
MET18 Connects the Cytosolic Iron-Sulfur Cluster Assembly Pathway to Active DNA Demethylation in
DNA cytosine methylation is a major epigenetic mark that confers transcriptional regulation. Active removal of DNA methylation is important for plants and mammals during development and in responses to various stress conditions. In the model plant species Arabidopsis thaliana, active DNA demethylation depends on a family of 5-methylcytosine DNA glycosylases/demethylases including ROS1, DME, and others. While the epigenetic function of this demethylase family is well-known, little is known about how their enzymatic activities may be regulated. In this report, we carried out a forward genetic screen for anti-silencing factors and identified MET18, a conserved component of cytosolic iron-sulfur cluster assembly (CIA) pathway in eukaryotes, as being required for the ROS1-dependent active DNA demethylation. Dysfunction of MET18 causes DNA hyper-methylation at thousands of genomic loci where DNA methylation is pruned by ROS1. In addition, ROS1 physically interacts with MET18 and other CIA pathway components; while a conserved iron-sulfur-binding motif is indispensable for ROS1 enzyme activity. Our results suggested that MET18 affects DNA demethylation by influencing ROS1 enzymatic activity via direct interaction with the iron-sulfur-binding motif of ROS1, highlighting a direct connection between iron-sulfur cluster assembly and active DNA demethylation.
Published in the journal:
. PLoS Genet 11(10): e32767. doi:10.1371/journal.pgen.1005559
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005559
Summary
DNA cytosine methylation is a major epigenetic mark that confers transcriptional regulation. Active removal of DNA methylation is important for plants and mammals during development and in responses to various stress conditions. In the model plant species Arabidopsis thaliana, active DNA demethylation depends on a family of 5-methylcytosine DNA glycosylases/demethylases including ROS1, DME, and others. While the epigenetic function of this demethylase family is well-known, little is known about how their enzymatic activities may be regulated. In this report, we carried out a forward genetic screen for anti-silencing factors and identified MET18, a conserved component of cytosolic iron-sulfur cluster assembly (CIA) pathway in eukaryotes, as being required for the ROS1-dependent active DNA demethylation. Dysfunction of MET18 causes DNA hyper-methylation at thousands of genomic loci where DNA methylation is pruned by ROS1. In addition, ROS1 physically interacts with MET18 and other CIA pathway components; while a conserved iron-sulfur-binding motif is indispensable for ROS1 enzyme activity. Our results suggested that MET18 affects DNA demethylation by influencing ROS1 enzymatic activity via direct interaction with the iron-sulfur-binding motif of ROS1, highlighting a direct connection between iron-sulfur cluster assembly and active DNA demethylation.
Introduction
DNA 5-cytosine methylation (5mC) is an epigenetic mark that is critical for maintaining genome integrity, regulating gene expression and responding to environmental stress in many higher eukaryotes [1–6]. A proper DNA methylation pattern is important for plant development and plant responses to various stress conditions [1,7]. In Arabidopsis, the DNA methylation pattern is established and maintained through different pathways by several DNA methyltransferases such as MET1, CMT3, DRM2 and CMT2 [1,8]. Active removal of 5mC also contributes to the steady state of DNA methylation levels [9]. Defects in active DNA demethylation can result in abnormal gene silencing due to increased DNA methylation in gene regulatory sequences [10,11].
In Arabidopsis, the ROS1/DME family of DNA glycosylases/lyases initiates active DNA demethylation by excising the 5mC and then cutting the phophodiester backbone through either β - or β, δ-elimination [2,6,10,12–14]. The β, δ-elimination generates a 3’ phosphate at the gap, which is then hydrolyzed by phosphatase ZDP [15], while β-elimination generates a 3’-phosphor-α, β - unsaturated aldehyde (3’-PUA), which is processed to a hydroxyl group by APE1L [16]. DNA polymerase and ligase fill the gap with an unmethylated cytosine through the base-excision repair pathway [2]. DNA demethylation by DME in the central cell contributes to parental specific expression of some imprinted genes in the endosperm, whereas ROS1 and other family members mainly affect the DNA methylation status in vegetative tissues [2]. The recruitment of ROS1 to the chromatin requires histone acetylation, which is catalyzed by IDM1 [11]. The in vivo activity of IDM1 and its targeting require other anti-silencing factors such as IDM2, IDM3 and MBD7 [17,18]. In addition, ROS3, which is an RNA-binding protein, has been suggested to mediate ROS1 recruitment at some loci because its mutation disrupts the subnuclear localization pattern of ROS1 and increases DNA methylation [19]. It is unclear, however, how the enzymatic activity of ROS1 may be regulated.
Many DNA-repair and -replication proteins contain iron-sulfur (Fe-S) clusters as cofactors [20,21], and such clusters are important for the enzymatic function and/or stability of the proteins. ROS1/DME DNA glycosylases are predicted Fe-S cluster-binding proteins since they have a conserved Fe-S-binding motif. A previous study has shown that the predicted Fe-S-binding motif is essential for DME enzymatic activity [22]. Eukaryotes have the ISC (iron–sulfur cluster assembly) apparatus in mitochondria, the SUF (sulfur mobilization) pathway in mitochondria and the CIA (cytosolic Fe-S assembly) pathway in cytoplasm for the assembly of Fe-S proteins in respective cellular compartments [23,24]. The CIA pathway is mainly responsible for the maturation of cytosolic and nuclear Fe-S proteins and depends on parts of the ISC pathway for a sulfur-containing component [23,24]. In the CIA pathway, two P loop NTPases, Cfd1 and Nbp35, form a protein complex which serves as a scaffold for Fe-S cluster assembly [25,26]. The Fe-S cluster is assembled to its binding protein by the CIA targeting complex, which consists of NAR1, CIA1, AE7, and MET18 in Arabidopsis [27]. In the complex, NAR1 shares sequence homology with Fe hydrogenases and binds to the Fe-S cluster [28]. CIA1 is a WD40 protein that functions as a scaffold for protein interactions, and AE7 is a DUF59 family protein [27,29]. MET18 is an ARM repeat-containing protein, which is a conserved component in the CIA pathway from yeast to plant to animal [21,23]. In yeast and animal cells, MET18 is the delivering protein that directly interacts with various Fe-S cluster-binding proteins [21,30,31].
Here, we identified MET18 as a component required for preventing transgene silencing as well as for normal DNA methylation patterns in Arabidopsis. We show that mutations in MET18 result in genome-wide DNA hypermethylation patterns similar to those in the ros1 mutant. We found a direct interaction between MET18 and ROS1, which suggests that MET18 is likely the end of the CIA pathway that transfers the Fe-S cluster to its apoprotein. These results also reveal for the first time a direct connection between the CIA pathway and DNA demethylation, and thus make a significant contribution to understanding how iron-sulfur cluster assembly affects epigenetic regulation.
Results
ASI3 is an anti-silencing factor
To search for anti-silencing factors in Arabidopsis, we previously developed a forward genetic screen system in Arabidopsis (Wang et al., 2013; Lei et al., 2014). In this system, the SUCROSE TRANSPORTER 2 (SUC2) gene driven by the cauliflower mosaic virus 35S promoter is overexpressed in Col-0 transgenic plants (referred to as 35S::SUC2). When grown on a medium containing sucrose, 35S::SUC2 seedlings produce short roots because the roots over-accumulate sucrose (Fig 1A). Both 35S::SUC2 and mutant plants grow normally on glucose-containing medium (Fig 1A). We generated an ethylmethane sulfonate (EMS)-mutagenized population and screened for mutants with long-root phenotype on sucrose-containing medium. With this strategy, many known components associated with DNA methylation were identified, including the loss-of-function of DNA demethylase ROS1 mutants (Fig 1A) and also RNA-directed DNA methylation (RdDM) mutants [32–34]. In this study, we identified a new recessive mutant named anti-silencing 3–1 (asi3-1) with a long-root phenotype (Fig 1A). We first examined the expression of the SUC2 transgene by real-time quantitative RT-PCR (qRT-PCR). Compared to its expression in 35S::SUC2 plants, SUC2 expression in asi3-1 plants was significantly reduced (Fig 1B). 35S::SUC2 plants also express two marker genes, neomycin phosphotransferase II (NPTII) and hygromycin phosphotransferase II (HPTII), driven by shorter forms of 35S promoter for transgenic plant selection. In contrast to 35S::SUC2 plants, which grew well on a kanamycin-containing medium, asi3-1 and ros1-13 were sensitive to kanamycin (Fig 1C). qPCR results indicated that the sensitivity was due to a reduction in NPTII transgene expression (Fig 1B). Moreover, HPTII expression was also down-regulated in the asi3-1 mutant (Fig 1B). The observation that ASI3 dysfunction leads to down-regulation of transgenes suggested that ASI3 may function as an anti-silencing factor that prevents transgene silencing in Arabidopsis. In addition to transgene silencing, the asi3-1 mutant also displayed morphological phenotypes such as reduced plant and leaf sizes (S1A Fig).

Through genetic mapping, the asi3-1 mutation was mapped to a narrow region between two simple sequence length polymorphism (SSLP) markers located on chromosome 5 : 19,050,000 and 20,070,000 (Fig 1D). By whole genome re-sequencing, we detected in this region a “CAA” to “TAA” mutation in the first exon of AT5G48120 gene, which encodes the Armadillo repeat motif (ARM)-containing protein MET18. This mutation causes an amino acid change from Q to a stop codon, likely leading to premature termination of translation (Fig 1E). To further confirm that the mutation in AT5G48120 caused the silencing phenotype, we cloned the wild-type genomic sequence of AT5G48120 and transformed it into the asi3-1 mutant. Like the 35S::SUC2, the transgenic plants exhibited a short-root phenotype (Fig 1F). We selected the transgenic lines with resistance to Basta and assessed the expression of the recombinant protein MET18-3FLAG-3HA (MET18-3FH) in the T1 generation by Western blotting (S1B Fig). The SUC2 transgene expression was recovered to the 35S::SUC2 level, indicating that the AT5G48120 gene complemented the transgene-silencing phenotype of the asi3-1 mutant (Fig 1G). Moreover, the reduced-plant size phenotype was rescued by the introduction of MET18 genomic DNA, indicating that this developmental defect was caused by MET18 dysfunction (S1B Fig). All of these results confirmed that AT5G48120 is the gene corresponding to the asi3-1 mutation. AT5G48120 (MET18) encodes an ARM repeat superfamily protein, which was previously identified as a homolog of yeast Met18 [27]. Because two T-DNA insertion alleles (met18-1 and met18-2) have been previously identified (Fig 1E) [27], we named the asi3-1 mutant met18-3 (Fig 1E).
MET18 dysfunction causes genome wide DNA hypermethylation
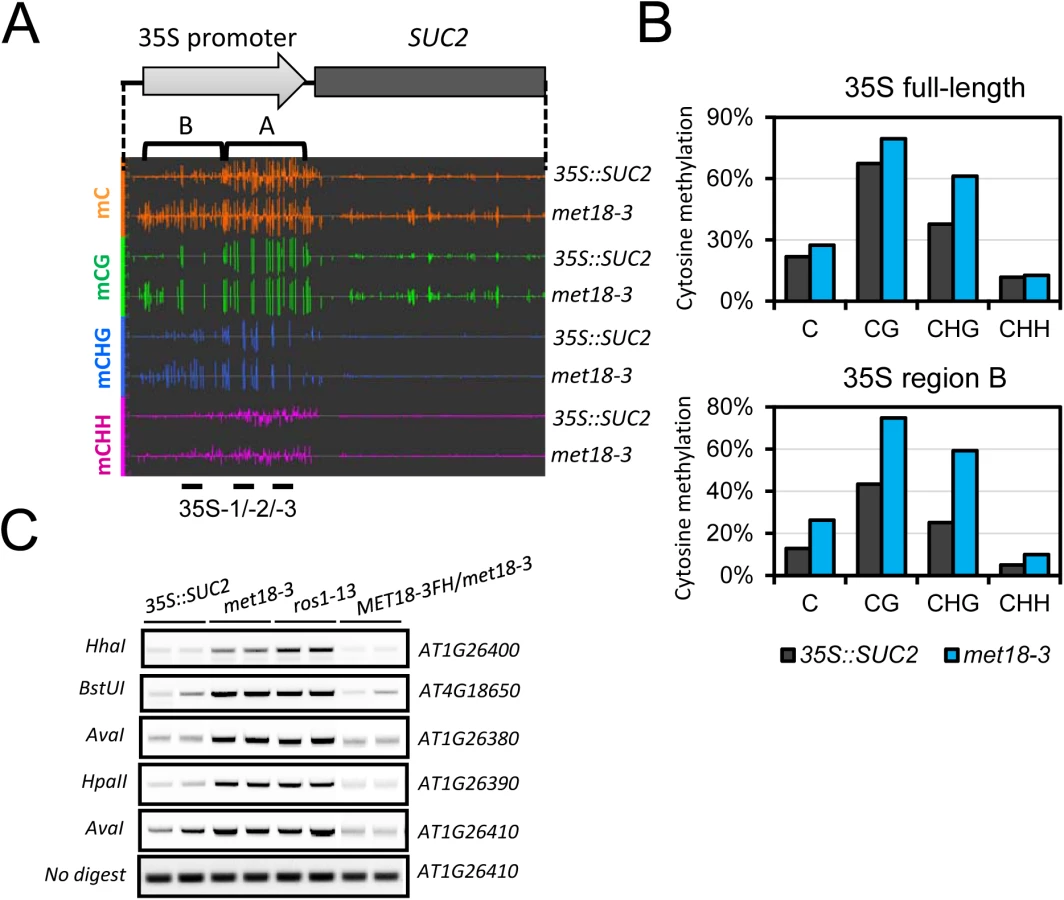
To investigate whether silencing of transgenes was associated with DNA methylation, we performed whole-genome bisulfite sequencing (WGBS) of 35S::SUC2 and met18-3 mutant seedlings. We examined the DNA methylation level in the 35S::SUC2 promoter region, which carries ~800 bp full-length 35S promoter different from the 35S promoters of NPTII and HPTII transgenes (~400bp containing two tandem repeats of 35S 3’ terminal sequence) (Fig 2A). The whole-genome methylation data showed clear increases in methylation in all cytosine contexts (CG, CHG, and CHH) in the met18-3 mutant compared to the 35S::SUC2, and especially in the upstream region (Region B) of the 35S promoter (Fig 2A and 2B). These results indicated that SUC2 transgene silencing was associated with DNA hypermethylation-mediated transcriptional repression. To confirm the DNA hypermethylation phenotype in met18-3, DNA methylation-sensitive PCR (Chop PCR) was performed. The results indicated that met18-3 displays a hypermethylation phenotype in all five examined loci (Fig 2C) that were previously identified as ROS1-target loci [11,33,34]. As a positive control, ros1-13 also showed hypermethylation at the examined loci (Fig 2C). As expected, introduction of MET18 genomic DNA rescued the DNA methylation phenotype in the met18-3 mutant, indicating that MET18 dysfunction is responsible for the hypermethylation phenotype in met18-3. Moreover, unlike RdDM mutants in which the transcript level of ROS1 gene is reduced [33,34], MET18 dysfunction did not affect ROS1 mRNA level (S2 Fig), suggesting that MET18 may be a new factor critical for DNA demethylation.
To further investigate the effects of MET18 dysfunction on whole genome DNA methylation level, we subjected two T-DNA insertion alleles, met18-1 and met18-2 [35](Fig 1E), for whole-genome bisulfite sequencing. Two biological replicates were sequenced for each allele (S3 Fig). Using the published Col-0 WGBS data as the control [11], we identified thousands of differentially methylated regions (DMRs) in the mutants. There are 4653/729, 3884/732, 4826/509 and 3687/730 hyper/hypo-DMRs in met18-1 replicate 1(rep.1), met18-1 rep.2, met18-2 rep.1 and met18-2 rep.2, respectively (S1 Table and Fig 3A and 3C). The predominance of hyper-DMRs in all 4 WGBS datasets strongly supports our hypothesis that MET18 has a negative effect on DNA methylation. We then compared the DMRs between different replicates and different mutant alleles. There are 2584 and 2406 overlapping hyper-DMRs between two technical replicates of met18-1 and met18-2, respectively (S4 Fig). The percentages of overlapping hyper-DMRs between replicates are about 56% (met18-1 rep.1) / 67% (met18-1 rep.2) and 50% (met18-2 rep.1) / 65% (met18-2 rep.2) (S4 Fig and S1 Table), respectively, indicating the majority of identified hyper-DMRs are reliable. Moreover, even for hyper-DMRs that appeared to be unique to one replicate, the DNA methylation level was also increased in the other replicate, compared to Col-0 control (S4B and S4D Fig), even though the increases in DNA methylation level were not enough to be considered as hyper-DMRs due to our stringent criteria. We then compared the hyper-DMRs between met18-1 and met18-2 and obtained 1254 common hyper-DMRs, which account for about 49% and 52% of total hyper-DMRs in met18-1 and met18-2, respectively (S4E and S5 Figs). Similarly, even for one allele-unique hyper-DMRs, DNA methylation level was also increased in the other allele, suggesting that more loci are overlapping between the two alleles than the Venn diagram showed (S4F Fig). Therefore, we used met18-1 (overlapping hyper-DMRs of met18-1 two replicates), met18-2 (overlapping hyper-DMRs of met18-2 two replicates) and met18 (overlapping hyper-DMRs of met18-1 and met18-2) hyper-DMRs for subsequent analysis.

MET18 acts in the same pathway with ROS1 in DNA demethylation
ROS1, DML2 and DML3 are the main 5mC DNA glycosylases/demethylases functioning in active DNA demethylation in vegetative tissues in Arabidopsis [2,10]. To investigate whether MET18 and ROS1 control a common set of target loci, we compared met18 DNA methylation data with two published DNA methylomes from the ROS1 mutant ros1-4 and a triple mutant of ROS1 and its two paralogs DML2, DML3, rdd [11,36]. We identified 6151 (5400 hyper - and 751 hypo-DMRs) and 9319 (8295 hyper - and 1024 hypo-DMRs) DMRs in ros1-4 and rdd mutants, respectively (S1 Table and Figs 3A and S5). Based on TAIR10 annotation, we analyzed the distribution of genomic features for the hyper-DMRs in met18, ros1-4 and rdd mutants. The results show that the distribution patterns are similar among met18, ros1-4 and rdd mutants (Fig 3B and 3C). We then compared the hyper-DMRs of met18 with those of ros1-4 and rdd mutants. As expected, the majority of the hyper-DMRs in ros1-4 (64%) overlapped with those in the rdd mutant (S6 Fig). For the met18-1 allele, there are 55% (1419/2584) and 59% (1535/2984) of hyper-DMRs overlapping with those of ros1-4 and rdd, respectively (Fig 4A and 4B, left panels). For the met18-2 allele, the percentages of overlapping hyper-DMRs with those of ros1-4 and rdd are 56% (1339/2412) and 58% (1400/2412), respectively (Fig 4A and 4B, left panels). For the overlapping hyper-DMRs between met18-1 and met18-2 (here refer to as met18), about 65% (814/1254) and 67% (837/1254) are overlapping with those of ros1-4 and rdd mutants, respectively (S6 Fig). These results indicate that MET18 and ROS1 control a common set of genomic loci to prevent their DNA hypermethylation. Moreover, the box plots based on hyper-DMR overlap of met18-ros1 and met18-rdd also indicated that even at ros1, rdd or ros1/rdd-unique loci, the average DNA methylation level was higher in met18 than in Col-0 (Fig 4A and4B, right panels), implying that there are more loci affected by MET18 than the Venn diagram showed (Fig 4A and 4B). Nevertheless, the partial-overlapping patterns also indicate a role of MET18 in controlling ROS1-independent, in addition to ROS1-dependent, DNA demethylation. The heatmaps based on hyper-DMR loci identified in met18, ros1 and rdd also suggested that hyper-DMR loci in met18 were also hypermethylated in ros1-4 and rdd mutants (S7 Fig). Collectively, these results indicate that hyper-DMRs in met18 highly overlap with those in ros1-4 and rdd, demonstrating that MET18 facilitates DNA demthylation mainly at ROS1-dependent loci.

The Fe-S motif is critical for ROS1 enzymatic activity
Proteins in the ROS1 DNA demethylase family contain a conserved Fe-S motif (Fig 5A) which was reported to be required for DME enzymatic activity [22]. To determine whether this motif is important for ROS1 enzymatic activity, we performed a DNA nicking assay as previously described [13]. We generated mutated forms of MBP-ROS1 fusion proteins by changing one of the four conserved cysteine to serine (C to S) and compared their enzymatic activities with that of the wild-type ROS1 protein. The MBP protein was used as a negative control. Coomassie Brilliant Blue (CBB) staining of purified proteins indicated that the mutated forms of ROS1 were properly expressed as wild-type ROS1 (Fig 5B lower panel). We incubated the purified recombinant proteins with either unmethylated or methylated DNA substrates. Any significant DNA nicking activity would result in the disappearance of the fast-migrating supercoiled DNA plasmid. Neither wild-type nor any of the mutated forms of ROS1 could cut the unmethylated substrate. However, wild-type but not the mutated ROS1 could cut the CG - or CHG-methylated substrates (Fig 5B, upper panel). The results show that mutating any of the four conserved cysteines reduces ROS1 activity, suggesting that all four cysteines in the Fe-S-binding motif are critical for ROS1 enzymatic activity (Fig 5B). Consistently, mutations of all 4 Fe-S-binding cysteines in DME were shown to impair DME activity [22].

The CIA components MET18 and AE7 physically interact with ROS1
We performed affinity purification of the MET18 complex using inflorescence tissues of Flag-tagged MET18 transgenic plants. The co-purified proteins were identified through mass spectrometric (MS) analyses (Fig 6A and S2 Table). Mass spectrometry showed that AE7 and CIA1, another two components functioning together with MET18 in the CIA pathway, were co-purified with MET18 (Fig 6A and S2 Table). Interestingly, ROS1 was also co-purified from MET18 transgenic plants but not from 35S::SUC2 control plant purification. These results suggest that CIA pathway components may interact with ROS1 in vivo.

To test for direct interactions between ROS1 and CIA components, yeast two-hybrid (Y2H) assays were performed. Yeast bearing MET18 in the AD vector and AE7 in the BD vector grew well in SD media lacking leucine, tryptophan, or histidine amino acids (SD-L/T/H) (Fig 6B), which is consistent with a previous report that MET18 directly interacts with AE7 [27]. The direct interaction of AE7 and MET18 was further confirmed by split luciferase assay in Arabidopsis protoplasts (Fig 6C). Yeast bearing ROS1 in the BD vector and MET18 in AD the vector also grew in the SD-L/T/H media, demonstrating that MET18 directly interacts with ROS1. Yeast bearing AE7 in the AD vector and ROS1 in the BD vector also grew in the SD-L/T/H media (Fig 6C), suggesting AE7 can also directly bind to ROS1. Moreover, the interactions of ROS1 with MET18 and AE7 were further confirmed by a split luciferase assay (Fig 6C). These results demonstrate that ROS1 physically interacts with the CIA components MET18 and AE7. To gain insights into the interaction domain of ROS1 with MET18, we generated truncated forms of ROS1, including the N terminal lysine-rich domain (NTD), the HhH-GPD motif-containing GPD domain and the C terminal domain (CTD)[37] (Fig 6D). The iron-sulfur motif is located in the overlapping region (100 amino acids) of GPD and CTD domain (Fig 6D). Split luciferase assay was performed to test the interactions between MET18 and the different forms of ROS1. The results showed that, besides full length ROS1, MET18 can also interact with ROS1GPD and ROS1CTD, but not with ROS1NTD (Fig 6D). This result suggests that the iron-sulfur motif may be the critical region for MET18-ROS1 interaction.
Discussion
DNA methylation and histone modifications are major epigenetic marks determining transcriptional activation or silencing of the associated genes [1,2]. In Arabidopsis, DNA demethylation mediated by the 5mC DNA glycosylase ROS1 prevents thousands of genomic loci from being hyper-methylated [11] and prevents transcriptional silencing at some loci [6,10,38,39]. In our previous searches for anti-silencing factors in a genetic screen, we identified many ros1 mutant alleles, illustrating that ROS1 is required to prevent the silencing of the 35S::SUC2 transgene [32–34]. Using the same system, we identified the asi3-1 mutant and mapped the mutation to the MET18 locus. Whole-genome bisulfite sequencing identified thousands of hyper-methylated loci in met18 mutants (Fig 3 and S1 Table), indicating that MET18 is a new factor controlling DNA demethylation. The comparison of DNA methylomes illustrated that hyper-DMRs in the met18 mutant highly overlap with those identified in ros1-4 and rdd mutants (Fig 4), supporting that MET18 functions in the ROS1/DML2/DML3-mediated DNA demethylation.
AtMET18 is a homolog of the yeast MET18 protein, which was identified as a component of the cytoplasmic Fe-S assembly machinery [27]. In Arabidopsis, AtMET18 forms a protein complex with other CIA components including AE7, CIA1, and NAR [27]. About 25 years ago, a DNA glycosylase functioning in base excision repair was found to contain an Fe-S cluster that was essential for its activity [40]. A recent study showed that mutation in the conserved Fe-S motif of DEMETER, a ROS1 paralogue in Arabidopsis, impaired the protein’s enzymatic activity in base excision repair [22]. Our site-directed mutagenesis analysis of ROS1 in a nicking assay showed that the iron-sulfur motif is also critical for ROS1 enzymatic activity (Fig 5). This result suggests that the enzymatic activity of ROS1 may require MET18-mediated Fe-S assembly. Interestingly, ROS1 was detected with other CIA components in the MET18-containing protein complex (Fig 6A). Our Y2H and split luciferase assays further confirmed that MET18 directly interacts with ROS1 and AE7 (Fig 6B and 6C). AE7 also directly interacted with ROS1. Thus, MET18 and AE7 may have partially redundant functions in delivering Fe-S clusters to target proteins. It is unclear how CIA components select target apoproteins because no conserved amino acid sequence was identified in the Fe-S proteins [41]. Our results are consistent with previous reports that CIA components target specific apoproteins via physical interaction [31,42]. Therefore, it is important that future studies determine the structural features that enable recognition between CIA components and the target apoproteins.
Biogenesis of Fe-S clusters is a multistep process that takes place in mitochondria and the cytoplasm, but how it is linked to nuclear Fe-S proteins is not known [30]. ROS1-mediated DNA demethylation is a nuclear process [10]. Our localization assay in tobacco leaves indicated that MET18 is mainly localized to the cytoplasm, although occasional localization in the nucleus was also observed (S8 Fig). ROS1 functions in the nucleus [10]. Luo et al. (2012) reported that the MET18-C terminus interacts with AE7 in both the nucleus and cytoplasm in bimolecular fluorescence complementation (BiFC) assays [27]. It is possible that the CIA machinery may facilitate ROS1 protein maturation in the cytoplasm before ROS1 is translocated into the nucleus.
Based on our results, a working model for the anti-silencing and DNA demethylation roles of MET18 can be proposed (S9 Fig). In this model, MET18 and other CIA components transfer an Fe-S cluster onto one of their apoproteins, ROS1, in the nucleus or cytoplasm. The mature ROS1 protein is then enzymatically active and recruited to the 35S::SUC2 promoter or other methylated genomic loci to remove methylated cytosines. The promoter is then demethylated, allowing for active transcription of the SUC2 transgene. The model helps to explain an earlier observation that a defect in AE7 causes DNA hypermethylation at two loci known to be ROS1 targets [27].
CIA proteins are important for the replication of DNA and the maintenance of genomic integrity [20,24,31,43–45]. Consistent with the importance of the CIA pathway in plants, both EMS and T-DNA insertion mutants of MET18 displayed a stunted phenotype (S1 Fig), indicating that MET18 is also indispensable for normal development. Unlike AE7, NAR1, and CIA1, whose homozygous mutants are lethal, the met18 mutant can grow and propagate normally, indicating that MET18 is less important than the other CIA components and there may be other proteins that are functionally redundant with MET18. Although there are no other MET18 homologs in Arabidopsis, other proteins in the CIA pathway such as AE7 may also be capable of delivering the Fe-S cluster to some apoproteins. This notion is consistent with our finding that not only MET18 but also AE7 could interact with ROS1 (Fig 6). Therefore, it can be speculated that, without MET18, some ROS1 protein probably can still retain Fe-S clusters and confer demethylation activity at some loci. This hypothesis can explain the observation that in some of the hyper-DMR loci met18 hypermethylation is not as strong as in the ros1 mutant.
The met18ros1 double mutant displayed the same phenotype of reduced size as the met18 single mutant but the ros1 single mutant or the rdd mutant displayed a normal growth phenotype (S1 Fig), indicating that MET18 participates in additional processes besides the ROS1-mediated DNA demethylation. Collectively, our study reveals that MET18 affects transgene silencing and DNA methylation by interacting with the DNA demethylase ROS1 for Fe-S delivery. Our study thereby demonstrates a direct linkage between the CIA pathway and ROS1-mediated active DNA demethylation. Through metabolites such as acetyl-CoA, SAM and SAH, which are important in histone and DNA modifications, the state of cellular metabolism is intimately connected to epigenetic modifications [46]. Knowledge of such connections is essential for our understanding of epigenetic regulation and how nutrition and metabolism affect development and diseases through epigenetic regulation [47]. In this regard, the linkage between the CIA pathway and active DNA demethylation represents an important but previously underappreciated connection between epigenetics and nutrition and metabolism.
Materials and Methods
Plant materials and growth conditions
All plants were grown under a long-day photoperiod (16-h light/8-h dark). 1/2-strength Murashige and Skoog (MS) medium containing 1% sucrose was used for studying root phenotype. All other experiments used growth medium with 1% glucose. Kanamycin of 100 mg/L was used for the kanamycin-sensitivity assay. Seedlings were photographed 14 days after germination.
Two other alleles of the met18 mutant, met18-1 (SALK_121963) and met18-2 (SALK_147068), were obtained from the Arabidopsis Biological Resource Center (ABRC) and confirmed by PCR-based genotyping [35]. MET18 genomic DNA including the upstream 2-kb promoter region was amplified by PCR and cloned into a modified pEarleyGate 302 vector [48], which bears three copies of FLAG and HA tags. The construct was introduced into the met18-3 mutant by Agrobacterium-mediated transformation via the floral dip method [49] All primers used in the study are listed in S3 Table.
Mutant screening and map-based cloning
The met18-3 mutant was obtained by a screen method described in our previous report [33,34]. In brief, EMS-treated M2 seeds were grown on ½ MS medium containing 1% sucrose. Seedlings with roots longer than the wild type plants were selected as potential transgene silencing mutants. To clone the ASI3 gene, the asi3-1 mutant was crossed with Landsberg erecta, and F1 plants were self-pollinated to obtain the F2 population. F2 seeds were planted on 1/2 MS medium containing 1% sucrose and 25 μg/mL hygromycin. Seedlings with normal, long roots were selected for genetic mapping in order to calculate asi3-1 linkage. The asi3-1 mutant genomic DNA was then re-sequenced to determine the location of the mutation in the mapping region.
Analysis of DNA methylation levels
DNA methylation-sensitive PCR was performed according to a previous report [34]. In brief, genomic DNA was extracted from 2-week-old seedlings using the DNeasy Plant Minikit (QIAGEN) and was quantified with NanoDrop 2000c UV-Vis Spectrophotometer (Thermo SCIENTIFIC). About 50 ng of DNA was subjected to DNA methylation-sensitive restriction enzyme digestion in a 50-μL reaction. The digested DNA was used as template for PCR, and the PCR products were subjected to agarose gel electrophoresis. The non-digested genomic DNA was used as control template for PCR.
For whole-genome bisulfite sequencing, DNA was extracted from 2-week-old seedlings. The DNA samples were submitted to BGI (Shenzhen, China) for bisulfite treatment and DNA sequencing. The bisulfite treatment and DNA methylation analysis were performed as previously described [50]. When counting the number of overlapping DMRs, if aregion in one mutant overlaps with several regions in another mutant, we calculated the number of overlapping regions from the perspective of met18, ros1-4, and rdd. For TE annotation, if a region overlaps with a TE or a transposable elemental gene, the region is classified as a TE region. Regions having no overlap with TE or transposable element gene and overlapping with protein-coding genes are classified as Gene regions.
Real-time quantitative RT-PCR
For real-time quantitative RT-PCR, total RNAs were extracted from 2-week-old seedlings using the RNeasy Plant Minikit (QIAGEN). After TURBO DNase I treatment (Ambion), 2 μg of RNA was subjected to RT reaction using the SuperScript III First-Strand Kit according to the manufacturer’s instructions (Invitrogen). The 1st-strand cDNAs were then amplified using IQ SYBR green supermix (BIO-RAD) with the CFX96 real-time PCR detection system (BIO-RAD).
ROS1 enzymatic activity assay
The MBP-ROS1 in the pMAL-c2x vector was constructed as previously reported [6,51]. Site-directed mutagenesis was performed via PCR to obtain the expression constructs of ROS1C1038S-, ROS1C1045S-, ROS1C1048S-, and ROS1C1054S-mutant proteins. All constructs were transformed into dcm- Codon Plus cells of E. coli strain BL21. The induction and purification of ROS1 and mutant proteins were performed as previously described [15]. The ROS1 enzymatic activity assay (the nicking assay) was performed as previously described [13]. In brief, plasmid pBluescript KS purified from the dcm- strain of E. coli BL21 (DE3) was methylated in vitro with MspI (mCHG) or SssI (mCG) methylases (NEB). The methylation status was confirmed by digestion with MspI and HpaII restriction endonucleases. The unmethylated plasmid was used as a control. The purified, closed-circular plasmids (250 ng) were then incubated with purified MBP-ROS1 protein or its mutated forms in nicking buffer containing 40 mM Hepes-KOH (pH 8.0), 0.1 M KCl, 0.5 mM EDTA, 0.5 mM DTT, and 0.2 mg/ml BSA at 37°C for 2 h. After adding stop solution (0.4 M EDTA and 1% SDS), the reaction mixtures were heated at 70°C for 5 min and subjected to 1% agarose gel electrophoresis.
Yeast two hybrid (Y2H), split luciferase assays and cellular localization
The full-length coding regions of MET18, ROS1, and AE7 were first individually cloned into the pENTR/D-TOPO directional cloning vector (Invitrogen) and then transferred into the destination vectors pDEST 22 (AD) and pDEST 32 (BD) (Invitrogen) via LR recombination using Gateway Clonase II Enzyme (Invitrogen).
For the split luciferase assay, constructs carrying full-length coding regions of MET18, AE7, and full-length/truncated forms of ROS1 fused with split luciferase in the pEarleyGate vector were co-transfected into Arabidopsis protoplast or Nicotiana Benthamiana for overnight incubation or 3-day-growth, respectively [48]. The empty vectors was used as a negative control. The luciferase activity was determined using CCD camera equipped with Winview software (Princeton instruments). Protoplast was prepared as previously reported [52]. For MET18-GFP cellular localization, Agrobacterium containing MET18-GFP construct driven by 35S promoter was infiltrated into Nicotiana benthamiana leaves. The cellular localization was examined by Zeiss Microscope at 3 day-post-infiltration.
Affinity purification and mass spectrometry
For affinity purification of MET18 and its associated proteins, 5 g of flower tissues were collected from MET18-3FLAG-3HA transgenic plants, and tissue from 35S::SUC2 plants was used as a negative control. Extraction of total proteins and affinity purification were performed as described previously [53] with minor modifications. Briefly, flower tissues were ground to fine powders in liquid nitrogen and suspended in 30 ml of lysis buffer (50 mM Tris pH 7.6, 150 mM NaCL, 5 mM MgCl2, 10% glycerol, 0.1% NP-40, 0.5 mM DTT, 1 mM PMSF, and 1 protease inhibitor cocktail tablet (Roche)). After further homogenization by douncing and centrifugation at 4°C, the supernatants were incubated with 120 μL of anti-FLAG M2 agarose beads (Sigma), which had been pre-equilibrated with lysis buffer. After incubation at 4°C with rotation for 2–3 hours, the agarose beads were washed three times for 5 minutes each time with 40 ml of lysis buffer, three times for 5 minutes each time with 1 mL of lysis buffer, and three times for 5 minutes each time with 1 ml of PBS buffer. The agarose beads were finally resuspended in 120 μL of PBS buffer for mass spectrometry according to a previous report [54].
Accession numbers
The raw WGBS sequencing dataset of met18-1 rep.1, met18-1 rep.2, met18-2 rep.1 and met18-2 rep.2 was deposited to the public database NCBI GEO (accession number GSE69281). The Col-0, ros1-4 and rdd whole genome bisulfite sequencing data were from GEO accession GSE33071 [11].
Supporting Information
Zdroje
1. Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11 : 204–220. doi: 10.1038/nrg2719 20142834
2. Zhu JK (2009) Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet 43 : 143–166. doi: 10.1146/annurev-genet-102108-134205 19659441
3. Zilberman D (2008) The evolving functions of DNA methylation. Curr Opin Plant Biol 11 : 554–559. doi: 10.1016/j.pbi.2008.07.004 18774331
4. Le TN, Schumann U, Smith NA, Tiwari S, Au PC, et al. (2014) DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol 15 : 458. doi: 10.1186/s13059-014-0458-3 25228471
5. Yu A, Lepere G, Jay F, Wang J, Bapaume L, et al. (2013) Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc Natl Acad Sci U S A 110 : 2389–2394. doi: 10.1073/pnas.1211757110 23335630
6. Morales-Ruiz T, Ortega-Galisteo AP, Ponferrada-Marin MI, Martinez-Macias MI, Ariza RR, et al. (2006) DEMETER and REPRESSOR OF SILENCING 1 encode 5-methylcytosine DNA glycosylases. Proc Natl Acad Sci U S A 103 : 6853–6858. 16624880
7. Le TN, Schumann U, Smith NA, Tiwari S, Au P, et al. (2014) DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol 15 : 458. doi: 10.1186/s13059-014-0458-3 25228471
8. Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, et al. (2013) The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153 : 193–205. doi: 10.1016/j.cell.2013.02.033 23540698
9. Zhang H, Zhu JK (2012) Active DNA demethylation in plants and animals. Cold Spring Harb Symp Quant Biol 77 : 161–173. doi: 10.1101/sqb.2012.77.014936 23197304
10. Gong Z, Morales-Ruiz T, Ariza RR, Roldan-Arjona T, David L, et al. (2002) ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 111 : 803–814. 12526807
11. Qian W, Miki D, Zhang H, Liu Y, Zhang X, et al. (2012) A histone acetyltransferase regulates active DNA demethylation in Arabidopsis. Science 336 : 1445–1448. doi: 10.1126/science.1219416 22700931
12. Ortega-Galisteo AP, Morales-Ruiz T, Ariza RR, Roldan-Arjona T (2008) Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol Biol 67 : 671–681. doi: 10.1007/s11103-008-9346-0 18493721
13. Agius F, Kapoor A, Zhu JK (2006) Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc Natl Acad Sci U S A 103 : 11796–11801. 16864782
14. Gehring M, Huh JH, Hsieh TF, Penterman J, Choi Y, et al. (2006) DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 124 : 495–506. 16469697
15. Martinez-Macias MI, Qian W, Miki D, Pontes O, Liu Y, et al. (2012) A DNA 3' phosphatase functions in active DNA demethylation in Arabidopsis. Mol Cell 45 : 357–370. doi: 10.1016/j.molcel.2011.11.034 22325353
16. Li Y, Cordoba-Canero D, Qian W, Zhu X, Tang K, et al. (2015) An AP Endonuclease Functions in Active DNA Dimethylation and Gene Imprinting in Arabidopsis. PLoS Genet 11: e1004905. doi: 10.1371/journal.pgen.1004905 25569774
17. Qian W, Miki D, Lei M, Zhu X, Zhang H, et al. (2014) Regulation of active DNA demethylation by an alpha-crystallin domain protein in Arabidopsis. Mol Cell 55 : 361–371. doi: 10.1016/j.molcel.2014.06.008 25002145
18. Lang Z, Lei M, Wang X, Tang K, Miki D, et al. (2015) The Methyl-CpG-Binding Protein MBD7 Facilitates Active DNA Demethylation to Limit DNA Hyper-Methylation and Transcriptional Gene Silencing. Mol Cell 57 : 971–983. doi: 10.1016/j.molcel.2015.01.009 25684209
19. Zheng X, Pontes O, Zhu J, Miki D, Zhang F, et al. (2008) ROS3 is an RNA-binding protein required for DNA demethylation in Arabidopsis. Nature 455 : 1259–1262. doi: 10.1038/nature07305 18815596
20. White MF, Dillingham MS (2012) Iron-sulphur clusters in nucleic acid processing enzymes. Curr Opin Struct Biol 22 : 94–100. doi: 10.1016/j.sbi.2011.11.004 22169085
21. Buzas DM, Nakamura M, Kinoshita T (2014) Epigenetic role for the conserved Fe-S cluster biogenesis protein AtDRE2 in Arabidopsis thaliana. Proc Natl Acad Sci U S A 111 : 13565–13570. doi: 10.1073/pnas.1404058111 25197096
22. Mok YG, Uzawa R, Lee J, Weiner GM, Eichman BF, et al. (2010) Domain structure of the DEMETER 5-methylcytosine DNA glycosylase. Proc Natl Acad Sci U S A 107 : 19225–19230. doi: 10.1073/pnas.1014348107 20974931
23. Couturier J, Touraine B, Briat JF, Gaymard F, Rouhier N (2013) The iron-sulfur cluster assembly machineries in plants: current knowledge and open questions. Front Plant Sci 4 : 259. doi: 10.3389/fpls.2013.00259 23898337
24. Netz DJ, Mascarenhas J, Stehling O, Pierik AJ, Lill R (2014) Maturation of cytosolic and nuclear iron-sulfur proteins. Trends Cell Biol 24 : 303–312. doi: 10.1016/j.tcb.2013.11.005 24314740
25. Roy A, Solodovnikova N, Nicholson T, Antholine W, Walden WE (2003) A novel eukaryotic factor for cytosolic Fe-S cluster assembly. EMBO J 22 : 4826–4835. 12970194
26. Hausmann A, Aguilar Netz DJ, Balk J, Pierik AJ, Muhlenhoff U, et al. (2005) The eukaryotic P loop NTPase Nbp35: an essential component of the cytosolic and nuclear iron-sulfur protein assembly machinery. Proc Natl Acad Sci U S A 102 : 3266–3271. 15728363
27. Luo D, Bernard DG, Balk J, Hai H, Cui X (2012) The DUF59 family gene AE7 acts in the cytosolic iron-sulfur cluster assembly pathway to maintain nuclear genome integrity in Arabidopsis. Plant Cell 24 : 4135–4148. doi: 10.1105/tpc.112.102608 23104832
28. Balk J, Pierik AJ, Netz DJ, Muhlenhoff U, Lill R (2004) The hydrogenase-like Nar1p is essential for maturation of cytosolic and nuclear iron-sulphur proteins. EMBO J 23 : 2105–2115. 15103330
29. Balk J, Aguilar Netz DJ, Tepper K, Pierik AJ, Lill R (2005) The essential WD40 protein Cia1 is involved in a late step of cytosolic and nuclear iron-sulfur protein assembly. Mol Cell Biol 25 : 10833–10841. 16314508
30. Gari K, Leon Ortiz AM, Borel V, Flynn H, Skehel JM, et al. (2012) MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science 337 : 243–245. doi: 10.1126/science.1219664 22678361
31. Stehling O, Vashisht AA, Mascarenhas J, Jonsson ZO, Sharma T, et al. (2012) MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 337 : 195–199. doi: 10.1126/science.1219723 22678362
32. Lei M, Zhang H, Julian R, Tang K, Xie S, et al. (2015) Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc Natl Acad Sci U S A 112 : 3553–3557. doi: 10.1073/pnas.1502279112 25733903
33. Lei M, La H, Lu K, Wang P, Miki D, et al. (2014) Arabidopsis EDM2 promotes IBM1 distal polyadenylation and regulates genome DNA methylation patterns. Proc Natl Acad Sci U S A 111 : 527–532. doi: 10.1073/pnas.1320106110 24248388
34. Wang X, Duan CG, Tang K, Wang B, Zhang H, et al. (2013) RNA-binding protein regulates plant DNA methylation by controlling mRNA processing at the intronic heterochromatin-containing gene IBM1. Proc Natl Acad Sci U S A 110 : 15467–15472. doi: 10.1073/pnas.1315399110 24003136
35. Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, et al. (2003) Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301 : 653–657. 12893945
36. Penterman J, Zilberman D, Huh JH, Ballinger T, Henikoff S, et al. (2007) DNA demethylation in the Arabidopsis genome. Proc Natl Acad Sci U S A 104 : 6752–6757. 17409185
37. Ponferrada-Marin MI, Parrilla-Doblas JT, Roldan-Arjona T, Ariza RR (2011) A discontinuous DNA glycosylase domain in a family of enzymes that excise 5-methylcytosine. Nucleic Acids Res 39 : 1473–1484. doi: 10.1093/nar/gkq982 21036872
38. Yamamuro C, Miki D, Zheng Z, Ma J, Wang J, et al. (2014) Overproduction of stomatal lineage cells in Arabidopsis mutants defective in active DNA demethylation. Nat Commun 5 : 4062. doi: 10.1038/ncomms5062 24898766
39. Zhu J, Kapoor A, Sridhar VV, Agius F, Zhu JK (2007) The DNA glycosylase/lyase ROS1 functions in pruning DNA methylation patterns in Arabidopsis. Curr Biol 17 : 54–59. 17208187
40. Cunningham RP, Asahara H, Bank JF, Scholes CP, Salerno JC, et al. (1989) Endonuclease III is an iron-sulfur protein. Biochemistry 28 : 4450–4455. 2548577
41. Rouault TA (2012) Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech 5 : 155–164. doi: 10.1242/dmm.009019 22382365
42. Papatriantafyllou M (2012) DNA Metabolism: MMS19: CIA agent for DNA-linked affairs. Nat Rev Mol Cell Biol 13 : 538. doi: 10.1038/nrm3411 22828929
43. Chanet R, Heude M (2003) Characterization of mutations that are synthetic lethal with pol3-13, a mutated allele of DNA polymerase delta in Saccharomyces cerevisiae. Curr Genet 43 : 337–350. 12759774
44. Kuo CF, McRee DE, Fisher CL, O'Handley SF, Cunningham RP, et al. (1992) Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science 258 : 434–440. 1411536
45. Boal AK, Genereux JC, Sontz PA, Gralnick JA, Newman DK, et al. (2009) Redox signaling between DNA repair proteins for efficient lesion detection. Proc Natl Acad Sci U S A 106 : 15237–15242. doi: 10.1073/pnas.0908059106 19720997
46. Keating ST, El-Osta A (2015) Epigenetics and metabolism. Circ Res 116 : 715–736. doi: 10.1161/CIRCRESAHA.116.303936 25677519
47. Carrer A, Wellen KE (2014) Metabolism and epigenetics: a link cancer cells exploit. Curr Opin Biotechnol 34 : 23–29. doi: 10.1016/j.copbio.2014.11.012 25461508
48. Earley KW, Haag JR, Pontes O, Opper K, Juehne T, et al. (2006) Gateway-compatible vectors for plant functional genomics and proteomics. Plant J 45 : 616–629. 16441352
49. Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16 : 735–743. 10069079
50. Duan C, Zhang H, Tang K, Zhu X, Qian W, et al. (2014) Specific but interdependent functions for Arabidopsis AGO4 and AGO6 in RNA-directed DNA methylation. EMBO J.
51. Ponferrada-Marin MI, Martinez-Macias MI, Morales-Ruiz T, Roldan-Arjona T, Ariza RR (2010) Methylation-independent DNA binding modulates specificity of Repressor of Silencing 1 (ROS1) and facilitates demethylation in long substrates. J Biol Chem 285 : 23032–23039. doi: 10.1074/jbc.M110.124578 20489198
52. Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2 : 1565–1572. 17585298
53. Law JA, Ausin I, Johnson LM, Vashisht AA, Zhu JK, et al. (2010) A protein complex required for polymerase V transcripts and RNA - directed DNA methylation in Arabidopsis. Curr Biol 20 : 951–956. doi: 10.1016/j.cub.2010.03.062 20409711
54. Wang P, Du Y, Hou Y, Zhao Y, Hsu C, et al. (2014) Nitric oxide negatively regulates abscisic acid signaling in guard cells by S-nitrosylation of OST1. Proc Natl Acad Sci U S A.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Single Strand Annealing Plays a Major Role in RecA-Independent Recombination between Repeated Sequences in the Radioresistant Bacterium
- The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals
- Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross
- DCA1 Acts as a Transcriptional Co-activator of DST and Contributes to Drought and Salt Tolerance in Rice
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy