
Parkinson's Disease–Associated Kinase PINK1 Regulates Miro Protein Level and Axonal Transport of Mitochondria
Mutations in Pten-induced kinase 1 (PINK1) are linked to early-onset familial Parkinson's disease (FPD). PINK1 has previously been implicated in mitochondrial fission/fusion dynamics, quality control, and electron transport chain function. However, it is not clear how these processes are interconnected and whether they are sufficient to explain all aspects of PINK1 pathogenesis. Here we show that PINK1 also controls mitochondrial motility. In Drosophila, downregulation of dMiro or other components of the mitochondrial transport machinery rescued dPINK1 mutant phenotypes in the muscle and dopaminergic (DA) neurons, whereas dMiro overexpression alone caused DA neuron loss. dMiro protein level was increased in dPINK1 mutant but decreased in dPINK1 or dParkin overexpression conditions. In Drosophila larval motor neurons, overexpression of dPINK1 inhibited axonal mitochondria transport in both anterograde and retrograde directions, whereas dPINK1 knockdown promoted anterograde transport. In HeLa cells, overexpressed hPINK1 worked together with hParkin, another FPD gene, to regulate the ubiquitination and degradation of hMiro1 and hMiro2, apparently in a Ser-156 phosphorylation-independent manner. Also in HeLa cells, loss of hMiro promoted the perinuclear clustering of mitochondria and facilitated autophagy of damaged mitochondria, effects previously associated with activation of the PINK1/Parkin pathway. These newly identified functions of PINK1/Parkin and Miro in mitochondrial transport and mitophagy contribute to our understanding of the complex interplays in mitochondrial quality control that are critically involved in PD pathogenesis, and they may explain the peripheral neuropathy symptoms seen in some PD patients carrying particular PINK1 or Parkin mutations. Moreover, the different effects of loss of PINK1 function on Miro protein level in Drosophila and mouse cells may offer one explanation of the distinct phenotypic manifestations of PINK1 mutants in these two species.
Published in the journal:
. PLoS Genet 8(3): e32767. doi:10.1371/journal.pgen.1002537
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002537
Summary
Mutations in Pten-induced kinase 1 (PINK1) are linked to early-onset familial Parkinson's disease (FPD). PINK1 has previously been implicated in mitochondrial fission/fusion dynamics, quality control, and electron transport chain function. However, it is not clear how these processes are interconnected and whether they are sufficient to explain all aspects of PINK1 pathogenesis. Here we show that PINK1 also controls mitochondrial motility. In Drosophila, downregulation of dMiro or other components of the mitochondrial transport machinery rescued dPINK1 mutant phenotypes in the muscle and dopaminergic (DA) neurons, whereas dMiro overexpression alone caused DA neuron loss. dMiro protein level was increased in dPINK1 mutant but decreased in dPINK1 or dParkin overexpression conditions. In Drosophila larval motor neurons, overexpression of dPINK1 inhibited axonal mitochondria transport in both anterograde and retrograde directions, whereas dPINK1 knockdown promoted anterograde transport. In HeLa cells, overexpressed hPINK1 worked together with hParkin, another FPD gene, to regulate the ubiquitination and degradation of hMiro1 and hMiro2, apparently in a Ser-156 phosphorylation-independent manner. Also in HeLa cells, loss of hMiro promoted the perinuclear clustering of mitochondria and facilitated autophagy of damaged mitochondria, effects previously associated with activation of the PINK1/Parkin pathway. These newly identified functions of PINK1/Parkin and Miro in mitochondrial transport and mitophagy contribute to our understanding of the complex interplays in mitochondrial quality control that are critically involved in PD pathogenesis, and they may explain the peripheral neuropathy symptoms seen in some PD patients carrying particular PINK1 or Parkin mutations. Moreover, the different effects of loss of PINK1 function on Miro protein level in Drosophila and mouse cells may offer one explanation of the distinct phenotypic manifestations of PINK1 mutants in these two species.
Introduction
PD is a neurodegenerative disorder characterized by the dysfunction and loss of dopaminergic (DA) neurons in the substantia nigra, although neurons in other brain regions are affected as well. Mutations in PINK1 and Parkin are linked to familial forms of early-onset PD [1], [2]. PINK1 encodes a Ser/Thr kinase with a mitochondrial targeting sequence, whereas Parkin encodes an E3 ubiquitin ligase. Studies in Drosophila first revealed that PINK1 and Parkin act in a common pathway to impact mitochondrial function and DA neuron maintenance [3]–[6], in part through the regulation of mitochondrial fission/fusion dynamics [7]–[11]. At least in primary cultured mammalian hippocampal neurons and DA neurons, PINK1 and Parkin have been shown to exert similar effects on mitochondrial dynamics as seen in Drosophila DA neurons [7], [12]. PINK1 and Parkin are also implicated in mitochondrial quality control [13]. Decreased mitochondrial membrane potential stabilizes the normally labile PINK1, which recruits Parkin to damaged mitochondria, leading to ubiquitination of mitochondrial proteins and marking damaged mitochondria for removal by autophagy [14]. Both mitochondrial fission/fusion dynamics and autophagy are considered important aspects of the mitochondrial quality control mechanism that mediates PINK1/Parkin function in DA neuron maintenance [11], [15]–[17].
In some PINK1 - or Parkin-linked PD patients, symptoms of peripheral neuropathy were also reported [18]–[20]. It is not clear whether this is caused by defects in the aforementioned functions or some other unknown function of PINK1/Parkin. Peripheral neuropathy is a clinical term used to describe various forms of damages to nerves of the peripheral nervous system by distinct mechanisms [21]. Many types of peripheral neuropathy are dependent on the length of neuronal axon, with neurons carrying long axons frequently affected. It is hypothesized that this is caused by defects in the axonal transport of key proteins and/or organelles such as mitochondria, which are critical for maintaining the axonal and synaptic physiology of those extremely polarized neurons [22]. This notion has gained significant support from recent studies of the inherited forms of peripheral neuropathies [22], [23]. Defective mitochondrial transport has also been considered a pathogenic event in other neurodegenerative diseases [22], [24], [25], including rodent models of PD [26], [27]. In primary cultured rat hippocampal neurons, overexpression of PINK1 has been shown to inhibit the lateral movement of photoactivated, mito-Dendra2-labelled mitochondria [12], raising the possibility that defects in the axonal transport of mitochondria may actively participate in PINK1-related PD pathogenesis.
A major aspect of axonal transport is mediated by motor proteins that travel on axonal microtubules, which are polarized and uniformly orientated, with their plus-ends pointing towards nerve terminals. The kinesin and dynein motors are involved in microtubule plus-end (anterograde) and minus-end directed (retrograde) transport, respectively [28]. Mitochondria are mainly produced in neuronal cell body and delivered to sites where metabolic demand is high, such as the synapses and nodes of Ranvier [29]. The functions of Mitochondrial Rho (Miro), a mitochondrial outer membrane GTPase [30], and the cytosolic protein Milton are critical for mitochondrial transport, as they serve to link mitochondria with kinesin motors and the microtubule cytoskeleton [31], [32]. In Drosophila Miro or Milton mutants, mitochondria accumulate in neuronal soma and fail to move into the axons [31], [32]. In cultured mammalian cells, overexpression of a constitutively active mutant of Miro was shown to induce cell death, suggesting that mitochondrial transport or some other aspect of Miro function is important for cell survival [30]. Whether this is relevant to in vivo conditions such as neurodegenerative disease settings is not known.
The fruit fly Drosophila melanogaster has served as an excellent model for studying neurodegenerative diseases [10]. It was in Drosophila that the in vivo function of PINK1 was first revealed [3]–[6]. PINK1 mutant flies exhibit abnormal wing postures, reduced flight ability and thoracic ATP level, degeneration of indirect flight muscle and DA neurons, and male sterility, which are caused by the accumulation of dysfunctional mitochondria, thus suggesting a role of PINK1 in mitochondrial function and/or quality control [3]–[6]. Further genetic studies in Drosophila have also uncovered important functions of PINK1 in regulating mitochondrial morphology and electron transport chain activity [7]–[9], [33].
The power of the Drosophila neurodegenerative disease models lies in the ability to facilitate unbiased genetic modifier screens to identify new players involved in the disease process. Using this approach, we show in this study that PINK1 genetically interacts with the mitochondrial transport machinery. Reduction of function in Miro, Milton, or kinesin heavy chain effectively rescued the PINK1 mutant phenotypes. On the other hand, overexpression (OE) of Miro led to the formation of enlarged mitochondria and resulted in DA neuron loss, thus phenocopying PINK1 mutants. By monitoring mitochondrial movement in live Drosophila larval motor neurons, which possesses long axons and could serve as a model system for studying peripheral neuropathy, we provide evidence that PINK1 directly regulates mitochondrial transport. The function of PINK1 in mitochondrial transport may contribute to PD pathogenesis in DA neurons and underlie the peripheral neuropathy symptoms associated with certain PINK1 mutations in some PD patients.
Our biochemical analysis demonstrated that overexpressed PINK1 in cooperation with Parkin could regulate Miro protein ubiquitination and stability, which might contribute to the regulatory effect of PINK1 on mitochondrial motility. While our paper was under review, it was suggested that PINK1 phosphorylates Miro at a conserved S156 residue, and that this phosphorylation event is required to activate proteasomal degradation of Miro in a Parkin-dependent manner [34]. However, our in vitro kinase assay using active recombinant PINK1 failed to show direct phosphorylation of Miro by PINK1. Moreover, a mutant form of hMiro1 with the S156 site mutated to Ala was equally susceptible to PINK1/Parkin-mediated degradation in HeLa cells. Thus, the exact molecular mechanism by which the PINK1/Parkin pathway regulates Miro protein level will require further investigation.
Results
Genetic interaction between PINK1 and the mitochondrial transport machinery
By taking advantage of the easily identifiable phenotype of abnormal wing posture induced by dPINK1 inactivation, we performed a genetic screen for modifiers of PINK1. The scheme was similar as described before [35]. In this screen, we identified components of the mitochondrial transport machinery as genetic modifier of PINK1. Knockdown of Miro, Milton or Kinesin heavy chain (Khc) each rescued the muscle phenotypes in PINK1B9 null mutant, including abnormal wing posture, decreased fly ability and ATP depletion (Figure 1A–1C). Conversely, overexpression (OE) of Miro and Khc enhanced such phenotypes (Figure 1A–1C). These results demonstrated strong genetic interaction between PINK1 and the mitochondrial transport machinery as a whole, supporting that mitochondrial transport is the underlying mechanism mediating their genetic interaction. In this study, we will focus our analysis on Miro, as a previous study in cultured cells suggested that mammalian Miro might physically interact with PINK1 [36].

Genetic interaction between Miro and PINK1 in the PD–relevant DA neurons
To test the relevance of the functional interaction between PINK1 and the mitochondrial transport machinery to PD pathogenesis, we examined their interaction in DA neurons, the disease-relevant cell type. As in the muscle, Miro-RNAi effectively rescued PINK1 mutant phenotypes in DA neurons, both in terms of mitochondrial aggregation (Figure 2A–2D) and DA neuron loss (Figure 2F). Moreover, Miro-OE alone, driven by the TH-Gal4 driver, caused aberrant mitochondrial aggregation (2E, 2G) and DA neuron loss (Figure 2F), thus phenocopying PINK1 loss-of-function effects (Figure 2B, 2F, 2G), although the Miro-OE effect was noticeably stronger than PINK1 mutant. It is worth noting that TH-Gal4-driven Miro-OE in PINK1 mutant background resulted in dramatically reduced viability (data not shown), although the surviving adults did not show further DA neuron loss than that induced by Miro-OE alone (Figure 2F). Together, these results demonstrate that PINK1 and Miro also exhibit strong genetic interaction in DA neurons, with decreased dMiro level/activity ameliorating the detrimental effects caused by the loss of dPINK1, whereas increased dMiro level or activity phenocopying dPINK1 mutants.

PINK1 regulates mitochondrial motility in Drosophila larval motor neurons
The strong genetic interaction between PINK1 and Miro raised the interesting possibility that PINK1 might directly regulate mitochondrial transport, the impairment of which might contribute to PINK1-related parkinsonism. This was further supported by the DA neuron loss induced by Miro-OE alone, which presumably acted by altering mitochondrial transport. To test this idea, we examined the effect of PINK1 on mitochondrial movement in Drosophila larval motor neurons, a system amenable to live imaging of mitochondrial transport. Mitochondrially-targeted GFP (mitoGFP) expressed specifically in motor neurons was used to track mitochondrial movement via live imaging in anesthetized third instar larvae (Figure 3A), using well-established procedures [37], [38]. To highlight the mitochondria undergoing active transport, a 61.5 µm-long segment of motor neuron was photobleached and the movement of fluorescently labeled mitochondria moving into the bleached area from both directions was recorded at 1 frame/2 s for 300 s (Figure 3B, Videos S1, S2, S3, S4, S5, S6, S7). From these videos, mitochondrial flux (the normalized number of mitochondria that passes certain point over time), mitochondrial net velocity (the normalized mitochondrial net displacement over time), and mitochondrial morphology (e.g. mitochondrial length) in different genetic backgrounds were analyzed. In general, mitochondrial net velocity is controlled primarily by the intrinsic properties and quantities of motor proteins associated with the mitochondria [38], while mitochondrial flux can also be significantly affected by mitochondrial morphology, as changes in mitochondrial morphological features such as length can increase or decrease the number of motile mitochondria.

We found that PINK1-OE decreased mitochondrial flux as well as net velocity in both anterograde and retrograde directions, similar to the effect of Miro-RNAi, although the PINK1-OE effect appeared to be slightly weaker (Figure 3B–3D; Videos S1, S2, S4). In contrast, PINK1-RNAi and Miro-OE both increased the net velocity of anterograde mitochondrial transport, with retrograde transport largely unaffected (Figure 3B, 3C; Videos S3, S5). PINK1-RNAi also increased anterograde mitochondrial flux (Figure 3C), while mitochondrial flux in Miro-OE background was reduced in both anterograde and retrograde directions (Figure 3C). The reduction of mitochondrial flux by Miro-OE could be partially explained by the formation of very long mitochondria in Miro-OE motor neurons (Figure 3E, 3F), as previously observed [38].
In addition to mitochondrial motility, PINK1 also affected mitochondrial length in motor neurons. PINK1-RNAi increased mitochondrial length in the axons of larval motor neurons as in Miro-OE case, although the effect of Miro-OE was much stronger. Conversely, PINK1-OE and Miro-RNAi both decreased mitochondrial length (Figure 3E, 3F). To address whether PINK1-induced mitochondrial motility change was due to its effect on mitochondrial length, we examined mitochondrial transport in genetic backgrounds where mitochondrial fusion/fission machinery was directly manipulated to alter mitochondrial length. Increasing mitochondrial fission by overexpression of the fission protein Fis1 or knockdown of the fusion protein Marf led to decreased mitochondrial length (Figure 3E, 3F), similar to the effects of Miro-RNAi or PINK1-OE. However, in contrast to the decreased mitochondrial flux and net velocity as observed in the Miro-RNAi or PINK1-OE backgrounds, Fis1-OE and Marf-RNAi both increased mitochondrial flux and net velocity in anterograde and retrograde directions (Figure 3B–3D; Videos S6, S7), suggesting that mitochondrial length and transport kinetics are not always directly correlated, which is consistent with a previous report [38]. Collectively, these results support the notion that PINK1 regulates mitochondrial transport and that its effect on mitochondrial motility is direct, rather than a secondary effect of mitochondrial length change.
Altered PINK1 activities affect mitochondrial distribution in motor neuron axons
In addition to mitochondrial motility, we also examined mitochondrial distribution at motor neuron nerve terminals at the larval neuromuscular junction (NMJ), which can be used as an indirect measure of mitochondrial motility. Consistent with a previous report [31], Miro-OE led to the accumulation of mitochondria in the most distal boutons, which is likely the consequence of net anterograde transport (Figure 4A, 4B). PINK1 knockdown showed similar effect (Figure 4A, 4B). Thus, the dysfunctional mitochondria in PINK1 mutant might gain longer retention time in the distal segment of motor neuron axons where synapses are formed. This finding may have clinical implications for PINK1 pathogenesis. In contrast, Miro-RNAi and PINK1-OE both led to decreased accumulation of mitochondria in the most distal boutons (Figure 4A, 4B). We conclude that PINK1 regulates mitochondrial distribution in motor neuron nerve terminals, likely through its effect on mitochondrial transport.

Overactivation of the PINK1/Parkin pathway reduces Miro protein level
Our results so far showed that PINK1 and Miro exert opposite effects on mitochondrial morphology, motility and distribution. We next explored the biochemical mechanisms underlying their negative genetic relationship. We first used HeLa cells to test whether Miro protein level might be regulated by PINK1 and possibly Parkin, which tends to work together with PINK1 in a common pathway [3]–[5], [39]. There are two Miro homologues in human cells, hMiro1 and hMiro2, that are ∼60% identical [30]. Overexpression of either hPINK1 or hParkin did not lead to obvious change of exogenous hMiro1 protein level under normal conditions, but a modest reduction of hMiro1 level was observed when hPINK1 and hParkin were co-expressed (Figure 5A, lane 5). A decline in mitochondrial membrane-potential induced by the mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP) was reported to activate the PINK1/Parkin pathway [13], [14], [39]. Under CCCP treatment condition, hPINK1 or hParkin each significantly stimulated hMiro1 ubiquitination (Figure 5A). Since HeLa cells express very little endogenous Parkin [13], the effect of hPINK1 alone on hMiro1 ubiquitination (Figure 5A, lane 8) suggested that other E3 ligase(s) might be recruited by hPINK1 to ubiquitinate hMiro1. However, this ubiquitination event did not appear to lead to destabilization of hMiro1 (Figure 5A, IB: Myc). In contrast, coexpression of hPINK1 and hParkin dramatically reduced hMiro1 level in the presence of CCCP (Figure 5A, lanes 9). Importantly, pathogenic mutations in hPINK1 or hParkin abolished this effect (Figure 5B, lanes 4 and 5 compared with lane 3, and lanes 9–11 compared with lane 8), indicating that functional hPINK1 and hParkin are both required in the destabilization of hMiro1. Previously, many outer mitochondrial membrane (OMM) proteins were shown to be degraded by the ubiquitin proteasome system (UPS) pathway in a PINK1/Parkin-dependent reaction at an early step of mitophagy, while other OMM proteins might be eliminated by subsequent autophagosome-dependent events [40], [41]. Thus, direct or indirect substrates of PINK1/Parkin could be distinguished by their degradation kinetics [40]. In our experiments, hMiro1 was more rapidly degraded than another OMM protein VDAC1 (Figure 5B, VDAC1), a reported Parkin substrate involved in mitophagy [14], supporting that hMiro1 is a direct substrate of Parkin.
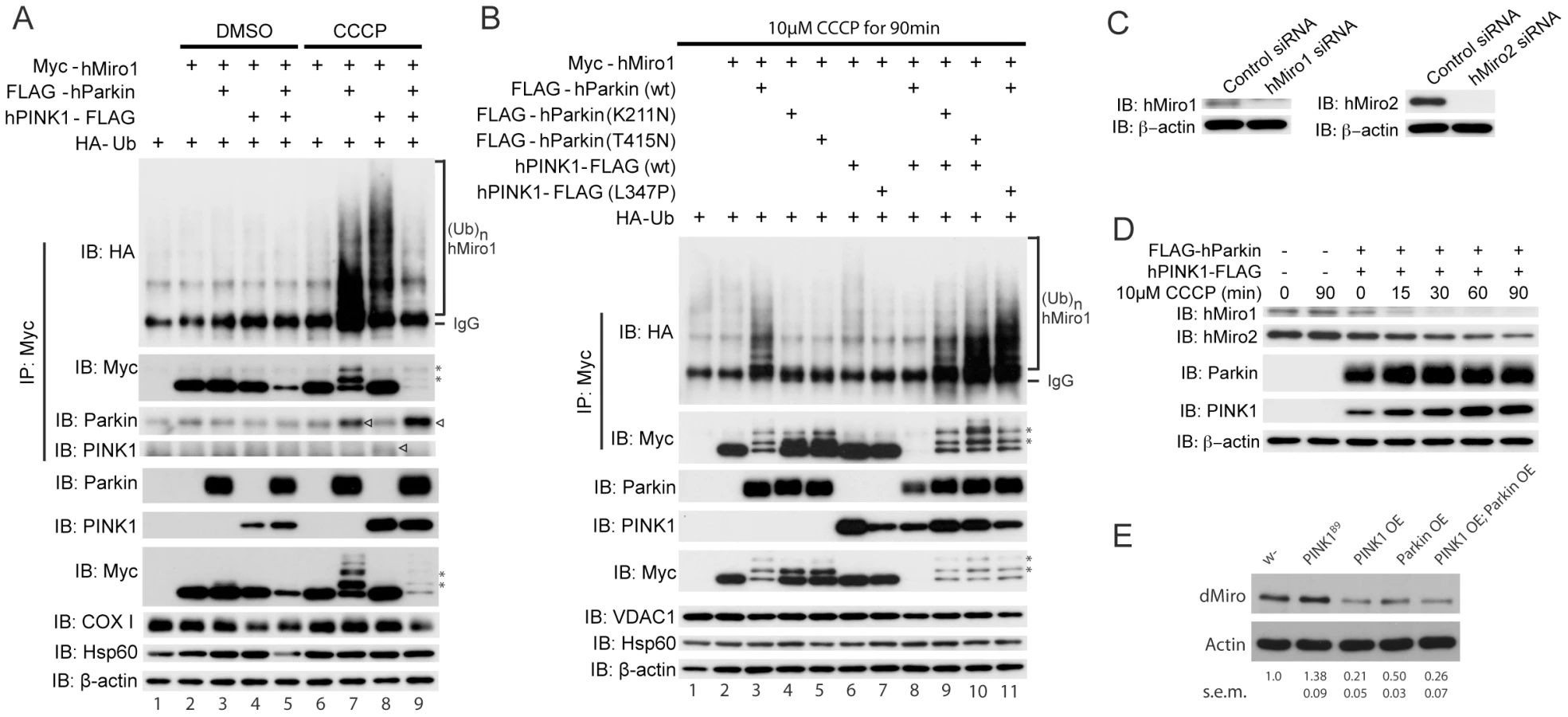
Similar to hMiro1, hMiro2 could also be ubiquitinated by PINK1 and Parkin co-expression or after CCCP treatment. However, the degradation of hMiro2 was at a much slower rate compared to hMiro1 (Figure S1), consistent with a previous result [40]. Furthermore, like exogenous hMiro1, endogenous hMiro1 was also rapidly degraded by PINK1/Parkin overexpression in HeLa cells and its level was dramatically reduced within 15 min of CCCP treatment. The degradation of endogenous hMiro2 was again at a much slower rate than that of hMiro1 (Figure 5C, 5D).
We next examined the effect of the PINK1/Parkin pathway on Miro protein level in an in vivo setting. Similar to the results in HeLa cells, Drosophila dMiro protein level was decreased in the brain extracts of PINK1 or Parkin overexpression adult flies (Figure 5E). Conversely, dMiro level was increased in PINK1B9 mutant brain extracts (Figure 5E). These results are consistent with dPINK1 negatively regulating dMiro protein level in vivo. It is worth noting that different from the effects seen in HeLa cells, overexpression of PINK1 or Parkin alone was sufficient to reduce dMiro level in adult Drosophila brain, and the co-expression of PINK1 and Parkin did not lead to much further reduction of dMiro level than PINK1-OE alone, suggesting that the endogenous levels or activities of PINK1 and Parkin are already sufficient to support each other's action in the Drosophila brain.
Knockdown of Miro promotes the removal of damaged mitochondria by Parkin-mediated mitophagy
Removal of damaged or dysfunctional mitochondria through mitophagy could be one mechanism by which the PINK1/Parkin pathway maintains mitochondrial health, at least under some conditions, and the accumulation of those abnormal mitochondria in PINK1 mutants could be the underlying cause of disease pathogenesis. Consistent with this notion, it was previously shown that enhancing autophagy could efficiently rescue dPINK1 mutant phenotypes [35]. To better understand the rescuing effect of Miro-RNAi in PINK1 mutant background, we examined the effect of Miro knockdown on mitophagy. We used CCCP treatment to induce mitochondrial damage in HeLa cells stably transfected with venus-Parkin, and subsequently monitored the removal of damaged mitochondria over time by examining the protein levels of mitochondrial markers on the inner/outer membrane or in the matrix and inter-membrane space. Simultaneous knockdown of hMiro1 and hMiro2 significantly accelerated the mitochondrial removal process, with all the mitochondrial markers disappearing faster in hMiro knockdown cells than in the control siRNA-treated cells (Figure 6A). This suggested that there was more active mitophagy after hMiro knockdown. To confirm this result, we monitored the mitochondrial network by immunofluorescence staining. Compared to the control siRNA-treated cells, knockdown of either hMiro1 or hMiro2 led to the accumulation of mitochondria in the perinuclear region, and knockdown of both hMiro1 and hMiro2 further enhanced this effect (Figure 6B). Fluorescence from the immunostaining of Tom20 (an OMM marker) but not HSP60 (a matrix marker) in hMiro1 and hMiro2 double knockdown cells was noticeably weaker than that in control siRNA treated cells at 3 h after CCCP treatment (Figure 6C), supporting the notion that hMiro knockdown facilitated an early event in Parkin-mediated mitophagy.

The conserved Ser-156 residue in hMiro1 is not required for PINK1/Parkin-mediated degradation under normal or CCCP treatment conditions in HeLa cells
We further investigated the molecular mechanisms by which the PINK1/Parkin pathway regulates Miro protein level or stability. While our paper was under review, a report showed that PINK1 phosphorylates Miro at a conserved S156 residue, and that this phosphorylation activates proteasomal degradation of Miro in a Parkin-dependent manner [34]. However, repeated in vitro kinase assays using an active GST-dPINK1 recombinant protein capable of efficient autophosphorylation [42] failed to show phosphorylation of GST-dMiroΔTM, a GST fusion protein of full-length dMiro with the transmembrane domain deleted (Figure 7A). Drosophila or mammalian PINK1 protein affinity purified from HEK293 cells by immunoprecipitation also failed to phosphorylate GST-dMiroΔTM in our assays (data not shown).

To further probe the significance of S156 phosphorylation in facilitating the proteasomal degradation of Miro promoted by the PINK1/Parkin pathway, we introduced S156A mutations into hMiro1 or hMiro2 and examined the stability of the mutant proteins in HeLa cells co-transfected with PINK1 and Parkin, under normal or CCCP treatment conditions. As shown in Figure 7B, the wild type and S156A mutant forms of hMiro1 were equally susceptible to PINK1/Parkin - mediated degradation under both conditions. The wild type and S156A mutant forms of hMiro2 behaved similarly as well (data not shown). Thus, the PINK1/Parkin pathway may regulate Miro protein level independent of S156 site phosphorylation under these experimental conditions in HeLa cells.
Effect of loss of PINK1 function on Miro protein level in mammalian cells
We also examined the effect of loss of PINK1 function on Miro protein level in mammalian cells. For this purpose, we used both HeLa cells with PINK1 knockdown and MEF cells derived from PINK1 (−/−) knockout mice. Surprisingly, unlike the situation in Drosophila, endogenous Miro1 or Miro2 protein levels were significantly reduced in PINK1 RNAi cells under normal or CCCP treatment conditions (Figure 7C, PINK1 RNAi in HeLa cells or Venus-Parkin stably transfected HeLa cells) and in PINK1 (−/−) MEF cells (Figure 7D). The introduction of Venus-Parkin resulted in CCCP/PINK1-dependent degradation of Miro1 (Figure 7C, Control RNAi in Venus-Parkin transfected HeLa cells). Thus, loss of PINK1 function in mammalian cells can lead to reduced expression of Miro1 and Miro2 proteins, presumably through mechanisms distinct from that operating under PINK1/Parkin co-overexpression condition.
Discussion
Mitochondrial dysfunction has long been implicated in the pathogenesis of PD. However, the exact mechanisms by which mitochondrial dysfunction arises in the disease process and how cells, particularly neurons, handle dysfunctional mitochondrial are not well understood. The identification of a mitochondrial quality control system involving two FPD genes, PINK1 and Parkin, has provided a much-needed point of entry to elucidate the role of mitochondria in the pathogenesis of PD. Here we showed that PINK1 directly regulates mitochondrial transport and that it affects the stability and/or activity of Miro, a mitochondrial Rho GTPase with a well-establish function in mitochondrial transport. Our conclusion is supported by the following evidence: 1) dMiro protein level is negatively regulated by PINK1 and Parkin in vivo in Drosophila; 2) Overexpressed PINK1 and Parkin act together to promote the ubiquitination and degradation of hMiro1 in HeLa cells; 3) Reduction of the activities of Miro or other components of the mitochondrial transport machinery effectively rescued dPINK1 mutant phenotypes. 4) Overexpression of dMiro in DA neurons phenocopied dPINK1 loss-of-function effects; 5) Manipulation of dPINK1 activity produced clear mitochondrial motility phenotypes opposite to that observed for dMiro manipulation in Drosophila larval motor neurons. Together, these results support that the mitochondrial transport defects caused by PINK1 inactivation represent one of the key pathogenic events that contribute to PD pathogenesis in the Drosophila model.
Neurons are highly polarized cells that rely heavily on axonal transport to distribute to axons and synapses critical proteins and organelles synthesized in the cell body, thereby maintaining neuronal function and health. Defects in axonal transport are often linked to diseases affecting peripheral neurons that tend to extend very long axons [21], [22], [29]. Although the symptoms of PD patients mainly arise from the loss of DA neurons, some PD patients carrying particular PINK1 and Parkin mutations developed peripheral neuropathy with unknown cause [18]–[20]. Our results showing the PINK1/Parkin pathway playing a critical role in regulating mitochondrial transport offers one potential explanation of the peripheral neuropathy symptoms observed in these PINK1 or Parkin-linked PD cases. It would be interesting to examine whether Miro protein level or activity is affected by these particular mutations in human cells. Moreover, we propose that defects in PINK1/Parkin-regulated mitochondrial transport may offer one explanation of the selective vulnerability of DA neurons observed in PD patients and animal models. DA neurons that make elaborate and long projections may be particularly vulnerable to impairment of the mitochondrial transport system.
Our results offer new insights into the mode of action of the PINK1/Parkin pathway in mitochondria quality control. We showed that, in Drosophila models, PINK1 OE led to decreased mitochondrial flux and net velocity, as observed in Miro knockdown background. In addition, we found that Miro knockdown could facilitate an early step of mitophagy in mammalian cells. These observations, together with the finding that the normally labile PINK1 protein is stabilized on damaged mitochondria [39], suggest a scenario whereby the accumulation of PINK1 on damaged mitochondria and the subsequent turnover of Miro could exert neuroprotection by (1) preventing damaged mitochondria from being anterogradely transported along the axons, thus increasing their chance of getting eliminated in the soma; and (2) promoting elimination of damaged mitochondria through mitophagy. This potentially explains the normal protective function of PINK1. When PINK1 function is impaired, however, on one hand mitochondria become dysfunctional as evidenced by morphology changes and impaired electron transport chain function [3]–[5], [33], [43]–[45], on the other hand, the anterograde mitochondrial transport is enhanced as shown in this study in Drosophila models. As a result, the dysfunctional mitochondria would have increased retention in the axons and synapses, resulting in increased reactive oxygen species (ROS) production, oxidative damage, and subsequent synaptic and axonal degeneration and eventual neuronal loss, at least in the Drosophila models. Many details of this model await further experimental validation. For example, it has been suggested that the reported effect of PINK1/Parkin on mitochondrial autophagy may not operate in the same manner in primary neurons as compared to cultured non-neuronal cells [46]. It also remains to be determined whether the effects of Miro on mitochondrial transport and mitophagy reflect a functional antagonism between these two processes, or two distinct functions of Miro in neuronal maintenance. In this respect, it is worth noting that the effect of Miro overexpression on cell survival in Drosophila is cell type-dependent: it causes DA neuron loss but has no obvious effect on muscle integrity (data not shown). It is possible that different tissues may have different sensitivities to impairments of Miro function. For example, muscle cells may be less susceptible to mitochondrial transport defects than neurons.
One interesting difference we observed between Drosophila and mouse systems was that although activation of the PINK1/Parkin pathway led to reduced Miro protein level in both systems, the loss of PINK1 in Drosophila resulted in increased steady-state Miro protein level, whereas its loss in mammalian cells as in PINK1 (−/−) MEF cells or PINK1 RNAi HeLa cells had the opposite effect. The mechanism of Miro downregulation in PINK1 loss-of-function mammalian cells is currently unknown, but it is presumably different from that used by activation of the PINK1/Parkin pathway. Since the upregulation of dMiro in dPINK1 mutant background is likely causal to DA neuron degeneration, as indicated by the rescue of DA neuron loss in dPINK1 mutant by dMiro-RNAi and the induction of DA neuron loss by dMiro-OE alone, it is tempting to speculate that the downregulation of Miro levels in PINK1 (−/−) mouse, as opposed to the dMiro upregulation in Drosophila PINK1 mutant, might contribute to the lack of DA neuron degeneration phenotype in the mouse PINK1 models [47]–[50]. Testing this hypothesis will require in vivo studies boosting Miro expression levels in wild type and PINK1 (−/−) mouse.
Our results also provide new insights into the process by which the PINK1/Parkin pathway promotes mitophagy. Previous studies suggested that upon recruitment to damaged mitochondria, Parkin activates the ubiquitin proteasome system to effect wide-spread degradation of OMM proteins in an autophagy-independent manner, and it was further proposed that this remodeling of OMM is important for a subsequent step of mitophagy [40]. The previously identified Parkin substrates, Mfn1 and Mfn2, although important for the effect of Parkin on mitochondrial fission/fusion dynamics, are not necessary for Parkin-induced mitophagy [40], [51]. Here we show that removal of Miro by the PINK1/Parkin pathway, in a presumably autophagy-independent but ubiquitination-dependent manner, facilitated mitophagy. Interestingly, knockdown of mammalian Miro itself promotes the formation of ring-like or round-shaped mitochondrial morphology, which is often observed in depolarized, mitophagy-ready mitochondria (Figure 6B; [13]). It is possible that the removal of Miro from OMM exposes certain recognition signals for the autophagy machinery, or that Miro/Milton/Kinesin-mediated mitochondrial transport may normally antagonize the mitophagy process. Supporting the latter scenario, an interaction between the BECLIN 1-interacting protein AMBRA1 and the dynein motor complex has been implicated in mammalian autophagy [52]. It also remains to be understood at the mechanistic level how PINK1 cooperates with Parkin to promote the ubiquitination and degradation of Miro. One attractive hypothesis is that PINK1 may directly phosphorylate Miro to promote its subsequent ubiquitination and degradation by Parkin, as suggested by a recent study [34]. However, our biochemical data have so far failed to support this hypothesis. It is possible that the divergent results are due to the different cell lines used or other experimental conditions. Alternatively, PINK1 may directly act on Parkin to promote Parkin's mitochondrial recruitment or activity in activating ubiquitin proteasome system-mediated ubiquitination and degradation of Miro. Further studies are needed to elucidate the molecular mechanisms of PINK1/Parkin action.
Finally, it is worth mentioning that studies in Drosophila models have identified a number of genetic modifiers of PINK1/Parkin [35], [53], [54]. While some of these genetic modifier genes may be directly related to the seemingly diverse biological activities of the PINK1/Parkin pathway, possibly mediated by distinct PINK1/Parkin substrates, others may reflect cellular compensatory responses to cope with the mitochondrial dysfunction caused by PINK1/Parkin inactivation. The fact that manipulations of each of these different cellular processes exert clear functional rescue of PINK1/Parkin mutant phenotypes suggests that there exists a signaling network linking the diverse activities of PINK1/Parkin in mitochondria biology with the nuclear-encoded cellular responses to mitochondrial dysfunction, and that many key players in this network represent novel and rational therapeutic targets.
Materials and Methods
Fly strains and reagents
Flies were raised according to standard procedures at indicated temperatures. Sources of fly strains and other reagents are as follows: dPINK1B9: Dr. J. Chung [3]; UAS-dMiro and anti-dMiro antibody: Dr. K. Zinsmaier [31]; TH-GAL4, UAS-PINK1, UAS-PINK1 RNAi and rabbit anti-Drosophila TH antibody: described before [5]; UAS-Miro RNAi106683: Vienna Drosophila RNAi Center; UAS-Miro RNAi27695, UAS-Milton RNAi28385 and UAS-Khc RNAi25898: Harvard Transgenic RNAi Project (TRiP) and Bloomington Drosophila Stock Center; all other fly lines: Bloomington Drosophila Stock Center; FLAG-hParkin mutants and HA-ubiquitin: Drs. N. Matsuda, K. Tanaka and S. Hatakeyama; Myc-hMiro1 and Myc-hMiro2 plasmids: Dr. P. Aspenström [30]; hPINK1 cDNAs were cloned into pcDNA3-FLAG vector.
Antibodies used in this study are as follows: anti-RHOT1/Miro1 (4H4, Abnova), anti-RHOT2/Miro2 (Protein technology Group), anti-PINK1 (Novus), anti-Parkin (PRK8, Santa Cruz Biotechnology), anti-Tom20 (FL-145, Santa Cruz Biotechnology), anti-VDAC1 (Abcam), anti-OXPHOS Complex IV subunit I/COX I (Invitrogen), anti-Tim23 (BD), anti-NDUFA9 (Invitrogen), anti-Cytochrome c (BD), anti-HtrA2/Omi (as described in [55]), anti-Hsp60 (BD), anti-PDHA1 (Abcam), anti-HA (3F10, Roche), anti-Myc (4A6, Millipore; #2272, Cell Signaling Technology), anti-β-actin (AC-15, Sigma-Aldrich), anti-α-tubulin (DM1A, Millipore), Peroxidase anti-Guinea Pig IgG antibody (Jackson ImmunoResearch), Texas Red-conjugated anti-HRP (Jackson ImmunoResearch), Alexa Fluor 488 nm-conjugated goat anti-chicken IgG (Invitrogen) and Alexa Fluor 594 nm-conjugated goat anti-rabbit IgG (Invitrogen).
Fly wing posture, behavior, ATP measurement, and immunohistochemistry
These assays were carried out essentially as described before [35]. The thoracic ATP level was measured using a luciferase based bioluminescence assay (ATP Bioluminescence Assay Kit HS II, Roche applied science) as described [35].
Whole-mount brain immunohistochemistry for TH and mitoGFP was performed as described previously [35]. For DA neuron mitochondrial morphology analysis, mitoGFP was expressed in Drosophila DA neurons using the TH-Gal4 driver. Brains from 3-day-old adult flies of the indicated genotypes were immunostained with the anti-TH antibody to label DA neuron and anti-GFP antibody to label mitochondria. For measurement of mitochondrial size distribution, the size of each mitochondrial aggregate was represented by the length of its longest axis. The percentage of DA neurons in the PPL1 cluster that have one or more mitochondria exceeding the indicated size was shown. Six flies of each genotype were used for the analysis.
Live imaging of mitochondrial movement
The motor neuron-specific OK6-Gal4 driver was used to express UAS-mitoGFP in the larval segment neurons, and 3rd instar male larvae raised at 29°C were used for live imaging. Larvae were briefly washed with water and anesthetized for 4 min in 1.5 ml eppendorf tubes containing 4 µl suprane (Baxter International Inc.), before being placed ventral side up into a small chamber. The chamber was created on glass slide with double-sided tape and cover glass. Additional suprane (4 µl) was introduced into the chamber before it was sealed with Valap (1∶1∶1 amount of vaseline, lanolin, parafin wax). Mitochondria were viewed with an upright Leica DM6000 B microscope equipped with a laser scanner and a 63× oil-immersion objective. Larvae were positioned to have their ventral ganglion (VG) appearing on the right of the acquired image and segmental nerves aligned horizontally across the image. Before recording mitochondrial movement, a centered region of 1024×200 pixel (61.5 µm×12.0 µm, 4× digital zoom) close to the VG was photobleached for 30 s with 488 nm excitation argon laser set at 80% output power. The viewing field was then zoomed out (2.5× digital zoom) and mitochondrial movement was immediately recorded by time-lapse video (2 s/frame, 300 s total) in a region of 1024×150 pixel (98.4 µm×14.4 µm) with the laser power reduced to 10% of the maximum output. The pinhole was set at 200 µm for all the experiments. Time-lapse images were acquired within 30 min of anesthetization. Mitochondrial flux was calculated by normalizing the number of mitochondria that passes certain point within the time frame examined. Mitochondrial net velocity was calculated by normalizing mitochondrial net displacement with time. At least 5 larvae of each genotype were analyzed.
Cell culture and transfection
HeLa cells were maintained at 37°C with 5% CO2 atmosphere in DMEM (Wako) supplemented with 10% FCS (GIBCO) and non-essential amino acids (Invitrogen). Plasmids and siRNA duplexes (Invitrogen) were transfected using Lipofectamine 2000 (Invitrogen) and Lipofectamine RNAiMAX (Invitrogen), respectively, according to manufacturer's instructions. To depolarize the mitochondria, HeLa cells were treated with 10 µM CCCP (Sigma-Aldrich) at 36 hr (for plasmids) or 72 hr (for siRNA) post-transfection.
The S156A mutations were introduced into human Miro1 and Miro2 by PCR-based mutagenesis using the following primers: For hMiro1, Forward primer: 5′ - GCA gag ctcttttatt acgcac -3′; Reverse primer: 5′ - tatg ttcttcaggt ttttcgc -3′. For hMiro2, Forward primer: 5′ - GCA gagct gttctactac gc -3′; Reverse primer: 5′ - ga tgttcctcag gttcttggc -3′. Full-length sequences of hMiro1 S156A and hMiro2 S156A were confirmed by sequencing.
Immunoblotting, immunoprecipitation, and in vitro phosphorylation
For brain extract preparation, 6 fly heads were quickly dissected and homogenized in 60 µl SDS-PAGE sample buffer. 5 µl brain extracts from each genotype were loaded onto SDS-PAGE gel. Guinea pig anti-dMiro antibody (1∶20000, from Dr. K. Zinsmaier) and Peroxidase Anti-Guinea Pig IgG antibody (1∶10000, Jackson ImmunoResearch Labs) were used for Western Blot. For HeLa cell-based biochemical analysis, cells were lysed in 1% Triton X-100 -based lysis buffer (10 mM Tris-HCl [pH 7.6], 120 mM NaCl, 5 mM EDTA, 1% Triton X-100 and protease inhibitor [Nacalai Tesuque]). Immunoprecipitation was performed using Immunoprecipitation Kit-Dynabeads Protein G (Invitrogen) according to manufacturer's instructions.
In vitro kinase assay was performed essentially as described [42], using a 2× GST-dPINK1 fusion protein with GST fused at both the N - and C-terminus of dPINK1 as the kinase source and a GST-dMiroΔTM fusion protein as the substrate. GST-dMiroΔTM covers amino acids 1–634 of the full-length dMiro protein. The GST-dMiro-ΔTM plasmid was constructed by amplifying a Myc-tagged dMiro fragment without the transmembrane domain from a pUAST-Myc-dMiro plasmid (obtained from Dr. K. Zinsmaier) using 5′-CGCCCG-GGTGAGCAGAAACTCATCTCTGAAGAAG-3′ and 5′-ATGCGGCCGCTACTTGGG-GTCCTCCGTC-ATC-3′ as primers. The amplified fragment was inserted into the SmaI and NotI cloning sites of the pGEX-6P-1 vector. Recombinant GST fusion proteins were purified from bacteria according to standard protocols.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.2% Triton X-100 (for mitophagy in Figure 6C) or 0.5% Triton X-100 (for mitochondrial morphology in Figure 6B) in PBS. Cells stained with the appropriated antibodies and counterstained with DAPI were imaged using a laser-scanning microscope (LMS510 META; Carl Zeiss, Inc.) with a Plan-Apochromat 63×NA1.4 or 100×/1.4 Oil differential interference contrast objective lens. Image contrast and brightness were adjusted in Image Browser (Carl Zeiss, Inc.)
Statistical analysis
Two-tailed Student's t tests were used for statistical analysis. p values of <0.05, <0.01, and <0.005 were indicated with one, two, and three asterisks (*), respectively.
Supporting Information
{kind=link}
Zdroje
1. KitadaTAsakawaSHattoriNMatsumineHYamamuraY 1998 Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392 605 608
2. ValenteEMAbou-SleimanPMCaputoVMuqitMMHarveyK 2004 Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304 1158 1160
3. ParkJLeeSBLeeSKimYSongS 2006 Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441 1157 1161
4. ClarkIEDodsonMWJiangCCaoJHHuhJR 2006 Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441 1162 1166
5. YangYGehrkeSImaiYHuangZOuyangY 2006 Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A 103 10793 10798
6. WangDQianLXiongHLiuJNeckameyerWS 2006 Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci U S A 103 13520 13525
7. YangYOuyangYYangLBealMFMcQuibbanA 2008 Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A 105 7070 7075
8. PooleACThomasREAndrewsLAMcBrideHMWhitworthAJ 2008 The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A 105 1638 1643
9. DengHDodsonMWHuangHGuoM 2008 The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A 105 14503 14508
10. LuBVogelH 2009 Drosophila models of neurodegenerative diseases. Annu Rev Pathol 4 315 342
11. WhitworthAJPallanckLJ 2009 The PINK1/Parkin pathway: a mitochondrial quality control system? J Bioenerg Biomembr 41 499 503
12. YuWSunYGuoSLuB 2011 The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet 20 3227 3240
13. NarendraDTanakaASuenDFYouleRJ 2008 Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183 795 803
14. GeislerSHolmstromKMSkujatDFieselFCRothfussOC 2010 PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12 119 131
15. YouleRJNarendraDP 2011 Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12 9 14
16. ChuCT 2010 A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum Mol Genet 19 R28 37
17. ImaiYLuB 2011 Mitochondrial dynamics and mitophagy in Parkinson's disease: disordered cellular power plant becomes a big deal in a major movement disorder. Curr Opin Neurobiol 21 935 941
18. TassinJDurrAde BrouckerTAbbasNBonifatiV 1998 Chromosome 6-linked autosomal recessive early-onset Parkinsonism: linkage in European and Algerian families, extension of the clinical spectrum, and evidence of a small homozygous deletion in one family. The French Parkinson's Disease Genetics Study Group, and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Am J Hum Genet 63 88 94
19. BonifatiVRoheCFBreedveldGJFabrizioEDe MariM 2005 Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65 87 95
20. AbbruzzeseGPigulloSSchenoneABelloneEMarcheseR 2004 Does parkin play a role in the peripheral nervous system? A family report. Mov Disord 19 978 981
21. BalohRH 2008 Mitochondrial dynamics and peripheral neuropathy. Neuroscientist 14 12 18
22. DuncanJEGoldsteinLS 2006 The genetics of axonal transport and axonal transport disorders. PLoS Genet 2 e124 doi:10.1371/journal.pgen.0020124
23. ZuchnerSVanceJM 2006 Mechanisms of disease: a molecular genetic update on hereditary axonal neuropathies. Nat Clin Pract Neurol 2 45 53
24. EbnethAGodemannRStamerKIllenbergerSTrinczekB 1998 Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol 143 777 794
25. MagraneJManfrediG 2009 Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal 11 1615 1626
26. Kim-HanJSAntenor-DorseyJAO'MalleyKL 2011 The Parkinsonian mimetic, MPP+, specifically impairs mitochondrial transport in dopamine axons. J Neurosci 31 7212 7221
27. SterkyFHLeeSWibomROlsonLLarssonNG 2011 Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A 108 12937 12942
28. HirokawaN 1998 Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 279 519 526
29. HollenbeckPJSaxtonWM 2005 The axonal transport of mitochondria. J Cell Sci 118 5411 5419
30. FranssonARuusalaAAspenstromP 2003 Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem 278 6495 6502
31. GuoXMacleodGTWellingtonAHuFPanchumarthiS 2005 The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47 379 393
32. StowersRSMegeathLJGorska-AndrzejakJMeinertzhagenIASchwarzTL 2002 Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36 1063 1077
33. LiuWAcin-PerezRGeghmanKDManfrediGLuB 2011 Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci U S A 108 12920 12924
34. WangXWinterDAshrafiGSchleheJWongYL 2011 PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 147 893 906
35. LiuSLuB 2010 Reduction of Protein Translation and Activation of Autophagy Protect against PINK1 Pathogenesis in Drosophila melanogaster. PLoS Genet 6 e1001237 doi:10.1371/journal.pgen.1001237
36. WeihofenAThomasKJOstaszewskiBLCooksonMRSelkoeDJ 2009 Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 48 2045 2052
37. PillingADHoriuchiDLivelyCMSaxtonWM 2006 Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell 17 2057 2068
38. RussoGJLouieKWellingtonAMacleodGTHuF 2009 Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci 29 5443 5455
39. NarendraDPJinSMTanakaASuenDFGautierCA 2010 PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol 8 e1000298 doi:10.1371/journal.pbio.1000298
40. ChanNCSalazarAMPhamAHSweredoskiMJKolawaNJ 2011 Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20 1726 37
41. YoshiiSRKishiCIshiharaNMizushimaN 2011 Parkin Mediates Proteasome-dependent Protein Degradation and Rupture of the Outer Mitochondrial Membrane. J Biol Chem 286 19630 19640
42. ImaiYKanaoTSawadaTKobayashiYMoriwakiY 2010 The loss of PGAM5 suppresses the mitochondrial degeneration caused by inactivation of PINK1 in Drosophila. PLoS Genet 6 e1001229 doi:10.1371/journal.pgen.1001229
43. GautierCAKitadaTShenJ 2008 Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105 11364 11369
44. SatoSHattoriN 2011 Genetic mutations and mitochondrial toxins shed new light on the pathogenesis of Parkinson's disease. Parkinsons Dis 2011 979231
45. ExnerNTreskeBPaquetDHolmstromKSchieslingC 2007 Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 27 12413 12418
46. Van LaarVSArnoldBCassadySJChuCTBurtonEA 2011 Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet 20 927 940
47. KitadaTPisaniAPorterDRYamaguchiHTscherterA 2007 Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A 104 11441 11446
48. Wood-KaczmarAGandhiSYaoZAbramovAYMiljanEA 2008 PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 3 e2455 doi:10.1371/journal.pone.0002455
49. WangHLChouAHWuASChenSYWengYH 2011 PARK6 PINK1 mutants are defective in maintaining mitochondrial membrane potential and inhibiting ROS formation of substantia nigra dopaminergic neurons. Biochimica et biophysica acta 1812 674 684
50. AkundiRSHuangZEasonJPandyaJDZhiL 2011 Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS ONE 6 e16038 doi:10.1371/journal.pone.0016038
51. TanakaAClelandMMXuSNarendraDPSuenDF 2010 Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191 1367 1380
52. Di BartolomeoSCorazzariMNazioFOliverioSLisiG 2010 The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 191 155 168
53. GreeneJCWhitworthAJAndrewsLAParkerTJPallanckLJ 2005 Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum Mol Genet 14 799 811
54. FernandesCRaoY 2011 Genome-wide screen for modifiers of Parkinson's disease genes in Drosophila. Mol Brain 4 17
55. SuzukiYImaiYNakayamaHTakahashiKTakioK 2001 A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 8 613 621
56. MatsudaNSatoSShibaKOkatsuKSaishoK 2010 PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189 211 221
57. PridgeonJWOlzmannJAChinLSLiL 2007 PINK1 Protects against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. PLoS Biol 5 e172 doi:10.1371/journal.pbio.0050172
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- PIF4–Mediated Activation of Expression Integrates Temperature into the Auxin Pathway in Regulating Hypocotyl Growth
- Metabolic Profiling of a Mapping Population Exposes New Insights in the Regulation of Seed Metabolism and Seed, Fruit, and Plant Relations
- A Splice Site Variant in the Bovine Gene Compromises Growth and Regulation of the Inflammatory Response
- Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson's Disease Genetics: The PDGene Database
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy