
The Genomic Aftermath of Hybridization in the Opportunistic Pathogen
Human pathogens belong to different phylogenetic clades and it is clear that the ability to infect humans emerged several times independently. The sequencing and comparison of genomes from pathogenic and non-pathogenic species and strains paves the way to identify what genomic changes underlie the emergence of virulence. In this study we sequenced 11 globally-distributed clinical isolates of Candida metapsilosis, an emerging fungal pathogen of growing concern. We found that all isolates were the result of a single hybridization between two unidentified species, which points to hybridization as a mechanism for the origin of a virulent lineage. We found that the hybrids likely originated by sexual reproduction as they were diploids and retained genomic regions of opposite mating types. We reconstructed the aftermath of the genome merging by identifying where recombination led to the removal of one of the parental subgenomes. We finally compare the newly-sequenced genome with those of other pathogens from the Candida clade and establish global trends, such an enriched repertoire in cell-wall proteins in more virulent species. Our results provide insight into how hybridization may play a key role in the emergence of novel pathogenic lineages.
Published in the journal:
. PLoS Genet 11(10): e32767. doi:10.1371/journal.pgen.1005626
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005626
Summary
Human pathogens belong to different phylogenetic clades and it is clear that the ability to infect humans emerged several times independently. The sequencing and comparison of genomes from pathogenic and non-pathogenic species and strains paves the way to identify what genomic changes underlie the emergence of virulence. In this study we sequenced 11 globally-distributed clinical isolates of Candida metapsilosis, an emerging fungal pathogen of growing concern. We found that all isolates were the result of a single hybridization between two unidentified species, which points to hybridization as a mechanism for the origin of a virulent lineage. We found that the hybrids likely originated by sexual reproduction as they were diploids and retained genomic regions of opposite mating types. We reconstructed the aftermath of the genome merging by identifying where recombination led to the removal of one of the parental subgenomes. We finally compare the newly-sequenced genome with those of other pathogens from the Candida clade and establish global trends, such an enriched repertoire in cell-wall proteins in more virulent species. Our results provide insight into how hybridization may play a key role in the emergence of novel pathogenic lineages.
Introduction
Hybridization between species is an important evolutionary mechanism that can drive the origin of novel lineages and adaptation to new environments. Hybridization results in the combination of two diverged genomes, which are subsequently shaped by processes of recombination, deletion, and other genomic rearrangements. Genomics have recently paved the way to investigate the stochastic and adaptive processes that follow genomic hybridization. As compared to metazoans or plants, fungi have lower prezygotic barriers and can reproduce clonally for long periods of time, thus hybridization is thought to have a large impact in the evolution of this clade. Consistently, the presence of hybrids in fungi have been extensively documented and an increasing number of cases are being described in the literature [1–8]. Hybridization has been proposed as a mechanism to drive the origin of new human fungal pathogens [8]. Indeed several hybrid species have been described among several human fungal pathogens [8–10], although in all these cases also non-hybrid strains of the parental species can infect humans. So far, only few studies have focused on population genomics of hybrid species. Here we report a population genomics analysis of an opportunistic pathogen that belongs to the Candida parapsilosis species complex.
This species complex comprises opportunistic pathogen species that cause serious infections in immunocompromised patients, and whose incidence has significantly increased in recent years [11]. Three distinct clades within this complex, formerly known as C. parapsilosis groups I, II and III, have been re-defined as different species: C. parapsilosis sensu stricto, C. orthopsilosis and C. metapsilosis, respectively [12]. These species differ in their degrees of prevalence and virulence, C. metapsilosis being the one with the lowest clinical prevalence, and accounting for only 1.1 to 8.4% of the infections caused by the complex [13]. In addition, the species differ in their degree of prevalence across different types of patients [14–15]. There are few studies investigating the virulence of C. parapsilosis sensu lato species, and particularly little is known about the rarer species C. orthopsilosis and C. metapsilosis. Nonetheless, results of several in vitro and in vivo studies suggest that, consistent with its lower prevalence, C. metapsilosis is the least virulent species of the complex [16–20]. A recent study involving 93 different C. parapsilosis sensu lato isolates, revealed that C. metapsilosis strains were unable to produce extracellular lipases and form pseudohyphae, attributes that are both recognized as important virulence factors for Candida spp. [18]. Furthermore, it has been shown that C. metapsilosis isolates are killed more efficiently by and are less cytotoxic to human primary macrophages [17,18]. Finally, other studies have focused on the antifungal susceptibility of different species within the complex showing that strain-to-strain differences within species are common [13,21,22].
The growing incidence of infections due to C. parapsilosis sensu lato spp. underlines the importance of studies investigating the virulence attributes and molecular genetics of these species. Furthermore, the C. parapsilosis species complex offers an exquisite system for the study of the evolution of pathogenic yeasts and their adaptation to the human host. The complex belongs to the broader CTG clade of Saccharomycetales, which include species that decode the CUG codon as serine instead of leucine. Although the CTG clade includes other clinically important species such as C. albicans and C. tropicalis [23], their phylogenetic position (see below) and particular virulence properties indicates that the species within the Candida parapsilosis species complex evolved pathogenesis towards humans independently of C. albicans and their closest relatives. The sequencing of reference strains for C. parapsilosis [24] and C. orthopsilosis [25] has been instrumental in assessing the main differences with other pathogens within the CTG clade, particularly with the model yeast pathogen C. albicans. Such analyses have revealed that, whereas most Candida species display a similar content of families represented by singleton genes, most of the variability is related to copy number differences in multi-gene families, with pathogens having increased number of members in families related to virulence [24]. For instance, initial comparisons found that C. parapsilosis has an expanded Hyr/Iff family of virulence-related cell wall genes relative to the less virulent species C. orthopsilosis [25]. Subsequent analyses have assessed the genomic diversity among C. parapsilosis [26] and C. orthopsilosis [8] isolates. These studies have enabled important discoveries, such as the realization of the existence of recombination between clinical and environmental lineages in C. parapsilosis, pointing to several recent and recurrent clinical outbreaks from the environment [26], or the discovery of hybrids between differentiated C. orthopsilosis subspecies, including the description of one virulent hybrid lineage isolated from two very distant locations [8]. In contrast to the growing awareness on the genomic diversity of other C. parapsilosis sensu lato species, for C. metapsilosis we lack both a reference genome and a comprehensive insight on the genomic diversity across isolates. This situation precludes understanding the emergence of virulence traits within this complex. To fill in this important gap we undertook the sequencing and analysis of the genomes of eleven C. metapsilosis clinical isolates. Unexpectedly, we found that all sequenced isolates, sampled from geographically distant locations, presented highly heterozygous genomes, which we show to result from a single hybridization event between two parental lineages differing by 4.5% at the nucleotide level.
Results and Discussion
Sequencing and assembly reveal a hybrid genome and a newly circularized mitochondrial genome
We used Illumina technology to sequence a panel of eleven C. metapsilosis clinical isolates from different geographical locations (Table 1). Initial attempts to assemble the individual strains using standard approaches proved unsuccessful, independently of sequencing coverage or the combined use of libraries of different read lengths and insert sizes (S1 Table). In particular, for the strain PL429 four different libraries were used at varying read lengths and insert sizes, totaling an overall coverage of 1,308x (Table 1), yet this still yielded a highly fragmented assembly with thousands of contigs. This elusive assembly for a relatively small genome was reminiscent of what we had previously observed in a highly heterozygous strain of C. orthopsilosis [8]. Indeed assemblies from highly heterozygous genomes are highly fragmented and result in a total genome size larger than expected [8,27]. This is because two alternative contigs are recovered for each heterozygous region, while a single, collapsed contig is recovered from each homozygous region. Such assemblies are difficult to scaffold further, as each homozygous contig could be joined, at each side, to either of the two heterozygous contigs. To generate a suitable reference assembled genome for C. metapsilosis, we used a previously developed heterozygous genome assembly strategy [8], which we describe here in greater detail (see Materials and Methods). To obtain an optimal assembly we had to apply this procedure to the combined data from two strains: the one with the highest sequencing coverage and the larger number of libraries, PL429 (SZMC1548), and the one producing the least fragmented de novo assembly, SZMC8094 (Table 1, S1 and S2 Tables). Thus the resulting reference assembly is chimeric in nature and comprises sequences from the two isolates. In addition, similar to previous highly heterozygous assemblies [8], only one of the haplotypes for each heterozygous region is represented in the final assembly. We then mapped heterozygous regions in each strain relative to this reference assembly.

The final assembly resulted in a total size of 13.3 Mb in seven putative chromosomes and two unplaced scaffolds, which is similar to the number of bands observed in a Pulsed Field Gel Electrophoresis (PFGE) (S1 Fig). The observed band patterning was also indicative of possible genomic rearrangements in several of the analyzed strains, including the two strains used in the reference assembly. Note that PFGE and short range pair-end reads provide information at different scales and thus integration of both sources of information is difficult. Predicted chromosome ends were enriched in C. metapsilosis telomere repeats (GGTTAGGATGTCCAAAGTATTGA), corresponding to the template domain of the telomerase (TER1) [28] in a region (827,407–829,461) of scaffold 2. The annotation of the genome (see Materials and Methods) resulted in 5,973 protein-coding genes in C. metapsilosis, which is roughly similar to the gene counts in C. parapsilosis (5,752) and C. orthopsilosis (5,784).
The mitochondrial genome was assembled in a single 21 kb-long contig [29]. Species from the Candida parapsilosis complex generally display linear mitochondrial chromosomes, but rare isolates presenting circularized mitochondrial DNA (mtDNA) have been identified in C. orthopsilosis and C. metapsilosis [23,29]. Our panel includes five strains whose mitochondrial architecture has been determined earlier (BP57, CP61, CP367, MCO448, PL448 and PL429), with PL448 being the only known case of a C. metapsilosis strain bearing a circular mitochondrial chromosome [30]. We here determined the architecture of the mitochondrial chromosomes of six additional strains from the genomic data (See Materials and Methods). Notably, we found a new case of circular mitochondrial chromosome in the strain SZMC21154, whereas the remaining mtDNAs of the newly-tested strains were predicted to be linear (S2A Fig). All these in-silico predictions were confirmed experimentally through PCR and PFGE tests (see Materials and Methods, Fig 1). As described in [31] the strain PL448 is a direct derivative of the clinical strain MCO448, and thus the two circularisation events must be necessarily independent evolutionary events. This is consistent with our phylogenetic analysis of the nuclear genomes of the strains (see below) and with the fact that, although the circularization results from end-to-end fusion events in both strains, it involves different specific sites in each case (S2B Fig). There is extremely low sequence variability of mtDNA among C. metapsilosis isolates. Hence, the mtDNA-based phylogeny cannot resolve this issue.

All sequenced C. metapsilosis clinical isolates result from a single hybrization event
We next mapped the reads obtained from the sequencing of each of the strains onto the above reference genome, which served to assess sequence variation (Fig 2 and S1 File). All sequenced strains were found to be highly heterozygous (22–26 heterozygous SNPs/kb, Table 1), with divergence between the alleles in heterozygous regions averaging approximately 4.5% (see Materials and Methods). This high divergence made it possible to delimit heterozygous and homozygous regions, and to calculate the fraction of the genome that was occupied by heterozygous blocks.

We will refer to blocks of homozygosity as loss of heterozygosity blocks (LOH, a genome track where heterozygosity has been lost). Such LOH blocks could be the result from several basic mechanisms, including mitotic recombination, break-induced replication or gene conversion [32]. Alternatively an homozygous track of a certain length can occur simply by chance as SNPs in heterozygous regions are not distributed uniformly, but the likelihood of this diminishes quickly with the size of the block. The definition of a threshold is challenging and any arbitrary value will produce false positives or false negatives at different rates. We explored this issue and opted for using a more relaxed (100 bp) and more stringent (200 bp) thresholds for the minimum gap between SNPs in a region designated as an LOH block (see Materials and Methods). Unless indicated otherwise the results at 100 bp threshold are indicated. The fraction of the genome occupied by heterozygous regions varied significantly across strains, ranging from 54.5% to 61.3% of the genome by length when the 100 bp threshold was applied (Table 1), and 63.4–68.5% at the 200 bp threshold. Overall 50% of the homozygous tracks in the genome were in LOH blocks larger than 3,626 bp (LOH-50). The fraction of heterozygous regions is much higher than the 17% found in a C. orthopsilosis hybrid (where a 100 bp threshold was used) [8]. This difference may indicate that the C. metapsilosis hybridization is a more recent event or, alternatively, that C. orthopsilosis lost heterozygosity more rapidly.
LOH tracts were generally short, with an average size of 535 bp (1,183 bp at 200 bp threshold) as compared to 2,151 bp in C. orthopsilosis (S3 Fig). This difference may be related to the lower level of heterozygosity in the latter, as multiple, partially overlapping or adjacent LOH events will be seen as a single longer homozygous blocks. Indeed in C. orthopsilosis 20% of the LOH blocks were longer than 1 kb, whereas in C. metapsilosis this fraction was only 7.2%. Interestingly, longer LOH blocks were enriched in sub-telomeric regions (Welch's t-test P<3.5e-06, S4 Fig). Importantly, all C. metapsilosis strains shared a significant fraction (~ 43%) of LOH blocks, with 1,581 LOH blocks having identical boundaries across all sequenced strains (out of an average of 10,920 LOH blocks, Table 1). Even at a stricter threshold of 200 bp, 125 blocks had identical boundaries in all 11 strains. Of these blocks, 29 were larger than 500bp, including 6 longer than 1kb. This indicates that all sequenced strains derive from the same primary hybridization event and shared a number of LOH events before they diverged. The alternative scenario that those regions with exact boundaries were identical in the parental strains differing on average 4.5% is very unlikely. Subsequently other LOH events were shared by only a fraction of the strains, with 19% of LOH blocks being strain-specific in a typical strain.
Contrary to the C. orthopsilosis case, where a completely homozygous strain (i.e. one of the putative hybrid parentals) exists [8], none of the sequenced C. metapsilosis strains is homozygous. Furthermore, both haplotypes in a given heterozygous region are roughly equally distant to either C. orthopsilosis or C. parapsilosis out-groups (0.35% and 0.51% difference between the two haplotypes, recpectively). This prevents assignment of each haplotype in a heterozygous region to a particular parent, and prevents phasing the homozygous regions (i.e. in Fig 2 we cannot say whether the grey homozygous regions come from one or the other parent). In the C. orthopsilosis hybrid both parents were found to be similarly represented among homozygous regions [8]. In the absence of an unequivocal parental mapping for C. metapsilosis, we approached the question of parental lineage representation using several indirect strategies. Firstly, our assembly process randomly incorporates one of the two haplotypes in a given heterozygous region. Thus, even if we cannot distinguish among parentals, we can arbitrarily name haplotype A the one included in the reference assembly, and assess for the remaining nine strains how often this or the alternative haplotype (haplotype B) is present in homozygous regions. We applied this procedure to scaffold6 from PL448, which underwent LOH of the entire chromosome (S1 File). This provided an overall estimate of 54.8% and 45.2% representation of the haplotypes A and B, respectively. We stress that these haplotypes do not involve any assignment to a given parental genome. Nevertheless, considering the random incorporation of parental haplotypes in the assembly, this result suggests an approximately balanced presence of both parentals in these homogenized regions. For the rest of the genome, haplotype B is highly underrepresented among LOHs, but this is the result of most LOH blocks being shared among most strains (S1 File).
Finally, to study the pattern of LOH on a local scale, we examined one specific region in four strains by PCR and re-sequencing. This 3.6 kb-long region around the gene g3863.t1 (coding for a protein with a DEAD-box RNA helicase domain), is interesting because it encompasses eight recent LOH blocks that are present specifically in 1 to 3 of the four strains. We can consider these eight LOH blocks to have originated independently and we can assess whether the same or a distinct parental haplotype was introgressed in each of these by selectively amplifying and sequencing each of the DNA molecules. Notably, in all cases LOHs present in a given strain resulted from haplotype A overwriting haplotype B (S5, S6 and S7 Figs). The probability that this occurred by chance assuming equal probability for both haplotypes is 0.0039, which suggests this local region has some preference to lose one of the haplotypes. Altogether these results suggest that, despite a bias towards preferential retention of one parental at the local level in some regions, there is no genome-wide preference for either of the two parental strains. This result is in line with that obtained for a C. orthopsilosis hybrid with similar divergence between parents [8]. This supports the idea that hybrids from less divergent parentals display more balanced inheritance due to a lower risk of genetic incompatibility (Bateson-Dobzhansky-Muller effect). Larger divergence such as the 10% found in Pichia sorbitophila has led to more unbalanced inheritance in that species [33].
Genome analyses suggest mating as the hybrid formation mechanism
The distribution of read counts at biallelic single nucleotide polymorphisms (SNPs) suggested a diploid state for the sequenced C. metapsilosis strains (S8 Fig). In addition, this analysis revealed partial aneuploidies in two strains, namely triploidy of scaffold5 in PL448 and partial triploidy of scaffold2 in PL448 and SZMC21154 (S8 Fig). These aneuploidies were independently confirmed by depth-of-coverage analyses (S3 Table). FACS (Fluorescence-Activated Cell Sorting) analyses were consistent with the predicted diploidy of 11 C. metapsilosis hybrid strains and C. orthopsilosis MCO456, although comparison of FACS results across species must be interpreted with caution (Materials and Methods, S9 Fig). Of note PL448 is the strain with more ploidy changes and the one that recently circularized its mitochondrial chromosome (see above). In addition some results suggested some level of heterogeneity in the samples of this strain (e.g. peak of biallelic counts closer to 40% rather than 50% in diploid chromosomes), which may be interpreted as an ongoing genomic instability.
The diploid state of the hybrids suggests mating between two haploid cells with genomes ~4.5% divergent as the probable mechanism of hybridization. It has been observed that Candida species respond to mating pheromones of different species within the clade [34]. Based on this, mating was suggested as the mechanism of formation of the previously reported C. orthopsilosis diploid hybrid [8]. However, the presence of a single mating type (MTL) idiomorph in the two sequenced C. orthopsilosis strains prevented a definitive conclusion. In the current study, however, we found that 10 out of the 11 sequenced C. metapsilosis genomes contain both MTLa and MTLa idiomorphs (Fig 3), albeit with MTLa incomplete (see below). This result is consistent with mating as the mechanism for hybrid formation and lends further support to the hypothesis that haploid forms and mating occurs in species of the C. parapsilosis clade, a clade which is generally considered asexual [24]. The parasexual cycle, as it occurs in C. albicans can also involve strains of opposite mating type, however in that case diploid state is achieved by concerted loss of chromosomes with little recombination between them [25] and thus most chromosomes would be homozygous for one or the other parental lineage. Of note, the proposal of mating as the origin of hybridization does not imply that the hybrid lineage is able to undergo a sexual cycle, and thus we cannot confidently assign the source of LOH to meiotic or mitotic recombination.

Remarkably, the genome sequences indicate that the MTLa idiomorph has partially overwritten the nonhomologous MTLa idiomorph in C. metapsilosis. Our reference genome assembly contained an MTLalpha idiomorph at the MTL locus (Fig 3A) on scaffold 5, with the MTLa genes assembled separately as a small contig (scaffold 28, Fig 3B). In species such as C. albicans [35] and C. orthopsilosis [36], the MTLalpha and MTLa idiomorphs are highly divergent in sequence (<50% identity) over a region of ~9 kb. In addition to containing the MTLalpha1/alpha2 or MTLa1/a2 genes, which are unrelated in sequence, this idiomorph-specific region also includes three divergent pairs of genes (OBPa/OBPalpha; PIKa/PIKalpha; PAPa/PAPalpha) that code for proteins that are not involved in mating and that appear to have diverged into separate alpha and a isoforms with low amino acid sequence identity because they became trapped in the non-recombining region of the mating-type locus millions of years ago [35]. These three genes are also arranged in a different order in the two idiomorphs. In 10 of the 11 C. metapsilosis strains, read-depth and mate-pair data indicated that MTLa haplotype appears to have the structure GAP1 –MTLalpha2 –OBPalpha–PIKhybrid–MTLa2 –MTLa1 - orf19.3202. In other words, a 6-kb region derived from MTLalpha and containing MTLalpha2, OBPalpha and part of PIKalpha (in that order) has overwritten a nonhomologous 6 kb region that is normally present in MTLa and contains PAPa, OBPa and PIKa (in that order). Consistent with this introgression happening after the single hybridization event, the divergence between alleles in the introgressed region is very low (0.025%). These results were confirmed by PCR amplification and re-sequencing of five representative strains (SZMC21154, SZMC8094, SZMC8095, DNS94 (CP61), DNS100 (CP376)). The resulting haplotype contains an intact copy of MTLalpha2 as well as MTLa2 and MTLa1, it has no PAP gene of any kind, and its PIK gene is chimeric (S10A and S10B Fig). Finally, SZMC21154 experienced an additional LOH removing the remaining MTLa cassette (MTLa1, MTLa2, PAPa), a situation which may be reminiscent of what may have occurred in the C. orthopsilosis hybrid. These results are in contrast with an earlier survey that reported that only the MTLα idiomorph was present in 18 isolates of C. metapsilosis [36] based on long-range PCR amplification. However, this discrepancy is likely to result from problems in the PCR in the former study, perhaps attributable to the introgression, because six of these 18 strains were fully sequenced in this study, and in all of them we found intact MTLa1 and MTLa2 (Fig 3).
Genomic variation within C. metapsilosis
Predicted duplications and deletions (copy number variations, CNVs) in sequenced strains were subjected to restrictive manual curation (see Materials and Methods), resulting in 84 deletions and 87 duplications relative to the reference assembly (S4 Table). Most CNVs affected coding regions, 71 deletions and 85 duplications, but we found no functional enrichment in deleted regions, while nucleic acid binding (GO:0003676) was enriched in duplicated regions. Nearly all CNVs are shared by more than one strain (82 deletions and 86 duplications) and 31 deletions and 15 duplications are shared by all strains, which further support a common origin for all strains. The largest duplication (DUP65, 20 kb), spanning 13 genes involved in DNA binding, transport and various enzymatic activities, is shared by eight strains. The largest deletion (DEL8, 13,976 bp) is heterozygous and is shared by all eleven strains. This suggests that the deletion was either present in one parental undergoing hybridization or happened subsequently but prior to the divergence of the strains considered.
To assess the population structure of the species and reconstruct the diversification history of the different C. metapsilosis clinical isolates we employed several alternative strategies. First, multiple correspondence analyses (MCA) was conducted using the SNPs (Fig 4). Secondly, SNP based trees were reconstructed using either all SNPs or homozygous SNPs exclusively (S11A, S11B and S11C Fig). Finally, we reconstructed the evolutionary relationships among the strains using LOH blocks and CNVs as characters (S11D and S11E Fig). All of these approaches resulted in a roughly similar clustering of the strains. Particularly, the MCA shows four major groups: SZMC8094 and CP367 from Italy appear close to each other and well separated from the rest; CP61 (Italy) and BP57 (Hungary) appear close together, two strains derived from a single isolate from Washington State (PL448 and MCO448) group together, whereas PL429 from Livermore, California (US) is rather distant from all the rest. The largest cluster is formed by the rest of the strains from various origins (Italy, Hungary, Spain and US), although the two US strains in this cluster appear somewhat more distant from the rest. These broad separations are also apparent in the phylogenetic trees, particularly when branch lengths are considered. The observed clustering is also consistent with earlier reported clusters based on Amplification Fragment Length Polymorphisms (AFLP), for the five strains common in both studies [37].

The significant differences between the hybrid isolates and the certain degree of geographical structure suggest that this lineage is relatively ancient and spread globally a long time ago. This is in stark contrast with what has been found for the C. orthopsilosis hybrid lineage, where the two sequenced isolates from distant locations were found to be nearly identical [8]. Given our lack of knowledge on mutation rates, generation time and life cycle of C. metapsilosis, we can only speculate on the relative time of when the hybridization occurred. Of note we can only measure divergence among the sequenced strains, while hybridization must have predated this time as indicated by the number of shared events. The low level of differences found between the two most diverged isolates (MCO448 and SZMC8094) suggests that a limited number of point mutations accumulated during this time: 3 SNPs in the mitochondrial genome (0.0124% divergence) and 53 SNPs in the longest common homozygous region (0.01767%).
Comparative genomics of the Candida parapsilosis species complex
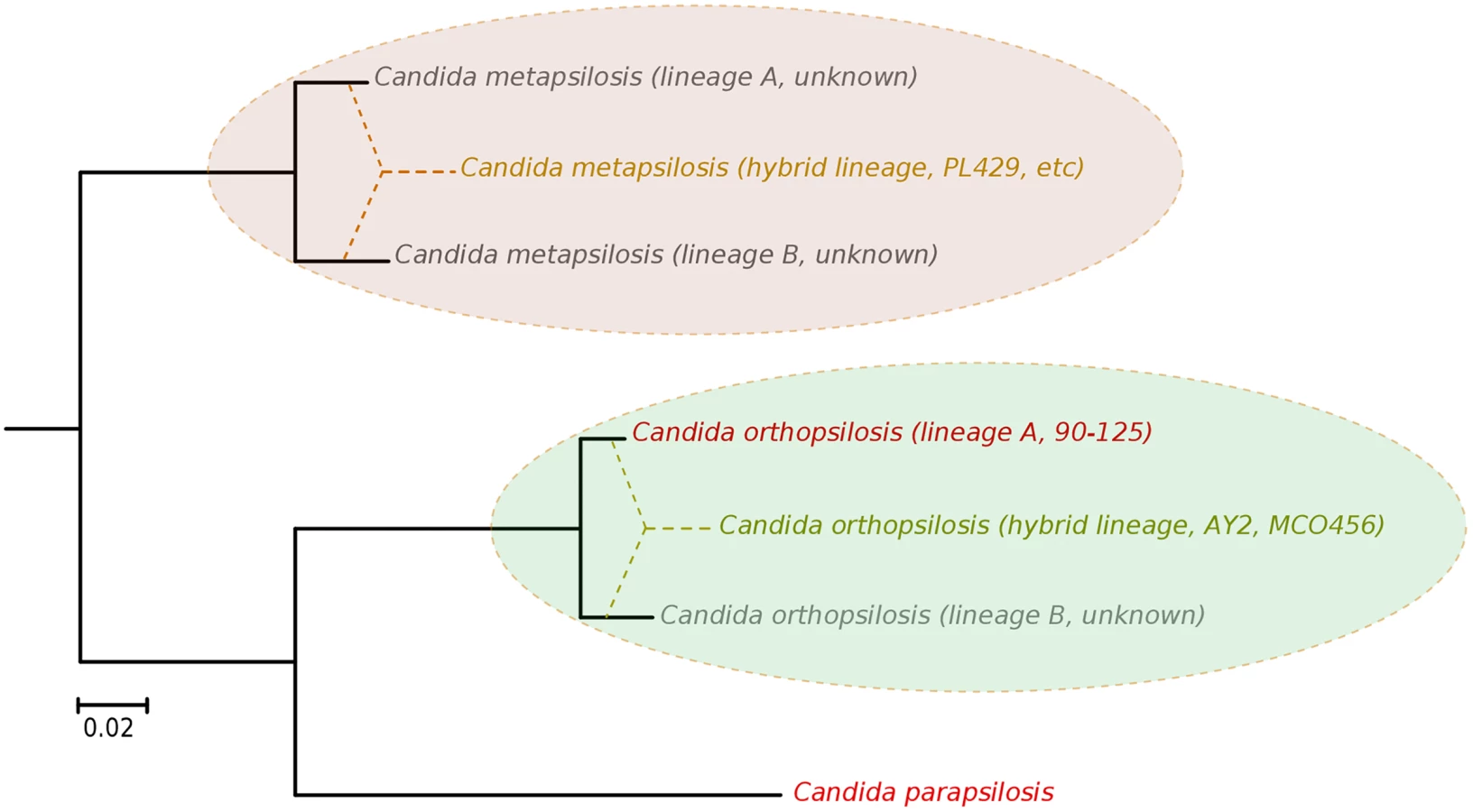
The availability of the genome sequence of C. metapsilosis allows, for the first time, to perform a comprehensive comparative genome analysis of the entire C. parapsilosis species complex. Whole genome alignments show that synteny is largely conserved among the three species of the complex (S12 Fig). Overall C. parapsilosis and C. orthopsilosis are closer to each other (98% of conserved synteny, 154 inversions) than either of them to C. metapsilosis (97%/231, and 96%/176 conserved synteny/inversions, respectively). We compared the gene content of the three species and compared them with other 23 sequenced Saccharomycetes by reconstructing the complete collection of evolutionary histories of the genes encoded in their genomes (i.e. the phylome) and establishing orthology and paralogy relationships among them [38]. The phylogenies, alignments and inferred homology relationships are available through phylomeDB [22]. We used 396 conserved, single-copy orthologs to reconstruct the evolutionary relationships among sequenced Candida species (Fig 5). This phylogeny was largely congruent with that of a super-tree derived from the whole phylome using a gene tree parsimony approach (S13 Fig). In contrast to earlier analyses based on an smaller sample of genes [25], but in line with the synteny analyses mentioned above and with an earlier phylogenetic analysis of mitochondrial genomes [29,31], our results support a basal position of C. metapsilosis to the exclusion of C. orthopsilosis and C. parapsilosis.

Predicted genes in the three species from the Candida parapsilosis species complex were grouped into 5,743 orthologous groups (including orthologs and in-paralogs), of which 5,045 (88%) are present across all three species. Of the widespread groups, 4,574 (91%) were present as one-to-one orthologs, while 226, 107 and 124 groups contained C. metapsilosis, C. orthopsilosis and C. parapsilosis specific paralogs, respectively. Differences in gene content for the three species are presented in (S5 Table). We here limit the discussion to several families considered relevant to explain virulence differences between species (Fig 5). The ability to form pseudohyphae has been associated to virulence in this clade [17]. Unlike C. parapsilosis and C. orthopsilosis, C. metapsilosis does not produce pseudohyphae [18], and this has been related to the lower virulence of the latter. Pseudohyphae production has been associated with two protein families: the cell-wall proteins Hyr/Iff and adhesion cell-surface glycoproteins ALS. Overall, C. metapsilosis encodes fewer members of Hyr/Iff family (13), than C. parapsilosis (17), but more than C. orthopsilosis (3 in 90–125 and 4 in MCO456) (see Phy00767CU tree in PhylomeDB). Interestingly, C. metapsilosis encodes one ALS gene, which is very close to ALS6 (CPAG_05054/CPAR2_404790), while the two C. orthopsilosis strains encode an ortholog closer to ALS7 (CPAG_05056/CPAR2_404800; see orf19.5736 in phylomeDB, phylome 464). Thus the lack of some ALS genes but not the number of members of Hyr/Iff family correlates with the inability to produce pseudohyphae and consequent lower virulence than in the remaining C. parapsilosis complex species.
We and others have previously shown that secreted lipases play an important role in the virulence of C. albicans and C. parapsilosis [18,40–42]. Despite earlier reports suggesting that C. metapsilosis strains were unable to produce extracellular lipases [19], we found that its genome actually codes for a similar number of secreted lipases (5) as C. parapsilosis and C. orthopsilosis (4). This indicates that the observed phenotypic differences may be due to different regulation of the lipase activity rather than to an inherent inability to produce secreted lipases. It has been demonstrated that individual lipase genes are differentially regulated in C. albicans during infection [43,44], and although we lack direct evidence on the function of C. metapsilosis lipase genes, it is appealing to speculate that a similar phenomenon exists in this species. Secreted aspartic proteases are also considered an important virulence factor in C. albicans and C. parapsilosis [45,46], and the other two species of the Candida parapsilosis complex have also been shown to exhibit this activity [18]. Interestingly, C. metapsilosis and C. parapsilosis encode more (14) secreted aspartic proteases (SAP) than any other Candida spp.: C. orthopsilosis (11 in MCO456 and 11 in 90–125) or C. albicans (10) (S5 Table and Phy0076724 in PhylomeDB). Although the capacity of C. orthopsilosis and C. metapsilosis SAPs to affect virulence has yet to be demonstrated, an extended SAP toolkit in species of the C. parapsilosis complex, which are not obligate commensals, may represent adaptation to both, environment and host. Similarly, C. metapsilosis, and C. parapsilosis encode more extracellular CFEM domain proteins (7), that are important for iron acquisition, than C. albicans (5). Overall, the broad functional class corresponding to cell wall and adhesion proteins seems to have a tendency to show larger numbers in pathogenic species as compared to less pathogenic ones, and is the only broad functional class that seems also expanded in non-CTG yeast pathogens, such as those in the Nakaseomyces clade [47].
Concluding remarks
Our results show compelling evidence that the opportunistic pathogen C. metapsilosis is a diploid hybrid species resulting from a single hybridization event, likely through mating, of two parental lineages that were ~4.5% divergent. This hybrid lineage expanded globally and currently isolated strains differ to a significant extent in their genomic background. Divergence has been mostly driven by differential LOH events but also by lineage-specific copy number variations, including large partial aneuploidies. Earlier studies based on AFLP have previously described a high degree of genetic heterogeneity among C. metapsilosis strains [37], which is consistent with our observations. In addition, our results provide a mechanistic basis for the source of this large heterogeneity: hybridization followed by differential LOH. It remains to be established, however, whether LOH is achieved mainly through mitotic recombination in clonal reproduction or whether sexual recombination or parasexual cycle also take place. In this respect most (10/11) of the strains harbor at least partial regions of both mating type loci, although in one of the analyzed strains the MTLa locus has completely overwritten MTLa. The levels of heterozygosity found for C. metapsilosis (22–26 SNPs/kb), are much larger than those described in C. albicans (2.5–3 SNPs/kb) [24]. However, considering the trend to lose heterozygosity with time, our findings open the question of whether the heterozygosity in C. albicans- and perhaps other highly heterozygous species in the CTG clade—may have originated via a similar process a longer time ago.
Our results argue for a re-definition of the species within the C. parapsilosis clade. We propose the existence of at least five different homozygous lineages and at least two hybrid lineages resulting from distinct combinations of the former. Two of the five homozygous lineages would be nowadays represented by C. parapsilosis and homozygous C. orthopsilosis Type 2 strains such as 90–125, whereas the remaining three are only partially represented by the sequences of C. orthopsilosis MCO456 and the C. metapsilosis sequences presented in this work (Fig 6). Whether homozygous strains for these three lineages are extinct or still exist remains to be determined. Considering the pervasiveness of hybrid strains across C. metapsilosis clinical isolates, it can be suspected that the corresponding homozygous, parental lineages are not able to infect humans. This would imply that hybridization has resulted in the evolutionary emergence of the ability to colonize and infect humans by combining characteristics from two parental species that are not able to do so. Hybridization is known to drive the adaptation to new niches and this work emphasizes the idea that new pathogenic lineages can emerge through hybridization of non-pathogenic parental species. Ability to colonize humans may not necessarily be the key advantageous trait that promoted the survival of a new hybrid lineage, but rather human can be a secondary niche that can be exploited opportunistically. Alternatively, a higher ability to persist in humans may promote the survival of hybrids between species that can only sporadically colonize humans. An interesting idea is whether the stress environment provided by humans to species that are not well adapted may promote activation of mating competence, which in turn may open the way for inter-species crossings. In this respect, metagenomics analyses have identified C. metapsilosis in the normal microbiota of one healthy individual, among 20 investigated [48], although in the absence of a genome sequence we do not know whether this commensal strain was homozygous or heterozygous. In addition, the relative divergence between C. metapsilosis hybrid strains seems to indicate that this lineage did not emerge recently, and that it did not expand as a clinical outbreak. Rather, recursive infections from an already diverged population seem more plausible. If this is the case, the presence of similar hybrid lineages in the natural environment of C. metapsilosis is expected. The earlier reported case of C. orthopsilosis hybrid seems to be different, as two distant isolates had nearly identical sequences. Resolving these open questions requires analysis of the diversity present in healthy individuals and environmental samples, a topic which is largely unexplored.
Materials and Methods
DNA extraction
C. metapsilosis cultures were grown overnight in an orbital shaker (200 rpm, 30°C) in 2 ml YPD medium (0.5% (w/v) yeast extract, 1% (w/v) peptone, 1% (w/v) glucose) supplemented with 100 unit/ml penicillin-streptomycin solution (Sigma). Subsequently, cells were centrifuged (850 xg, 5 minutes) and were washed twice with 1x sterile PBS. The pellet was resuspended in 500 μl lysis buffer (1% (w/v) SDS, 50 mM EDTA, 100 mM TRIS pH = 8), 500 μl glass bead was added to the cells and were disrupted by using a vortex for 3 minutes. 275 μl 7M ammonium-acetate was added (65°C, 5 min) and the samples were then cooled on ice for 5 minutes. 500 μl of chloroform-isoamylalcohol (24 : 1) was added to the mixture, and the samples were centrifuged for 10 minutes at 16000 xg. The upper phase was transferred to a new microcentrifuge tube, and the previous step was repeated. 500 μl isopropanol was mixed with the upper phase in a new microcentrifuge tube, and the mixture was held in a refrigerator at -20°C for 5 minutes. The solution was centrifuged at 16000 xg for 10 minutes. The supernatant was discarded, and the pellet was washed twice with 500 μl 70% ethanol. After the second washing step the pellet was dried, and resuspended in 100 μl sterile bi-distilled water containing 250 μg/ml RN-ase (Sigma).
Genome sequencing
The genome sequences for the 11 strains were obtained at the Ultra-sequencing core facility of the CRG, using Illumina GAIIx, HiSeq2000 and MiSeq sequencing machines. For paired-end libraries, DNA was fragmented by nebulization or in Covaris to a size ~300 bp, ~400 bp, ~600 bp. The ends of the DNA fragments were blunted with T4 DNA polymerase and Klenow fragment (New England Biolabs), after shearing. DNA was purified with a QIAquick PCR purification kit (Qiagen). 3’-adenylation was performed by incubation with dATP and 3’-5’-exo - Klenow fragment (New England Biolabs). DNA was purified using MinElute spin columns (Qiagen) and double-stranded Illumina paired-end adapters were ligated to the DNA using rapid T4 DNA ligase (New England Biolabs). After another purification step, adapter-ligated fragments were enriched, and adapters were extended by selective amplification in an 18-cycle PCR reaction using Phusion DNA polymerase (Finnzymes). Libraries were quantified and loaded into Illumina flow-cells at concentrations of 7–20 pM. Cluster generation was performed in an Illumina cluster station. Sequence runs of 2x50, 2×76, 2x100 or 2x250 cycles were performed on the sequencing instrument. For the preparation of mate-pair libraries 15 micrograms of genomic DNA were sheared using a covaris instrument to the desired size range of 2.5 or 5 kb, respectively. Following size selection on a 0.8% agarose gel, the size fraction of interest was recovered, and library preparation was performed using a modification of the Illumina mate-pair preparation protocol, whereby a biotinylated double-stranded adapter was included in the circularisation reaction [49]. After circularisation, linear background was removed by exonuclease digestion, and the sample further fragmented in the covaris. Fragments that included the biotinylated adapter were enriched using streptavidin beads, and used to prepare an Illumina library. Sequencing was performed on an Illumina HiSeq 2000 sequencer using a 2 x 50 nt paired-end sequencing protocol. Base calling was performed using Illumina pipeline software. In multiplexed libraries, we used 4 bp internal indices (5’ indexed sequences). De-convolution was performed using the CASAVA software (Illumina).
Genome assembly
Reads were pre-processed before assembly to trim at the first undetermined base or at the first base having PHRED quality below 10. We filtered out pairs with one (or both) reads shorter than 31 bases after trimming. SOAPdenovo2 [50] was used to assemble paired-end reads into supercontigs with K-mer ranging from 31 to 91. As the initial assembly was very fragmented (2–4k scaffolds) and nearly twice as large (21–22 Mb) as other C. parapsilosis complex species (~13 Mb in 7–8 chromosomes), we assumed the presence of heterozygous regions in our strains. Heterozygous regions (9 Mb in 861 contigs) were removed by Haplomerger version 20120810 [51]. Subsequently, the remaining supercontigs were further scaffolded by SSPACE2 [52] and gaps were filled using GapCloser from the SOAPdenovo2 package. The random incorporation into the assembly of alternative heterozygous contigs was tested by repeating the whole procedure several times, starting from reads that were randomly re-ordered.
Genome annotation
Genes were predicted using Augustus version 2.5.5 [53] and C. parapsilosis CDC317 gene models for training [24]. Predicted gene models were curated using RNA-Seq reads to find evidence for exon-intron boundaries and exon skipping. Subsequently, we grouped predicted genes into orthologous groups and transferred functional annotation from one-to-one orthologs in model species i.e. Candida albicans or Saccharomyces cerevisiae, based on predictions from the MetaPhORs approach [54]. Finally, genes were further annotated using InterProScan 5RC4 [55].
Detection of SNPs, loss of heterozygosis, and divergence estimates
Genomic reads were aligned onto C. metapsilosis assembly using Bowtie2 with “very sensitive local alignment” mode [56]. SNPs and INDELs were called using GATK version 2.1–13 [57]. We filtered out clusters of five variants within 20 bases and low quality variants, as described in GATK documentation (QD < 2.0 || MQ < 40 || FS > 60.0 || HaplotypeScore > 13.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0). Subsequently, we divided the genome into three categories: unknown, heterozygous, and homozygous. Firstly, regions having lower (<75%) or higher (>125%) coverage than expected were assigned as unknown. Then, we marked heterozygous regions as those having two or more heterozygous sites closer than 100 bases. The remaining regions of the genome were considered homozygous, thus loss of heterozygosity (LOH) regions (100 bp threshold). A stricter threshold (200 bp) was established by considering LOH blocks shorter than 200 bp as heterozygous regions. Note that the two methods differ in expected number of false positive and false negatives. At 4.5% divergence the probability of having by chance a stretch of 100 bp with no heterozygous SNPs is 0.01 (0.955100) and the expected number of LOH blocks of this length or longer in a 13.6 Mb genome is approximately 5,854 blocks, these numbers vary dramatically to 0.0001 and 58 when a 200 bp threshold is applied (Expected number is approximated as E = Npqk, where N is genome size in basepairs, p and q are the probabilities of having a SNP or not, respectively and k is the required size of the block). We compared the number of expected blocks of a given size or longer with the observed sizes in the real strains (S14 Fig). This made clear that a large number of short apparent LOH blocks (up to ~59%) may be artefactual when a threshold of 100 bp is applied (~2.9% at 200 bp). However, the stricter threshold would conversely discard many true LOH blocks (up to 97.1% of the discarded blocks would be real if we consider the observed–expected). Given that most differences affect blocks of short length, the effects of the total fraction of the genome assigned to homozygous or heterozygous regions is affected to a lesser extend. We further assessed the accuracy of our method by simulating 20 fully heterozygous genomes (13.6 Mb; 4.5% divergence between haplotypes) to estimate false positive of LOH detection (how often we would detect LOH in perfectly heterozygous genome using current settings). Heterozygous genomes were simulated using fasta2diverged.py v1.0 (https://github.com/Gabaldonlab/ngs_public) with the homozygous chromosome set of C. metapsilosis assuming 4.5% divergence between haplotypes. LOH regions (at least 100bp regions having less than 2 SNPs) appear by chance in all simulations and on average sum up 782,850 bp (5.84% of the genome). Thus we estimate that less than 6% of the regions detected as LOH in our analysis correspond to false positives. Increasing LOH length cut-off from 100bp to 200bp decreases false positive rate to 0.13%, however. Throughout the manuscript we provide estimates based on both of these thresholds. The sequence divergence between the parental haplotypes was calculated as the number of heterozygous sites found in heterozygous regions divided by the cumulative length of heterozygous regions, using a 100 bp threshold.
Discrimination of linear and circular mitochondrial chromosomes
Two main characteristics allow in silico differentiation of linear and circular mtDNA chromosomes in C. metapsilosis. First, due to the presence of telomeres, consisting of tandem arrays of 620 bp repeat, linear chromosomes are expected to be longer (24,152 bp, NC_006971) than circular ones (22,175 bp, AY391853) [29]. Second, paired-end reads resulting from circular mitochondrial genomes when aligned to the termini of the linear mitochondrial chromosome reference assembly should have their partners aligned in the opposite end of the chromosome with disconcordant orientation (FF or RR instead for FR). To check this, genomic reads were aligned on the linear mitochondrial chromosome reference of C. metapsilosis MCO448 (NC_006971) as indicated above.
Experimental analysis of the mitochondrial genome topology
Mitochondrial DNAs in C. metapsilosis isolates were analysed by PFGE and PCR essentially as described in [30,31]. Briefly, whole-cell DNA samples were prepared in agarose blocks and separated in a 1.5% (w/v) agarose gel using a CHEF Mapper XA Chiller System (Biorad) with pulse switching set at 5 to 20 seconds (linear ramping) and 120o angle for 42 hours at 5 V/cm and 10oC (Fig 1A) or in a 1.0% (w/v) agarose gel in a Pulsaphor apparatus (LKB) in contour-clamped homogeneous electric field (CHEF) configuration with pulse switching from 5 to 50 seconds (interpolation) for 24 hours at 150 V and 9oC (Fig 1B). All separations were performed in 0.5x TBE buffer (45 mM Tris-borate, 1 mM EDTA, pH 8.0). Southern blots were hybridized with radioactively labeled probes derived from the mitochondrial genes cox2 and nad4. For PCR analysis, the reactions contained diluted total cell DNA, 0.5 μM upstream and downstream primers (5’-ATTGTTGCTTTTGTTGTTGA-3’ and 5’-TTAGCTGTTGTTGCTATTACT-3’ derived from the subtelomeric genes nad3 and atp6, respectively), 0.2 mM dNTPs each, 1× reaction buffer with 2 mM MgCl2, and 1 U of Taq DNA polymerase (Invitrogen). The amplification was performed using the following cycler profile: 3 min at 95°C; 25× (1 min at 94°C, 45 seconds at 56°C, 1 min at 68°C); 3 min at 72°C. The primers bind to the opposite subterminal regions of the linear mitochondrial genome and allow amplification of about 0.95 kb long fragment derived from the end-to-end junction of circularized genome forms (Fig 1C).
Detection of structural variants
Structural variants were detected using a methodology described and experimentally validated elsewhere [54], which we have implemented in bam2sv.py python script v1.0 (available at https://github.com/Gabaldonlab/ngs_public/). bam2sv.py v1.0 detects duplications, deletion and inversions by means of insert size deviations and incongruent read pairing between paired-end reads. In addition, duplications and deletions are detected from deviations from mean depth of coverage. All detected variants were manually curated. In addition, we generated genome graphs for all chromosomes illustrating copy number variation, as well as, heterozygous and homozygous regions (S1 File).
Experimental validation of a LOH region and MAT locus introgression
To validate the two parental sequences, we performed PCR and Sanger sequencing of the region scaffold2|size2959145 : 1,570,477–1,574,155 from C. metapsilosis genome in four different strains: BP57 (DNS25), CP61 (DNS94), CP376 (DNS100) and PL429. Four PCR primers were designed using Primer3 v4 webtool [58], namely two forward primers and two reverse primers, with four and three different bases among them, respectively, corresponding to allelic differences in each parental sequence (S5 Fig).
FWD_1 : 5’-TTGACTGCTGAAGCTGTCTTGG-3’;
FWD_2 : 5’-TTAACCGCTGAAGTTGTCTTTGA-3’;
REV_1 : 5’-ATTCCATCTTGGCGCATCTT-3’;
REV_2 : 5’-GACTCCATCTTGACGAATCTTGG-3’.
Thus, four touchdown PCR reactions (FWD_1+REV_1, FWD_1+REV_2, FWD_2+REV_1 and FWD_2+REV_2) were carried out using Expand Long Range, dNTPack kit (Roche) according to manufacturer’s instructions. Briefly, each reaction included primer concentration of 0.3 μM, 10 μl of 5X Buffer with MgCl2 (final MgCl2 concentration of 2.5mM), 2.5 μl of PCR Nucleotide Mix, 3% (v/v) of DMSO, 100 ng of DNA and 3.5 U of Enzyme Mix in a final volume of 50 μl. Cycling condition began with a warm-up step of 2 min at 92°C, followed by 15 cycles of 10 seconds at 92°C, 15 seconds at the corresponding annealing temperature per each pair of primers (decreasing 0.5°C each cycle) and 4 min at 68°C. The initial annealing temperatures were: 61.6°C, 64.1°C, 60°C, and 62.5°C for FWD_1+REV_1, FWD_1+REV_2, FWD_2+REV_1 and FWD_2+REV_2, respectively. Then, other 20 cycles of 10 seconds at 92°C, 15 seconds at the corresponding annealing temperature and 4 min at 68°C (increasing the extension time 20 seconds at each cycle) were set up, with a final extension step at 68°C for 7 minutes. PCR products were confirmed by 1.5% agarose gel electrophoresis and were then purified using QIAquick PCR Purification Kit (QIAGEN). Specific PCR products were only obtained when combining the primer sets FWD_1+REV_2, and FWD_2+REV_1 (amplicon size of 3678 bp), while no product was obtained when combining FWD_1+REV_1 or FWD_2+REV_2 primers (S6 Fig). The purified PCR products were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) and sequencing products were precipitated and purified using EDTA 125mM, sodium acetate 3M and 100% ethanol. To cover the entire length of the products, apart from the PCR primers described above, further internal primers common to both parental sequences were designed and used for direct sequencing:
INT_FWD: 5’ - AGTTTGGAAATTGTCATCTTGGAT-3’;
INT_REV: 5’ - ACTTCTTTAAAATACTAACCACATCCATCTC-3’.
The MTL introgression was checked by means of PCR and Sanger sequencing in following strains of C. metapsilosis: SZMC21154, SZMC8092, SZMC8094, SZMC8095, CP61 (DNS94), CP367 (DNS100). We have designed 8 different primers:
MTLα1_1F: 5’-GCCGCCTGAGAAGTATGAAG-3’;
MTLα1_1R: 5’-TTGGTGACCACAGGAAAACA-3’;
MTLα2_1F: 5’-GTGCTCCTCAAGCACAATCA-3’;
MTLα2_1R: 5’-GTCGCCCAGACAACTCTAGC-3’;
MTLa1_1F: 5’-GCTTGAGTGGGGATTGAGTC-3’;
MTLa1_1R: 5’-AATCGTTTTCGGGGTTTTCT-3’;
MTLa2_1F: 5’-TCTCCGGATTCGTTCAATTC-3’;
MTLa2_1R: 5’-CTTGACCCCAAAGCTTTCAA-3’.
The introgression was validated by 4 different PCRs (MTLα1_1F –MTLα1_1R, MTLα2_1F –MTLα2_1R, MTLa1_1F –MTLa1_1R and MTLa2_1F –MTLa2_1R) using Pfu DNA polymerase from PROMEGA. The reaction mixture consisted of 5μl of Buffer 10X with MgSO4 and1μl of 10 mM dNTPs, both provided by the manufacturer, 2μl 10 μM of both forward and reverse primers, 0.4μl of Pfu DNA polymerase 3 U/μl and water filled up to 50μl. The PCR started with initial denaturation at 95°C for 2min, this was followed 30 cycles of 30 seconds at 92°C, 30 seconds at 61.0°C, 30 seconds at 72°C, it was finished with final extension for 5min at 72°C and cooled to 4oC. PCR products were confirmed by 1.5% agarose gel electrophoresis and were then purified using QIAquick PCR Purification Kit (QIAGEN).
Finally, in order to confirm linearity of MTL after introgression, we have amplified and Sanger sequenced genomic regions flanking introgression site. We have designed 6 primers:
P1f: 5’-TATCGGGTTTGAAGCTGCTC -3’;
P1rα: 5’-GCTGAATGCTGGTTTTTGGT -3’;
P2ra: 5’-TACTCCACGTTGTTTTGTTAAAG-3’;
P2fα: 5’-TTATGATTGGGATGGGTTGG -3’;
P2fa: 5’-CACATTTGAATGGACGTTGG -3’;
P2r: 5’-CGATAAATCAGCGCAACAAT-3’.
The linearity of introgression was validated by 4 different PCRs (P1f ‐ P1rα, P1f—P2ra, P2fα—P2r and P2fa—P2r) using Pfu DNA Polymerase from PROMEGA. The reaction mixture consisted of 5μl of Buffer 10x with MgSO4 and1μl of 10 mM dNTPs, both provided by the manufacturer, 2μl 10 μM of both forward and reverse primers, 0.4μl of Pfu DNA polymerase 3 U/μl and water filled up to 50μl. The PCR started with initial denaturation at 95°C for 2min, this was followed 30 cycles of 30 seconds at 92°C, 30 seconds at either 56.2°C (P1f ‐ P1rα), 57.2°C (P1f –P2ra) or 54.2°C (P2fα–P2r and P2fa–P2r); 30 seconds at 72°C, it was finished with final extension for 5 min at 72°C and cooled to 4oC. PCR products were confirmed by 1.5% agarose gel electrophoresis and were then purified using QIAquick PCR Purification Kit (QIAGEN). Sanger sequencing was performed using an ABI Prism 3730xl DNA Analyzer (Applied Biosystems).
Pulsed-field gel electrophoresis
Yeasts cells were cultured for 24 hours, at 30°C, in an oxygen rich environment, in 5ml YPD (0.5% (w/v) yeast extract, 1% (w/v) peptone, 1% (w/v) dextrose) supplemented with 100 unit/ml penicillin-streptomycin (Sigma) in an orbital shaker (180 rpm). After 24 hours 100 μl suspension was transferred to 5 ml YPD and the cells were incubated for 24 hours again, under the same conditions. Agarose blocks containing the intact chromosomes were prepared using the method of Schwartz & Cantor [59] with the following modifications. 1 ml of the yeast suspension was transfered in a sterile microcentrifuge tube, and was washed two times with 4°C 0.05 M EDTA (pH = 7.5) (Sigma) (2000 xg, 3 min). 1.3x108 cells were resuspended in Isotonic Buffer (0.1 M phosphate–citrate buffer, equipped with 0.7 M sorbitol, 0.3 M mannitol, 0.001 M EDTA pH = 5.8) containing 1 M potassium-thioglycolate (Reanal), and it was incubated for 1 hour, at 30°C in an orbital shaker (180 rpm). The suspension was washed once with Isotonic Buffer (2000 xg, 3 min). The pellet was resuspended in 5 ml Isotonic Buffer containing 3% (w/v) Helicase and 0.5% (w/V) NovoZym 234 (Novo BioLabs), and it was incubated at 30°C, in a sterile 15 ml Falcon tube, overnight, in an orbital shaker (180 rpm). The spheroplasts were collected and washed once with Isotonic Buffer (300 xg, 5 min). The pellet was resuspended in 42°C 0.125 M EDTA, mixed immediately with prewarmed (42°C) 2% (w/v) low-melting-point agarose (Sigma), then placed into a mould chamber. After solidification, inserts were incubated in 2 ml NDS buffer (1% (w/v) N-laurylsarcosine in 0.5 M EDTA, pH = 9.5) supplemented with 1 mg/ml Proteinase K (Sigma) at 50°C. During the two days of incubation the NDS-Proteinase K solution was replaced once. The inserts were washed once with 0.5 M EDTA (pH = 8) overnight, then were stored in 0.5 M EDTA (pH = 8) at 4°C until usage. Finally, yeast chromosomes were separated by CHEF [60] method by using Bio-Rad CHEF-DR II Drive Module, Bio-Rad Pulsefield 760 Modul and Bio-Rad Power Supply. The chromosomal DNA plugs were placed and separated in 0.9% (w/v) agarose gel (Sigma) prepared with filtered 0.5x TBE buffer with the following settings: 60–450 sec switching time, 90 V voltage, 168 h running time. 0.5x TBE was used as a running buffer and the temperature was kept at 10°C during the whole procedure. The buffer was replaced to fresh one 3 times during the running process. The gel was stained for 30 min in a 0.1% ethidium-bromide solution and was destained in distilled water overnight, at 4°C. The results were documented by using UVP Bio-Doc-It System.
Fluorescence-activated cell sorting (FACS) analysis
Ploidy analyses were confirmed using FACS for 12 Candida samples. Cells were grown in YPD medium at 30°C (overnight, 200 rpm), harvested, resuspended in deionized distilled water and fixed in ethanol at a 107 cells/ml concentration (overnight, 4°C). For the staining of cells with SYBR Green I, cells were first washed and resuspended in 750 μl of 50 mM sodium citrate buffer, treated with 250 μl of 1mg/ml RNase A solution for 1 hour at 50°C and finally with 50 μl of 20 mg/ml proteinase K solution for 1 hour at 50°C. Then, 20 μl of SYBR Green I (Life Technologies, diluted 1 : 10 th in Tris-EDTA buffer, pH 8.0) were added to the samples and they were stained overnight at 4°C protected from light. Triton X-100 was added at a final concentration of 0.25% (v/v) and samples were vortex. Finally, samples were sonicated (3 consecutive ultrasound pulsed at 30 W for 2 seconds with intervals of 2 seconds between each pulse) to eliminate most of the cell clumps before FACS analysis with FACScan at the FACS Unit from CRG/UPF.
Synteny analysis
Chromosomes/scaffolds of C. parapsilosis CDC317, C. orthopsilosis 90–125 and C. metapsilosis SZMC8094 were aligned with LAST aligner v189 [61]. Alignments shorter than 1kb were filtered out. Syntenic blocks were defined as contiguous regions in the same chromosome/scaffold of both genomes aligning over 10 kb, percentage of synteny is computed as the total length of syntenic blocks over the length of the genome. Synteny breaks were inferred if fragments longer than 10 kb from a contiguous region in one genome was aligning to at least two different chromosomes in the other species. Inversions in the query sequence were called when alignment direction changed within a given synteny block.
Phylome reconstruction
The evolutionary histories of all C. metapsilosis protein-coding genes were reconstructed in the context of 27 Saccharomycotina species (S6 Table), using the PhylomeDB pipeline [62]. In brief this pipeline proceeds as follows: first, homologs were retrieved using Smith-Waterman [63] with E-value cut-off of 1e-05 and considering only sequences that aligned with at least 50% of their length. Subsequently, homologs were aligned using three programs: MUSCLE v3.8 [64], MAFFT v6.712b [65] and KALIGN v2.04 [66] in two directions: forward and reverse. The six resulting alignments were combined with M-COFFEE [67] and finally trimmed using trimAl v1.3 [68] applying consistency cutoff of 0.1667 and a gap score cutoff of 0.1. Neighbour Joining trees were reconstructed and the likelihood of obtained topology was computed, allowing branch-length optimisation, using seven different models (JTT, LG, WAG, Blosum62, MtREV, VT and Dayhoff), as implemented in PhyML 3.0 [69]. One evolutionary model best fitting the data was determined for each alignment by comparing the likelihood of the used models according to the AIC criterion. Finally, Maximum Likelihood (ML) trees were inferred for selected models. In all cases a discrete gamma-distribution model with four rate categories plus invariant positions was used, the gamma parameter and the fraction of invariant positions were estimated from the data. Branch support was computed using an aLRT (approximate likelihood ratio test) parametric test based on a chi-square distribution, as implemented in PhyML. All alignments and trees generated have been deposited in PhylomeDB with ID 243 [39]. Orthologs predicted for C. metapsilosis genes can be retrieved from MetaPhOrs database [54].
Reconstruction of evolutionary relationships among strains
Five separate analyses were used to reconstruct the evolutionary relationships among the sequenced strains. Alignments were reconstructed from SNP information in three different types of regions: (i) Patterns from homozygous SNPs present in the longest LOH block that is shared by all the strains; (ii) SNP patterns from the whole genome. Heterozygous SNPs were encoded using the base which is alternative to that in the reference, and choosing randomly one of the alternative bases in heterozygous SNPs if both were different from the reference; and (iii) Haplotype patterns based on three-state haplotype (heterozygous, hapA, hapB) assignment in windows of 1 kb, resulting in 1,798 phylogenetically informative patterns; Separately, character-based matrices were reconstructed using (iv) 127 ploidy patterns from 84 deletions and 87 duplications (S4 Table); ploidy state was coded as 0 for null deletion, 1 for heterozygous deletion, 2 for wild-type (no deletion and duplication), 3 for duplication (3 copies of given locus), 4 for duplication (4 copies of given locus), and so on; and (v) 587 LOH presence and absence patterns. Maximum Likelihood phylogenetic trees were reconstructed from these alignments using RAxML 7.2.8 using GTRCAT for sequence alignments (datasets i to iii), and GTRGAMMA for multi-state character matrices (datasets iv and v) [70]. Finally, multiple correspondence analysis (MCA, an extension of Principal Component Analysis to categorical data) of 618,120 SNPs with at least 10x coverage was performed using ade4 package from R [71].
Reconstruction of species phylogeny
Two different approaches were used to reconstruct a species tree of Candida species and relatives. First a parsimony-based super-tree was reconstructed using duptree v1.48 [72] from the 5,780 gene trees from C. metapsilosis phylome available at PhylomeDB id 243 [39]. Then a Maximum Likelihood (ML) tree was reconstructed with PhyML v3.0 [69] and the Jones-Taylor-Thornton (JTT) evolutionary model, based on an amino acid super-matrix of 232,760 columns resulting from concatenating the alignments of 396 one-to-one orthologs. The two species trees are nearly identical with Robinson-Foulds symmetric distance of 6, meaning both trees share 50 out of 56 possible splits. All phylogenies were visualised with ETE [73].
Data deposition
Sequencing data, genome assembly and annotation have been deposited to EBI-ENA (accession: PRJNA238968). Reconstructed trees and alignments were deposited to PhylomeDB (http://phylomedb.org) as PhylomeDB ID 243.
Supporting Information
Zdroje
1. Morales L, Dujon B. Evolutionary role of interspecies hybridization and genetic exchanges in yeasts. Microbiol Mol Biol Rev MMBR. 2012;76 : 721–739. doi: 10.1128/MMBR.00022-12 23204364
2. González SS, Barrio E, Gafner J, Querol A. Natural hybrids from Saccharomyces cerevisiae, Saccharomyces bayanus and Saccharomyces kudriavzevii in wine fermentations. FEMS Yeast Res. 2006;6 : 1221–1234. 17156019
3. Louis VL, Despons L, Friedrich A, Martin T, Durrens P, Casarégola S, et al. Pichia sorbitophila, an Interspecies Yeast Hybrid, Reveals Early Steps of Genome Resolution After Polyploidization. G3 Bethesda Md. 2012;2 : 299–311.
4. Xu J, Luo G, Vilgalys RJ, Brandt ME, Mitchell TG. Multiple origins of hybrid strains of Cryptococcus neoformans with serotype AD. Microbiol Read Engl. 2002;148 : 203–212.
5. Baker E, Wang B, Bellora N, Peris D, Hulfachor AB, Koshalek JA, et al. The Genome Sequence of Saccharomyces eubayanus and the Domestication of Lager-Brewing Yeasts. Mol Biol Evol. 2015;
6. Wendland J. Lager yeast comes of age. Eukaryot Cell. 2014;13 : 1256–1265. doi: 10.1128/EC.00134-14 25084862
7. Marcet-Houben M, Gabaldón T. Beyond the Whole-Genome Duplication: Phylogenetic Evidence for an Ancient Interspecies Hybridization in the Baker’s Yeast Lineage. PLoS Biol. 2015;13: e1002220. doi: 10.1371/journal.pbio.1002220 26252497
8. Pryszcz LP, Németh T, Gácser A, Gabaldón T. Genome comparison of Candida orthopsilosis clinical strains reveals the existence of hybrids between two distinct subspecies. Genome Biol Evol. 2014;6 : 1069–1078. doi: 10.1093/gbe/evu082 24747362
9. Bovers M, Hagen F, Kuramae EE, Diaz MR, Spanjaard L, Dromer F, et al. Unique hybrids between the fungal pathogens Cryptococcus neoformans and Cryptococcus gattii. FEMS Yeast Res. 2006;6 : 599–607. 16696655
10. Hagen F, Khayhan K, Theelen B, Kolecka A, Polacheck I, Sionov E, et al. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet Biol FG B. 2015;78 : 16–48. doi: 10.1016/j.fgb.2015.02.009 25721988
11. Trofa D, Gácser A, Nosanchuk JD. Candida parapsilosis, an emerging fungal pathogen. Clin Microbiol Rev. 2008;21 : 606–625. doi: 10.1128/CMR.00013-08 18854483
12. Tavanti A, Davidson AD, Gow NAR, Maiden MCJ, Odds FC. Candida orthopsilosis and Candida metapsilosis spp. nov. to replace Candida parapsilosis groups II and III. J Clin Microbiol. 2005;43 : 284–292. 15634984
13. Cantón E, Pemán J, Quindós G, Eraso E, Miranda-Zapico I, Álvarez M, et al. Prospective multicenter study of the epidemiology, molecular identification, and antifungal susceptibility of Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis isolated from patients with candidemia. Antimicrob Agents Chemother. 2011;55 : 5590–5596. doi: 10.1128/AAC.00466-11 21930869
14. Lockhart SR, Messer SA, Pfaller MA, Diekema DJ. Geographic distribution and antifungal susceptibility of the newly described species Candida orthopsilosis and Candida metapsilosis in comparison to the closely related species Candida parapsilosis. J Clin Microbiol. 2008;46 : 2659–2664. doi: 10.1128/JCM.00803-08 18562582
15. Oliveira VKP, Paula CR, Colombo AL, Merseguel KB, Nishikaku AS, Moreira D, et al. Candidemia and death by Candida orthopsilosis and Candida metapsilosis in neonates and children. Pediatr Neonatol. 2014;55 : 75–76. doi: 10.1016/j.pedneo.2013.07.006 24113226
16. Gácser A, Schäfer W, Nosanchuk JS, Salomon S, Nosanchuk JD. Virulence of Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis in reconstituted human tissue models. Fungal Genet Biol FG B. 2007;44 : 1336–1341. 17391997
17. Gago S, García-Rodas R, Cuesta I, Mellado E, Alastruey-Izquierdo A. Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis virulence in the non-conventional host Galleria mellonella. Virulence. 2014;5 : 278–285. doi: 10.4161/viru.26973 24193303
18. Németh T, Tóth A, Szenzenstein J, Horváth P, Nosanchuk JD, Grózer Z, et al. Characterization of virulence properties in the C. parapsilosis sensu lato species. PloS One. 2013;8: e68704. doi: 10.1371/journal.pone.0068704 23874732
19. Orsi CF, Colombari B, Blasi E. Candida metapsilosis as the least virulent member of the “C. parapsilosis” complex. Med Mycol. 2010;48 : 1024–1033. doi: 10.3109/13693786.2010.489233 20507266
20. Bertini A, De Bernardis F, Hensgens LAM, Sandini S, Senesi S, Tavanti A. Comparison of Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis adhesive properties and pathogenicity. Int J Med Microbiol IJMM. 2013;303 : 98–103. doi: 10.1016/j.ijmm.2012.12.006 23403338
21. Garcia-Effron G, Canton E, Pemán J, Dilger A, Romá E, Perlin DS. Epidemiology and echinocandin susceptibility of Candida parapsilosis sensu lato species isolated from bloodstream infections at a Spanish university hospital. J Antimicrob Chemother. 2012;67 : 2739–2748. doi: 10.1093/jac/dks271 22868644
22. Bonfietti LX, Martins M dos A, Szeszs MW, Pukiskas SBS, Purisco SU, Pimentel FC, et al. Prevalence, distribution and antifungal susceptibility profiles of Candida parapsilosis, Candida orthopsilosis and Candida metapsilosis bloodstream isolates. J Med Microbiol. 2012;61 : 1003–1008. doi: 10.1099/jmm.0.037812-0 22493277
23. Nosek J, Holesova Z, Kosa P, Gacser A, Tomaska L. Biology and genetics of the pathogenic yeast Candida parapsilosis. Curr Genet. 2009;55 : 497–509. doi: 10.1007/s00294-009-0268-4 19662416
24. Butler G, Rasmussen MD, Lin MF, Santos MAS, Sakthikumar S, Munro CA, et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459 : 657–662. doi: 10.1038/nature08064 19465905
25. Riccombeni A, Vidanes G, Proux-Wéra E, Wolfe KH, Butler G. Sequence and analysis of the genome of the pathogenic yeast Candida orthopsilosis. PloS One. 2012;7: e35750. doi: 10.1371/journal.pone.0035750 22563396
26. Pryszcz LP, Németh T, Gácser A, Gabaldón T. Unexpected genomic variability in clinical and environmental strains of the pathogenic yeast Candida parapsilosis. Genome Biol Evol. 2013;5 : 2382–2392. doi: 10.1093/gbe/evt185 24259314
27. Safonova Y, Bankevich A, Pevzner PA. dipSPAdes: Assembler for Highly Polymorphic Diploid Genomes. In: Sharan R, editor. Research in Computational Molecular Biology. Springer International Publishing; 2014. pp. 265–279. Available: http://link.springer.com/chapter/10.1007/978-3-319-05269-4_21
28. Gunisova S, Elboher E, Nosek J, Gorkovoy V, Brown Y, Lucier J-F, et al. Identification and comparative analysis of telomerase RNAs from Candida species reveal conservation of functional elements. RNA. 2009;15 : 546–559. doi: 10.1261/rna.1194009 19223441
29. Valach M, Pryszcz LP, Tomaska L, Gacser A, Gabaldón T, Nosek J. Mitochondrial genome variability within the Candida parapsilosis species complex. Mitochondrion. 2012;12 : 514–519. doi: 10.1016/j.mito.2012.07.109 22824459
30. Rycovska A, Valach M, Tomaska L, Bolotin-Fukuhara M, Nosek J. Linear versus circular mitochondrial genomes: intraspecies variability of mitochondrial genome architecture in Candida parapsilosis. Microbiol Read Engl. 2004;150 : 1571–1580.
31. Kosa P, Valach M, Tomaska L, Wolfe KH, Nosek J. Complete DNA sequences of the mitochondrial genomes of the pathogenic yeasts Candida orthopsilosis and Candida metapsilosis: insight into the evolution of linear DNA genomes from mitochondrial telomere mutants. Nucleic Acids Res. 2006;34 : 2472–2481. 16684995
32. Bennett RJ, Forche A, Berman J. Rapid mechanisms for generating genome diversity: whole ploidy shifts, aneuploidy, and loss of heterozygosity. Cold Spring Harb Perspect Med. 2014;4.
33. Louis VL, Despons L, Friedrich A, Martin T, Durrens P, Casarégola S, et al. Pichia sorbitophila, an Interspecies Yeast Hybrid, Reveals Early Steps of Genome Resolution After Polyploidization. G3 Bethesda Md. 2012;2 : 299–311.
34. Alby K, Bennett RJ. Interspecies pheromone signaling promotes biofilm formation and same-sex mating in Candida albicans. Proc Natl Acad Sci U S A. 2011;108 : 2510–2515. doi: 10.1073/pnas.1017234108 21262815
35. Hull CM, Johnson AD. Identification of a mating type-like locus in the asexual pathogenic yeast Candida albicans. Science. 1999;285 : 1271–1275. 10455055
36. Sai S, Holland LM, McGee CF, Lynch DB, Butler G. Evolution of mating within the Candida parapsilosis species group. Eukaryot Cell. 2011;10 : 578–587. doi: 10.1128/EC.00276-10 21335529
37. Hensgens LAM, Tavanti A, Mogavero S, Ghelardi E, Senesi S. AFLP genotyping of Candida metapsilosis clinical isolates: evidence for recombination. Fungal Genet Biol FG B. 2009;46 : 750–758. doi: 10.1016/j.fgb.2009.06.006 19559094
38. Gabaldón T. Large-scale assignment of orthology: back to phylogenetics? Genome Biol. 2008;9 : 235. doi: 10.1186/gb-2008-9-10-235 18983710
39. Huerta-Cepas J, Capella-Gutiérrez S, Pryszcz LP, Marcet-Houben M, Gabaldón T. PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 2014;42: D897–902. doi: 10.1093/nar/gkt1177 24275491
40. Gácser A, Trofa D, Schäfer W, Nosanchuk JD. Targeted gene deletion in Candida parapsilosis demonstrates the role of secreted lipase in virulence. J Clin Invest. 2007;117 : 3049–3058. 17853941
41. Trofa D, Soghier L, Long C, Nosanchuk JD, Gacser A, Goldman DL. A rat model of neonatal candidiasis demonstrates the importance of lipases as virulence factors for Candida albicans and Candida parapsilosis. Mycopathologia. 2011;172 : 169–178. doi: 10.1007/s11046-011-9429-3 21667319
42. Gácser A, Stehr F, Kröger C, Kredics L, Schäfer W, Nosanchuk JD. Lipase 8 affects the pathogenesis of Candida albicans. Infect Immun. 2007;75 : 4710–4718. 17646357
43. Schofield DA, Westwater C, Warner T, Balish E. Differential Candida albicans lipase gene expression during alimentary tract colonization and infection. FEMS Microbiol Lett. 2005;244 : 359–365. 15766791
44. Stehr F, Felk A, Gácser A, Kretschmar M, Mähnss B, Neuber K, et al. Expression analysis of the Candida albicans lipase gene family during experimental infections and in patient samples. FEMS Yeast Res. 2004;4 : 401–408. 14734020
45. Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev MMBR. 2003;67 : 400–428, table of contents. 12966142
46. Horváth P, Nosanchuk JD, Hamari Z, Vágvölgyi C, Gácser A. The identification of gene duplication and the role of secreted aspartyl proteinase 1 in Candida parapsilosis virulence. J Infect Dis. 2012;205 : 923–933. doi: 10.1093/infdis/jir873 22301631
47. Gabaldón T, Martin T, Marcet-Houben M, Durrens P, Bolotin-Fukuhara M, Lespinet O, et al. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics. 2013;14 : 623. doi: 10.1186/1471-2164-14-623 24034898
48. Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, et al. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010;6: e1000713. doi: 10.1371/journal.ppat.1000713 20072605
49. Dohm JC, Minoche AE, Holtgräwe D, Capella-Gutiérrez S, Zakrzewski F, Tafer H, et al. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature. 2014;505 : 546–549. doi: 10.1038/nature12817 24352233
50. Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1 : 18. doi: 10.1186/2047-217X-1-18 23587118
51. Huang S, Chen Z, Huang G, Yu T, Yang P, Li J, et al. HaploMerger: reconstructing allelic relationships for polymorphic diploid genome assemblies. Genome Res. 2012;22 : 1581–1588. doi: 10.1101/gr.133652.111 22555592
52. Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinforma Oxf Engl. 2011;27 : 578–579.
53. Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34: W435–439. 16845043
54. Pryszcz LP, Huerta-Cepas J, Gabaldón T. MetaPhOrs: orthology and paralogy predictions from multiple phylogenetic evidence using a consistency-based confidence score. Nucleic Acids Res. 2011;39: e32. doi: 10.1093/nar/gkq953 21149260
55. Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33: W116–120. 15980438
56. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9 : 357–359. doi: 10.1038/nmeth.1923 22388286
57. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20 : 1297–1303. doi: 10.1101/gr.107524.110 20644199
58. Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40: e115. 22730293
59. Schwartz DC, Cantor CR. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell. 1984;37 : 67–75. 6373014
60. Chu G, Vollrath D, Davis RW. Separation of large DNA molecules by contour-clamped homogeneous electric fields. Science. 1986;234 : 1582–1585. 3538420
61. Frith MC, Hamada M, Horton P. Parameters for accurate genome alignment. BMC Bioinformatics. 2010;11 : 80.: doi: 10.1186/1471-2105-11-80 20144198
62. Huerta-Cepas J, Capella-Gutierrez S, Pryszcz LP, Denisov I, Kormes D, Marcet-Houben M, et al. PhylomeDB v3.0: an expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions. Nucleic Acids Res. 2011;39: D556–560. doi: 10.1093/nar/gkq1109 21075798
63. Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147 : 195–197. 7265238
64. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32 : 1792–1797. 15034147
65. Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9 : 286–298. doi: 10.1093/bib/bbn013 18372315
66. Lassmann T, Frings O, Sonnhammer ELL. Kalign2: high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 2009;37 : 858–865. doi: 10.1093/nar/gkn1006 19103665
67. Wallace IM, O’Sullivan O, Higgins DG, Notredame C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 2006;34 : 1692–1699. 16556910
68. Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinforma Oxf Engl. 2009;25 : 1972–1973.
69. Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59 : 307–321. doi: 10.1093/sysbio/syq010 20525638
70. Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinforma Oxf Engl. 2005;21 : 456–463.
71. Dray S., Dufour A.B. The ade4 package: implementing the duality diagram for ecologists. J Stat Softw. 2007;22 : 1–20.
72. Wehe A, Bansal MS, Burleigh JG, Eulenstein O. DupTree: a program for large-scale phylogenetic analyses using gene tree parsimony. Bioinforma Oxf Engl. 2008;24 : 1540–1541.
73. Huerta-Cepas J, Dopazo J, Gabaldón T. ETE: a python Environment for Tree Exploration. BMC Bioinformatics. 2010;11 : 24. doi: 10.1186/1471-2105-11-24 20070885
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Single Strand Annealing Plays a Major Role in RecA-Independent Recombination between Repeated Sequences in the Radioresistant Bacterium
- The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals
- Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross
- DCA1 Acts as a Transcriptional Co-activator of DST and Contributes to Drought and Salt Tolerance in Rice
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy