
The Ciliopathy Protein CC2D2A Associates with NINL and Functions in RAB8-MICAL3-Regulated Vesicle Trafficking
Ciliopathies are a group of disorders caused by dysfunction of primary cilia, ubiquitous organelles involved in signal transduction. Mutations in CC2D2A cause two ciliopathies, Joubert and Meckel syndromes, and result in loss of ciliary protein localization. The mechanism by which CC2D2A, located at the ciliary transition zone, controls ciliary protein composition and its link to vesicular trafficking of incoming cargo remain largely unknown. Here, we identify a series of physical interactions linking CC2D2A to vesicular trafficking controlled by the small GTPase RAB8, suggesting a new model, whereby CC2D2A provides a specific docking point for ciliary-bound vesicles at the entrance to the ciliary compartment. We first identify NINL as a physical and genetic interaction partner of CC2D2A, show that both proteins co-localize at the entrance to the cilium and demonstrate that absence of Ninl or Cc2d2a result in similar retinal phenotypes in zebrafish, including mislocalization of Rab8. We further identify MICAL3, a protein known to bind RAB8, as another NINL interaction partner, thus linking CC2D2A to RAB8A-controlled trafficking. Finally, we describe an individual with Joubert syndrome, in whom combined CC2D2A and NINL mutations result in an enhanced phenotype, illustrating the impact of the detected interaction on the disease.
Published in the journal:
. PLoS Genet 11(10): e32767. doi:10.1371/journal.pgen.1005575
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005575
Summary
Ciliopathies are a group of disorders caused by dysfunction of primary cilia, ubiquitous organelles involved in signal transduction. Mutations in CC2D2A cause two ciliopathies, Joubert and Meckel syndromes, and result in loss of ciliary protein localization. The mechanism by which CC2D2A, located at the ciliary transition zone, controls ciliary protein composition and its link to vesicular trafficking of incoming cargo remain largely unknown. Here, we identify a series of physical interactions linking CC2D2A to vesicular trafficking controlled by the small GTPase RAB8, suggesting a new model, whereby CC2D2A provides a specific docking point for ciliary-bound vesicles at the entrance to the ciliary compartment. We first identify NINL as a physical and genetic interaction partner of CC2D2A, show that both proteins co-localize at the entrance to the cilium and demonstrate that absence of Ninl or Cc2d2a result in similar retinal phenotypes in zebrafish, including mislocalization of Rab8. We further identify MICAL3, a protein known to bind RAB8, as another NINL interaction partner, thus linking CC2D2A to RAB8A-controlled trafficking. Finally, we describe an individual with Joubert syndrome, in whom combined CC2D2A and NINL mutations result in an enhanced phenotype, illustrating the impact of the detected interaction on the disease.
Introduction
Primary cilia are microtubule-based organelles protruding from the apical surface of most differentiated vertebrate cell types where they play a crucial role in transduction of extra-cellular signals to the cell [1]. Cilia achieve this function by concentrating and regulating receptors and channels that are required for sensing these signals in their membrane domain. Consequently, the ciliary membrane has a distinct composition from that of the adjacent plasma membrane, despite them being continuous with each other [2]. The tight regulation required to maintain the specificity of the ciliary membrane composition is achieved by complex trafficking and sorting mechanisms at the entry point to the ciliary compartment, as well as by a diffusion barrier present at the base of the cilium [3,4]. The transition zone, at the base of the ciliary axoneme, plays a crucial role in this sorting mechanism [5,6]. Indeed, dysfunction of proteins normally localized at the transition zone leads to both abnormal access to the ciliary compartment for proteins that should not localize there and loss of normal localization for ciliary proteins [5,7]. The actual mechanism, by which these transition zone proteins contribute to this sorting of ciliary proteins, remains however largely unknown.
Mutations in transition zone proteins in humans lead to several ciliopathies such as Joubert syndrome. Ciliopathies are a group of human disorders caused by dysfunction of primary cilia and characterized by overlapping genetics and phenotypes [8]. As cilia are present on most vertebrate cells, their dysfunction can manifest as a wide array of phenotypic features affecting most organs systems [9]. Retinal dystrophy is a common finding in ciliopathies given that retinal photoreceptor outer segments, which are the site of the phototransduction cascade, are highly specialized primary cilia [10]. Joubert syndrome (JBTS) (OMIM 213300) is a prototypical ciliopathy with a phenotypic spectrum that can encompass most of the typical ciliopathy phenotypes [11,12]. It is characterized by a specific hindbrain malformation termed the molar tooth sign (MTS), in addition to which affected individuals may have retinal dystrophy, tubulo-interstitial kidney disease, liver fibrosis, skeletal dysplasia and polydactyly [13–15]. To date, mutations in over 27 different genes have been reported as an underlying cause for JBTS [12,16–20]. Most of these genes encode proteins associated in multi-protein complexes localized at the transition zone of the primary cilium [7,21].
Mutations in CC2D2A (Coiled-coil and C2-domains containing protein 2A) are the second most common genetic cause for JBTS, accounting for almost 9% of affected individuals [12,22]. Moreover, mutations in CC2D2A can also result in the genetically related and more severe Meckel syndrome, which is a perinatal-lethal disorder characterized by encephalocele, polydactyly, cystic kidneys and liver fibrosis [23]. CC2D2A is part of one of the ciliary transition zone complexes with several other JBTS proteins [7,21]. Two Cc2d2a mouse mutants have been described, presenting with severe brain malformation (holoprosencephaly), microphthalmia, curved body axis and randomized left-right axis, all typical ciliopathy-associated phenotypes [7,24]. Interestingly, mouse embryonic fibroblasts from one of the reported Cc2d2a-/- mice appear to lack cilia entirely [24] whereas disruption of CC2D2A function in the other reported mutant does not compromise ciliogenesis in mouse embryonic fibroblasts [7]. Instead, the ciliary localization of several proteins (including ARL13B, Adenylyl Cyclase III, Smoothened and Polycystin2) is lost, suggesting that presence of CC2D2A at the transition zone is required for appropriate targeting of proteins to the ciliary compartment [7]. The zebrafish cc2d2a mutant sentinel demonstrates a curved body axis, pronephric cysts and a striking retinal phenotype with short and dysmorphic photoreceptor outer segments [25]. In addition, the photoreceptors of cc2d2a mutants also show mislocalization of opsins in the cell body and cytoplasmic accumulation of vesicles in the apical portion of the cells and around the connecting cilium (equivalent of the transition zone in photoreceptors), suggesting a defect in opsin trafficking. Opsins are the photosensitive pigment molecules concentrated at high levels in the outer segments and required for sensing the light signal. Trafficking of opsins from the cell body towards the ciliary compartment is (at least in part) controlled by the small GTPase Rab8, which coats rhodopsin-carrier vesicles allowing their docking and fusion at the ciliary base [26]. Expression of a dominant-negative form of Rab8a leads to accumulation of rhodopsin-containing vesicles in photoreceptors [27]. In addition, RAB8A is also involved in ciliary membrane biogenesis in other cell types and thus appears to play a general role in orchestrating trafficking towards the ciliary compartment [28,29]. The trafficking defect observed in cc2d2a-/- photoreceptors appears to be mediated by loss of normal Rab8 localization [25] but the precise mechanism by which loss of this transition zone protein affects the localization of Rab8 and the trafficking of ciliary-directed opsins remains unclear.
In the current work, we identify a chain of physical interactions linking CC2D2A to RAB8A through NINL and MICAL3. Using a zebrafish model, we demonstrate that loss of Ninl function leads to a similar retinal phenotype as loss of Cc2d2a, including short outer segments, mislocalization of opsins and accumulation of vesicles. Based on the physical and genetic interactions that we identify, we propose a model in which CC2D2A provides a docking point at the photoreceptor ciliary base, allowing RAB8A-positive vesicles to bind through a series of interactions involving CC2D2A-NINL-MICAL3-RAB8A.
Results
CC2D2A associates with NINL
In order to shed light on the function of CC2D2A we performed a dedicated ciliary yeast two-hybrid assay with different fragments of CC2D2A against a panel of 164 proteins, containing most of the ciliopathy-associated proteins [30]. A direct binary interaction between CC2D2A and both isoforms (A and B) of the centrosome - and basal body-associated protein NINL (Ninein-like protein) was identified (Fig 1a). NINL isoforms A and B are distinguished by the fact that isoform B is 349 amino acids shorter due to skipping of the large exon 17 (S1a Fig). Both isoforms share predicted EF-hand domains in the N-terminal region as well as coiled-coil domains in the more C-terminal portion. Both isoforms display similar broad expression patterns, with the strongest expression patterns in cochlea, brain, testis, kidney and retina [31].

By generating deletion constructs for CC2D2A and subsequent evaluation of the interaction with NINL, we could pinpoint the interaction to the two predicted coiled-coil domains (433-637aa) present in CC2D2A (Fig 1a). Since CC2D2A and NINL isoform B demonstrated the strongest interaction (Fig 1a), we focused on NINL isoform B (NINLisoB) for confirmation and further investigation of this interaction. Co-immunoprecipitation assays performed using full-length tagged-constructs for NINLisoB and CC2D2A, showed co-precipitation of the two proteins. FLAG-tagged LRRK2 that was used as a negative control did not co-precipitate with HA-tagged NINLisoB, which confirmed the specificity of the interaction between NINLisoB and CC2D2A in this assay (Fig 1b). A reciprocal co-immunoprecipitation experiment confirmed the interaction between NINLisoB and CC2D2A (Fig 1c).
CC2D2A and NINL co-localize at the base of cilia independently of each other
To further validate the interaction between CC2D2A and NINLisoB in ciliated mammalian cells, we transfected hTERT-RPE1 cells (human telomerase reverse transcriptase retinal pigment epithelium cells) with expression-constructs of wild-type mRFP-tagged NINLisoB, eCFP-tagged CC2D2A or a combination of both. When expressed alone, eCFP-tagged CC2D2A localizes to the ciliary base (basal body, accessory centriole) and also (partly) to the ciliary transition zone, which was visualized using anti-RPGRIP1L as a marker (Fig 2a and 2b). mRFP-tagged NINL isoform B was localized at the ciliary base adjacent to the ciliary transition zone (Fig 2c and 2d). When co-expressed, NINLisoB and CC2D2A co-localized at the base of the primary cilium (Fig 2e-e”).

To investigate the localization and function of endogenous Ninl, we turned to the zebrafish model. The zebrafish genome harbors a single ninl orthologue (Genbank NP_001268727) that has 45% similarity with human NINL. Conserved domains include the predicted EF-hand domains and multiple coiled-coil domains. Cloning of zebrafish ninl from whole embryo mRNA at 5dpf revealed that all identified zebrafish transcripts lack the large exon 17 which is present only in human NINL isoform A but not in isoform B (S1A Fig). Therefore, zebrafish ninl is most similar to the shorter human NINL isoform B. RNA in situ hybridization with two different antisense probes derived from the 5’-end and the 3’-end of the zebrafish ninl transcript revealed broad expression at 14–18 somites in neural tube, inner ear, developing eye and pronephros. Expression persists in the retina at least up to 6 dpf (days post fertilization; last developmental stage assessed) (S1B Fig). Antibody staining showed punctate localization of endogenous Ninl in zebrafish retina at the base of the cilium in 4dpf larvae (Fig 2g). While co-staining with Cc2d2a antibodies was not possible due to different fixation conditions, co-staining of serial sections with anti-centrin and anti-Ninl or anti-Cc2d2a antibodies respectively revealed that both endogenous proteins partially co-localize at the base of the photoreceptor cilium of 4 dpf old zebrafish larvae (Fig 2f and 2g). We observed that Cc2d2a localizes slightly more apically with respect to Centrin than Ninl, consistent with Cc2d2a localization at the connecting cilium, while Ninl localization is overlapping more broadly with the basal body (Fig 2f and 2g, schematized in j).
In order to determine whether Cc2d2a localization is dependent on the presence of Ninl, we performed morpholino-induced knockdown studies in zebrafish. Injection of 2 ng/nl of ninl translation-blocking morpholino (atgMO) led to efficient knockdown of Ninl, as demonstrated by substantially decreased antibody staining in cryosections through morphant retina (S2a and S2b” Fig). On Western blots of whole 5dpf larval extracts, a single strong band of 80 kDa is present in wild-type fish (S2d Fig), which is consistent with results from immunoprecipitation from retinal bovine extracts with a previously published antibody against human NINL (S2e Fig [31]). This band is strongly reduced in ninl atgMO injected larvae (S2d Fig), supporting the specificity of the morpholino and of the antibody.
Ninl knockdown led to typical ciliopathy-associated phenotypes, including curved body shape, enlarged brain ventricle and pronephric cysts (S3a–S3g Fig). The specificity of the observed phenotype was confirmed by rescue experiments, in which co-injection of 2 ng/nl ninl MO with capped MO-resistant human NINL-mRNA reduced the prevalence of the curved body phenotype in a dose-dependent manner (curved body shape in 71% of ninl atgMO injected larvae (n = 207) versus 36% in ninl atgMO + ninl mRNA injected larvae (n = 203), data pooled from 2 biological replicates, P<0.0001, two-tailed Fisher’s exact test; S4a–S4d Fig). Finally, the specificity of the observed phenotypes was further confirmed by a second morpholino against ninl targeting the splice site at the intron14/exon15 junction and thus causing aberrant splicing with premature truncation (S5c Fig). This splice morpholino led to similar phenotypes as the atgMO, including ventriculomegaly and abnormal photoreceptor outer segments (S5a and S5b Fig). The body curvature phenotype was absent in the splice morphants, which may be explained either by rescue of this early phenotype by maternal ninl mRNA, which remains unaffected by splice morpholinos (as seen in some ciliopathy zebrafish mutants such as talpid3 where only the maternal zygotic mutants have a curved body shape [32]), or by less efficient gene knockdown with this morpholino, as normal transcript persists in addition to the aberrant transcript (S5c Fig). Indeed, using the anti-NINL antibody, we observed a milder decrease of Ninl protein on Western blots and on immuno-histochemistry of retinal cryosections at 5dpf for the ninl ex15 spMO as compared to the atgMO (S2c and S2d Fig).
Localization of Cc2d2a at the connecting cilium, shown by anti-Cc2d2a immunostaining, was unaffected by Ninl knockdown (Fig 2h). Conversely, immunostainings using anti-Ninl antibodies revealed no clear mislocalization of Ninl in the retina of cc2d2a-/- larvae (Fig 2i). Taken together, these data indicate that Cc2d2a and Ninl co-localize at the ciliary base independently of each other.
NINL knockdown in zebrafish leads to outer segment loss, opsin mislocalization and vesicle accumulation
Since cc2d2a-/- zebrafish have prominent retinal abnormalities [25], we focused our phenotypic analysis on the retina of ninl morphants. Retinal lamination was unaffected in ninl morphants (Fig 3a and 3b). In contrast, photoreceptors demonstrated shortened axonemes and abnormal outer segments, as seen on retinal cryosections at 4 dpf stained with boron-dipyrromethene (bodipy) to mark the outer segment membrane disks (Fig 3c and 3d’) and anti-acetylated alpha-tubulin and anti-Ift88 antibodies to mark the axoneme (Fig 3e and 3f). Measurement of outer segment (OS) length of early 4dpf larvae, performed in a blinded manner as to injection status, revealed a significant shortening (mean OS length 1.6 +/ - 0.26 μm in ninl atgMO morphants compared to 3.9 +/ - 0.32 μm in wild-type, P<0.0001, unpaired Student’s t-test, n>10 larvae from each group in each of 2 biological replicates; S4e Fig). This retinal phenotype was observed with both the ninl translation-blocking and the splice-blocking morpholinos (S5b’ Fig). Little to no photoreceptor cell death was observed with TUNEL assay on cryosections of 4dpf morphant larvae (S3i and S3j Fig) compared to ift88-/ - retinas that are known to display prominent photoreceptor cell death at the same stage (and were thus used as positive controls; S3k Fig). In general, minimal cell death was observed at 4dpf throughout the embryo, including in brain of larvae with overt ventriculomegaly (S3l Fig). Co-injection of 150pg of capped human NINL mRNA with 2ng/nl ninl atgMO restored normal outer segment length (mean OS length in rescued larvae 3.8 +/ - 0.25 μm, P<0.0001, unpaired Student’s t-test, n = 10 larvae; S4a’–S4c’ and S4e Fig).
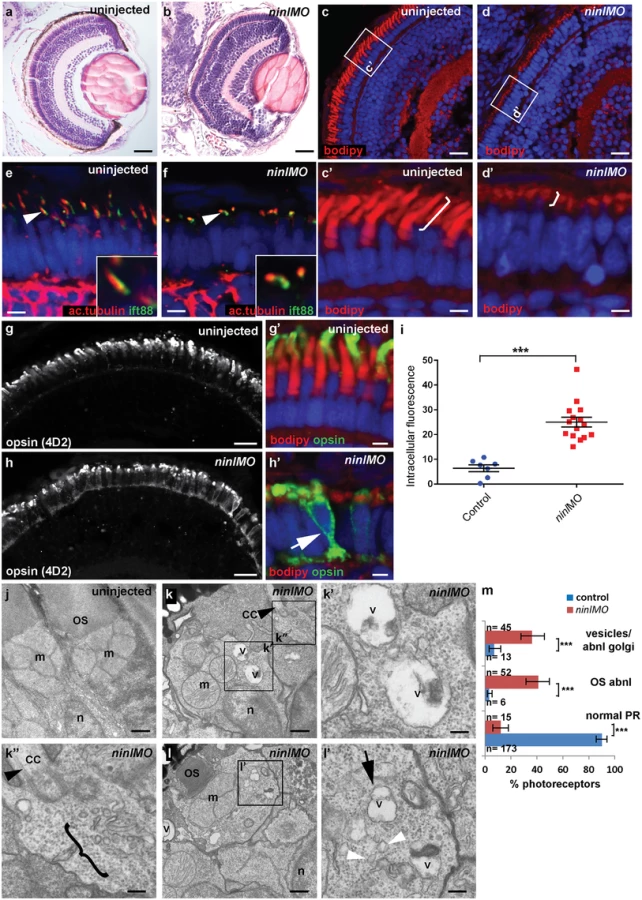
Immuno-staining with anti-opsin antibodies (4D2 antibody) demonstrated significant accumulation of opsins in the inner segment and throughout the cell body of ninl-depleted photoreceptors (Fig 3g–3i; mean intracellular fluorescence was significantly increased in ninl morphants compared to controls, P<0.0001, unpaired Student’s t-test, n = 15 morphant larvae and 7 control larvae, 2 replicate experiments). At the ultra-structural level, two types of abnormal membrane-bound structures were observed by transmission electron microscopy in ninl morphants: large vacuole-like structures were present in the cell body and small vesicular structures accumulated around the Golgi complex and below the connecting cilium (Fig 3j–3m; vacuolar and/or vesicular structures were present in 45/112 photoreceptors from 6 morphant eyes compared to 13/192 photoreceptors from 4 uninjected and 4 Control Oligo injected eyes; P<0.0001, Fisher’s exact test). These phenotypes are partially reminiscent of those observed in cc2d2a-/- embryos [25], supporting a common or coordinated function for Cc2d2a and Ninl in the process of vesicular trafficking towards the ciliary compartment.
Ninl genetically interacts with cc2d2a and may act as a genetic modifier for CC2D2A-associated Joubert Syndrome
To further delineate the relationship between cc2d2a and ninl, we tested whether a synergistic effect was detectable between the two genes by using partial ninl knockdown in the cc2d2a mutant background. We observed that injection of a sub-phenotypic dose of ninl MO (0.75 ng/nl), which causes no discernible phenotype in wild-type larvae, significantly increased the penetrance and severity of pronephric cysts in cc2d2a mutants: 89% of ninlMO-cc2d2a-/- zebrafish developed cysts compared to 40% of uninjected cc2d2a-/- larvae (p<0.0001, Fisher’s exact test) and the size of these cysts was significantly increased (as measured by the area of the dilated glomerulus and proximal tubules: 0.044 +/-0.004 mm2 for cc2d2a-/ - + ninlMO (n = 16) as compared to 0.016 +/-0.002 mm2 for uninjected cc2d2a-/ - (n = 8, P<0.0001, unpaired Student’s t-test) (Fig 4a–4d). Importantly, the cc2d2a+/ - and cc2d2a+/+ siblings from the same injection clutch did not develop pronephric cysts at these sub-phenotypic ninl MO doses (Fig 4a and 4d). In the retina, the opsin mislocalization phenotype in cc2d2a-/- larvae (4 dpf) was enhanced by the addition of the same sub-phenotypic dose of ninlMO (Fig 4e–4h; P<0.0001, Student’s t-test, n = 19 cc2d2a-/ - + ninlMO and n = 16 cc2d2a-/ - uninjected, 2 replicates). These findings support a genetic interaction between cc2d2a and ninl and suggest that NINL could be a genetic modifier for CC2D2A-caused disorders or even contribute to the genetic spectrum underlying Joubert/Meckel syndrome. Following this rationale, we sequenced NINL in a cohort of 346 individuals with Joubert syndrome (from 291 families) using a molecular inversion probes (MIPs) capture method followed by next-generation sequencing [33] but did not identify any individuals carrying bi-allelic rare deleterious NINL variants. We did however find 3 individuals with heterozygous NINL mutations predicted to be deleterious. Individual UW48-3 carried the homozygous missense CC2D2A mutation c. 3364C>T (p.P1122S), previously shown to be causal for Joubert syndrome, and a heterozygous NINL frameshift mutation leading to a stop codon after 43 amino acids (c.3020delC, p.P1007Lfs*43) (Fig 4i) (and no other rare deleterious variant in any of the known JS genes). Phenotypically, this subject had a severe form of JBTS with retinal dystrophy, hearing loss, ventriculomegaly in addition to the MTS and renal failure leading to death at age 7 years. In comparison, subject UW 36–3 carried the same homozygous CC2D2A c.3364C>T (p.P1122S) mutation but no additional NINL variants (or rare deleterious variants in other JBTS genes) and presented with the “pure JBTS” phenotype, consisting only of the MTS with associated ataxia, developmental delay and respiratory rhythm disturbance (Fig 4j). Subject UW07-3 carried a heterozygous NINL nonsense mutation (c.2446 G>A, p.R816X) in addition to causal, compound heterozygous C5ORF42 frameshift mutations (c.8726delG; p.A2909Qfs*4 and c.493delA, p.I165Yfs*17). This subject presented a classical Joubert phenotype without extra-neuronal manifestations, suggesting that the additional NINL frameshift had no effect on the clinical manifestations (Fig 4k). Finally, subject UW57-3 carried a heterozygous NINL missense mutation (c.1631A>T, p.E544V), predicted to be deleterious by Polyphen2, along with bi-allelic causal TMEM67 mutations (c.2825T>G, p.F942C and c.978+3 A>G). This individual had Joubert syndrome with coloboma but no retinal, renal or hepatic involvement (Fig 4l). Given the known association between TMEM67 mutations and coloboma [12,34], this additional feature is most likely explained by the causal gene mutations, while the additional NINL variant appears to have no obvious effect on the phenotype in individual UW57-3. While it remains possible that additional sequence variants in non-JBTS genes also contributed to the enhanced phenotype in individual UW48-3 and while our findings from a large human cohort remain of anecdotal nature given the rarity of this highly heterogeneous genetic disorder, taken together with the zebrafish experiments, they suggest that NINL may act as a genetic modifier specifically for CC2D2A-caused Joubert syndrome.

Ninl is required for correct Rab8a localization
Previous work on the cc2d2auw38 zebrafish mutant demonstrated that loss of Cc2d2a leads to abnormal Rab8a localization in retinal photoreceptors [25]. Given the opsin mislocalization and vesicle accumulation phenotypes observed in ninl morphants, the known role of Rab8a in opsin trafficking [27,35,36] and the interaction with cc2d2a demonstrated here, we next determined whether loss of Ninl function also had an effect on Rab8a localization. For this purpose, we used a transgenic construct that drives expression of mCherry-tagged Rab8a in wild-type zebrafish photoreceptors in a punctate manner [25]. When expressed in ninl morphants (atgMO), mCherry-tagged Rab8a localized in significantly fewer puncta than when expressed in controls (42% of expressing photoreceptors displayed Rab8 puncta in ninl morphants (n = 38/87 from 14 larvae) compared to 73% in uninjected controls (n = 48/66 from 13 larvae), p = 0.0005, two-tailed Fisher’s exact test; Fig 5a–5c). Instead, expression of the transgene was mostly diffuse throughout the photoreceptor cell body of ninl morphants. A similar result was obtained using an anti-Rab8a antibody that recognizes endogenous small Rab8a puncta, which are found throughout the cell body, concentrated at the synapse and in the inner and outer segments in controls (Fig 5d-d’). In ninl-knockdown larvae, the number of endogenous Rab8a puncta was significantly reduced (Fig 5e-e’ and quantification in f: the average number of puncta per μm2 was reduced to 0.04 +/ - 0.01 (or 1 puncta per 25 μm2) in ninl morphants as compared to 0.09 +/ - 0.01 (or 1 puncta per 11 μm2) in uninjected wild-type, P = 0.01, unpaired Student’s t-test), supporting a role for Ninl in Rab8 localization.

MICAL3 associates with NINL and is mislocalized in NINL and CC2D2A-depleted cells
In order to unravel the underlying molecular cause of the observed vesicle accumulation and to identify proteins that interact with NINL, we next generated N-terminal Strep/FLAG-tagged fusion proteins of NINL isoA and isoB. A single-step affinity purification combined with quantification by stable isotope labeling of amino acids in cell culture (SILAC) and tandem affinity purification (TAP) [37] were applied to isolate the protein complexes in their native functional states from human embryonic kidney 293T (HEK293T) cells. The complexes were subsequently analyzed by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). The identified interactome consisted of 174 unique proteins (Fig 6a, S1 table). An important association was found with multiple subunits of the cytoplasmic dynein 1-dynactin motor complex (DYNC1H1, DYNC1LI1, DYNC1LI2, DYNCI2, DYNLRB1, DCTN1-4, and DCTN6) which is involved in minus end–directed microtubule-associated transport. In addition, six actin-binding proteins (ARP1, ARP1B, ARP10, CAPZA1, CAPZA2 and CAPZB) and three subunits of Ca2+/calmodulin-dependent protein kinase II (CaMKII) (CAMK2A, CAMK2D, and CAMK2G), involved in non-canonical Wnt5a signaling, synaptic plasticity and kidney development [38], were found to associate with NINL. An additional relevant NINL interaction partner identified was MICAL3 (Microtubule-associated Monooxygenase, Calponin and LIM domain containing 3 protein), which is known to participate in a protein complex with RAB6 and RAB8 that is involved in the fusion of exocytotic vesicles [39], a process that appears to be deficient in the retina of cc2d2a mutants and ninl morphants. We validated the interaction between NINLisoB and MICAL3 by reciprocal co-immunoprecipitations (Fig 6b) and confirmed that endogenously expressed MICAL3 is present at the photoreceptor connecting cilium in rat retina (P20), partially overlapping with the cilium and basal body marker polyglutamylated tubulin (Fig 7b–7d’). In hTERT-RPE1 cells, mRFP-tagged NINLisoB (Fig 7e-e”) and eCFP-tagged CC2D2A (Fig 7f-f”) partially overlapped with tagged MICAL3.


To evaluate the role of NINL and CC2D2A in MICAL3 localization, we silenced the expression of NINL and CC2D2A in ciliated hTERT-RPE1 cells using siRNA, which was quantified by qPCR analysis (Fig 7j and 7k). Subsequent immunohistochemical stainings showed predominant MICAL3 localization at the ciliary base in non-targeting siRNA-treated cells (Fig 7g) whereas silencing of NINL expression resulted in a dispersed distribution of MICAL3 throughout the cell body (Fig 7h). Downregulation of CC2D2A expression in hTERT-RPE1 cells had a less pronounced effect on MICAL3 localization, resulting in partial mislocalization to the cell body (Fig 7i). These findings support a link between CC2D2A and MICAL3-RAB8-mediated vesicle trafficking/fusion through NINL.
Discussion
Dysfunction of transition zone proteins causes several ciliopathies such as Joubert syndrome, Meckel syndrome, nephronophthisis or Usher syndrome [16,21,40–43]. Previous work suggests that transition zone proteins in general, and CC2D2A in particular, are required for correct localization of transmembrane proteins to the ciliary membrane [7,25],. The mechanism by which transition zone proteins exert this function and the link to upstream ciliary-directed vesicular trafficking mechanisms remain however largely unknown. In this work, we identify NINL as a novel physical interaction partner for the transition zone protein CC2D2A and propose a model linking CC2D2A to RAB8A-controlled vesicle trafficking through a dual role for NINL in microtubule-based vesicle transport (Fig 8). The association of NINL with both the cytoplasmic dynein 1-dynactin motor complex (Dona et al, co-submitted manuscript) and MICAL3 supports a role for NINL in the initial transport of trans-Golgi network-derived RAB8A-MICAL3 coated vesicles towards the base of the photoreceptor cilium, while the association of NINL with CC2D2A provides a docking point for these incoming vesicles at the entrance of the ciliary compartment.

Ciliary transmembrane proteins are synthesized in the cell body and travel from the Golgi towards the cilium in vesicles which move along microtubules using a cytoplasmic dynein motor [44]. Once at the entrance of the ciliary compartment, these vesicles must dock and fuse with the periciliary membrane to deliver their cargo into the ciliary membrane [45]. This path has been particularly well studied in photoreceptors, where large quantities of opsins and membrane continuously have to replenish the disks which constitute this photo-sensitive structure [26,46,35]. Opsin trafficking is severely affected in both zebrafish cc2d2a mutants and ninl morphants, suggesting that both proteins play an important role in this transport which is crucial for the correct morphogenesis and homeostasis of the outer segments. Their co-localization at the base of the photoreceptor cilium could suggest that Cc2d2a and Ninl play a similar or combined role in opsin transport or that one protein is required to localize the other. However, since we found that each protein localizes independently of the other, and that ninl knockdown enhances the cc2d2a null mutant phenotype, the relationship between them is likely more complex than a simple linear pathway. At the ultrastructural level, the loss of function phenotypes of these two proteins also slightly diverge from each other: although vesicles accumulate in both cases in the affected photoreceptors, small vesiculo-tubular structures accumulate mostly apically around the connecting cilium in cc2d2a mutants [25], while this work shows that small vesicles and larger vacuoles are also present more basally and closer to an abnormal Golgi apparatus in ninl morphants. This suggests that both proteins are important for vesicular trafficking but play different roles in this process.
NINL has been previously shown to bind several other ciliopathy proteins present at the base of cilia, specifically LCA5 and USH2A [31], suggesting that it may play a more pivotal role in vesicular trafficking in photoreceptors than CC2D2A. Given that the zebrafish ninl morphant phenotype is more severe than the cc2d2a mutant phenotype, this further suggests a more central role for Ninl than for Cc2d2a in cilium-directed trafficking. This hypothesis is also supported by the lack of bi-allelic NINL mutations in a large human cohort of Joubert syndrome. Indeed, this may be interpreted as lack of tolerance to loss-of-function mutations in NINL, as these would lead to more severe phenotypes or early embryonic lethality. The direct interaction between NINL and the dynein 1-dynactin complex [47] which we confirmed and expanded in the associated study by Dona et al, suggests that NINL might be involved in minus end–directed microtubule-associated transport of organelles and cargo towards the base of the cilium. An appealing model would thus propose that NINL functions both more upstream in ciliary-directed vesicular trafficking than CC2D2A as well as at the base of the cilium where it interacts with several different proteins including CC2D2A.
While no bi-allelic rare deleterious NINL variants were identified in our JBTS cohort, we did find heterozygous NINL mutations in individuals with Joubert syndrome. Interestingly, only the individual with causal bi-allelic CC2D2A mutations and a heterozygous truncating NINL mutation had a severe phenotype with retinal and terminal renal disease. In comparison, the individuals with causal mutations in other JBTS genes and a heterozygous deleterious NINL mutation (or with the same causal CC2D2A mutation alone) had the classical “pure Joubert” phenotype without retinal or renal involvement. While bi-allelic CC2D2A mutations can result in a wide range of JBTS-associated phenotypes, the majority of individuals with causal CC2D2A mutations and JBTS display the “pure JBTS” phenotype [22]. The more severe phenotype only of the individual carrying causal CC2D2A mutations and an additional NINL truncating variant suggests that deleterious variants in NINL may act as genetic modifiers specifically of CC2D2A-caused ciliopathies such as Joubert syndrome. Unfortunately, the rarity of this disorder and its prominent genetic heterogeneity with over 27 associated genes prevent identification of multiple individuals sharing the same combination of causal and additional genetic variants, precluding identification of a statistically significant effect of rare variants as genetic modifiers using human genetics alone. Our findings from a large Joubert cohort therefore remain of anecdotal nature. However, the physical and genetic interaction in zebrafish identified in this work substantially strengthen the significance of this finding and suggest that deleterious variants in NINL may indeed enhance the retinal and renal phenotype in individuals with CC2D2A-associated Joubert syndrome. The effect on the retinal phenotype may be explained by the importance of NINL function in photoreceptors as highlighted in the present study. Enhancement of the renal phenotype by the additional NINL mutation may be explained by the association identified in this study between NINL and the PKD2-target CaMKII, which is important for renal development [38].
The identification of MICAL3 as an interaction partner for NINL is of particular relevance in the context of vesicular trafficking given that MICAL3 binds RAB8A and plays a role in exocytotic vesicle fusion [39]. MICAL3 is part of the MICAL family of flavoprotein monooxygenases which regulate the actin cytoskeleton by disassembling the actin filaments. The redox function of MICAL3 is required to promote vesicle fusion, possibly by destabilizing protein complexes and remodeling the docking-fusion complexes in which it is engaged [39]. The role of RAB8A in vesicle fusion at the ciliary base has been abundantly documented in various cell types including photoreceptors [27,28]. While RAB8A was found to bind several ciliopathy proteins directly including CEP290 and RGPR [48,49], no direct interaction has been demonstrated between CC2D2A and RAB8A, despite a functional interaction in zebrafish photoreceptors and a requirement for CC2D2A in RAB8A localization in mouse embryonic fibroblasts [24,25]. Our findings now provide a model explaining the link between CC2D2A and RAB8A (Fig 8): RAB8A-coated vesicles destined to the ciliary compartment are bound by MICAL3 which in turn binds NINL that is associated to the cytoplasmic dynein 1 motor complex (Dona et al, companion manuscript), allowing the movement along the microtubules. Once at the base of the cilium, NINL interacts with CC2D2A, providing the specificity of the docking point at the entrance to the ciliary compartment. Finally, the redox activity of MICAL3 promotes remodeling of the complex allowing fusion of the vesicle and release of cargo into the peri-ciliary membrane.
A role for CC2D2A in promoting the assembly of ciliary subdistal appendages was recently suggested whereby CC2D2A would be required for docking of transport vesicles [24]. This is compatible with our model which also provides a possible mechanism to explain how transition zone proteins may regulate ciliary protein composition by providing specific docking points at the entry to the ciliary compartment. Dysfunction of transition zone proteins can lead to a variety of ciliopathies and it is likely that abnormal ciliary protein composition is at least in part responsible for the observed disease phenotypes even in the absence of ciliogenesis defects. This provides an opportunity for the development of pathway-specific therapies aiming at modulating trafficking routes and restoring normal ciliary protein content. In this perspective, unraveling the cell biological function of disease genes such as CC2D2A as presented in the current study is a prerequisite for the future development of pharmacological treatments for patients with ciliopathies.
Materials and Methods
Ethics statement
All animal protocols were in compliance with Swiss legal ethical guidelines and the European Union Regulatory Agency guidelines for the use of fish in biomedical research and experiments and were approved by the local authorities (Veterinäramt Zürich TV4206). Human Subject Research Procedures were approved by the Institutional Review Boards at the University of Washington and Seattle Children’s Hospital (IRB-UW # 28853), and all participants or their legal representatives provided written informed consent.
Zebrafish
Zebrafish (Danio Rerio) were maintained as described [50]. The cc2d2aw38/sentinel mutant (referred to as cc2d2a mutant or cc2d2a-/-) was previously described [25,51,52]. The transgenic Tg(wt1b:EGFP) line was previously described [53]. Embryos were raised at 28°C in embryo medium and pigment development was inhibited by phenylthiourea as described in Westerfield [50]. ninl translation-blocking (5’-CATCCTCGTCCATCCCACCACATAC-3’) morpholino (MO) and splice blocking (5’-CCCAACACTAAAGAGATACACCAAT-3’) morpholinos were designed by Gene Tools Inc. (USA) and 1nl was injected into zebrafish embryos at the one-cell stage. After a titration curve, we established that 2ng/nl was the optimal phenotypic dose consistently causing the major phenotypes without significant cell death, while at the low dose of 0.75ng/nl, no phenotypes were observed (therefore called the “sub-phenotypic dose”). For the splice morpholino, the optimal phenotypic dose was 4ng/nl. For rescue experiments, cDNA encoding full length human NINL isoform B was cloned into a pCS2+ vector made compatible with the Gateway system (Invitrogen, USA), pCS2+/DEST, and subsequently transcribed with the SP6 Message Machine kit (Ambion, USA) according to manufacturer’s instructions. The cherry-Rab8a construct was previously described [25]. All quantifications were performed blinded as to injection status. All animal protocols were in compliance with internationally recognized guidelines for the use of fish in biomedical research and experiments and were approved by the local authorities (Veterinäramt Zürich TV4206).
Plasmids
pDONR201 vectors containing cDNA encoding human NINL isoform A and B as well as aa 1–998, aa 433–637, aa 992–1177 of human CC2D2A were previously described [31,52]. Using Gateway cloning technology, cDNA fragments encoding aa 992–1620 and aa 1171–1620 of human CC2D2A (NM_001080522) were cloned in pDONR201 according to manufacturer’s instructions. pEGFP-C1-MICAL3 was kindly provided by Dr. A. Akhmanova (Utrecht University, The Netherlands).
Yeast two-hybrid interaction assay
The direct interaction between CC2D2A and other ciliary proteins was tested using a GAL4-based yeast two-hybrid system (Hybrizap, Stratagene, USA) as previously described [30]. The DNA binding domain (GAL4-BD) fused to full length CC2D2A was used as a bait to test the interaction with previously described ciliopathy and cilium-associated proteins fused to an activation domain (GAL4-AD). Constructs encoding GAL4-BD and GAL4-AD fusion proteins were co-transformed in yeast strain PJ69-4A. The direct interaction between baits and preys induced the activation of the reporter genes, resulting in the growth of yeast colonies on selective media (deficient of histidine and adenine) and induction of α-galactosidase and β-galactosidase colorimetric reactions [54].
Affinity purification of protein complexes
HEK293T cells transiently expressing the SF-TAP tagged NINLisoB were grown in SILAC DMEM (PAA) supplemented with 3 mM l-glutamine (PAA), 10% dialyzed fetal bovine serum (PAA), 0.55 mM lysine, and 0.4 mM arginine. Light SILAC medium was supplemented with 12C6,14N2 lysine and 12C6,14N4 arginine. Heavy SILAC medium was supplemented with either 13C6 lysine and 13C6,15N4 arginine or 13C6,15N2 lysine and 13C6,15N4 arginine. 0.5 mM proline was added to all SILAC media to prevent arginine-to-proline conversion [55]. All amino acids were purchased from Silantes. For one-step Strep purifications, SF-TAP–tagged proteins and associated protein complexes were purified essentially as described previously [37,56]. HEK293T cells transiently expressing the SF-TAP tagged constructs were lysed in lysis buffer containing 0.5% Nonidet-P40, protease inhibitor cocktail (Roche), and phosphatase inhibitor cocktails I and II (Sigma-Aldrich) in TBS (30 mM Tris-HCl, pH 7.4, and 150 mM NaCl) for 20 minutes at 4°C. After sedimentation of nuclei at 10,000 g for 10 minutes, the cleared lysates were transferred to Strep-Tactin-Superflow beads (IBA) and incubated for 1 hour before the resin was washed 3 times with wash buffer (TBS containing 0.1% NP-40 and phosphatase inhibitor cocktails I and II). The protein complexes were eluted by incubation for 10 minutes in Strep-elution buffer (IBA). After purification, the samples were precipitated with chloroform and methanol and subjected to in-solution tryptic cleavage as described previously [57]. LC-MS/MS analysis was performed on an Ultimate3000 nano HPLC system (Dionex) coupled to a LTQ OrbitrapXL mass spectrometer (Thermo Fisher Scientific) by a nanospray ion source. The raw data were analyzed using Sequest (Thermo Fisher Scientific) or Mascot and Scaffold (Proteome Software) as described previously [57]. Proteins were considered to be specific protein complex components if they were not detected in the control and were detected at least twice with two or more peptides (peptide probability >80%) in three experiments. The protein probability threshold was set to 99%.
Knockdown of NINL and CC2D2A in cultured hTERT-RPE1 cells by RNAi
Three Silencer Select siRNAs targeting NINL and CC2D2A were purchased from Life Technologies (targeting sequences are listed in S2 Table). For transfection, a pool of three siRNAs per gene (45 nM final concentration) were plated in MW12 plates with or without glass slides. Lipofectamine RNAiMax (LifeTechnologies) and Opti-MEM (LifeTechnologies) were added to the duplexes and incubated for 10–20 minutes according to manufacturer’s protocol to allow the formation of transfection complexes. Human telomerase reverse transcriptase-transformed retinal pigment epithelium (hTERT-RPE1) cells from American Type Culture Collection (ATCC) were then plated in MW12 plates. Per plate, non-targeting Silencer Select duplexes (LifeTechnologies) were included as negative controls. After 24 hours of transfection, cells were serum-starved to induce ciliogenesis. After 72 hours of transfection, knockdown-efficiency was determined by isolating total RNA from one 12-well with Trizol (Invitrogen, USA), followed by first-strand cDNA synthesis (iScript; Bio-Rad, USA). Quantitative PCRs using GoTaq (Promega), with validated NINL-, CC2D2A- and GUSB-specific primers (sequences are listed in S3 Table), were performed as previously described [31]. The second 12-well of cells were fixed with 2% paraformaldehyde, permeabilized with 1% Triton-X-100/PBS and stained with anti-MICAL3 antibodies (kindly provided by Dr. A. Akhmanova). Images were taken with an Axioplan2 Imaging fluorescence microscope (Zeiss, Germany) equipped with a DC350FX camera (Zeiss, Germany).
Co-immunoprecipitation in HEK293T cells
HA-tagged NINL isoform B was expressed by using the mammalian expression vector pcDNA3-HA/DEST, FLAG-tagged CC2D2A, LRRK2 and STRAD by using p3xFLAG-CMV/DEST and strep-FLAG-tagged NINL isoform B by using pNTAPe5/DEST from the Gateway cloning system (Invitrogen, USA). eGFP and eGFP-tagged MICAL3 were expressed from pEGFP-C1 (Clontech, USA). All plasmids contain a CMV promoter. HEK293T cells were co-transfected using Effectene (Qiagen, USA) according to manufacturer’s instructions. Twenty-four hours after transfection cells were washed with PBS and subsequently lysed on ice in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton-X-100 supplemented with complete protease inhibitor cocktail (Roche, Germany)). HA-tagged NINL isoform B was immunoprecipitated from cleared lysates overnight at 4°C by using rat monoclonal anti-HA-beads (Roche, Germany), while FLAG-tagged CC2D2A, LRRK2, STRAD and NINL isoform B were immunoprecipitated by using monoclonal anti-FLAG M2 Agarose beads (Sigma, Germany) and eGFP-tagged MICAL3 was immunoprecipitated using anti-GFP polyclonal antibodies (Abcam) coupled to ProtA/G beads (Santa Cruz, USA). After 4 washes in lysis buffer, the protein complexes were analyzed on immunoblots using the Odyssey Infrared Imaging System (LI-COR, USA). Tagged molecules were detected by anti-HA, anti-FLAG or anti-GFP mono - or polyclonal antibodies. As secondary antibody IRDye800 goat-anti-mouse IgG (Rockland Antibodies and Assays) and Alexa Fluor 680 goat-anti-rabbit IgG (Life Technologies) were used.
Immunohisto - and immunocytochemistry
Zebrafish larvae were fixed in 4% paraformaldehyde (PFA) overnight at 4°C, embedded in OCT and cryosectioned following standard protocols. Sections were blocked using PBDT (PBS, 1% DMSO, 0.1% Triton X, 2mg/ml BSA) with 10% goat serum for 30 minutes at RT before incubation with primary antibodies overnight. Primary antibodies were mouse monoclonal anti-acetylated alpha tubulin (1 : 500, clone 6-11B-1, Sigma), mouse monoclonal anti-polyglutamylated tubulin GT335 (1 : 500, gift from C. Janke, Institut Curie, France), mouse anti-zebrafish Cc2d2a (1 : 20, [25]), rabbit anti-NINL (1 : 100; LSBio Cat# LS-C201509), mouse anti-pan centrin 20H5 (1 : 200, clone 20H5 Millipore), mouse anti-Rab8a (1 : 100, clone 3G1 Novus Biologicals), mouse anti-opsin 4D2 (1 : 100, gift from R. Molday, University of British Columbia) and rabbit anti-Ift88 (gift from B. Perkins [58], Cleveland Clinic), mouse monoclonal anti-FLAG (1 : 1000, Sigma), rabbit polyclonal anti-human MICAL3 [39]. Secondary antibodies were Alexa Fluor goat anti-rabbit or goat anti-mouse IgG (Life Technologies) used at 1 : 300. Bodipy (1 : 300, Invitrogen) was applied for 20 minutes after the secondary antibodies and nuclei were counterstained with DAPI. Rab8 puncta detected by immuno-staining using the mouse anti-Rab8 antibody were analyzed blinded as to injection status in ImageJ. A region of interest was manually determined on single confocal sections and was thresholded (allways with the same parameters); the “analyze particles” function of ImageJ was then used to determine the number of puncta per μm2. For quantification of intracellular fluorescence after 4D2 (opsin) immuno-staining, a region of interest including 10–15 photoreceptor cell bodies was determined on single confocal sections using ImageJ and the mean grey value was measured. For quantification of the proximal pronephric area, the fluorescent region corresponding to the glomerulus and the proximal tubules up to the curved part of the tubule was outlined manually in ImageJ and the “measure” function was used to determine the area of the outlined region. All quantifications were performed blinded as to injection status. Confocal imaging was performed on a Leica HCS LSI.
Paraffin sections and Transmission Electron Microscopy
For paraffin sections, 4 dpf old ninl morphant larvae were fixed in 4% PFA overnight at 4°C, embedded in paraffin and sectioned following standard protocols. For Transmission Electron Microsopy, ninl morphant and control larvae were fixed overnight at 4°C in a freshly prepared mixture of 2,5% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodiumcacodylate buffer (pH 7.4). After rinsing in buffer, specimens were post-fixed in a freshly prepared mixture, containing 1% osmiumtetroxide and 1% potassiumferrocyanide in 0.1 M sodiumcacodylate buffer (pH 7.4), during 2 h at room temperature. After rinsing, tissues were dehydrated through a graded series of ethanol and embedded in epon. Ultrathin (rostrocaudally) sections (70nm), comprising zebrafish eyes at the optic nerve level, were collected on formvar coated grids, subsequently stained with 2% uranyl acetate and Reynold’s lead citrate, and examined with a Jeol1010 electron microscope.
Sequencing of NINL in a cohort of Joubert syndrome patients
346 individuals (from 291 families) with Joubert syndrome (JBTS) from the University of Washington Joubert Research Center were examined for mutations in NINL. Minimal enrollment criteria included clinical findings of JBTS (intellectual impairment, hypotonia, ataxia) and diagnostic or supportive brain imaging findings, or presence of a sibling with JBTS along with supportive clinical or imaging features. Procedures were approved by the Institutional Review Boards at the UW and Seattle Children’s Hospital, and all participants or their legal representatives provided written informed consent. Genomic DNA from peripheral blood or saliva was extracted and all NINL exons were captured by Molecular Inversion Probes (MIPS) [33]. Captured DNA was PCR amplified and sequenced on either the Illumina HiSeq or MiSeq platform. Sequence reads were mapped using the Burrows-Wheeler Aligner (BWA v.0.5.9). Variants were called using the Genome Analysis Tookit (GATK v2.5–2) and annotated with SeattleSeq (http://snp.gs.washington.edu/SeattleSeqAnnotation138/). Minimal quality criteria for analyzed variants were DP (Depth) ≥ 8, QD (Quality by Depth) > 5, and ABHet (Heterozygous Allele Balance) <0.8. The variant list was then filtered for rare and deleterious variants. Only variants with minor allele frequency of <1% were considered given the rarity of JBTS (estimated prevalence 1/80’000 [11]). All non-sense, frameshift and canonical splice-site variants, as well as missense variants with Polyphen2 scores >0.8 were considered deleterious. Selected variants were Sanger confirmed.
Statistical analyses
For all quantifications of zebrafish experiments, the Graphpad Prism6 software (http://www.graphpad.com/scientific-software/prism/) was employed to generate scatter plots, calculate mean values and SEM values, and perform statistical tests. Continuous data was analyzed using two-tailed, unpaired Student’s t-test and categorical data was analyzed using Fisher’s exact test.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010; 11 (5): 331–344. doi: 10.1038/nrg2774 20395968
2. Tyler KM, Fridberg A, Toriello KM, Olson CL, Cieslak JA et al. Flagellar membrane localization via association with lipid rafts. J Cell Sci. 2009; 122 (6): 859–866.
3. Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV et al. A Septin Diffusion Barrier at the Base of the Primary Cilium Maintains Ciliary Membrane Protein Distribution. Science. 2010; 329 (5990): 436–439. doi: 10.1126/science.1191054 20558667
4. Nachury MV, Seeley ES, Jin H. Trafficking to the Ciliary Membrane: How to Get Across the Periciliary Diffusion Barrier. Annu Rev Cell Dev Biol. 2010; 26 (1): 59–87.
5. Williams CL, Li C, Kida K, Inglis PN, Mohan S et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011; 192 (6): 1023–1041. doi: 10.1083/jcb.201012116 21422230
6. Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO reports. 2012; 13 (7): 608–618. doi: 10.1038/embor.2012.73 22653444
7. Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011; 43 (8): 776–784. doi: 10.1038/ng.891 21725307
8. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011; 364 (16): 1533–1543. doi: 10.1056/NEJMra1010172 21506742
9. Badano JL, Mitsuma N, Beales PL, Katsanis N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu Rev Genom Human Genet. 2006; 7 (1): 125–148.
10. Insinna C, Besharse JC.Intraflagellar transport and the sensory outer segment of vertebrate photoreceptors. Dev Dyn. 2008; 237 (8): 1982–1992. doi: 10.1002/dvdy.21554 18489002
11. Parisi MA, Glass I. Joubert syndrome and related disorders. Genereviews. 2003. http://www.ncbi.nlm.nih.gov/boooks/NBK1325/
12. Bachmann-Gagescu R, Dempsey JC, Phelps IG, O'Roak BJ, Knutzen DM et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015; 52 (8): 514–22. doi: 10.1136/jmedgenet-2015-103087 26092869
13. Maria BL, Hoang KBN, Tusa RJ, Mancuso AA, Hamed LM et al. "Joubert Syndrome" Revisited: Key Ocular Motor Signs With Magnetic Resonance Imaging Correlation. J Child Neurol. 1997; 12 (7): 423–430. 9373798
14. Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013; 12 (9): 894–905. doi: 10.1016/S1474-4422(13)70136-4 23870701
15. Doherty D. Joubert Syndrome: Insights Into Brain Development, Cilium Biology, and Complex Disease. Semin Pediatr Neurol. 2009; 16 (3): 143–154. doi: 10.1016/j.spen.2009.06.002 19778711
16. Romani M, Micalizzi A, Valente EM Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013; 12 (9): 894–905. doi: 10.1016/S1474-4422(13)70136-4 23870701
17. Romani M, Micalizzi A, Kraoua I, Dotti M, Cavallin M et al. Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J Rare Dis. 2014;9 (1): 72.
18. Tuz K, Bachmann-Gagescu R, O’Day DR, Hua K, Isabella CR et al. Mutations in CSPP1 Cause Primary Cilia Abnormalities and Joubert Syndrome with or without Jeune Asphyxiating Thoracic Dystrophy. Am J Hum Genet. 2014; 94 (1): 62–72. doi: 10.1016/j.ajhg.2013.11.019 24360808
19. Bachmann-Gagescu R, Phelps IG, Dempsey JC, Sharma VA, Ishak GE, et al. KIAA0586 is mutated in Joubert Syndrome.Hum Mutat. 2015; 36 (9): 831–5. doi: 10.1002/humu.22821 26096313
20. Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M et al. A Homozygous PDE6D Mutation in Joubert Syndrome Impairs Targeting of Farnesylated INPP5E Protein to the Primary Cilium. Hum Mutat. 2014; 35 (1): 137–146. 24166846
21. Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ et al. Mapping the NPHP-JBTS-MKS Protein Network Reveals Ciliopathy Disease Genes and Pathways. Cell. 2011; 145 (4): 513–528. doi: 10.1016/j.cell.2011.04.019 21565611
22. Bachmann-Gagescu R, Ishak GE, Dempsey JC, Adkins J, O'Day D et al. Genotype–phenotype correlation in CC2D2A-related Joubert syndrome reveals an association with ventriculomegaly and seizures. J Med Genet. 2012; 49 (2): 126–137. doi: 10.1136/jmedgenet-2011-100552 22241855
23. Mougou-Zerelli S, Thomas S, Szenker E, Audollent S, Elkhartoufi N et al. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype–phenotype correlation. Hum Mutat. 2009; 30 (11): 1574–1582. doi: 10.1002/humu.21116 19777577
24. Veleri S, Manjunath SH, Fariss RN, May-Simera H, Brooks M et al. Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis. Nat Commun. 2014; 20 (5):4207.
25. Bachmann-Gagescu R, Phelps IG, Stearns G, Link BA, Brockerhoff SE et al. The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum Mol Genet. 2011; 20 (20): 4041–4055. doi: 10.1093/hmg/ddr332 21816947
26. Deretic D, Wang J. Molecular assemblies that control rhodopsin transport to the cilia. Vision Res. 2012; 75 (0): 5–10.
27. Moritz OL, Tam BM, Hurd LL, Peränen J, Deretic D et al. Mutant rab8 Impairs Docking and Fusion of Rhodopsin-bearing Post-Golgi Membranes and Causes Cell Death of TransgenicXenopus Rods. Mol Biol Cell. 2001; 12 (8): 2341–2351. 11514620
28. Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J et al. A Core Complex of BBS Proteins Cooperates with the GTPase Rab8 to Promote Ciliary Membrane Biogenesis. Cell. 2007; 129 (6): 1201–1213. 17574030
29. Westlake CJ, Baye LM, Nachury MV, Wright KJ, Ervin KE et al. Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. PNAS. 2011; 108 (7): 2759–2764. doi: 10.1073/pnas.1018823108 21273506
30. Cevik S, Sanders AAWM, van Wijk E, Boldt K, Clarke L et al. Active Transport and Diffusion Barriers Restrict Joubert Syndrome-Associated ARL13B/ARL-13 to an Inv-like Ciliary Membrane Subdomain. PLoS Genet. 2013; 9 (12): e1003977 EP. doi: 10.1371/journal.pgen.1003977 24339792
31. van Wijk E, Kersten FFJ, Kartono A, Mans DA, Brandwijk K et al. Usher syndrome and Leber congenital amaurosis are molecularly linked via a novel isoform of the centrosomal ninein-like protein. Hum Mol Genet. 2009; 18 (1): 51–64. doi: 10.1093/hmg/ddn312 18826961
32. Ben J, Elworthy S, Ng ASM, van Eeden F, Ingham PW. Targeted mutation of the talpid3 gene in zebrafish reveals its conserved requirement for ciliogenesis and Hedgehog signalling across the vertebrates. Development. 2011; 138 (22): 4969–4978. doi: 10.1242/dev.070862 22028029
33. O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science. 2012; 338 (6114): 1619–1622. doi: 10.1126/science.1227764 23160955
34. Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet. 2010; 47 (1): 8–21. doi: 10.1136/jmg.2009.067249 19574260
35. Wang J, Deretic D. Molecular complexes that direct rhodopsin transport to primary cilia. Prog Retin Eye Res. 2014; 38 (0): 1–19.
36. Wang J, Morita Y, Mazelova J, Deretic D. The Arf GAP ASAP1 provides a platform to regulate Arf4 ‐ and Rab11–Rab8‐mediated ciliary receptor targeting. EMBO J. 2012; 31 (20): 4057–4071. doi: 10.1038/emboj.2012.253 22983554
37. Gloeckner CJ, Boldt K, Schumacher A, Roepman R, Ueffing M. A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics. 2007; 7 (23): 4228–4234. 17979178
38. Rothschild SC, Francescatto L, Drummond IA, Tombes RM. CaMK-II is a PKD2 target that promotes pronephric kidney development and stabilizes cilia. Development. 2011; 138 (16): 3387–3397. doi: 10.1242/dev.066340 21752935
39. Grigoriev I, Yu KL, Martinez-Sanchez E, Serra-Marques A, Smal I et al. Rab6, Rab8, and MICAL3 Cooperate in Controlling Docking and Fusion of Exocytotic Carriers. Curr Biol. 2011; 21 (11): 967–974. doi: 10.1016/j.cub.2011.04.030 21596566
40. Wolfrum U1 Liu X, Schmitt A, Udovichenko IP, Williams DS. Myosin VIIa as a common component of cilia and microvilli. Cell Motil Cytoskeleton. 1998; 40 (3): 261–271. 9678669
41. Maerker T, van Wijk E, Overlack N, Kersten FFJ, McGee J et al. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum Mol Genet. 2008; 17 (1): 71–86. 17906286
42. van Wijk E, van der Zwaag B, Peters T, Zimmermann U, te Brinke H et al. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum Mol Genet. 2006; 15 (5): 751–765. 16434480
43. Kremer H, van Wijk E, Märker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006; 15 (suppl 2): R262.
44. Madhivanan K, Aguilar RC. Ciliopathies: The Trafficking Connection. Traffic. 2014; 15 (10): 1031–1056. doi: 10.1111/tra.12195 25040720
45. Hsiao Y, Tuz K, Ferland R (2012) Trafficking in and to the primary cilium. Cilia 1 (1): 4. doi: 10.1186/2046-2530-1-4 23351793
46. Kennedy B, Malicki J. What drives cell morphogenesis: A look inside the vertebrate photoreceptor. Dev Dyn. 2009; 238 (9): 2115–2138. doi: 10.1002/dvdy.22010 19582864
47. Casenghi M, Barr FA, Nigg EA. Phosphorylation of Nlp by Plk1 negatively regulates its dynein-dynactin-dependent targeting to the centrosome. J Cell Sci. 2005; 118 (21): 5101–5108.
48. Kim J, Krishnaswami SR, Gleeson JG, CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet. 2008; 17 (23): 3796–3805. doi: 10.1093/hmg/ddn277 18772192
49. Murga-Zamalloa CA, Atkins SJ, Peranen J, Swaroop A, Khanna H. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum Mol Genet. 2010; 19 (18): 3591–3598. doi: 10.1093/hmg/ddq275 20631154
50. Westerfield M The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio). University of Oregon Press, Eugene, OR; 2000.
51. Owens KN, Santos F, Roberts B, Linbo T, Coffin AB et al. Identification of Genetic and Chemical Modulators of Zebrafish Mechanosensory Hair Cell Death. PLoS Genet. 2008; 4 (2): e1000020 EP. doi: 10.1371/journal.pgen.1000020 18454195
52. Gorden NT, Arts HH, Parisi MA, Coene KLM, Letteboer SJF et al. CC2D2A Is Mutated in Joubert Syndrome and Interacts with the Ciliopathy-Associated Basal Body Protein CEP290. Am J Hum Genet. 2008; 83 (5): 559–571. doi: 10.1016/j.ajhg.2008.10.002 18950740
53. Perner B, Englert C, Bollig F. The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev Bio. 2007; 309 (1): 87–96.
54. Letteboer SF, Roepman R (2008) Versatile Screening for Binary Protein-Protein Interactions by Yeast Two-Hybrid Mating. In: Thompson J, Ueffing M, Schaeffer-Reiss C, editors. Functional Proteomics: Humana Press. pp. 145–159; 2008.
55. Bendall SC, Hughes C, Stewart MH, Doble B, Bhatia M et al. Prevention of Amino Acid Conversion in SILAC Experiments with Embryonic Stem Cells. Mol Cell Proteomics. 2008; 7 (9): 1587–1597. doi: 10.1074/mcp.M800113-MCP200 18487603
56. den Hollander AI, Koenekoop RK, Mohamed MD, Arts HH, Boldt K et al. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat Genet. 2007; 39 (7): 889–895. 17546029
57. Gloeckner CJ, Boldt K, Ueffing M. Strep/FLAG Tandem Affinity Purification (SF-TAP) to Study Protein Interactions. Current Protocols in Protein Science: John Wiley & Sons Inc.; 2001.
58. Krock BL, Perkins BD. The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle–kinesin-II dissociation in vertebrate photoreceptors. J Cell Sci. 2008; 121 (11): 1907–1915.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Single Strand Annealing Plays a Major Role in RecA-Independent Recombination between Repeated Sequences in the Radioresistant Bacterium
- The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals
- Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross
- DCA1 Acts as a Transcriptional Co-activator of DST and Contributes to Drought and Salt Tolerance in Rice
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy