008.06.013 18694564
15. Serpe M, Umulis D, Ralston A, Chen J, Olson DJ, Avanesov A, et al. The BMP-binding protein Crossveinless 2 is a short-range, concentration-dependent, biphasic modulator of BMP signaling in Drosophila. Developmental cell. 2008;14(6):940–53. doi: 10.1016/j.devcel.2008.03.023 18539121
16. Zhang JL, Patterson LJ, Qiu LY, Graziussi D, Sebald W, Hammerschmidt M. Binding between Crossveinless-2 and Chordin von Willebrand factor type C domains promotes BMP signaling by blocking Chordin activity. PloS one. 2010;5(9):e12846. doi: 10.1371/journal.pone.0012846 20886103
17. Chen J, Honeyager SM, Schleede J, Avanesov A, Laughon A, Blair SS. Crossveinless d is a vitellogenin-like lipoprotein that binds BMPs and HSPGs, and is required for normal BMP signaling in the Drosophila wing. Development. 2012;139(12):2170–6. doi: 10.1242/dev.073817 22573617
18. Matsuda S, Blanco J, Shimmi O. A feed-forward loop coupling extracellular BMP transport and morphogenesis in Drosophila wing. PLoS genetics. 2013;9(3):e1003403. doi: 10.1371/journal.pgen.1003403 23555308
19. Waddington CH. The genetic control of wing development in Drosophila. J Genet. 1940;41 : 75–139.
20. Fristrom D, Wilcox M, Fristrom J. The distribution of PS integrins, laminin A and F-actin during key stages in Drosophila wing development. Development. 1993;117(2):509–23. 8330522
21. Fristrom D, Gotwals P, Eaton S, Kornberg TB, Sturtevant M, Bier E, et al. Blistered: a gene required for vein/intervein formation in wings of Drosophila. Development. 1994;120(9):2661–71. 7956840
22. Murray MA, Fessler LI, Palka J. Changing distributions of extracellular matrix components during early wing morphogenesis in Drosophila. Developmental biology. 1995;168(1):150–65. 7883070
23. Brabant MC, Fristrom D, Bunch TA, Brower DL. Distinct spatial and temporal functions for PS integrins during Drosophila wing morphogenesis. Development. 1996;122(10):3307–17. 8898242
24. Urbano JM, Torgler CN, Molnar C, Tepass U, Lopez-Varea A, Brown NH, et al. Drosophila laminins act as key regulators of basement membrane assembly and morphogenesis. Development. 2009;136(24):4165–76. doi: 10.1242/dev.044263 19906841
25. Wang X, Harris RE, Bayston LJ, Ashe HL. Type IV collagens regulate BMP signalling in Drosophila. Nature. 2008;455(7209):72–7. doi: 10.1038/nature07214 18701888
26. Bunt S, Hooley C, Hu N, Scahill C, Weavers H, Skaer H. Hemocyte-secreted type IV collagen enhances BMP signaling to guide renal tubule morphogenesis in Drosophila. Developmental cell. 2010;19(2):296–306. doi: 10.1016/j.devcel.2010.07.019 20708591
27. Sawala A, Sutcliffe C, Ashe HL. Multistep molecular mechanism for Bone morphogenetic protein extracellular transport in the Drosophila embryo. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(28):11222–7. doi: 10.1073/pnas.1202781109 22733779
28. Winstanley J, Sawala A, Baldock C, Ashe HL. Synthetic enzyme-substrate tethering obviates the Tolloid-ECM interaction during BMP gradient formation. eLife. 2015;4.
29. Lindner JR, Hillman PR, Barrett AL, Jackson MC, Perry TL, Park Y, et al. The Drosophila Perlecan gene trol regulates multiple signaling pathways in different developmental contexts. BMC developmental biology. 2007;7 : 121. 17980035
30. Herranz H, Weng R, Cohen SM. Crosstalk between epithelial and mesenchymal tissues in tumorigenesis and imaginal disc development. Current biology: CB. 2014;24(13):1476–84. doi: 10.1016/j.cub.2014.05.043 24980505
31. Liu W, Yoon J, Burg M, Chen L, Pak WL. Molecular characterization of two Drosophila guanylate cyclases expressed in the nervous system. The Journal of biological chemistry. 1995;270(21):12418–27. 7759483
32. McNeil L, Chinkers M, Forte M. Identification, characterization, and developmental regulation of a receptor guanylyl cyclase expressed during early stages of Drosophila development. The Journal of biological chemistry. 1995;270(13):7189–96. 7706258
33. Morton DB, Hudson ML. Cyclic GMP regulation and function in insects. Adv Insect Physiol. 2002;29 : 1–54.
34. Ayoob JC, Yu HH, Terman JR, Kolodkin AL. The Drosophila receptor guanylyl cyclase Gyc76C is required for semaphorin-1a-plexin A-mediated axonal repulsion. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24(30):6639–49.
35. Overend G, Cabrero P, Guo AX, Sebastian S, Cundall M, Armstrong H, et al. The receptor guanylate cyclase Gyc76C and a peptide ligand, NPLP1-VQQ, modulate the innate immune IMD pathway in response to salt stress. Peptides. 2012;34(1):209–18. doi: 10.1016/j.peptides.2011.08.019 21893139
36. Davies SA, Overend G, Sebastian S, Cundall M, Cabrero P, Dow JA, et al. Immune and stress response 'cross-talk' in the Drosophila Malpighian tubule. Journal of insect physiology. 2012;58(4):488–97. doi: 10.1016/j.jinsphys.2012.01.008 22306292
37. Patel U, Davies SA, Myat MM. Receptor-type guanylyl cyclase Gyc76C is required for development of the Drosophila embryonic somatic muscle. Biology open. 2012;1(6):507–15. doi: 10.1242/bio.2012943 23213443
38. Patel U, Myat MM. Receptor guanylyl cyclase Gyc76C is required for invagination, collective migration and lumen shape in the Drosophila embryonic salivary gland. Biology open. 2013;2(7):711–7. doi: 10.1242/bio.20134887 23862019
39. Chak K, Kolodkin AL. Function of the Drosophila receptor guanylyl cyclase Gyc76C in PlexA-mediated motor axon guidance. Development. 2014;141(1):136–47. doi: 10.1242/dev.095968 24284209
40. Potter LR. Guanylyl cyclase structure, function and regulation. Cellular signalling. 2011;23(12):1921–6. doi: 10.1016/j.cellsig.2011.09.001 21914472
41. Osborne KA, Robichon A, Burgess E, Butland S, Shaw RA, Coulthard A, et al. Natural behavior polymorphism due to a cGMP-dependent protein kinase of Drosophila. Science. 1997;277(5327):834–6. 9242616
42. Gurjar MV, Sharma RV, Bhalla RC. eNOS gene transfer inhibits smooth muscle cell migration and MMP-2 and MMP-9 activity. Arteriosclerosis, thrombosis, and vascular biology. 1999;19(12):2871–7. 10591663
43. Jurasz P, Sawicki G, Duszyk M, Sawicka J, Miranda C, Mayers I, et al. Matrix metalloproteinase 2 in tumor cell-induced platelet aggregation: regulation by nitric oxide. Cancer research. 2001;61(1):376–82. 11196190
44. Zaragoza C, Balbin M, Lopez-Otin C, Lamas S. Nitric oxide regulates matrix metalloprotease-13 expression and activity in endothelium. Kidney international. 2002;61(3):804–8. 11849429
45. Tsuruda T, Boerrigter G, Huntley BK, Noser JA, Cataliotti A, Costello-Boerrigter LC, et al. Brain natriuretic Peptide is produced in cardiac fibroblasts and induces matrix metalloproteinases. Circulation research. 2002;91(12):1127–34. 12480813
46. Vellaichamy E, Khurana ML, Fink J, Pandey KN. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. The Journal of biological chemistry. 2005;280(19):19230–42. 15710627
47. Krejci P, Masri B, Fontaine V, Mekikian PB, Weis M, Prats H, et al. Interaction of fibroblast growth factor and C-natriuretic peptide signaling in regulation of chondrocyte proliferation and extracellular matrix homeostasis. J Cell Sci. 2005;118(Pt 21):5089–100. 16234329
48. Li P, Oparil S, Novak L, Cao X, Shi W, Lucas J, et al. ANP signaling inhibits TGF-beta-induced Smad2 and Smad3 nuclear translocation and extracellular matrix expression in rat pulmonary arterial smooth muscle cells. Journal of applied physiology. 2007;102(1):390–8. 17038494
49. Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, et al. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circulation research. 2008;102(2):185–92. 17991884
50. Schwappacher R, Weiske J, Heining E, Ezerski V, Marom B, Henis YI, et al. Novel crosstalk to BMP signalling: cGMP-dependent kinase I modulates BMP receptor and Smad activity. The EMBO journal. 2009;28(11):1537–50. doi: 10.1038/emboj.2009.103 19424179
51. Gong K, Xing D, Li P, Hilgers RH, Hage FG, Oparil S, et al. cGMP inhibits TGF-beta signaling by sequestering Smad3 with cytosolic beta2-tubulin in pulmonary artery smooth muscle cells. Molecular endocrinology. 2011;25(10):1794–803. doi: 10.1210/me.2011-1009 21868450
52. Schwappacher R, Kilic A, Kojonazarov B, Lang M, Diep T, Zhuang S, et al. A Molecular Mechanism for Therapeutic Effects of cGMP-elevating Agents in Pulmonary Arterial Hypertension. The Journal of biological chemistry. 2013;288(23):16557–66. doi: 10.1074/jbc.M113.458729 23612967
53. Golic KG. Site-specific recombination between homologous chromosomes in Drosophila. Science. 1991;252(5008):958–61. 2035025
54. Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117(4):1223–37. 8404527
55. Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Developmental biology. 1975;42(2):211–21. 1116643
56. Christoforou CP, Greer CE, Challoner BR, Charizanos D, Ray RP. The detached locus encodes Drosophila Dystrophin, which acts with other components of the Dystrophin Associated Protein Complex to influence intercellular signalling in developing wing veins. Developmental biology. 2008;313(2):519–32. 18093579
57. Cooper MT, Conant AW, Kennison JA. Molecular genetic analysis of Chd3 and polytene chromosome region 76B-D in Drosophila melanogaster. Genetics. 2010;185(3):811–22. doi: 10.1534/genetics.110.115121 20439780
58. Parks AL, Cook KR, Belvin M, Dompe NA, Fawcett R, Huppert K, et al. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat Genet. 2004;36(3):288–92. 14981519
59. Potter LR. Regulation and therapeutic targeting of peptide-activated receptor guanylyl cyclases. Pharmacology & therapeutics. 2011;130(1):71–82.
60. Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52. 3291115
61. Klaiber M, Dankworth B, Kruse M, Hartmann M, Nikolaev VO, Yang RB, et al. A cardiac pathway of cyclic GMP-independent signaling of guanylyl cyclase A, the receptor for atrial natriuretic peptide. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(45):18500–5. doi: 10.1073/pnas.1103300108 22027011
62. Thompson DK, Garbers DL. Dominant negative mutations of the guanylyl cyclase-A receptor. Extracellular domain deletion and catalytic domain point mutations. The Journal of biological chemistry. 1995;270(1):425–30. 7814405
63. Kalderon D, Rubin GM. cGMP-dependent protein kinase genes in Drosophila. The Journal of biological chemistry. 1989;264(18):10738–48. 2732245
64. Morrison DK, Murakami MS, Cleghon V. Protein kinases and phosphatases in the Drosophila genome. The Journal of cell biology. 2000;150(2):F57–62. 10908587
65. Tiong SY, Keizer C, Nash D, Bleskan J, Patterson D. Drosophila purine auxotrophy: new alleles of adenosine 2 exhibiting a complex visible phenotype. Biochem Genet. 1989;27(5–6):333–48. 2803228
66. Tiong SY, Nash D. Genetic analysis of the adenosine3 (Gart) region of the second chromosome of Drosophila melanogaster. Genetics. 1990;124(4):889–97. 2108904
67. Clark DV. Molecular and genetic analyses of Drosophila Prat, which encodes the first enzyme of de novo purine biosynthesis. Genetics. 1994;136(2):547–57. 8150282
68. O'Donnell AF, Tiong S, Nash D, Clark DV. The Drosophila melanogaster ade5 gene encodes a bifunctional enzyme for two steps in the de novo purine synthesis pathway. Genetics. 2000;154(3):1239–53. 10757766
69. Day JP, Dow JA, Houslay MD, Davies SA. Cyclic nucleotide phosphodiesterases in Drosophila melanogaster. Biochem J. 2005;388(Pt 1):333–42. 15673286
70. Day JP, Houslay MD, Davies SA. A novel role for a Drosophila homologue of cGMP-specific phosphodiesterase in the active transport of cGMP. Biochem J. 2006;393(Pt 2):481–8. 16232123
71. Day JP, Cleghon V, Houslay MD, Davies SA. Regulation of a Drosophila melanogaster cGMP-specific phosphodiesterase by prenylation and interaction with a prenyl-binding protein. Biochem J. 2008;414(3):363–74. doi: 10.1042/BJ20080560 18503409
72. Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76 : 481–511. 17376027
73. Arora K, Sinha C, Zhang W, Ren A, Moon CS, Yarlagadda S, et al. Compartmentalization of cyclic nucleotide signaling: a question of when, where, and why? Pflugers Arch. 2013;465(10):1397–407. doi: 10.1007/s00424-013-1280-6 23604972
74. Yu K, Sturtevant MA, Biehs B, Francois V, Padgett RW, Blackman RK, et al. The Drosophila decapentaplegic and short gastrulation genes function antagonistically during adult wing vein development. Development. 1996;122(12):4033–44. 9012523
75. Sotillos S, de Celis JF. Regulation of decapentaplegic expression during Drosophila wing veins pupal development. Mech Dev. 2006;123(3):241–51. 16423512
76. Molnar C, Lopez-Varea A, Hernandez R, de Celis JF. A gain-of-function screen identifying genes required for vein formation in the Drosophila melanogaster wing. Genetics. 2006;174(3):1635–59. 16980395
77. Friedrich MV, Schneider M, Timpl R, Baumgartner S. Perlecan domain V of Drosophila melanogaster. Sequence, recombinant analysis and tissue expression. European journal of biochemistry / FEBS. 2000;267(11):3149–59. 10824099
78. Egoz-Matia N, Nachman A, Halachmi N, Toder M, Klein Y, Salzberg A. Spatial regulation of cell adhesion in the Drosophila wing is mediated by Delilah, a potent activator of betaPS integrin expression. Developmental biology. 2011;351(1):99–109. doi: 10.1016/j.ydbio.2010.12.039 21215259
79. Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. The Journal of biological chemistry. 1996;271(17):10079–86. 8626565
80. Llano E, Pendas AM, Aza-Blanc P, Kornberg TB, Lopez-Otin C. Dm1-MMP, a matrix metalloproteinase from Drosophila with a potential role in extracellular matrix remodeling during neural development. The Journal of biological chemistry. 2000;275(46):35978–85. 10964925
81. Llano E, Adam G, Pendas AM, Quesada V, Sanchez LM, Santamaria I, et al. Structural and enzymatic characterization of Drosophila Dm2-MMP, a membrane-bound matrix metalloproteinase with tissue-specific expression. The Journal of biological chemistry. 2002;277(26):23321–9. 11967260
82. Page-McCaw A, Serano J, Sante JM, Rubin GM. Drosophila matrix metalloproteinases are required for tissue remodeling, but not embryonic development. Developmental cell. 2003;4(1):95–106. 12530966
83. Deady L, Shen W., Mosure S., Spradling A. C., Sun J. Matrix metalloproteinase 2 is required for ovulation and corpus luteum formation in Drosophila. PLoS genetics. 2015.
84. Uhlirova M, Bohmann D. JNK - and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. Embo Journal. 2006;25(22):5294–304. 17082773
85. Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochimica et biophysica acta. 2010;1803(1):55–71. doi: 10.1016/j.bbamcr.2010.01.003 20080133
86. Wei S, Xie Z, Filenova E, Brew K. Drosophila TIMP is a potent inhibitor of MMPs and TACE: similarities in structure and function to TIMP-3. Biochemistry. 2003;42(42):12200–7. 14567681
87. Srivastava A, Pastor-Pareja JC, Igaki T, Pagliarini R, Xu T. Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(8):2721–6. 17301221
88. Pastor-Pareja JC, Xu T. Shaping cells and organs in Drosophila by opposing roles of fat body-secreted Collagen IV and perlecan. Developmental cell. 2011;21(2):245–56. doi: 10.1016/j.devcel.2011.06.026 21839919
89. Zang Y, Wan M, Liu M, Ke H, Ma S, Liu LP, et al. Plasma membrane overgrowth causes fibrotic collagen accumulation and immune activation in Drosophila adipocytes. eLife. 2015;4:e07187. doi: 10.7554/eLife.07187 26090908
90. Cho JY, Chak K, Andreone BJ, Wooley JR, Kolodkin AL. The extracellular matrix proteoglycan perlecan facilitates transmembrane semaphorin-mediated repulsive guidance. Genes & development. 2012;26(19):2222–35.
91. Ruka KA, Miller AP, Blumenthal EM. Inhibition of diuretic stimulation of an insect secretory epithelium by a cGMP-dependent protein kinase. American journal of physiology Renal physiology. 2013;304(9):F1210–6. doi: 10.1152/ajprenal.00231.2012 23445619
92. Zars T. Short-term memories in Drosophila are governed by general and specific genetic systems. Learning & memory. 2010;17(5):246–51.
93. Kanao T, Sawada T, Davies SA, Ichinose H, Hasegawa K, Takahashi R, et al. The Nitric Oxide-Cyclic GMP Pathway Regulates FoxO and Alters Dopaminergic Neuron Survival in Drosophila. PloS one. 2012;7(2).
94. Junger MA, Rintelen F, Stocker H, Wasserman JD, Vegh M, Radimerski T, et al. The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. Journal of biology. 2003;2(3):20. 12908874
95. Dolez M, Nicolas JF, Hirsinger E. Laminins, via heparan sulfate proteoglycans, participate in zebrafish myotome morphogenesis by modulating the pattern of Bmp responsiveness. Development. 2011;138(1):97–106. doi: 10.1242/dev.053975 21115608
96. Wang X, Page-McCaw A. A matrix metalloproteinase mediates long-distance attenuation of stem cell proliferation. The Journal of cell biology. 2014;206(7):923–36. doi: 10.1083/jcb.201403084 25267296
97. Zhang J, Schulze KL, Hiesinger PR, Suyama K, Wang S, Fish M, et al. Thirty-one flavors of Drosophila rab proteins. Genetics. 2007;176(2):1307–22. 17409086
98. Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(26):15050–5. 11742088
99. Zhai RG, Hiesinger PR, Koh TW, Verstreken P, Schulze KL, Cao Y, et al. Mapping Drosophila mutations with molecularly defined P element insertions. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(19):10860–5. 12960394
100. Verleyen P, Baggerman G, Wiehart U, Schoeters E, Van Lommel A, De Loof A, et al. Expression of a novel neuropeptide, NVGTLARDFQLPIPNamide, in the larval and adult brain of Drosophila melanogaster. Journal of neurochemistry. 2004;88(2):311–9. 14690519
101. Tanaka T, Nakamura A. The endocytic pathway acts downstream of Oskar in Drosophila germ plasm assembly. Development. 2008;135(6):1107–17. doi: 10.1242/dev.017293 18272590
102. Satoh AK, O'Tousa JE, Ozaki K, Ready DF. Rab11 mediates post-Golgi trafficking of rhodopsin to the photosensitive apical membrane of Drosophila photoreceptors. Development. 2005;132(7):1487–97. 15728675
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Single Strand Annealing Plays a Major Role in RecA-Independent Recombination between Repeated Sequences in the Radioresistant Bacterium
- The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals
- Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross
- DCA1 Acts as a Transcriptional Co-activator of DST and Contributes to Drought and Salt Tolerance in Rice
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy