
A Systems Approach Identifies Essential FOXO3 Functions at Key Steps of Terminal Erythropoiesis
Red blood cells (RBCs) are highly specialized cells that transport oxygen throughout the body and are essential for survival. However, RBCs have a limited lifespan and need to be replenished continuously by stem cells in the bone marrow. Mammalian RBCs are unique in that in order to fully mature they exclude their nucleus and other organelles. Mechanisms involved in these processes are not well understood at the molecular level. Defects in any of the these processes may lead to red blood cell defects, a decreased capacity to transport oxygen and/or a block in red blood cell production in vitro. Therefore, understanding how these processes are regulated at the molecular level can lead to promising new therapies for red blood cell defects and improved methods of generating red blood cells in a dish. Here, using an integrated computational and experimental biology approach, we found that the nuclear factor FOXO3 is a crucial regulator of red blood cell production by coordinating the expression of many of the genes specific for terminal maturation of red blood cells. Furthermore we found that FOXO3 can even increase the production of normal red blood cells in culture raising the possibility that enhancing FOXO3 may have a therapeutic use. Our studies identify FOXO3 as a novel regulator of RBC enucleation and terminal erythropoiesis.
Published in the journal:
. PLoS Genet 11(10): e32767. doi:10.1371/journal.pgen.1005526
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005526
Summary
Red blood cells (RBCs) are highly specialized cells that transport oxygen throughout the body and are essential for survival. However, RBCs have a limited lifespan and need to be replenished continuously by stem cells in the bone marrow. Mammalian RBCs are unique in that in order to fully mature they exclude their nucleus and other organelles. Mechanisms involved in these processes are not well understood at the molecular level. Defects in any of the these processes may lead to red blood cell defects, a decreased capacity to transport oxygen and/or a block in red blood cell production in vitro. Therefore, understanding how these processes are regulated at the molecular level can lead to promising new therapies for red blood cell defects and improved methods of generating red blood cells in a dish. Here, using an integrated computational and experimental biology approach, we found that the nuclear factor FOXO3 is a crucial regulator of red blood cell production by coordinating the expression of many of the genes specific for terminal maturation of red blood cells. Furthermore we found that FOXO3 can even increase the production of normal red blood cells in culture raising the possibility that enhancing FOXO3 may have a therapeutic use. Our studies identify FOXO3 as a novel regulator of RBC enucleation and terminal erythropoiesis.
Introduction
Erythropoiesis ensures the daily production of over 200 billion RBCs whose main function is to carry oxygen. Decreased production of RBCs is associated with many human disorders involving impaired erythroblast maturation. The generation of RBCs in vitro from embryonic stem cells or human-induced pluripotent stem cells (iPS cells) has been proposed to provide a cost-effective and safe blood supply. Despite recent development [1] achieving efficient production of functional RBCs has been hindered by incomplete knowledge of terminal erythroblast maturation.
Generation of RBCs involves the differentiation of hematopoietic stem cells into common megakaryocyte and erythroid progenitors, which give rise to lineage-restricted erythroid progenitors, erythroblasts, and ultimately erythrocytes. During the final stages of erythropoiesis, proliferation of erythroblasts is coupled with differentiation as terminally differentiating erythroblasts accumulate hemoglobin, reduce cell size, and condense their nuclei. Following enucleation, reticulocytes remodel their membrane and clear mitochondria and remaining organelles to transition into fully mature erythrocytes [2]. This complex process is controlled by integration of erythropoietin receptor (EpoR) signaling with the function of erythroid lineage-specific transcription factors including GATA–1, KLF–1 and TAL–1 (SCL) and their cofactors [3]. Despite recent progress [4–6], many questions remain unanswered regarding whether these factors function alone or together to control enucleation and/or to remove organelles, including mitochondria, during terminal erythroblast maturation. Increasing evidence suggest that FOXO3 cooperates with these factors and their requisite coregulators to control specific molecular/cellular steps that drive terminal erythroid maturation [4–6].
FOXO3 belongs to the FOXO family of Forkhead transcription factors composed in mammals of the highly related members FOXO1, FOXO3, FOXO4 and FOXO6. FOXOs are homeostatic maintaining factors implicated in many diseases including cancer, diabetes, and erythroid disorders [7–10]. FOXOs integrate fundamental biological processes through the regulation of cell cycle, oxidative stress, DNA damage responses, apoptosis, inflammatory responses, and metabolism [7,11]. FOXO genes have evolutionary conserved functions in stem cell maintenance and longevity [12–24]. Emerging evidence suggests that FOXO may also play a key role in tissue-tissue communication [7,25–27]. Among FOXO factors, FOXO3 is critical for normal and stress erythropoiesis [8–10,28–31]. This is evident as Foxo3 mutant mice die rapidly when exposed to acute erythroid oxidative challenge [29]. Notably, FOXO3 expression and function increase progressively with erythroblast maturation [29,32,33]. Despite these findings, whether FOXO3 has any function in the regulation of terminal erythroblast maturation remains unknown.
Using an integrated systems and experimental biology approach, we demonstrate that FOXO3 is critical for the correct temporal expression of at least one third of the genes differentially expressed in normal maturing erythroblasts. Dysregulation of this subset of genes due to FOXO3 loss led to defects at distinct stages of terminal erythroblast maturation and RBC production. Our data demonstrate that FOXO3 is critical for erythroblast enucleation through polarization of the nucleus expulsion direction and is required for mitochondrial clearance. The transcriptomic analyses also revealed that increasingly maturing primary erythroblasts express immune-related transcripts whose expression is highly modulated upon loss of FOXO3. Collectively, these findings demonstrate that FOXO3 is an essential component of the transcriptional program that regulates terminal erythroblast maturation and required for the erythroblast enucleation process.
Results
Comparative transcriptomic analysis reveals potential new functions for FOXO3 in terminally maturing erythroblasts
To investigate the FOXO3-regulated transcriptional program during erythroblast maturation we compared the transcriptome of adult bone marrow erythroid precursor populations of Foxo3-/- mice to that of wild type (WT) mice. Since immature erythroblasts accumulate in Foxo3 mutant bone marrow [29], we reasoned that the relative accumulation of immature erythroblasts due to FOXO3 deletion might reflect a block in their terminal maturation. Immature erythroblasts were isolated using flow cytometry relying on the immunophenotype and forward scatter properties of erythroblasts according to Chen et al. [34]. The erythroid specific marker TER 119 was combined with CD44 and forward scatter, which both decrease during maturation, to resolve the progressive stages of erythroblast differentiation [34] (Fig 1A). This approach enabled the distinction and purification of three consecutive stages of maturation of erythroid populations (pro-, basophilic and polychromatophilic erythroblasts) as defined by gates I, II and III respectively (Fig 1A). Relatively pure subpopulations of erythroblasts as shown by morphological analysis (S1A Fig) were isolated using this gating strategy. RNA was isolated from the first three gates (Fig 1A, depicted in red) and deep sequencing analysis was conducted to compare wild type and Foxo3 mutant erythroblast transcriptomes. In the morphological analysis and subsequent validation experiments, Gate IV cells (depicted in black) encompassing mainly reticulocytes that are enucleated cells preceeding mature red blood cells were also included (Fig 1A).

Erythroid-specific genes of wild type erythroblasts were grouped according to low, intermediate, or high levels of expression that was validated by qRT-PCR, which faithfully reproduced the levels and patterns of gene expression in each group (S1B Fig and S1 Table). Expression of globin genes was used as a control to confirm the identity of erythroblasts. Although the expression was too high to be reliably quantified by cufflinks software used for RNA-Seq analysis, qRT-PCR analysis demonstrated globin gene upregulation during erythroid maturation (S1C Fig and S1 Table). Analysis of wild-type erythroblasts revealed 5514 genes (S2 Table) with at least two fold differential expression between gates. These genes were grouped into eight clusters using k-means clustering (Fig 1B). Using Gene Ontology (GO) term analysis with FuncAssociate 2.0, signaling pathways and biological processes enriched in each of the clusters were delineated (S3 Table). The chromatin immunoprecipitation (ChIP) enrichment analysis tool, ChEA, [35,36] was used to identify potential transcription factors that may occupy the genes within each cluster (S4 Table). Cluster A, which grouped 600 genes that were continuously downregulated from Gates I to III, is enriched in inflammatory and apoptotic genes. In agreement with RNA-Seq analysis of human erythroblasts, genes continuously up-regulated during terminal erythroid maturation and clustered in G and H (801 and 446 genes respectively) were enriched for autophagy-related genes underscoring the importance of autophagy during erythroblast maturation [4,33,37]. Genes initially upregulated and then down-regulated were enriched for cell cycle-related processes, chromatin remodeling, DNA repair and mitotic genes (Clusters E and F, 867 and 865 genes respectively) (Fig 1B and S3 Table). Erythroid-specific genes including heme biosynthetic enzymes, iron metabolism, and erythroid membrane genes were distributed in clusters F, G, and H (S3 Table). Consistent with these results, known erythroid transcription factors including GATA–1, KLF–1, TAL–1, and the transcriptional co-activator EP300 (E1A binding protein p300) were associated with the sustained up-regulation in clusters G and H (S4 Table). Expression of Clusters E and F genes which were downregulated at Gate III, was associated with E2F4, E2F1, c-MYC transcription factors, and Cyclin D1 (CCND1), all known for regulating cell cycle progression. These data support previous findings [38] that erythroblasts continue to cycle at late stages of their maturation.
Immune-related genes were enriched in clusters B and C (1355 and 295 genes respectively) which include genes initially downregulated and then slightly upregulated in gate III (Fig 1B). As anticipated many inflammation-related genes, such as S100 Calcium Binding Protein A11 (s100a11), interleukin 17 receptor α (Il17ra), and immediate early response 2 (Ier2) were downregulated with maturation (S2A Fig). Furthermore, ChEA analysis implicated MYB and PU.1, known repressors of terminal erythroid maturation, as transcription factors regulating B and C gene clusters [39–41] (S4 Table). Accordingly, mRNA expression of MYB and PU.1 decreased during erythroid maturation (S2B Fig). Interestingly and consistent with previous findings [42,43], genes linked to several immune pathways, including several genes involved in the interferon response and lymphocyte activation pathways, were upregulated upon terminal erythroid maturation (clusters G and H) (Fig 1B). Expression of interferon-related genes including interferon regulatory factor (Irf7) and radical S-adenosyl methionine domain containing 2 (Rsad2) was validated by qRT-PCR which found these to be upregulated 2–15 fold upon maturation of bone marrow erythroblasts (S2C Fig). The corresponding proteins were also expressed in TER119+ erythroblasts that are negative for CD45 (TER119+CD45-) confirming the specificity of their expression and lack of non-erythroid cell contamination (S2D Fig). These observations suggest that these immune-related genes may have a specific function in terminal erythroid maturation.
FOXO3 is a critical determinant of the erythroid cell transcriptome
For comparing WT and Foxo3 mutant erythroblast expression profiles, genes were re-clustered. Genes with at least a two-fold differential expression between the same gate of WT and Foxo3 mutant erythroblasts were reclustered for further analysis. Expression of 3906 genes (S5 Table), approximately 35% of the total expressed genes, was strikingly altered in Foxo3-/- erythroid precursors (Gates I to III). In agreement with the progressive increase in the expression and function of FOXO3 with erythroid maturation [29,31], the majority of differences in gene expression among gates I to III of wild type and Foxo3-/- erythroblasts were detected in Gate III (S3A and S3B Fig). Loss of FOXO3 led to both repression (clusters Q and R, 926 and 509 genes respectively) and induction (clusters I and J, 444 and 1094 genes respectively) of gene expression during erythroid maturation (Fig 1C). Immune-related pathways, including macrophage and neutrophil activation pathways enriched in clusters I and J, fell within programs that are normally repressed, but were aberrantly upregulated in the absence of FOXO3 (Figs 1C and S3C and S6 Table). These results indicate that loss of FOXO3 may enhance the expression of many inflammatory-related genes in maturing erythroblasts (S3C Fig), consistent with the anti-inflammatory function of FOXO3 [27,44]. The greatest impact of FOXO3 loss was exemplified by clusters Q and R, which consist of genes normally upregulated during erythroid maturation. Cluster R was enriched for autophagy and catabolic processes, while Cluster Q was enriched for heme biosynthesis, erythroid differentiation, nucleosome assembly, and DNA packaging-related processes (Fig 1C and S6 Table). Both clusters Q and R designate genes that are continuously up-regulated from Gates I to III. However, differences arise between the clusters from the loss of FOXO3. In the absence of FOXO3, the progressive upregulation of genes between Gates I to III is abrogated in cluster R. In contrast genes in cluster Q are not upregulated from Gates II to III. The stalled gene activation in Cluster Q suggests that FOXO3 is required for the transition to the gene expression profile characteristic of Gate III erythroblasts. The distinct pattern of clusters Q and R may reflect distinct modes of FOXO3 action for each respective gene cohort. Since 40% of the genes in both clusters are established targets of GATA–1, TAL–1, and/or KLF–1 transcription factors (S7 Table) [3], these results, consistent with previous findings [6], raise the possibility that FOXO3 cooperates with these factors to sustain gene transcription during terminal erythroid maturation.
Defective autophagy in Foxo3-/- erythroblasts impairs reticulocyte maturation
Autophagy was one of the main pathways highly up-regulated upon erythroblast maturation (Fig 1B, clusters G and H). However, autophagy-related genes were greatly dysregulated in Foxo3-/- mutant erythroblasts (Fig 1B, and S6 Table cluster R). Autophagy (or macro-autophagy) serves as a homeostatic mechanism that mediates the consumption of damaged or old cellular components, as well as the cellular remodeling that is associated with cell differentiation [45]. In erythroblasts, autophagy is implicated specifically in the clearance of mitochondria (mitophagy or selective mitochondrial autophagy) during terminal erythroid maturation [37,46,47].
To evaluate the role of FOXO3 in regulating autophagy in primary erythroblasts, we validated expression of autophagy-related genes differentially expressed between wild type and Foxo3 mutant erythroblasts (Figs 2A and S4A). The expression pattern of some of these genes, including Nix (Bnip3l) and Ulk1 (Fig 2A), was upregulated 15 - to over 40-fold during late stages of bone marrow erythroblast maturation and reticulocyte formation. This finding is supported by the known function of these genes in mitochondrial removal [46–48]. The similar pattern of expression of other autophagy-related genes, including Gabarapl2 (Gate–16), p62, Atg14, and Pink1 (Figs 2A and S4A) suggests their involvement in erythroid maturation. In addition, autophagy genes, including Gabarapl1 and Map1lc3b lie within the highly expressed gene cohort at all stages of erythroblast maturation indicating these genes may be involved in the homeostatic control of erythroid maturation (S1 Table). Consistent with this, the core autophagy genes Atg5 and Atg7 were expressed at similar levels throughout erythroid maturation relative to other autophagy genes examined (S4B Fig). However, expression of several autophagy-related gene transcripts was profoundly compromised in Foxo3 mutant erythroblasts (Figs 2A and S4A), suggesting that these genes are potential direct targets of FOXO3 in erythroblasts. ChIP analysis revealed occupancy at Btg1, a known FOXO3 direct target [28] and several autophagy-related genes including Nix, Gabarapl2 and Ulk1 in wild-type, but not in Foxo3-deficient erythroblasts. Thus, FOXO3 occupied regulatory regions of these genes in primary erythroblasts in vivo (Fig 2B). Occupancy was not detected at upstream sequences lacking FOXO3 binding sites in these regulatory regions. Several of the genes shown in Figs 2A and S4A are direct FOXO3 transcriptional targets, including Map1lc3b and Atg14 in non hematopoietic cells [49–51], while others including Gabarapl2 were not known to be regulated by FOXO3 in any system. Interestingly, Nix and Ulk1 are known regulators of erythroblast mitochondrial removal [47] [48].

We reasoned that the failure to upregulate autophagy gene expression might decrease autophagy during differentiation of Foxo3-mutant erythroblasts and compromise their terminal maturation. To test this, we measured the conversion of the soluble free form of Microtubule-associated protein 1 light chain 3B (LC3B-I) to the lipidated LC3B-II form, an essential step in autophagosome activation. Autophagosomes are generated as a result of sequential assembly and activation of autophagy-related proteins [45]. Once activated, they engulf the damaged organelle or proteins and fuse with lysosomes to degrade their cargo. The LC3B-II/LC3B-I ratio, an important indicator of autophagy [52], was decreased significantly in freshly isolated primary Foxo3-mutant erythroblasts under homeostatic conditions (Fig 3A), suggesting that autophagy was compromised in these cells. Further gate-by-gate analysis suggested that defective autophagy was mainly observed within Foxo3-/- gate III and IV erythroblasts (Fig 3B). To further ensure that these results reflected impairment in autophagosome formation and not abnormalities related to events upstream or downstream [53], we evaluated the autophagic flux in erythroblasts using an autophagosome-specific fluorescent probe [54]. Addition of chloroquine to bone marrow cultures blocked degradation and induced accumulation of autophagosomes in wild type and Foxo3-/- erythroblasts, enabling their measurement over time by flow cytometry and western blotting for LC3B-II (Fig 3C and 3D). Foxo3-deficient erythroblasts exhibited significantly reduced autophagic flux as compared to their wild type counterparts specifically in Gate IV erythroblasts that are highly enriched in reticulocytes (Fig 3C). The LC3B-II accumulation was also delayed in chloroquine-treated Foxo3-/- relative to wild type erythroblasts (Fig 3D). The reduced autophagy flux was mainly detected in late stage Foxo3-deficient erythroblasts, consistent with the alteration of autophagy-related gene expression in these cells (S4A Fig). Collectively, these results are consistent with the notion that FOXO3 controls autophagy at late, but not early, stages of erythroid maturation (Fig 3B and 3C).

We considered whether low levels of autophagy that are characteristic of Foxo3-/- erythroblasts suffice to support mitochondria removal during reticulocyte maturation. The frequency of circulating reticulocytes in Foxo3-mutant mice is increased to compensate for increased RBC destruction [29]. However, in Foxo3-mutants a significant fraction of mature RBCs are devoid of the transferrin receptor CD71 but remain positive for the mitochondria-specific MitoTracker Red probe (Fig 3E). This was further confirmed using a distinct mitochondria-specific probe MitoTracker Green (S4C Fig). These results indicate that despite their increased numbers in Foxo3 mutant mice [29] reticulocytes retain mitochondria, delaying the complete maturation into RBCs. This increase is consistent with the failure to induce multiple autophagy-related genes in Foxo3-/- erythroblasts, which have been implicated specifically in mitophagy (Figs 2A and S4A). Nonetheless, the impact of impaired autophagy on mitochondrial removal in Foxo3-mutant reticulocytes appeared to be relatively modest, suggesting that compensatory mechanisms might be involved. Alternatively these results might reflect the contribution of distinct autophagy pathways controlling mitochondrial removal in erythroid cells [55–57].
FOXO3 is essential for erythroblast enucleation
Key steps in RBC formation include chromatin condensation and expulsion of the nucleus (enucleation) from late-stage erythroblasts [2] [58] [59]. We discovered that many genes involved in nucleosome assembly and DNA packaging-related processes were downregulated in Foxo3-mutant erythroblasts (Fig 1C, cluster Q, S6 Table), raising the possibility that FOXO3 controls chromatin condensation and/or enucleation. Using DRAQ5, a fluorescent probe that binds DNA in vivo, we found that there were significantly fewer enucleated Foxo3-mutant bone marrow erythroblasts than wild type cells (Figs 4A and S5A). QRT-PCR expression analysis of cluster Q showed that genes implicated in chromatin condensation and/or enucleation Mxi1, Riok3, Smarca4, Trim58, Rac GTPase I and II [60–63] were all reduced at distinct stages of Foxo3-mutant erythroblast maturation (Figs 4B and S5B). Among these, Riok3 and Trim58 transcripts were upregulated 60 - and over 120-fold, respectively, during differentiation of normal bone marrow erythroblasts, suggesting these genes may function beyond enucleation during terminal erythroblast maturation (S5B Fig). In contrast, expression of RacGTPase I and II decreased with maturation of normal bone marrow erythroblasts. ChIP of endogenous FOXO3 in wild type, but not in Foxo3 mutant, bone marrow erythroblasts revealed occupancy at regulatory regions of Mxi1 and Riok3 (Fig 4C). Thus, these genes similar to the autophagy genes examined earlier may also be directly regulated by FOXO3 in erythroblasts.

We used imaging flow cytometry to develop mechanistic insights into the potential enucleation defect in Foxo3 mutant erythroblasts. As described previously, [58,64] TER119+ erythroblasts can be segregated according to their cell and nuclear size into progressive stages of maturation (pro-, basophilic, polychromatic and orthochromatic erythroblasts) (Fig 5A). While this analysis confirmed the increase in erythroid precursors described previously in Foxo3 mutant bone marrow (S5C Fig) [29], the analysis also revealed that the accumulation occurs at the orthochromatic stage of Foxo3 mutant erythroblast maturation (Figs 5A and S5C). Prior to enucleation, late-stage erythroblasts displace the nucleus from its central location. The percentage of orthochromatic erythroblasts containing an asymmetrically positioned nucleus was quantified using the delta centroid, a measure of the distance between the center of the cell body and center of the DRAQ5-stained nucleus [58]. This analysis revealed a lower percentage of enucleating erythroblasts in the Foxo3-mutant versus wild-type bone marrow (Fig 5B).

To investigate how the FOXO3 loss impacts enucleation, we conducted a high-resolution morphological analysis of bone marrow erythroblasts. As shown in Fig 6A, confocal analysis revealed wild-type enucleating erythroblasts by the presence of a gap in the bright TER119 membrane staining as described previously [65]. Wild type enucleating erythroblasts displayed a dumbbell-shaped nucleus, with a neck located at the TER119 sorting boundary of the nascent reticulocyte. This ensures that cells use a single direction for nuclear extrusion. In contrast, Foxo3 mutant erythroblasts exhibited multiple nuclear necks accompanied by multiple sorting boundaries with each lobe extruding in a different direction away from the nascent reticulocyte. This unique pattern suggested defective polarization of Foxo3 mutant erythroblasts during enucleation (Fig 6A). Accordingly, 48% of the Foxo3-/- enucleating erythroblasts exhibited abnormal enucleation morphologies. These results were verified by imaging flow cytometric analysis by distinguishing orthochromatic erythroblasts with tri-lobular nuclei from normal extruding nuclei (Fig 6A–6C). This analysis demonstrated that erythroblasts with tri-lobular nuclei were remarkably more frequent in Foxo3-/- versus wild type bone marrow (Fig 6B amd 6C). These results indicate that FOXO3 is critical for enucleation and may control erythroblast polarization/nuclear positioning as a prelude to enucleation. Consistent with this notion, several genes implicated in cytoskeleton organization, cell polarization and cytokenesis, are deregulated in Foxo3-/- erythroblasts (Figs 6D, S5C and S6A). Notably, the small RhoGTPase CDC42-related gene cluster that regulates cytoskeleton organization and cell polarization [66,67] is aberrantly upregulated in Foxo3-/- erythroblasts (Fig 6D).

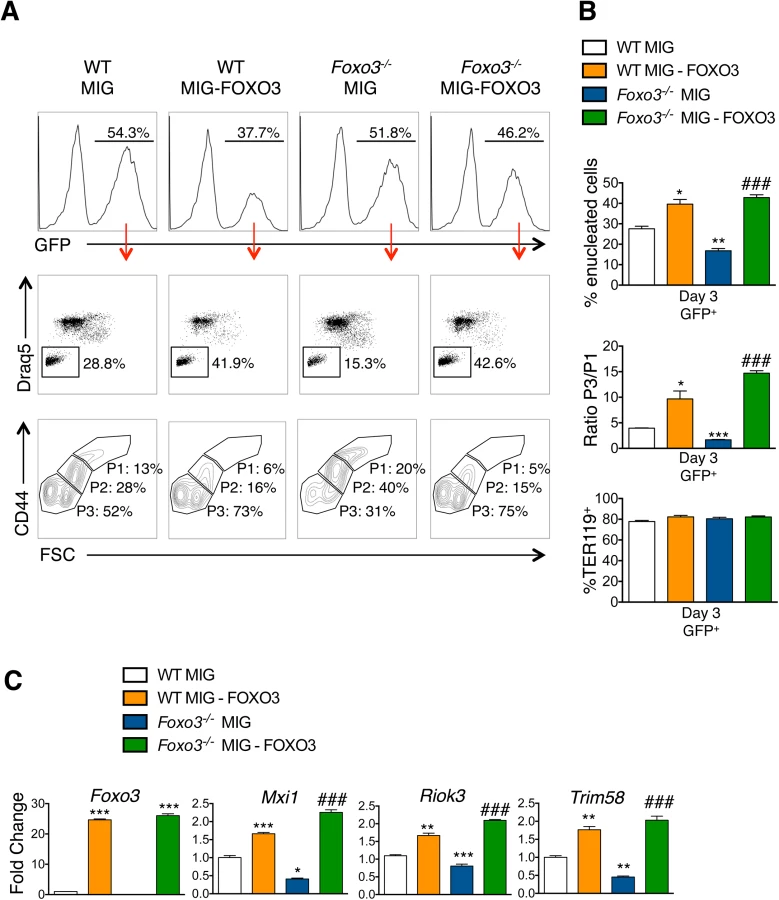
To investigate whether FOXO3 might directly regulate enucleation, we examined the capacity of FOXO3 to rescue enucleation in Foxo3 mutant erythroblasts. MSCV-IRES-GFP (MIG) or MIG-FOXO3 were ectopically expressed in wild type and Foxo3-/- bone marrow-derived erythroid progenitors and subjected to in vitro maturation by adapting a previously established protocol, for bone marrow erythroblasts [68] (Fig 7A). Gene expression patterns of Gata1, Pu.1, and Hbb-b2, were examined over 3 days of maturation and validated that they resemble in vivo erythroblast maturation patterns (S7A Fig). Flow cytometry analysis enabled resolution of the most immature erythroblast populations (ex vivo P1), exhibiting high CD44 levels and large size, compared to the most mature erythroblast population (ex vivo P3), exhibiting low CD44 levels and small size. The ratio of P3 to P1, termed maturation index here, reveals the extent of maturation at day 3 of culture. Foxo3-/- erythroblasts show an overall decrease in their ability to mature (Fig 7A and 7B) [29]. Importantly, ectopic expression of FOXO3 in mutant erythroblasts rescued their defective maturation without modulating the total number of TER119 cells (Fig 7A–7C). In addition, the last stage of maturation was specifically impaired in Foxo3 mutant erythroblasts (P3/P2) at ∼ 50% (± 0.046, n = 3) of that of wild type (P3/P2) cells and rescued by ectopic expression of FOXO3 (Fig 7A, bottom panels). Furthermore, a similar degree of rescue by ectopic FOXO3 expression was observed when comparing the very last stages, S2 (enucleated) and S1 (nucleated) mature erythroblasts in culture, where the majority of enucleation occurs (S7B Fig). In fact, overexpression of FOXO3 rescued the enucleation capacity of Foxo3-/- erythroblasts to levels similar or higher than that of wild type controls in ex vivo maturation cultures (Fig 7A and 7B). In support of a potential direct FOXO3 control of enucleation the ectopic expression of FOXO3 increased the expression of Mxi1, Riok3, and Trim58 (Figs 7C and S7C). Notably, overexpression of FOXO3 further improved enucleation, the maturation index, and expression of enucleation related genes in WT erythroblasts (Figs 7A–7C and S7B). Ectopic expression of FOXO3 also improved expression of autophagy-related genes in Foxo3-/- erythroblasts (S7C Fig).
Discussion
Our studies show that FOXO3 has critical physiological functions at key steps of terminal erythroid maturation. While FOXO3 is required for enucleation and for subsequent mitochondrial clearance in reticulocytes, FOXO3 loss impacts more profoundly upon the enucleation process. Enucleation is a critical limiting step in erythroid maturation [2]. The removal of the nucleus results from the culmination of a multi-step process that starts early in differentiation and encompasses chromatin condensation, cell polarization, formation of contractile actin ring and ultimate enucleation [2]. Our studies demonstrate that FOXO3 is essential for normal enucleation to occur. FOXO3 directly controls the expression of several critical regulators of enucleation, including Riok3 and Xpo7, important regulators of chromatin condensation [49,60,69], as well as Mxi1, implicated in contractile actin ring formation [60]. FOXO3 also potentially controls Trim58 expression that is a gene critical for erythroblast nuclear polarization and/or extrusion [63] (Figs 4 and S5B). Loss of FOXO3 severely compromised erythroblast polarization and resulted in multilobed nucleated erythroblasts in agreement with FOXO3 regulation of genes implicated in cell polarity and cytoskeleton organization (Figs 4–6, S5 and S6). The aberrant gene expression of the CDC42 network suggests that FOXO3 may mediate polarization or the cytoskeleton organization of enucleating erythroblasts, which are potentially important for the chromatin condensation and/or nuclear expulsion (Figs 4–6). These abnormalities are likely to be direct consequence of FOXO3 loss and not due to processes resulting from developmental lack of FOXO3, as reintroduction of FOXO3 normalized the defective enucleation within a short period of time in culture (Fig 7). Future studies will address the precise mechanism of FOXO3 function in this process. PI3-kinase, an upstream negative regulator of FOXO3 in erythroblasts [70], is required for erythroblast maturation [71,72] and polarization during enucleation in vitro [73]. However, signals other than PI3-kinase/AKT are likely to override the PI3-kinase repression of FOXO3 activity during erythroblast enucleation in vivo [18]. It is therefore probable that both PI3-kinase and FOXO3 control erythroblast terminal maturation and enucleation.
FOXO3 is an established regulator of autophagy [25,74–76]. Here we demonstrated that FOXO3 is critical for removal of mitochondria during terminal erythroid maturation. We found that FOXO3 regulates transcription of autophagy-related genes in maturing erythroblasts and FOXO3 loss compromised autophagosome formation and autophagy, reduced autophagic flux in maturing erythroblasts and impaired mitochondrial clearance in RBCs (Figs 2 and 3). Despite the profound reduction in autophagy gene expression, specifically at late stages of erythroblast maturation, and the negative impact on autophagosome formation, the influence of FOXO3 loss on mitochondria clearance was relatively modest (Fig 3E). Together these findings indicate that under homeostatic conditions, the relatively low autophagy gene expression is sufficient to maintain clearance of mitochondria in FOXO3 mutant erythroblasts. These results support the notion that multiple autophagy pathways might regulate mitochondrial clearance in erythroid cells [55–57].
In erythroid cells, FOXO3 amplifies the capacity of GATA–1 to induce autophagy gene expression [4,5]. Consistent with this, loss of FOXO3 led to notable decrease in the expression of many of autophagy-related genes in primary maturing erythroblasts (Fig 2) despite endogenous GATA–1 expression (S7A Fig). These results indicate that GATA–1 induction of autophagy gene expression in terminally maturing primary erythroblasts may be compromised in the absence of FOXO3.
Our unbiased genome-wide analysis revealed that, in addition to the approximately 600 genes that have been categorized as erythroid specific [6], more than 1000 genes, including genes involved in cell cycle, autophagy and immune-modulation are upregulated during terminal erythroid maturation. Loss of FOXO3 impaired significantly the expression of over 65% of these genes. Furthermore, FOXO3 loss limited to various degrees (clusters Q and R) the increase in expression of several GATA1, TAL1 and/or KLF–1 targets during terminal erythroid maturation. These results (Fig 1C) combined with the ChEA prediction (S7 Table), support a model in which FOXO3 is required to activate and/or enhance (cluster R), or sustain (cluster Q) gene expression during terminal erythroid maturation (Fig 8). As further support for this potential role of direct FOXO3 transcriptional activation of Cluster Q and R genes, we analyzed a list of direct FOXO3 targets determined by ChIP-seq [77] in a colon carcinoma cell-line. This comparison shows a significant number of overlap with cluster Q and R genes, suggesting that these genes are direct FOXO3 targets (S8A Fig, S8 Table). A sizeable number of genes do not overlap; this is most likely due to a difference in cell type and species. Consistent with previous findings [4–6,77–80] these results support a model (Fig 8) in which erythroid transcription factor complexes induce Foxo3 gene expression in immature erythroblasts which in turn cooperates with these factors to sustain and/or enhance the erythroid transcriptional program during terminal maturation. A similar model has been proposed for the induction of expression of interferon regulatory factor IRF2 and IRF6, which subsequently cooperate with GATA–1 and TAL1 transcription factors to establish the adult human erythroid program [81]. Collectively, these findings raise the possibility that this mode of regulation of gene expression might be a common mechanism during erythroid differentiation and maturation. Cooperation of these transcription factors with FOXO3 is likely to be critical for terminal erythroblast maturation. Nonetheless, in the absence of ChIP-Seq data, we are unable to determine with certainty all genes that are direct products of FOXO3 cooperation with other transcription factors in maturing erythroblasts.

Taken together our findings demonstrate that FOXO3 activity critically regulates progressive stages of terminal primary erythroblast maturation. As FOXO3 activity is critical for the correct temporal gene expression in maturing erythroblasts, we predict that abnormal FOXO3 expression/function may significantly influence erythroid disorders as has been reported specifically for hemoglobinopathies [8–10]. Since post-translational modifications of FOXO3 are the main determinants of FOXO3 output, analysis of FOXO3 function in addition to its expression will be critical in the context of disease. Future studies should elucidate whether modulations of FOXO3 activity influence the efficiency of RBC production in vitro or in disorders of erythroid cells in vivo.
Materials and Methods
Mice
The generation and genotyping of mice (on C57Bl6 genetic background) were performed as previously described [14,31]. WT and Foxo3-/- mouse littermates (8 to 12 weeks old) of Foxo3 heterozygous intercrosses were used in all experiments. Protocols were approved by the Institutional Animal Care and Use Committee of Mount Sinai School of Medicine.
Library preparation and mRNA sequencing
Total bone marrow cells from WT and Foxo3-/- mice (n = 3) were collected in IMDM supplemented with 15% FBS and each genotype combined. Cells from the two samples were pre-incubated with 10% rat serum, stained for TER119 and CD44 cell surface markers and three erythroid populations comprising proerythroblasts (Gate I), basophilic erythroblasts (Gate II) and polychromatic erythroblasts (Gate III) were FACS sorted as previously described [34]. Total RNA was isolated using the RNeasy Micro Kit (Qiagen) and mRNAseq libraries were prepared using the Illumina True Seq RNA prep kit following manufacturer’s instructions. Samples were sequenced in parallel lanes in a Hi Seq™ 2000 platform (Illumina) to obtain more than 107 single end 100bp reads per sample.
RNA-seq read processing
The RNA-seq reads were mapped to mouse genome build mm9 using Tophat v2.0.3 [82] with default parameters. The mapped reads were processed to calculate the FPKM (fragments per kilo-base per million reads) and identify differentially expressed genes using Cufflinks v1.3.0 [83]. The statistics for data processing is shown in (S9 Table).
Pathway analysis of differentially expressed genes at distinct maturation stages
For WT analysis, only genes that fulfilled both of the following criteria were selected: (1) to show a FPKM > 1 in at least one of the three populations (pro-, baso - or polychromatic erythroblasts) and (2) to have an amplitude equal or above two. Amplitude was calculated as the maximum difference in expression between all three gates (e.g. Gate I vs Gate II, Gate II vs Gate III or Gate I vs Gate III). The resulted 5514 genes were then normalized using Z-score and clustered (Cluster 3.0 software k-means). For the comparison between WT and Foxo3-/- samples we followed the same strategy as stated above. Only those genes that displayed a FPKM ≥ 1 in at least one of the six samples and an amplitude ≥ 2 were considered as differentially expressed between WT and Foxo3-/- samples. In this case, amplitude was calculated by computing gene expression differences between WT and Foxo3-/- at each particular gate (e.g. WT Gate I vs. Foxo3-/- Gate I) and only genes with a two-fold difference in at least one of the comparisons were selected. The selected 3904 genes were then normalized using Z-score and clustered as indicated above.
RNA Isolation and qRT–PCR
FACS-sorted cells were washed once in PBS and directly diluted in RLT buffer after centrifugation. Total RNA was isolated using the RNeasy Micro and Mini kit (Qiagen) same method used for RNA-seq. First-strand cDNA was synthesized using the SuperScript IITM Reverse Transcriptase (Invitrogen). Quantitative RT-PCR was performed using SYBR Premix Ex TaqTm II (Tli RNase H Plus) (Takara, #RR820A) with technical duplicates using an ABI Prism 7900 HT Cycler (Applied Biosystems). Gene-specific primers spanning intron-exon boundaries were designed using Primer-Blast (NCBI). Primers used in Fluidigm microfluidics technology experiments were designed by Fluidigm. The PCR cycle parameters were as follows: 95°C for 10’ followed by 40 cycles at 95°C for 15” and 60°C for 1’. Results were obtained with the Sequence Detection System 4.2 software (Applied Biosystems) and further analyzed by the 2-ΔΔCt method. β actin was used as a loading control. Results are shown as fold-change relative to wild type controls. Primer specific sequences are listed in S10 Table.
Flow cytometry
Bone marrow single cell suspensions were obtained and washed in IMDM + 15% FBS. Cells were pre-incubated with 10% rat serum and stained with anti-CD44-APC (#559250 BD) and anti-TER119-PE (#553673 BD) antibodies. For DRAQ5 (#DR50200 Biostatus) analysis cells were stained for CD44-PE (#553134 BD) antibody and TER119-Biotin (#553672 BD) followed by Streptavidin-PE-Cy7 (#557598 BD). After washing, cells were incubated with DRAQ5 DNA binding dye (1/1000 dilution in PBS + 2% FBS) at 37°C and washed. Samples were analyzed in a FacsCanto flow cytometer (BD). Data was analyzed by FlowJo software (Treestar).
Analysis of mitochondrial clearance
For MitoTracker Green or Red (#M7514, #M7512 Molecular Probes) analyses cells were pre-incubated with 10% rat serum, stained with anti-CD71-APC (#17–0711 eBiosciences) and incubated with mitotracker (20nM for 20 min at 37°C). Cells were then washed and analyzed by FACS.
Autophagy flux
Autophagy flux was measured by flow cytometry using the Cyto-ID autophagy detection kit (Enzo Life Sciences). Briefly, WT and Foxo3-/- bone marrow cells were preincubated with 10% rat serum and stained with TER119-PE and CD44-APC antibodies on ice. After washing, cells were resuspended at 106 cells/ml with 0.5 ml of IMDM supplemented with 15% FBS, 2 U/ml EPO and 50 ng/ml SCF in 24 well plates. Cells were cultured for 2 h at 37C° and chloroquine (50 μM) was added for 0, 10, 20, 40, 60, or 80 min. Cells were then kept on ice, washed twice in cold PBS + 5% FBS and resuspended in Cyto-ID solution consisting of 0.5 ml of PBS + 5% FBS + 0.25 μl of Cyto-ID. Cells were incubated for 30 min at room temperature in the dark, washed once with cold PBS + 5% FBS. Autophagosome content was determined by measuring the Cyto-ID (FITC) geometrical mean for each erythroid subpopulation. Autophagy flux was calculated by subtracting time 0 value from each of the other time points. All time points were analyzed for each bone marrow sample.
Imaging flow cytometry
Bone marrow cells were pre-incubated with 10% rat serum and stained with TER119-FITC, CD71-PE, GR-1-EF450, and DRAQ5 (all from ebioscience). After washing, cells were fixed with 4% paraformaldehyde for 20 min at room temperature. Cells were centrifuged (30 seconds at 2000g), washed with PBS and centrifuged again. Images were acquired with an ImageStreamX and analyzed using IDEAS 6.0 software (Amnis/EMDmilipore). Single, focused, TER119+, Gr1- cells were further analyzed for cells size (area TER119 morphology mask), nuclear size (area DRAQ5 morphology mask). Enucleating cells were enumerated from TER119+ small cell and nuclear size cells (orthochromatic cells - See Fig 3D) by increased asymmetry of nucleus (delta centroid X/Y of TER119 morphology mask and DRAQ5 morphology mask) and a boundary of low signal between the emerging pyrenocyte and reticulocyte parts of the enucleating cell (TER119 intensity in Valley mask). Lobular shaped nuclei were identified using the 3-fold symmetry feature on the DRAQ5 morphology mask.
Fluidigm—96.96 Dynamic Array IFC
For Fluidigm dynamic array performance, specific target amplification (STA) was performed according to the manufacturer´s protocol (PN 100–3488 B1). Briefly, cDNA was pre-amplified using the TaqMan PreAmp Master Mix (Applied Biosystems) for the 96 genes of interest. The amplification parameters were as follows: 95°C for 10’, followed by 12 cycles at 95°C for 15” and 60°C for 4’. After STA, Exonuclease I treatment was performed as recommended by the manufacturer. Briefly, Exonuclease I and Exonuclease I buffer (New England Biolabs) were added to the STA samples, and samples were then incubated for 30’ at 37°C, followed by the enzyme inactivation at 80°C for 15’. Finally, to load the dynamic array IFC, samples were prepared with the SsoFast EvaGreen Supermix with Low ROX (Bio-Rad) and 20x DNA Binding Dye Sample Loading Reagent (Fluidigm). Primers were diluted with Assay Loading Reagent (Fluidigm) and DNA Suspension Buffer (Teknova). After priming the 96x96 chip in the IFC Controller MX, samples and primers were loaded into their respective inlets. The chip was then loaded by the IFC Controller MX. The chip was run following the GE 96x96 PCR+Melt v2.pcl protocol in the Biomark using the Data Collection Software (Fluidigm). Results were obtained with the Fluidigm Real-Time PCR Analysis software (Fluidigm) and further analyzed by the 2-ΔΔCt method. β actin was used as a loading control. Results shown as fold-change relative to Gate I wild type controls. Primer specific sequences are listed in S10 Table.
Confocal fluorescence microscopy
Total bone marrow cells were fixed in 4% paraformaldehyde (PFA) overnight on ice, washed with PBS, permeabilized with 0.3% Triton–100 for 20 min, and blocked in 3–4% BSA, 1% goat serum in PBS overnight at 4°C. Cells were then stained in solution as described previously [65] with anti-TER119-Alexa488, Rhodamine - Phalloidin and Hoechst. After washing in PBS, cells were cytospun into slides, coverslipped and images acquired on a Zeiss LSM780 confocal fluorescence microscope with a 100X/1.4 N.A. objective using zoom 1 or 2. The entire cytospin area was systematically scanned to avoid bias in collection. Images were processed using Volocity 6.1.1 and Adobe Photoshop, and images constructed in Adobe Illustrator.
Western blot analysis
Equal WT and Foxo3-/- cell numbers were resuspended directly in Laemmli sample buffer with DTT. Samples were then boiled at 95°C for 10 min and kept at -80°C until used. Electrophoresis was performed on a 15% SDS-PAGE and transferred to a PVDF Immobilon-P membrane (Millipore). Membranes were blocked with 5% BSA in PBS 0.1% Tween–20 and incubated overnight at 4°C in 1% BSA in PBS + 0.1% Tween–20 with either anti-LC3B (#3868 Cell Signaling) or anti-actin (sc–1616 Santa Cruz) antibodies and then incubated with the appropriate secondary antibodies conjugated to HRP at 1/5000 for 1 h at room temperature. Membranes were washed and developed using the ECL reagents (Pierce) with Blue sensitive films (Crystalgen). Films were then scanned and measurements were made using the Multi-Gauge software from Fujifilm following the manufacturer’s instructions. For flux measurements, TER119+ cells were isolated and plated in IMDM supplemented with 15% FBS, 2 U/ml EPO and 50 ng/ml SCF and kept in culture for 2 h. Chloroquine (50 μM) was then added to the cultures for the indicated time. Cells were then collected, washed in PBS and resuspended directly in Laemmli sample buffer and analyzed by Western blot.
Chromatin Immunoprecipitation (ChIP)
ChIP was carried out in WT total bone marrow TER119+ cells as previously described [17]. Briefly, cells were cross-linked in 0.4% formaldehyde in PBS, and lysed (10 mM Tris-HCl pH8.0, 10 mM NaCl, 0.2% NP40). Lysate was sonicated for 30 cycles of 30 s on/30 s off at 4C° using a Bioruptor Standard sonication device (Diagenode). The cell lysate was then diluted in ChIP dilution buffer (20 mM Tris-HCl, pH 8.0, 2 mM EDTA 150 mM NaCl, 1% Triton, 0.01% SDS) and incubated at 4C° overnight with anti-FOXO3a antibody (Millipore #07–702) and Magna ChIPTM Protein A+G magnetic beads (Millipore #16–663). Beads were then washed (20 mM Tris-HCl, pH 8.0, 2 mM EDTA, 50 mM NaCl, 1% Triton, 0.1% SDS) and recovered. The antibody-protein-DNA complexes were reverse cross-linked for DNA isolation and quantitative PCR (qPCR) analysis. Foxo3-/- TER119+ cells were used as negative controls. Putative binding sites were located using MatInspector from Genomatix (http://www.genomatix.de/). Primer specific sequences are listed in S11 Table. See S12 Table for all antibodies.
Erythroblast expansion and differentiation culture
Bone marrow cultures were performed using a modified protocol of [68]. Briefly, WT and Foxo3-/- bone marrow cells were enriched for hematopoietic progenitors using the EasySep Mouse Hematopoietic Progenitor Cell Enrichment Kit (#19756 Stemcell Technologies) and expanded at < 2x106 cells/ml in non-treated 24-well plates with erythroid expansion medium. Erythroid expansion media consists of Stem Span SFEM (StemCell Technologies) supplemented with 2 U/ml human recombinant EPO (Amgen), 100 ng/ml SCF (PreproTech), 40 ng/ml insulin-like growth factor–1 (PreproTech), 10−6 M dexamethasone (D2915; Sigma), 0.4% cholesterol mix (Gibco) and 1% penicillin/streptomycin (Gibco). Two days later, cells were washed twice with PBS and plated at a concentration <1x106 cells/mL in erythroid differentiation medium consisting of IMDM supplemented with 2 U/ml EPO, 100 ng/ml SCF, 10% Serum replacement (Invitrogen), 5% Platelet-Derived Serum, glutamine, MTG (1.27 μl in 10 ml of 1 : 10 dilution) and 10% Protein-Free Hybridoma Media. Cells were incubated in maturation medium for 3 days, with additional media added at day 3.
Retroviral production and transduction
Retroviral supernatants of MIG and MIG-FOXO3 were produced as previously described [29,72]. Bone marrow expanding erythroid progenitors were resuspended in retroviral supernatants (multiplicity of infection of 10) on retronectin-coated dishes in expansion medium for 12 h. Suspension cells were then removed and only attached cells were collected by incubation with PBS and 5 mM EDTA at 37C° for 15 min. Cells were then washed, resuspended in expansion medium and incubated for another 24 h. Cells were then washed twice in PBS and cultured with erythroid differentiation medium as detailed above.
Supporting Information
Zdroje
1. Lapillonne H, Kobari L, Mazurier C, Tropel P, Giarratana MC, et al. (2010) Red blood cell generation from human induced pluripotent stem cells: perspectives for transfusion medicine. Haematologica 95 : 1651–1659. doi: 10.3324/haematol.2010.023556 20494935
2. Ji P, Murata-Hori M, Lodish HF (2011) Formation of mammalian erythrocytes: chromatin condensation and enucleation. Trends Cell Biol 21 : 409–415. doi: 10.1016/j.tcb.2011.04.003 21592797
3. Kerenyi MA, Orkin SH (2010) Networking erythropoiesis. J Exp Med 207 : 2537–2541. doi: 10.1084/jem.20102260 21098097
4. Kang YA, Sanalkumar R, O'Geen H, Linnemann AK, Chang CJ, et al. (2012) Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol 32 : 226–239. doi: 10.1128/MCB.06166-11 22025678
5. McIver SC, Kang YA, DeVilbiss AW, O'Driscoll CA, Ouellette JN, et al. (2014) The exosome complex establishes a barricade to erythroid maturation. Blood 124 : 2285–2297. doi: 10.1182/blood-2014-04-571083 25115889
6. Li L, Freudenberg J, Cui K, Dale R, Song SH, et al. (2013) Ldb1-nucleated transcription complexes function as primary mediators of global erythroid gene activation. Blood 121 : 4575–4585. doi: 10.1182/blood-2013-01-479451 23610375
7. Eijkelenboom A, Burgering BM (2013) FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 14 : 83–97. doi: 10.1038/nrm3507 23325358
8. Pourfarzad F, von Lindern M, Azarkeivan A, Hou J, Kia SK, et al. (2013) Hydroxyurea responsiveness in beta-thalassemic patients is determined by the stress response adaptation of erythroid progenitors and their differentiation propensity. Haematologica 98 : 696–704. doi: 10.3324/haematol.2012.074492 23100274
9. Franco SS, De Falco L, Ghaffari S, Brugnara C, Sinclair DA, et al. (2014) Resveratrol accelerates erythroid maturation by activation of FoxO3 and ameliorates anemia in beta-thalassemic mice. Haematologica 99 : 267–275. doi: 10.3324/haematol.2013.090076 23975182
10. Sheehan VA, Crosby JR, Sabo A, Howard TA, Muzny DM, et al. (2013) FOXO3 Variants Are Associated With Lower Fetal Hemoglobin Levels In Children With Sickle Cell Disease Blooda 122.
11. Salih DA, Brunet A (2008) FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 20 : 126–136. doi: 10.1016/j.ceb.2008.02.005 18394876
12. Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, et al. (2007) Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1 : 101–112. doi: 10.1016/j.stem.2007.02.001 18371339
13. Tothova Z, Gilliland DG (2007) FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell 1 : 140–152. doi: 10.1016/j.stem.2007.07.017 18371346
14. Yalcin S, Zhang X, Luciano JP, Mungamuri SK, Marinkovic D, et al. (2008) Foxo3 Is Essential for the Regulation of Ataxia Telangiectasia Mutated and Oxidative Stress-mediated Homeostasis of Hematopoietic Stem Cells. J Biol Chem 283 : 25692–25705. doi: 10.1074/jbc.M800517200 18424439
15. Paik JH, Ding Z, Narurkar R, Ramkissoon S, Muller F, et al. (2009) FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 5 : 540–553. doi: 10.1016/j.stem.2009.09.013 19896444
16. Renault VM, Rafalski VA, Morgan AA, Salih DA, Brett JO, et al. (2009) FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 5 : 527–539. doi: 10.1016/j.stem.2009.09.014 19896443
17. Zhang X, Yalcin S, Lee DF, Yeh TY, Lee SM, et al. (2011) FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat Cell Biol 13 : 1092–1099. doi: 10.1038/ncb2293 21804543
18. Rimmele P, Bigarella CL, Liang R, Izac B, Dieguez-Gonzalez R, et al. (2014) Aging-like Phenotype and Defective Lineage Specification in SIRT1-Deleted Hematopoietic Stem and Progenitor Cells. Stem Cell Reports 3 : 44–59. doi: 10.1016/j.stemcr.2014.04.015 25068121
19. Giannakou ME, Goss M, Junger MA, Hafen E, Leevers SJ, et al. (2004) Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science 305 : 361. 15192154
20. Partridge L, Bruning JC (2008) Forkhead transcription factors and ageing. Oncogene 27 : 2351–2363. doi: 10.1038/onc.2008.28 18391977
21. Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA (2003) Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science 301 : 215–218. 12855809
22. Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, et al. (2011) AKT/FOXO Signaling Enforces Reversible Differentiation Blockade in Myeloid Leukemias. Cell 146 : 697–708. doi: 10.1016/j.cell.2011.07.032 21884932
23. Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, et al. (2010) TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 463 : 676–680. doi: 10.1038/nature08734 20130650
24. Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, et al. (2008) FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A 105 : 13987–13992. doi: 10.1073/pnas.0801030105 18765803
25. Demontis F, Perrimon N (2010) FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 143 : 813–825. doi: 10.1016/j.cell.2010.10.007 21111239
26. Zhang P, Judy M, Lee SJ, Kenyon C (2013) Direct and indirect gene regulation by a life-extending FOXO protein in C. elegans: roles for GATA factors and lipid gene regulators. Cell Metab 17 : 85–100. doi: 10.1016/j.cmet.2012.12.013 23312285
27. Lee JC, Espeli M, Anderson CA, Linterman MA, Pocock JM, et al. (2013) Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell 155 : 57–69. doi: 10.1016/j.cell.2013.08.034 24035192
28. Bakker WJ, Blazquez-Domingo M, Kolbus A, Besooyen J, Steinlein P, et al. (2004) FoxO3a regulates erythroid differentiation and induces BTG1, an activator of protein arginine methyl transferase 1. J Cell Biol 164 : 175–184. 14734530
29. Marinkovic D, Zhang X, Yalcin S, Luciano JP, Brugnara C, et al. (2007) Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Invest 117 : 2133–2144. 17671650
30. Yu D, dos Santos CO, Zhao G, Jiang J, Amigo JD, et al. (2010) miR–451 protects against erythroid oxidant stress by repressing 14-3-3zeta. Genes Dev 24 : 1620–1633. doi: 10.1101/gad.1942110 20679398
31. Zhang X, Camprecios G, Rimmele P, Liang R, Yalcin S, et al. (2014) FOXO3-mTOR metabolic cooperation in the regulation of erythroid cell maturation and homeostasis. Am J Hematol 89 : 954–963. doi: 10.1002/ajh.23786 24966026
32. Wong P, Hattangadi SM, Cheng AW, Frampton GM, Young RA, et al. (2011) Gene induction and repression during terminal erythropoiesis are mediated by distinct epigenetic changes. Blood 118: e128–138. doi: 10.1182/blood-2011-03-341404 21860024
33. An X, Schulz VP, Li J, Wu K, Liu J, et al. (2014) Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 123 : 3466–3477. doi: 10.1182/blood-2014-01-548305 24637361
34. Chen K, Liu J, Heck S, Chasis JA, An X, et al. (2009) Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci U S A 106 : 17413–17418. doi: 10.1073/pnas.0909296106 19805084
35. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, et al. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14 : 128. doi: 10.1186/1471-2105-14-128 23586463
36. Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, et al. (2010) ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26 : 2438–2444. doi: 10.1093/bioinformatics/btq466 20709693
37. Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, et al. (2009) Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood 114 : 157–164. doi: 10.1182/blood-2008-04-151639 19417210
38. Peslak SA, Wenger J, Bemis JC, Kingsley PD, Koniski AD, et al. (2012) EPO-mediated expansion of late-stage erythroid progenitors in the bone marrow initiates recovery from sublethal radiation stress. Blood 120 : 2501–2511. 22889760
39. Pereira R, Quang CT, Lesault I, Dolznig H, Beug H, et al. (1999) FLI–1 inhibits differentiation and induces proliferation of primary erythroblasts. Oncogene 18 : 1597–1608. 10102630
40. Rekhtman N, Radparvar F, Evans T, Skoultchi AI (1999) Direct interaction of hematopoietic transcription factors PU.1 and GATA–1: functional antagonism in erythroid cells. Genes Dev 13 : 1398–1411. 10364157
41. Stadhouders R, Thongjuea S, Andrieu-Soler C, Palstra RJ, Bryne JC, et al. (2012) Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. EMBO J 31 : 986–999. doi: 10.1038/emboj.2011.450 22157820
42. Morera D, Roher N, Ribas L, Balasch JC, Donate C, et al. (2011) RNA-Seq reveals an integrated immune response in nucleated erythrocytes. PLoS One 6: e26998. doi: 10.1371/journal.pone.0026998 22046430
43. Greenfest-Allen E, Malik J, Palis J, Stoeckert CJ Jr. (2013) Stat and interferon genes identified by network analysis differentially regulate primitive and definitive erythropoiesis. BMC Syst Biol 7 : 38. doi: 10.1186/1752-0509-7-38 23675896
44. Litvak V, Ratushny AV, Lampano AE, Schmitz F, Huang AC, et al. (2012) A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature 490 : 421–425. doi: 10.1038/nature11428 22982991
45. Yang Z, Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22 : 124–131. doi: 10.1016/j.ceb.2009.11.014 20034776
46. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, et al. (2007) NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A 104 : 19500–19505. 18048346
47. Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, et al. (2008) Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112 : 1493–1502. doi: 10.1182/blood-2008-02-137398 18539900
48. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, et al. (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454 : 232–235. doi: 10.1038/nature07006 18454133
49. Bakker WJ, van Dijk TB, Parren-van Amelsvoort M, Kolbus A, Yamamoto K, et al. (2007) Differential regulation of Foxo3a target genes in erythropoiesis. Mol Cell Biol 27 : 3839–3854. 17353275
50. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, et al. (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6 : 458–471. 18054315
51. Xiong X, Tao R, DePinho RA, Dong XC (2012) The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem 287 : 39107–39114. doi: 10.1074/jbc.M112.412569 22992773
52. Klionsky DJ (2012) A human autophagy interaction network. Autophagy 8 : 439–441. doi: 10.4161/auto.19926 22781101
53. Mizushima N, Yoshimori T (2007) How to interpret LC3 immunoblotting. Autophagy 3 : 542–545. 17611390
54. Oeste CL, Seco E, Patton WF, Boya P, Perez-Sala D (2013) Interactions between autophagic and endo-lysosomal markers in endothelial cells. Histochem Cell Biol 139 : 659–670. doi: 10.1007/s00418-012-1057-6 23203316
55. Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, et al. (2014) Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun 5 : 4004. doi: 10.1038/ncomms5004 24895007
56. Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, et al. (2009) Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461 : 654–658. doi: 10.1038/nature08455 19794493
57. Mortensen M, Ferguson DJ, Simon AK (2010) Mitochondrial clearance by autophagy in developing erythrocytes: clearly important, but just how much so? Cell Cycle 9 : 1901–1906. 20495377
58. Konstantinidis DG, Pushkaran S, Johnson JF, Cancelas JA, Manganaris S, et al. (2012) Signaling and cytoskeletal requirements in erythroblast enucleation. Blood 119 : 6118–6127. doi: 10.1182/blood-2011-09-379263 22461493
59. Keerthivasan G, Wickrema A, Crispino JD (2011) Erythroblast enucleation. Stem Cells Int 2011 : 139851. doi: 10.4061/2011/139851 22007239
60. Zhang L, Flygare J, Wong P, Lim B, Lodish HF (2011) miR–191 regulates mouse erythroblast enucleation by down-regulating Riok3 and Mxi1. Genes Dev 25 : 119–124. doi: 10.1101/gad.1998711 21196494
61. Ji P, Jayapal SR, Lodish HF (2008) Enucleation of cultured mouse fetal erythroblasts requires Rac GTPases and mDia2. Nat Cell Biol 10 : 314–321. doi: 10.1038/ncb1693 18264091
62. Ji P, Lodish HF (2010) Rac GTPases play multiple roles in erythropoiesis. Haematologica 95 : 2–4. doi: 10.3324/haematol.2009.015511 20065075
63. Thom CS, Traxler EA, Khandros E, Nickas JM, Zhou OY, et al. (2014) Trim58 Degrades Dynein and Regulates Terminal Erythropoiesis. Dev Cell.
64. McGrath KE, Kingsley PD, Koniski AD, Porter RL, Bushnell TP, et al. (2008) Enucleation of primitive erythroid cells generates a transient population of "pyrenocytes" in the mammalian fetus. Blood 111 : 2409–2417. 18032705
65. Sui Z, Nowak RB, Bacconi A, Kim NE, Liu H, et al. (2014) Tropomodulin3-null mice are embryonic lethal with anemia due to impaired erythroid terminal differentiation in the fetal liver. Blood 123 : 758–767. doi: 10.1182/blood-2013-03-492710 24159174
66. Goehring NW, Grill SW (2013) Cell polarity: mechanochemical patterning. Trends Cell Biol 23 : 72–80. doi: 10.1016/j.tcb.2012.10.009 23182746
67. Wang L, Yang L, Filippi MD, Williams DA, Zheng Y (2006) Genetic deletion of Cdc42GAP reveals a role of Cdc42 in erythropoiesis and hematopoietic stem/progenitor cell survival, adhesion, and engraftment. Blood 107 : 98–105. 16174757
68. England SJ, McGrath KE, Frame JM, Palis J (2011) Immature erythroblasts with extensive ex vivo self-renewal capacity emerge from the early mammalian fetus. Blood 117 : 2708–2717. doi: 10.1182/blood-2010-07-299743 21127173
69. Hattangadi SM, Martinez-Morilla S, Patterson HC, Shi J, Burke K, et al. (2014) Histones to the cytosol: Exportin 7 is essential for normal terminal erythroid nuclear maturation. Blood.
70. Ghaffari S, Jagani Z, Kitidis C, Lodish HF, Khosravi-Far R (2003) Cytokines and BCR-ABL mediate suppression of TRAIL-induced apoptosis through inhibition of forkhead FOXO3a transcription factor. Proc Natl Acad Sci U S A 100 : 6523–6528. 12750477
71. Ghaffari S, Kitidis C, Zhao W, Marinkovic D, Fleming MD, et al. (2006) AKT induces erythroid-cell maturation of JAK2-deficient fetal liver progenitor cells and is required for Epo regulation of erythroid-cell differentiation. Blood 107 : 1888–1891. 16254141
72. Zhao W, Kitidis C, Fleming MD, Lodish HF, Ghaffari S (2006) Erythropoietin stimulates phosphorylation and activation of GATA–1 via the PI3-kinase/AKT signaling pathway. Blood 107 : 907–915. 16204311
73. Wang J, Ramirez T, Ji P, Jayapal SR, Lodish HF, et al. (2012) Mammalian erythroblast enucleation requires PI3K-dependent cell polarization. J Cell Sci 125 : 340–349. doi: 10.1242/jcs.088286 22331356
74. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, et al. (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin–1 and cause skeletal muscle atrophy. Cell 117 : 399–412. 15109499
75. Zhao J, Brault JJ, Schild A, Cao P, Sandri M, et al. (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6 : 472–483. 18054316
76. Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, et al. (2013) FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature.
77. Eijkelenboom A, Mokry M, de Wit E, Smits LM, Polderman PE, et al. (2013) Genome-wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Mol Syst Biol 9 : 638. doi: 10.1038/msb.2012.74 23340844
78. Eijkelenboom A, Mokry M, Smits LM, Nieuwenhuis EE, Burgering BM (2013) FOXO3 selectively amplifies enhancer activity to establish target gene regulation. Cell Rep 5 : 1664–1678. doi: 10.1016/j.celrep.2013.11.031 24360957
79. Li L, Jothi R, Cui K, Lee JY, Cohen T, et al. (2011) Nuclear adaptor Ldb1 regulates a transcriptional program essential for the maintenance of hematopoietic stem cells. Nat Immunol 12 : 129–136. doi: 10.1038/ni.1978 21186366
80. Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, et al. (2011) Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144 : 296–309. doi: 10.1016/j.cell.2011.01.004 21241896
81. Xu J, Shao Z, Glass K, Bauer DE, Pinello L, et al. (2012) Combinatorial assembly of developmental stage-specific enhancers controls gene expression programs during human erythropoiesis. Dev Cell 23 : 796–811. doi: 10.1016/j.devcel.2012.09.003 23041383
82. Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25 : 1105–1111. doi: 10.1093/bioinformatics/btp120 19289445
83. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7 : 562–578. doi: 10.1038/nprot.2012.016 22383036
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Single Strand Annealing Plays a Major Role in RecA-Independent Recombination between Repeated Sequences in the Radioresistant Bacterium
- The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals
- Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross
- DCA1 Acts as a Transcriptional Co-activator of DST and Contributes to Drought and Salt Tolerance in Rice
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy