
Variation in Modifies Risk of Neonatal Intestinal Obstruction in Cystic Fibrosis
Meconium ileus (MI), a life-threatening intestinal obstruction due to meconium with abnormal protein content, occurs in approximately 15 percent of neonates with cystic fibrosis (CF). Analysis of twins with CF demonstrates that MI is a highly heritable trait, indicating that genetic modifiers are largely responsible for this complication. Here, we performed regional family-based association analysis of a locus that had previously been linked to MI and found that SNP haplotypes 5′ to and within the MSRA gene were associated with MI (P = 1.99×10−5 to 1.08×10−6; Bonferroni P = 0.057 to 3.1×10−3). The haplotype with the lowest P value showed association with MI in an independent sample of 1,335 unrelated CF patients (OR = 0.72, 95% CI [0.53–0.98], P = 0.04). Intestinal obstruction at the time of weaning was decreased in CF mice with Msra null alleles compared to those with wild-type Msra resulting in significant improvement in survival (P = 1.2×10−4). Similar levels of goblet cell hyperplasia were observed in the ilea of the Cftr−/− and Cftr−/−Msra−/− mice. Modulation of MSRA, an antioxidant shown to preserve the activity of enzymes, may influence proteolysis in the developing intestine of the CF fetus, thereby altering the incidence of obstruction in the newborn period. Identification of MSRA as a modifier of MI provides new insight into the biologic mechanism of neonatal intestinal obstruction caused by loss of CFTR function.
Published in the journal:
. PLoS Genet 8(3): e32767. doi:10.1371/journal.pgen.1002580
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002580
Summary
Meconium ileus (MI), a life-threatening intestinal obstruction due to meconium with abnormal protein content, occurs in approximately 15 percent of neonates with cystic fibrosis (CF). Analysis of twins with CF demonstrates that MI is a highly heritable trait, indicating that genetic modifiers are largely responsible for this complication. Here, we performed regional family-based association analysis of a locus that had previously been linked to MI and found that SNP haplotypes 5′ to and within the MSRA gene were associated with MI (P = 1.99×10−5 to 1.08×10−6; Bonferroni P = 0.057 to 3.1×10−3). The haplotype with the lowest P value showed association with MI in an independent sample of 1,335 unrelated CF patients (OR = 0.72, 95% CI [0.53–0.98], P = 0.04). Intestinal obstruction at the time of weaning was decreased in CF mice with Msra null alleles compared to those with wild-type Msra resulting in significant improvement in survival (P = 1.2×10−4). Similar levels of goblet cell hyperplasia were observed in the ilea of the Cftr−/− and Cftr−/−Msra−/− mice. Modulation of MSRA, an antioxidant shown to preserve the activity of enzymes, may influence proteolysis in the developing intestine of the CF fetus, thereby altering the incidence of obstruction in the newborn period. Identification of MSRA as a modifier of MI provides new insight into the biologic mechanism of neonatal intestinal obstruction caused by loss of CFTR function.
Introduction
Cystic fibrosis (CF; MIM 219700, http://www.omim.org) is an autosomal recessive condition caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR; MIM 602421) gene [1]. The earliest manifestation of CF is meconium ileus (MI), a prenatal obstruction of the small intestine at the ileocecal junction. Meconium, the intestinal contents of the developing gut that form the first bowel movement, has an abnormally high protein content in CF neonates thought to be due to defective proteolysis [2]–[4]. Impaction of the tenacious meconium results in intestinal obstruction in approximately 15% of CF newborns. This complication presents as abdominal distention, failure to pass meconium, and vomiting and was near universally fatal in CF newborns until effective treatment (enema or surgery) was developed. The long term effects of MI have been a matter of debate as some investigators have reported worse outcomes while others observed no significant differences from CF subjects without MI [5]–[7].
Genetic modifiers have been implicated in the development of MI for over 40 years as recurrence risk for this complication in siblings with CF (∼0.25) has consistently been shown to be higher than that in unrelated individuals with CF (∼0.15) [8]–[12]. Concordance analysis in monozygous and dizygous twins demonstrated that the heritability of MI approaches 1.0, confirming that modifier genes play a substantial role in MI [12]. CFTR also contributes to risk as MI almost exclusively manifests in CF subjects with exocrine pancreatic insufficiency (PI), which is highly correlated with CFTR genotype [13], [14]. Furthermore, the incidence of MI appears to vary among CFTR mutations that confer PI. For example, the amino acid substitution p.Gly551Asp (“G551D”) has been associated with reduced risk of MI compared to the most common CFTR mutation that causes a deleterious in-frame deletion of one amino acid (p.Phe508del, “delta F508”) [15], [16].
Initial attempts to identify MI modifier genes in humans utilized localization results from mouse studies. Mice with disruption of Cftr present with an MI-like phenotype; however, it differs from human MI in several respects. First, intestinal obstruction in CF mice causes mortality shortly after birth and at the time of weaning with the introduction of solid food [17]–[19]. Life-threatening obstruction in humans occurs in the perinatal period, while episodes of variable severity termed distal intestinal obstruction syndrome (DIOS) can also occur throughout life (5 to 12 episodes per 1,000 patient-years), especially in adults with PI and as a post-operative complication of surgical intervention, particularly organ transplantation [20], [21]. Second, obstructive lesions in CF mice have been observed in the jejunum, ileum and colon, compared to predominantly ileo-colic localization in humans [22]. Third, pancreatic exocrine disease is much less prominent in mouse models of CF [17], [23], [24]. On the other hand, there are instructive genetic similarities between mice and humans with CF. Cftr alleles influence the rate and severity of murine intestinal obstruction [17], [25]–[28] and strain-specific differences in the penetrance of intestinal obstruction indicate that different modifier genes underlie obstruction at birth and at weaning [19]. Candidate gene approaches in CF mice have revealed that decreased expression of the sodium hydrogen exchanger 3 (Nhe3) or mucin 1 (Muc1) or over-expression of the chloride calcium channel activated 3 protein (Clca3/Gob5) can reduce intestinal obstruction at weaning [29]–[31].
Newer animal models of CF have provided additional clues in the search for modifiers of MI. CFTR knock-out ferrets and pigs have been shown to develop intestinal obstruction that is anatomically and temporally equivalent to that observed in humans; however, the phenotype is highly penetrant in these animals (75% and 100%, respectively) [32], [33]. Animal models of CF and heritability studies in humans suggest that intestinal obstruction due to loss of CFTR function is a consistent feature, and that the incomplete penetrance of this trait in humans with CF is likely due to the presence of genetic modifiers. We present here the results of a regional association analysis of a linked locus on chromosome 8 [12], and report the identification and functional confirmation of a modifier gene for MI in humans and mice with CF.
Results
Regional association analysis of CF patients implicates variants near and within MSRA as modifiers of MI
To investigate a region on chromosome 8 that had previously shown linkage to MI [12], transmission analysis of SNPs was performed using families from the Cystic Fibrosis Twin and Sibling Study (TSS). As MI is associated with an increased recurrence risk among siblings, we enriched for genes that modify this phenotype by analyzing 133 families in which at least one subject was affected with MI (26 MI concordant and 91 discordant pairs, 2 concordant and 8 discordant sets of 3, and 6 singletons; Table 1). Since MI rarely occurs in the absence of PI, individuals were excluded if their CFTR genotypes were associated with exocrine pancreatic sufficiency (PS) (n = 7) or if they clinically demonstrated PS (n = 5). All SNPs on the Illumina 610-Quad genotyping panel that passed rigorous quality control (2,896 SNPs) [34] were included from an approximately 9 Mb region within the linkage locus where the SNP LOD score exceeded 1.0 (boundaries: rs2945913-rs2285274; green shaded area in linkage plot inset, Figure 1). Parental genotypes were utilized to test the transmission of SNP alleles using pedigree-based association testing (PBAT), an extension of the transmission disequilibrium test that is robust against population stratification [35], [36]. A cluster of SNPs proximal to and within MSRA (MIM 601250) showed evidence of association with MI when an additive mode of inheritance was assumed (Figure 1). One SNP within this cluster 5′ to MSRA, rs614197, exceeded the threshold for region-wide significance (P = 8.35×10−6, Bonferroni corrected P = 0.024). However, this SNP was not associated with MI in a sample of unrelated CF patients from the Canadian Consortium for Genetic Studies (CGS; Table 1).
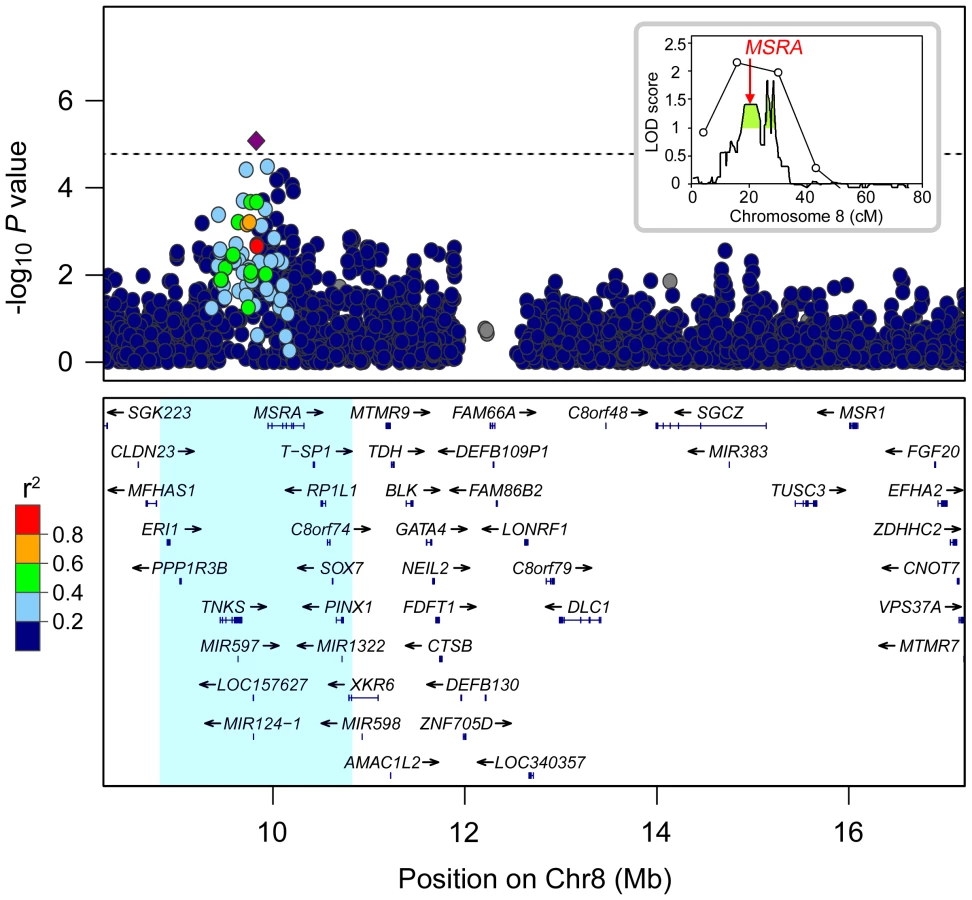

Lack of significant association between MI and rs614197 in the CGS sample led us to question whether the initial observation in the TSS families was spurious, or if detection of association could be confounded by interacting loci [37] or by heterogeneity of effect of alleles at the locus [38], [39]. To test the latter concept, we used haplotype analysis to search for additional genetic variation associated with MI in the region surrounding MSRA. Haplotypes derived from a sliding window of three consecutive SNPs across a 2 Mb region centered at rs614197 (boundaries: rs17700611-rs4240673) were tested for transmission distortion under an additive genetic model (Figure S1). We used combinations of three SNPs as a compromise between the number of haplotypes generated and the penalty incurred by multiple testing. Two haplotypes exceeded the threshold for significant regional association after Bonferroni correction for 2,890 different haplotypes observed in the 133 families studied (Table 2). The combination of rs10903323 T, rs4840475 G and rs17151637 A (T-G-A) spanning a 3.5 kb region in intron 3 of MSRA (Figure 2) was significantly over-transmitted to individuals without MI, or in other words had a protective effect on MI (54 informative families, raw P = 1.08×10−6; Bonferroni P = 3.13×10−3). In the entire TSS cohort, the T-G-A haplotype frequency was 14.9% (13.0% in subjects with MI, 25.0% in those without MI). A second, overlapping haplotype (Figure 2) containing two of the same SNP alleles as the T-G-A haplotype (rs4840475 G, rs17151637 A, plus rs6601427 C; Table 2) similarly demonstrated association with the absence of MI (48 informative families, raw P = 1.17×10−5; Bonferroni P = 0.034). Another haplotype located just 5′ to MSRA showed nearly significant over-transmission to individuals with MI, indicating that it conferred risk for MI (59 informative families, raw P = 1.99×10−5; Bonferroni P = 0.057). Interestingly, this haplotype included rs614197, the SNP that was associated with MI in the initial single marker analysis (rs586123 G, rs614197 G and rs2055729 C; Table 2). The SNPs comprising these three haplotypes displayed weak linkage disequilibrium (LD) with the exception of the second and third SNP in the 5′ haplotype (r2 = 0.55; Figure 2). Thus, the haplotypes are likely detecting additional genetic variation that cannot be assessed by the tagging SNPs on this array platform.


To evaluate whether the MSRA alleles identified here by association could explain the former linkage signal on chromosome 8, the frequency of the T-G-A haplotype that showed the strongest association signal was calculated in siblings with CF that were concordant for MI. Sixteen sib-pairs contributed positively to the LOD score while the remaining 14 sib-pairs had a negative LOD score. The frequency of the T-G-A MSRA haplotype was lower in siblings that contributed to linkage (5 of 64 chromosomes, 7.8%) compared to siblings that did not (12 of 44 chromosomes, 21.4%; P = 0.033). Given that the T-G-A haplotype is associated with a decreased rate of MI, it appears that the observed linkage with risk for MI in our previous study was due to the increased sharing of other MSRA haplotypes that confer a higher risk of MI.
A replication study was performed using CF patients recruited by the CGS. The CGS is representative of the general CF population in Canada and comprises approximately 70% of all Canadian CF patients [40]. The overall rate of MI in this group was 15.9% (n = 220 with MI, 1,163 without MI), which is consistent with the incidence of MI reported in numerous Caucasian CF populations [8], [10], [16]. The frequency of the T-G-A haplotype in the 1,335 CGS subjects in whom haplotypes were ascertained was similar (15.4%) to that observed in the TSS (14.9%; Table 2). The T-G-A haplotype showed association with MI in the CGS sample under an additive model (OR = 0.72, 95% CI [0.53–0.98], P = 0.04), in agreement with the initial finding in the TSS. The incidence of MI in subjects with 0 copies of the T-G-A haplotype was 17.3% (165/954 subjects), 13.1% (46/351) in those with 1 copy and 10% (3/30) in those with 2 copies.
The concept that variation in CFTR influences the risk of MI is evident from the observation that a PI state (primarily determined by CFTR genotype) is required for the development of MI [14]. However, there is a finer correlation between CFTR genotype and MI as two CFTR mutations that are highly correlated with PI, p.Gly551Asp and p.Gly542X, have been shown to decrease or increase MI risk, respectively, from that conferred by the common mutation p.Phe508del [14]–[16]. As p.Gly551Asp is present at a relatively high frequency among European CF alleles (∼2% [41]), we evaluated the association between this mutation and MI in the TSS and CGS cohorts. The incidence of MI in p.Gly551Asp-bearing subjects was 7.8% in the TSS (n = 51) and 5.9% in the CGS (n = 51), compared to 20.5% (n = 507) and 17.9% (n = 851) in p.Phe508del homozygotes (P = 0.026, 0.033), respectively. Combining these two samples of CF subjects demonstrated that the odds of MI in subjects with p.Gly551Asp was about a third of that in p.Phe508del homozygotes (OR = 0.32, 95% CI [0.13–0.76]; P = 0.010), comparable to the report by Hamosh, et al [15]. The finding that variation in the disease-causing gene alters the incidence of MI even among PI subjects suggested that the relationship between MI and the MSRA haplotype could be confounded by variation in CFTR. To control for genetic heterogeneity in CFTR, we tested for association between the T-G-A haplotype and MI by evaluating 1,017 subjects with the same genotype (p.Phe508del homozygotes) drawn from the TSS MI families (n = 166) and CGS studies (n = 851). A meta-analysis conducted using logistic regression coefficients and standard errors [42] from individual TSS and CGS analyses revealed that the T-G-A haplotype retained an additive protective effect (P = 0.001), indicating that MSRA modifies MI independently of variation in CFTR.
Msra modifies intestinal obstruction and survival in CF mice
Intestinal obstruction at the time of weaning is the primary cause of death in mouse models of CF [22], [43]. As the effect of the MI-associated haplotypes upon MSRA expression was unknown, we elected to introduce null alleles of Msra into mouse models of CF with high and low rates of mortality due to intestinal obstruction to detect whether loss of Msra expression reduced or exacerbated the rate of obstruction. Intestinal obstruction in a null CF mouse model (C57BL/6J Cftr−/−) leads to high mortality (>80% [17]) by 40 days of age while lower rates of mortality [44] occur in a CF mouse model (C57BL/6JR117H/R117H) with a targeted knock-in of a missense mutation (p.Arg117His) associated with residual CFTR function [45], [46] and very low rates of MI in humans with CF [47]. Mice heterozygous for the Cftr null (Cftr+/−) or p.Arg117His allele (Cftr+/R117H) were crossed to mice with one or two Msra null alleles to produce CF mice (Cftr−/− or CftrR117H/R117H) with wild-type (+/+), heterozygous (+/−), and null (−/−) Msra genotypes. As expected, Cftr−/− mice displayed a sharp drop in survival around the time of weaning when solid food is introduced to the diet (Figure 3). The median survival of the Cftr−/− mice was 22 days, consistent with the high rates of mortality due to intestinal obstruction reported in other CF ‘null’ mice [17], [18], [24], [48], all of which would have been homozygous for wild-type Msra (i.e. Msra+/+). However, survival was significantly improved in Msra+/− and Msra−/− CF mice compared to their Msra+/+ littermates (P = 0.022 and P = 1.2×10−4, respectively, by log-rank test; Figure 3A). At the end of the follow-up period, 61% of Msra−/− and 42% of Msra+/− mice were still living compared to 17% of Msra+/+ mice. The increasing trend in survival across genotypes (Ptrend = 1.3×10−4) mirrors the additive effect of the MSRA haplotype observed in humans. As anticipated, CftrR117H/R117H mice displayed reduced mortality, notably through the weaning period, compared to Cftr-null mice (64.3% of Msra+/+ mice alive at 40 days, n = 14; Figure 3B). However, there was no difference in the rate of survival between Msra+/+ mice and mice with one Msra null allele (Msra+/−: 76.5% alive at 40 days, n = 51; P = 0.33, log-rank test) or two null alleles (Msra−/−: 70.6% alive at 40 days, n = 51; P = 0.51). Mortality due to intestinal obstruction was confirmed in all animals for which the carcass was identified intact, and these were primarily animals that succumbed after weaning. Thus, loss of Msra expression increased survival in Cftr−/− mice by reducing the rate of fatal intestinal obstruction.

Excessive mucus accumulation in the crypts and lumen along with goblet cell hyperplasia are characteristic findings in the small and large intestine of Cftr−/− mice [17], [22], [43]. As goblet cells are the primary source for mucus in the intestine, we sought to determine if the goblet cell content of villi observed in the ileum of 15 day old mice is affected by Msra expression (Figure 4A). In wild-type (WT; i.e. non-CF) mice, goblet cell counts per villus ranged from 7.6% to 19.7% with a median of 11.1% (Figure 4B). The fraction of goblet cells in Msra−/− mice was similar to WT, ranging from 5.8% to 19.9% with a median of 13.0%, while the fraction of goblet cells in Cftr−/− mice had a wider range, 12.4% to 90.1%, and higher median (23.7%) than WT or Msra−/− mice (Figure 4B). The fraction of goblet cells in WT and Msra−/− mice and the increased proportion in Cftr−/− mice is comparable to the numbers reported in other studies [29], [31]. Like the Cftr−/− mice, goblet cell fraction in the ileum of Cftr−/−Msra−/− mice varied widely, ranging from 16.3% to 90.1% with a median of 25.9% (Figure 4B). Cftr−/− and Cftr−/−Msra−/− mice displayed considerable heterogeneity in the fraction of goblet cells per section with both groups having a subset of villi where ∼90% of cells were goblet cells (two sections in Cftr−/− and four in Cftr−/−Msra−/−).

As goblet cell fractions were not normally distributed for the Cftr−/− and Cftr−/−Msra−/− mice, we evaluated these differences using the non-parametric Mann-Whitney test. WT and Msra−/− mice had similar distributions (P = 0.97) whereas both differed significantly from Cftr−/− (WT: P = 7.6×10−5; Msra−/−: P = 2.6×10−4). Similarly, Cftr−/−Msra−/− mice differed from WT (P = 2.4×10−5) and Msra−/− mice (P = 3.8×10−5). However, the distributions in Cftr−/− and Cftr−/−Msra−/− did not differ when all observations were included (P = 0.67) or when the sections with goblet cell fractions exceeding 89% were excluded (median 22.2% vs. 22.3%, respectively; P = 0.26). Thus, loss of Msra expression does not appear to affect goblet cell hyperplasia in the ileum of CF mice despite reducing intestinal obstruction and increasing survival.
Discussion
Neonatal intestinal obstruction (also known as meconium ileus or MI) has incomplete penetrance (15%) and high heritability (∼1.0) suggesting a prominent role for modifier genes in this complication of CF [12]. Both candidate gene and genome-wide studies indicate that multiple genetic modifiers of low effect contribute to this trait in humans and in mice [12], [19], [49], [50]. The polygenic etiology of MI combined with its low incidence in CF present a substantial challenge to identifying the responsible genetic modifiers. However, by employing both linkage and transmission methods in a family-based study followed by replication in an unrelated sample of CF patients, we were able to implicate the MSRA gene on chromosome 8. To test whether manipulation of Msra expression modified intestinal obstruction in the CF mouse, we elected to use a null allele of Msra to avoid temporal or spatial issues that might have complicated a transgenic over-expression strategy. As we did not know if loss of expression would increase or decrease the rate of obstruction, we employed two CF mouse models with different rates of mortality due to intestinal obstruction at the time of weaning. Our hypothesis was that the CF null model (with high rates of obstruction) would reveal whether loss of Msra function decreased obstruction while the p.Arg117His model (with lower rates of obstruction) would reveal whether loss of Msra function increased obstruction. Indeed, reduction of Msra expression in the null CF mouse model resulted in a significant decrease in intestinal obstruction. The lack of effect in the p.Arg117His CF mice suggested that the modifying effect of Msra did not exceed the reduction in obstruction conferred by residual function of CFTR bearing p.Arg117His. Together, the two CF mouse models indicated that loss of Msra afforded protection from intestinal obstruction during the time of weaning.
The Msra null allele was generated from 129-derived embryonic stem cells, and mortality from obstruction is nearly 100% in mice on the 129 background [17]. Thus, it is unlikely that variation in the 129-derived region surrounding Msra is responsible for the reduced intestinal obstruction. Furthermore, the Msra region in mice displays minimal synteny with the region surrounding MSRA on chromosome 8 in humans. For example, tankyrase (TNKS), a gene adjacent to the 5′ end of MSRA in humans is not adjacent to Msra on chromosome 14 in the mouse but is located on mouse chromosome 8. The mouse studies provide compelling evidence supporting the contention that MSRA modulates MI in humans. Interestingly, non-CF Msra+/− mice have previously been shown to have reduced lifespan thought to be the consequence of enhanced vulnerability to oxidative stress compared to wild-type animals [51]. In contrast, we observed longer survival in CF mice lacking Msra. Our opposing finding suggests that in the disease context of CF, having less Msra affords a protective benefit.
The initial starting point in our modifier gene search was a 9 Mb region on chromosome 8 within a region linked to risk for MI in 30 concordant sibling pairs [12]. Association analysis of 133 families using transmission disequilibrium testing identified a single SNP (rs614197) that achieved region-wide significance. Lack of significant association between MI and this SNP in the CGS sample motivated us to search for evidence of untyped alleles using haplotype analysis. The association of both ‘protective’ and ‘risk’ haplotypes with MI led us to surmise that alleles of different effect exist in or near MSRA. We then tested whether the ‘protective’ MSRA haplotype had a similar influence on MI in an independent CF sample. While the TSS sample was potentially subject to ascertainment bias due to its recruitment criteria (i.e. having a sibling with CF), the CGS sample was ideal for replication as recruitment was based only on a diagnosis of CF, and the sample comprises 70% of the patients with CF in Canada. The significant correlation between the ‘protective’ haplotype and reduced incidence of MI in the CGS sample and conformity with an additive model provided reassurance that variants in MSRA modified MI risk. Finally, as several studies, including the present study, have indicated that the CFTR genotype can affect the rate of MI [14]–[16], we evaluated whether CFTR allelic variation contributed to the association between the MSRA haplotype and MI. The T-G-A haplotype associated with MI in an additive fashion in individuals with identical CFTR genotypes (p.Phe508del homozygotes), thereby demonstrating that MSRA modifies MI independently of variation in CFTR, the disease-causing gene.
Heterogeneity of effect appears to explain the observed linkage of risk for MI to the region encompassing MSRA. As noted above, the T-G-A haplotype demonstrating robust association with MI conferred protection from MI rather than risk for MI. However, this is not the most common haplotype (∼15%); thus most siblings carry other haplotypes derived from the alleles of the three SNPs that, by definition, confer higher risk for MI than the protective T-G-A haplotype. As predicted by this assumption, the T-G-A haplotype was significantly underrepresented in siblings concordant for MI who contributed to the linkage signal on chromosome 8. By the same token, the T-G-A haplotype was over represented in siblings who did not contribute to linkage. Hence, the observed modest linkage in siblings concordant for MI was the result of sharing of neutral MSRA haplotypes or haplotypes conferring risk for MI, and lack of sharing of alleles associated with protection from MI. Sequencing of the coding regions of MSRA in three CF subjects with MI and no T-G-A haplotypes and in three subjects without MI and two T-G-A haplotypes did not identify any plausible causative variants (data not shown). Thus, we conclude that the haplotypes are tagging as yet unidentified genetic variation within or near MSRA.
Central roles for luminal hydration and mucus production in intestinal obstruction in CF mice at the time of weaning have been supported by the manipulation of expression of two genes. In one study, reduced expression of the sodium hydrogen exchanger (Nhe3) led to decreased intestinal sodium absorption, thereby increasing the hydration of luminal contents, alleviating obstruction, and improving survival [29]. Similarly, knock-out of the mucin gene Muc1 in CF mice improved survival due to reduced intestinal mucus content and less obstruction at the time of weaning [30]. As noted by many others [22], [43] as well as in this study, goblet cell hyperplasia is a consistent histologic feature in the small and large intestines of CF mice. However, the role of goblet cells in intestinal obstruction in CF mice is not clear [43]. Reduced expression of Nhe3 relieved obstruction and eliminated goblet cell hyperplasia [29] while increased expression of the goblet cell marker Clca3 (Gob5) relieved obstruction and increased goblet cell hyperplasia in CF mice [31]. Rozmahel and colleagues suggested that Clca3 may reduce mucin release from goblet cells, given the observation of increased goblet cell size, thereby reducing luminal mucus content and intestinal obstruction in the CF mice [31]. Evidence of association between MI and the p.Ser357Asn variant in CLCA1, the human ortholog of the murine Clca3, in 682 European CF subjects suggests that this goblet cell marker may also contribute to intestinal obstruction in humans [52]. Thus, alteration of mucus content in the CF intestine, either by reduction in goblet cell numbers or down-regulation of mucus release, appears to affect the rate of intestinal obstruction. Our analysis indicated that loss of Msra expression did not affect goblet cell hyperplasia in the Cftr−/− ileum despite mitigating intestinal obstruction; thus the link between Msra and intestinal obstruction in the context of CF is not immediately clear.
In humans, loss of CFTR function leads to a combination of impaired pancreatic secretion of proteolytic enzymes and deficient luminal hydration of meconium [53], [54]. Consequently, meconium from CF neonates is abnormally proteinaceous compared to that of normal neonates, and it has been proposed that the altered viscoelastic properties of meconium predispose fetuses and neonates with CF to intestinal obstruction [55]. In young CF mice, pancreatic exocrine function is relatively preserved [17]. However, inadequate hydration and excessive mucus secretion leads to distension of the crypts of Lieberkühn in the ileum and colon and formation of concretions that appear to play a role in obstruction [17], [22], [23], [56]. MSRA encodes an antioxidant enzyme that modifies the activity of certain proteins by reducing methionine residues [57]. It is expressed in the intestine and other tissues, particularly the liver, kidney, and brain [58]–[60]. A possible link between MSRA and MI may be that MSRA can modulate the activity of proteolytic enzymes [61]. Maximizing the activity of any residual enzymes produced by the fetal pancreas would likely contribute to the breakdown of intestinal proteins in utero, thereby reducing the risk of obstruction. Evidence that MSRA modifies intestinal obstruction provides new opportunities to investigate the above concepts and the mechanisms underlying intestinal obstruction in mice and in humans lacking CFTR.
Materials and Methods
Ethics statement
This study was approved by the institutional/ethical review boards of all participating institutions. Written, informed consent or assent was obtained from all subjects before enrollment in the study. Experiments on mice were approved by the Case Western Reserve University Institutional Animal Care and Use Committee.
Study population and phenotype description
Study subjects were derived from the North American CF Modifier Gene Consortium, which is comprised of three independent collections of CF subjects. Subjects in the discovery sample were part of the Cystic Fibrosis Twin and Sibling Study (TSS) at Johns Hopkins University (n = 1,125 subjects). Enrollment was based on conclusive diagnosis of CF [62]. Methods for isolation of patient DNA [63] and identification of CFTR mutations [64], [65] have been previously described. The diagnosis of MI was based on the presence of the following features in the newborn period: lack of passage of stool within 24 hours after birth, evidence of intestinal obstruction on abdominal radiograph (ground-glass appearance of intestine, air-fluid levels, and/or intra-abdominal calcifications), evidence of colonic abnormality (microcolon on radiograph), and treatment for obstruction (enema or surgery). Individuals with clinically defined PS, a CFTR mutation associated with PS, or unknown pancreatic status were excluded (n = 143), as was one subject with unknown MI status. The primary analysis was conducted in 133 “MI families” in which at least one sibling was affected with MI (270 subjects, 169 parents). Case/control analyses were restricted to persons of self-reported European descent to minimize the potential for spurious associations due to race-related differences in allele frequencies; whereas the primary family-based transmission analysis, which was robust against population stratification, included an additional 23 individuals of non-European or mixed descent.
Findings from the primary analysis were tested in an independent sample which has been described elsewhere [34]. The replication population consisted of 1,573 CF subjects from the CGS [40]. All subjects were defined as having PI. Exclusion of non-whites yielded 1,383 subjects (including 56 sib-ships) for analysis. Rates of MI in subjects carrying the CFTR p.Gly551Asp mutation (c.1652G>A) or who were homozygous for p.Phe508del were evaluated in the entire TSS sample and the CGS sample. Subjects with p.Gly551Asp carried this mutation in trans with another PI-associated mutation: p.Cys343X (c.1029delC), c.1585-1G>A, p.Lys1177SerfsX15 (c.3528delC), c.489+1G>T, p.Glu585X (c.1753G>T), p.Phe508del, p.Gly542X (c.1624G>T), p.Gly551Asp, p.Asn1303Lys (c.3909C>G), p.Arg553X (c.1657C>T), p.Val520Phe (c.1558G>T), or p.Trp1282X (c.3846G>A). Mutation legacy names can be found in the Cystic Fibrosis Mutation Database (http://www.genet.sickkids.on.ca).
Genotyping
Linkage disequilibrium between SNPs was assessed using Haploview (http://www.broad.mit.edu/mpg/haploview) [66]. DNA extracted from either whole blood or transformed lymphocyte cell lines was hybridized to the Illumina Infinium 610-Quad SNP array platform for whole genome genotyping at the McGill University and Génome Québec Innovation Centre. Genotyping was performed and stringent quality control measures were employed simultaneously in both cohorts, and the quality of SNP calls was deemed to be very high (0.004% discordance between replicate samples). SNPs were excluded from analysis in all cohorts if the call rate was <90%, if the minor allele frequency was <1%, or if the Mendelian error rate was >1%. For family-based studies, any marker displaying non-Mendelian inheritance was dropped from analysis for any family with the error. A detailed description of additional quality control measures can be found in Wright, et al [34].
Statistical analysis
For the primary study, family-based association testing of SNPs and haplotypes was performed using the PBAT module [67] implemented within the Golden Helix HelixTree software package (Golden Helix, Inc. Bozeman, MT, USA; http://www.goldenhelix.com). A sliding-window approach was employed to test the transmission of haplotypes composed of three adjacent SNPs (frequency >1%). A 2 Mb region was selected for haplotype testing as the number of possible unique haplotypes would be nearly equal to the number of SNPs tested in the initial analysis. Therefore, to be considered significant, a haplotype would have to reach the same Bonferroni-corrected threshold (or higher) that was set for SNPs in the initial analysis. An additive genetic model was applied under a null hypothesis of linkage and no association, and standard phenotypic residuals were used as offsets to increase the power of the test statistic. Bonferroni correction was applied by multiplying nominal P values by the total number of SNPs or haplotypes tested. The SNP association plot was generated using LocusZoom 1.0 (http://csg.sph.umich.edu/locuszoom).
For case/control analyses, haplotypes were derived in the primary and replication populations using an expectation-maximization (EM) algorithm implemented in Golden Helix. Statistical analysis was performed in Stata10 (StataCorp, College Station, TX, USA). Comparison of MI status to the number of copies of haplotypes was performed using Fisher's exact test (using only subjects with haplotypes that could be determined with 100% posterior probability). Odds ratios comparing the odds of MI in subjects with 0, 1 or 2 copies of the chr8 haplotype, thus assuming an additive model, were estimated using logistic regression. For subjects with more than one haplotype assignment, EM probability estimates were used to weight haplotypes. For TSS subjects, parental and sibling genotypes were utilized when possible to resolve phase. For studies including siblings (TSS and CGS), empiric standard errors account for the possibility of sib-pair correlation [68].
Mouse studies
Mice heterozygous for a Cftr null allele, B6.129P2-Cftrtm1Unc [17] or for the Cftr missense allele p.Arg117His, B6.129S6-Cftrtm2Uth [44], and either homozygous or heterozygous for a null allele of Msra [51] were generated as breeders to produce CF mice carrying the three genotypes of Msra (+/+, +/−, and −/−) used in this study. Mice were housed at constant temperature (22°C) on a 12 hour light/dark cycle. Cages were checked daily for births and for monitoring health and survival of animals to 40 days. Mice were weaned at 21 days of age onto an enriched diet (9F Sterilizable Rodent Diet 7960, Harlan Teklad, Madison, WI) and provided water ad libitum. Mice were of mixed background, but predominantly C57BL/6J (>87%; crossed a minimum of two generations to C57BL/6J and some animals up to ten generations).
Genotyping was carried out on 7 to 10-day old animals and if death occurred before this point, genotypes were determined from carcasses. Cftr genotyping was performed as previously described [44]. Msra genotyping was carried out using two primer sets to generate a 579-bp product for the wild-type allele (forward 5′-GTGTGAGAATAAACAGATGTTCTATGC-3′ and reverse 5′-GGGTTGAGTACACTCCTTTCA-3′) or a 320-bp product for the mutant null allele (forward 5′-AAAGCGCCTCCCTACCCG-3′ and reverse 5′-ACTGTGCCCAGTTTAGTCCGTG-3′). Samples were amplified by an initial denaturation for 5 min at 95°C followed by 35 cycles of 95°C for 30 sec, 59°C for 30 sec, and 72°C for 30 sec. PCR products were fractionated on a 1% agarose gel. Kaplan-Meier survival curves were plotted and differences in survival were analyzed by the non-parametric test of trends and log-rank test of equality using Stata10.
For histology, freshly harvested tissues were fixed in 10% formalin. Tissue was embedded in paraffin, sectioned at 5 µm thickness on a microtome, and mounted on glass slides for microscopy. Deparaffinized slides were stained with Alcian blue and periodic acid Schiff (PAS) stains, then counterstained with acidified Harris hematoxylin. Comparison of goblet cell proportions between groups was performed using the non-parametric Mann-Whitney test.
Supporting Information
{kind=link}
Zdroje
1. WelshMJRamseyBWAccursoFJCuttingGR 2001 Cystic Fibrosis. ScriverCRBeaudetALValleDSlyWS The Metabolic and Molecular Bases of Inherited Disease New York McGraw-Hill, Inc 5121 5188
2. SchuttWHIslesTE 1968 Protein in meconium from meconium ileus. Arch Dis Child 43 178 181
3. BrockDJBarronL 1986 Biochemical analysis of meconium in fetuses presumed to have cystic fibrosis. Prenat Diagn 6 291 298
4. HsiehMCBerryHK 1988 Protease inhibitor and defective proteolysis in cystic fibrosis. Dig Dis Sci 33 282 288
5. EvansAKFitzgeraldDAMcKayKO 2001 The impact of meconium ileus on the clinical course of children with cystic fibrosis. Eur Respir J 18 784 789
6. LaiHJChengYChoHKosorokMRFarrellPM 2004 Association between initial disease presentation, lung disease outcomes, and survival in patients with cystic fibrosis. Am J Epidemiol 159 537 546
7. KapplerMFeilckeMSchroterCKraxnerAGrieseM 2009 Long-term pulmonary outcome after meconium ileus in cystic fibrosis. Pediatr Pulmonol 44 1201 1206
8. DonnisonABShwachmanHGrossRE 1966 A review of 164 children with meconium ileus seen at the Children's Hospital Medical Center, Boston. Pediatrics 37 833 850
9. AllanJRRobbieMPhelanPDDanksDM 1981 Familial occurence of meconium ileus. Eur J Ped 135 291 292
10. KeremECoreyMKeremB-SDuriePTsuiLC 1989 Clinical and genetic comparisons of patients with cystic fibrosis, with or without meconium ileus. J Pediatr 114 767 773
11. PicardEAviramMYahavYRivlinJBlauH 2004 Familial concordance of phenotype and microbial variation among siblings with CF. Pediatr Pulmonol 38 292 297
12. BlackmanSMDeering-BroseRMcWilliamsRNaughtonKColemanB 2006 Relative contribution of genetic and nongenetic modifiers to intestinal obstruction in cystic fibrosis. Gastroenterology 131 1030 1039
13. KeremBRommensJMBuchananJAMarkiewiczDCoxTK 1989 Identification of the cystic fibrosis gene: genetic analysis. Science 245 1073 1080
14. KristidisPBozonDCoreyMMarkiewiczDRommensJ 1992 Genetic determination of exocrine pancreatic function in cystic fibrosis. Am J Hum Genet 50 1178 1184
15. HamoshAKingTMRosensteinBJCoreyMLevisonH 1992 Cystic fibrosis patients bearing the common missense mutation Gly->Asp at codon 551 and the deltaF508 are indistinguishable from deltaF508 homozygotes except for decreased risk of meconium ileus. Am J Hum Genet 51 245 250
16. FeingoldJGuilloud-BatailleM 1999 Genetic comparisons of patients with cystic fibrosis with or without meconium ileus. Clinical Centers of the French CF Registry. Ann Genet 42 147 150
17. SnouwaertJNBrigmanKKLatourAMMaloufNNBoucherRC 1992 An animal model for cystic fibrosis made by gene targeting. Science 257 1083 1088
18. RatcliffREvansMJCuthbertAWMacVinishLJFosterD 1993 Production of a severe cystic fibrosis mutation in mice by gene targeting. Nat Genet 4 35 41
19. RozmahelRWilschanskiMMatinAPlyteSOliverM 1996 Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nature Genet 12 280 287
20. HouwenRHvan der DoefHPSermetIMunckAHauserB 2010 Defining DIOS and constipation in cystic fibrosis with a multicentre study on the incidence, characteristics, and treatment of DIOS. J Pediatr Gastroenterol Nutr 50 38 42
21. ColomboCEllemunterHHouwenRMunckATaylorC 2011 Guidelines for the diagnosis and management of distal intestinal obstruction syndrome in cystic fibrosis patients. J Cyst Fibros 10 Suppl 2 S24 S28
22. GrubbBRBoucherRC 1999 Pathophysiology of gene-targeted mouse models for cystic fibrosis. APS Conf 79 S193 S214
23. DorinJRDickinsonPAltonEWFWSmithSNGeddesDM 1992 Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature 359 211 215
24. ColledgeWHAbellaBSSouthernKWRatcliffRJiangC 1995 Generation and characterization of a deltaF508 cystic fibrosis mouse model. Nature Genet 10 445 452
25. DorinJRStevensonBJFlemingSAltonEWDickinsonP 1994 Long-term survival of the exon 10 insertional cystic fibrosis mutant mouse is a consequence of low level residual wild-type CFTR gene expression. Mamm Genome 5 465 472
26. van DoorninckJHFrenchPJVerbeekEPetersRHPCMorreauH 1995 A mouse model for the cystic fibrosis deltaF508 mutation. EMBO J 14 4403 4411
27. DelaneySJAltonESmithSLunnDFarleyD 1996 Cystic fibrosis mice carrying the missense mutation G551D replicate human genotype-phenotype correlations. EMBO J 15 955 963
28. DickinsonPKimberWLKilanowskiFMWebbSStevensonBJ 2000 Enhancing the efficiency of introducing precise mutations into the mouse genome by hit and run gene targeting. Transgenic Res 9 55 66
29. BradfordEMSartorMAGawenisLRClarkeLLShullGE 2009 Reduced NHE3-mediated Na+ absorption increases survival and decreases the incidence of intestinal obstructions in cystic fibrosis mice. Am J Physiol Gastrointest Liver Physiol 296 G886 G898
30. ParmleyRRGendlerSJ 1998 Cystic fibrosis mice lacking Muc1 have reduced amounts of intestinal mucus. J Clin Invest 102 1798 1806
31. YoungFDNewbiggingSChoiCKeetMKentG 2007 Amelioration of cystic fibrosis intestinal mucous disease in mice by restoration of mCLCA3. Gastroenterology 133 1928 1937
32. SunXSuiHFisherJTYanZLiuX 2010 Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest 120 3149 3160
33. MeyerholzDKStoltzDAPezzuloAAWelshMJ 2010 Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am J Pathol 176 1377 1389
34. WrightFAStrugLJDoshiVKCommanderCWBlackmanSM 2011 Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nature Genet 43 539 546
35. LangeCLairdNM 2002 On a general class of conditional tests for family-based association studies in genetics: the asymptotic distribution, the conditional power, and optimality considerations. Genet Epidemiol 23 165 180
36. BremerLABlackmanSMVanscoyLLMcDougalKEBowersA 2008 Interaction between a novel TGFB1 haplotype and CFTR genotype is associated with improved lung function in cystic fibrosis. Hum Mol Genet 17 2228 2237
37. GreeneCSPenrodNMWilliamsSMMooreJH 2009 Failure to replicate a genetic association may provide important clues about genetic architecture. PLoS ONE 4 e5639 doi:10.1371/journal.pone.0005639
38. GalarneauGPalmerCDSankaranVGOrkinSHHirschhornJN 2010 Fine-mapping at three loci known to affect fetal hemoglobin levels explains addtional genetic variation. Nature Genet 42 1049 1051
39. WoodARHernandezDGNallsMAYaghootkarHGibbsJR 2011 Allelic heterogeneity and more detailed analyses of known loci explain additional phenotypic variation and reveal complex patterns of association. Hum Mol Genet 20 4082 4092
40. DorfmanRLiWSunLLinFWangY 2009 Modifier gene study of meconium ileus in cystic fibrosis: statistical considerations and gene mapping results. Hum Genet 126 763 778
41. WatsonMSCuttingGRDesnickRJDriscollDAKlingerK 2004 Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med 6 387 391
42. van HouwelingenHCArendsLRStijnenT 2002 Advanced methods in meta-analysis: multivariate approach and meta-regression. Stat Med 21 589 624
43. WilkeMBuijs-OffermanRMAarbiouJColledgeWHSheppardDN 2011 Mouse models of cystic fibrosis: Phenotypic analysis and research applications. Journal of Cystic Fibrosis 10 S152 S171
44. Van HeeckerenAMSchluchterMDDrummMLDavisPB 2004 Role of Cftr genotype in the response to chronic Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol 287 L944 L952
45. SheppardDNRichDPOstedgaardLSGregoryRJSmithAE 1993 Mutations in CFTR associated with mild-disease-form Cl - channels with altered pore properties. Nature 362 160 164
46. CarrollTPMcIntoshIEganMEZeitlinPLCuttingGR 1994 Transmembrane mutations alter the channels characteristics of the cystic fibrosis transmembrane conductance regulator expressed in Xenopus oocytes. Cell Physiol Biochem 4 10 18
47. The Cystic Fibrosis Genotype-Phenotype Consortium 1993 Correlation between Genotype and Phenotype in Patients with Cystic Fibrosis. N Engl J Med 329 1308
48. HodgesCACottonCUPalmertMRDrummML 2008 Generation of a conditional null allele for Cftr in mice. Genesis 46 546 552
49. ZielenskiJCoreyMRozmahelRMarkiewiczDAznarezI 1999 Detection of a cystic fibrosis modifier locus for meconium ileus on human chromosome 19q13. Nature Genet 22 128 129
50. NorkinaODe LisleRC 2005 Potential genetic modifiers of the cystic fibrosis intestinal inflammatory phenotype on mouse chromosomes 1, 9, and 10. BMC Genet 6 29
51. MoskovitzJBar-NoySWilliamsWMRequenaJBerlettBS 2001 Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci U S A 98 12920 12925
52. van der DoefHPSliekerMGStaabDAlizadehBZSeiaM 2010 Association of the CLCA1 p.S357N variant with meconium ileus in European patients with cystic fibrosis. J Pediatr Gastroenterol Nutr 50 347 349
53. BerschneiderHMKnowlesMRAzizkhanRGBoucherRCTobeyNA 1988 Altered intestinal chloride transport in cystic fibrosis. FASEB J 2 2625 2629
54. TaylorCJBaxterPSHardcastleJHardcastlePT 1988 Failure to induce secretion in jejunal biopsies from children with cystic fibrosis. Gut 29 957 962
55. GreenMNClarkeJTShwachmanH 1958 Studies in cystic fibrosis of the pancreas; protein pattern in meconium ileus. Pediatrics 21 635 641
56. O'NealWKHastyPMcCrayPBJrCaseyBRivera-PerezJ 1993 A severe phenotype in mice with a duplication of exon 3 in the cystic fibrosis locus. Hum Mol Genet 2 1561 1569
57. OienDBMoskovitzJ 2008 Substrates of the methionine sulfoxide reductase system and their physiological relevance. Curr Top Dev Biol 80 93 133
58. KuschelLHanselASchonherrRWeissbachHBrotN 1999 Molecular cloning and functional expression of a human peptide methionine sulfoxide reductase (hMsrA). FEBS Lett 456 17 21
59. MoskovitzJWeissbachHBrotN 1996 Cloning the expression of a mammalian gene involved in the reduction of methionine sulfoxide residues in proteins. Proc Natl Acad Sci U S A 93 2095 2099
60. MoskovitzJJenkinsNAGilbertDJCopelandNGJurskyF 1996 Chromosomal localization of the mammalian peptide-methionine sulfoxide reductase gene and its differential expression in various tissues. Proc Natl Acad Sci U S A 93 3205 3208
61. WeissbachHResnickLBrotN 2005 Methionine sulfoxide reductases: history and cellular role in protecting against oxidative damage. Biochim Biophys Acta 1703 203 212
62. RosensteinBJCuttingGR 1998 The diagnosis of cystic fibrosis: A consensus statement. J Pediatr 132 589 595
63. CuttingGRAntonarakisSEBuetowKHKaschLMRosensteinBJ 1989 Analysis of DNA polymorphism haplotypes linked to the cystic fibrosis locus in North American Black and Caucasian families supports the existence of multiple mutations of the cystic fibrosis gene. Am J Med Genet 44 307 318
64. WangXMyersASaikiRKCuttingGR 2002 Development and Evaluation of a PCR-based, Line Probe Assay for the Detection of 58 Alleles in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Gene. Clin Chem 48 1121 1123
65. GromanJDMeyerMEWilmottRWZeitlinPLCuttingGR 2002 Variant cystic fibrosis phenotypes in the absence of CFTR mutations. N Engl J Med 347 401 407
66. BarrettJCFryBMallerJDalyMJ 2005 Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263 265
67. LangeCDeMeoDSilvermanEKWeissSTLairdNM 2004 PBAT: tools for family-based association studies. Am J Hum Genet 74 367 369
68. WilliamsRL 2000 A note on robust variance estimation for cluster-correlated data. Biometrics 56 645 646
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- PIF4–Mediated Activation of Expression Integrates Temperature into the Auxin Pathway in Regulating Hypocotyl Growth
- Metabolic Profiling of a Mapping Population Exposes New Insights in the Regulation of Seed Metabolism and Seed, Fruit, and Plant Relations
- A Splice Site Variant in the Bovine Gene Compromises Growth and Regulation of the Inflammatory Response
- Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson's Disease Genetics: The PDGene Database
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy