
-by- Regulatory Divergence Causes the Asymmetric Lethal Effects of an Ancestral Hybrid Incompatibility Gene
The Dobzhansky and Muller (D-M) model explains the evolution of hybrid incompatibility (HI) through the interaction between lineage-specific derived alleles at two or more loci. In agreement with the expectation that HI results from functional divergence, many protein-coding genes that contribute to incompatibilities between species show signatures of adaptive evolution, including Lhr, which encodes a heterochromatin protein whose amino acid sequence has diverged extensively between Drosophila melanogaster and D. simulans by natural selection. The lethality of D. melanogaster/D. simulans F1 hybrid sons is rescued by removing D. simulans Lhr, but not D. melanogaster Lhr, suggesting that the lethal effect results from adaptive evolution in the D. simulans lineage. It has been proposed that adaptive protein divergence in Lhr reflects antagonistic coevolution with species-specific heterochromatin sequences and that defects in LHR protein localization cause hybrid lethality. Here we present surprising results that are inconsistent with this coding-sequence-based model. Using Lhr transgenes expressed under native conditions, we find no evidence that LHR localization differs between D. melanogaster and D. simulans, nor do we find evidence that it mislocalizes in their interspecific hybrids. Rather, we demonstrate that Lhr orthologs are differentially expressed in the hybrid background, with the levels of D. simulans Lhr double that of D. melanogaster Lhr. We further show that this asymmetric expression is caused by cis-by-trans regulatory divergence of Lhr. Therefore, the non-equivalent hybrid lethal effects of Lhr orthologs can be explained by asymmetric expression of a molecular function that is shared by both orthologs and thus was presumably inherited from the ancestral allele of Lhr. We present a model whereby hybrid lethality occurs by the interaction between evolutionarily ancestral and derived alleles.
Published in the journal:
. PLoS Genet 8(3): e32767. doi:10.1371/journal.pgen.1002597
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002597
Summary
The Dobzhansky and Muller (D-M) model explains the evolution of hybrid incompatibility (HI) through the interaction between lineage-specific derived alleles at two or more loci. In agreement with the expectation that HI results from functional divergence, many protein-coding genes that contribute to incompatibilities between species show signatures of adaptive evolution, including Lhr, which encodes a heterochromatin protein whose amino acid sequence has diverged extensively between Drosophila melanogaster and D. simulans by natural selection. The lethality of D. melanogaster/D. simulans F1 hybrid sons is rescued by removing D. simulans Lhr, but not D. melanogaster Lhr, suggesting that the lethal effect results from adaptive evolution in the D. simulans lineage. It has been proposed that adaptive protein divergence in Lhr reflects antagonistic coevolution with species-specific heterochromatin sequences and that defects in LHR protein localization cause hybrid lethality. Here we present surprising results that are inconsistent with this coding-sequence-based model. Using Lhr transgenes expressed under native conditions, we find no evidence that LHR localization differs between D. melanogaster and D. simulans, nor do we find evidence that it mislocalizes in their interspecific hybrids. Rather, we demonstrate that Lhr orthologs are differentially expressed in the hybrid background, with the levels of D. simulans Lhr double that of D. melanogaster Lhr. We further show that this asymmetric expression is caused by cis-by-trans regulatory divergence of Lhr. Therefore, the non-equivalent hybrid lethal effects of Lhr orthologs can be explained by asymmetric expression of a molecular function that is shared by both orthologs and thus was presumably inherited from the ancestral allele of Lhr. We present a model whereby hybrid lethality occurs by the interaction between evolutionarily ancestral and derived alleles.
Introduction
Species can be isolated from one another by a variety of reproductive barriers. One widely observed barrier is hybrid incompatibility (HI), the inviability or sterility of interspecies offspring. The key premise of the Dobzhansky-Muller (D-M) model explaining the evolution of HI is that genetic changes fixed in one population need not be compatible with changes fixed in a different population [1], [2]. This is most commonly illustrated as two independently evolving populations that each diverge from the ancestral state and fix new alleles. Hybridization between the two populations brings together the independently derived alleles, thereby generating a genotype unscreened by natural selection. This genotype may suffer from an incompatible interaction between the derived alleles, resulting in developmental breakdown of the hybrid progeny. A key feature of this model is that HI alleles have diverged in sequence and function (perhaps extensively) from their ancestral states. A second important prediction of the model is asymmetry: Gene “A” from species one may interact with gene “B” from species two to cause HI, but not vice-versa [3]. Questions fundamental to understanding speciation then are: What molecular divergence between the ancestral and derived alleles is causing HI? Is this divergence at the level of regulatory or structural changes? What are the evolutionary forces causing this divergence?
One unifying emerging trend is that HI loci often show high levels of divergence caused by natural selection [3], [4]. These findings are exciting, because if molecular divergence created by selection is causing HI, then the phenotypic target of selection is, at least in part, the evolutionary basis of speciation. A major goal then is to understand the role of selection in the evolution of incompatible divergence. Interestingly, studies on several recently characterized HI genes implicate divergence of heterochromatin and heterochromatin-binding proteins as the cause of incompatibility [5], [6]. As heterochromatin is the graveyard of selfish genetic elements, this functional divergence could be the legacy of genetic conflicts between the host species and the invasion of selfish DNAs such as transposable elements and satellite DNAs [7], [8].
A variation of the D-M model suggests that HI can also be caused by interactions between alleles that have not diverged from the ancestral state and derived alleles that have diverged in only one lineage [9]. If an HI allele has not diverged from its ancestral state, then this model predicts that its HI effects will be symmetrical, with orthologs from both species contributing to HI. Several examples of ancestral-derived incompatibilities have been discovered, and consistent with expectations the HI genes, when known, have experienced limited sequence divergence [10]–[12].
On the other hand, the expectation of a strict dichotomy between ancestral and derived HI alleles may reflect an over-simplified view of HI. Hybrids are the sum of two independently evolving genomes and thus suffer from multiple suboptimal interactions [3]. For example, species-specific divergence at cis and trans-regulatory elements is associated with widespread transcriptional dysregulation in hybrids [13], [14]. This creates a genetic background distinct from either parental species, and several well-studied HI genes have genetic properties in hybrids that are significantly different from or even opposite to their intraspecific roles [3].
Crosses between D. melanogaster females and D. simulans males produce inviable hybrid sons and sterile hybrid daughters [15]. The incompatible D-M interaction in hybrid males can in part be explained by the interaction between two genes, Hybrid male rescue (Hmr) on the D. melanogaster X-chromosome and Lhr on the D. simulans 2nd chromosome [16]. A loss of function mutation in either Hmr or Lhr alone is sufficient to suppress the lethality of hybrid sons [16]–[19]. Thus it is the activity of these genes that causes hybrid breakdown.
Lhr (also known as HP3) encodes a protein that localizes to heterochromatin by directly binding to Heterochromatin Protein 1 (HP1) [16], [20], [21]. Population genetic analyses demonstrated that the Lhr protein coding sequence (CDS) has diverged extensively between D. melanogaster and D. simulans under positive selection, leading to the suggestion that Lhr has co-evolved with species-specific heterochromatin sequences [16]. If this co-evolution reflects a history of genetic conflict then one might predict that hybrid lethality is caused by defects in heterochromatin structure or maintenance, and that Lhr orthologs have functionally diverged in their heterochromatin localization properties such that they would mislocalize in the presence of heterochromatin from different species.
The hybrid lethality gene Lhr appeared initially to be a clear example of a derived D-M hybrid incompatibility locus. Consistent with the expectation of functional divergence, we previously found that the rescue of hybrid lethality via Lhr is asymmetric; removal of D. simulans Lhr (sim-Lhr) rescues lethal hybrid sons but removal of D. melanogaster Lhr (mel-Lhr) does not [16]. Surprisingly, however, Lhr orthologs from D. melanogaster, D. simulans and the outgroup species D. yakuba all have hybrid lethal activity when overexpressed in hybrids [21]. LHR proteins from these species also retain heterochromatic localization when expressed in polytenized salivary-gland cells, demonstrating that natural selection has not caused a wholesale change in Lhr function. This set of results suggests either that functional divergence is not an all-or-none property, or that Lhr is an ancestral HI locus, rather than a derived one.
To distinguish between these two possibilities, and to uncover the functional divergence underlying the asymmetric rescue properties of Lhr orthologs, we developed a native-promoter driven transgenic system that allows a sensitive comparison of the functions and localization properties of D. simulans and D. melanogaster Lhr orthologs. Using this system, we have compared Lhr function in both pure species and hybrids using three sets of experiments: (1) genetic tests for hybrid lethal activity and interaction with its D-M partner, Hmr; (2) detailed cytological mapping of the heterochromatic localization of LHR and its association with hybrid lethality, and (3) expression analysis comparing transcriptional levels of the Lhr orthologs.
Results
Both D. simulans and D. melanogaster Lhr have hybrid lethal activity under native expression conditions
We generated parallel strains of D. melanogaster containing either D. simulans Lhr (sim-Lhr) or D. melanogaster Lhr (mel-Lhr) transgenes using the φC31 site-specific integration system [22]. Each Lhr ortholog was C-terminally tagged with an HA epitope and was expressed under the control of its native regulatory sequences (Figure 1). The transgenic constructs contained the eye-color marker white+ and were each integrated into the attP2 site on the third chromosome. We tested the transgenes for wild type activity by assaying for complementation of the D. simulans Lhr1 hybrid rescue mutation. D. simulans Lhr1 is a loss-of-function mutation that acts as a dominant suppressor of hybrid lethality [16], [18]. Complementation here means that the transgene provides sufficient wild type Lhr activity to suppress rescue by the Lhr1 mutation, thus causing hybrid male inviability.
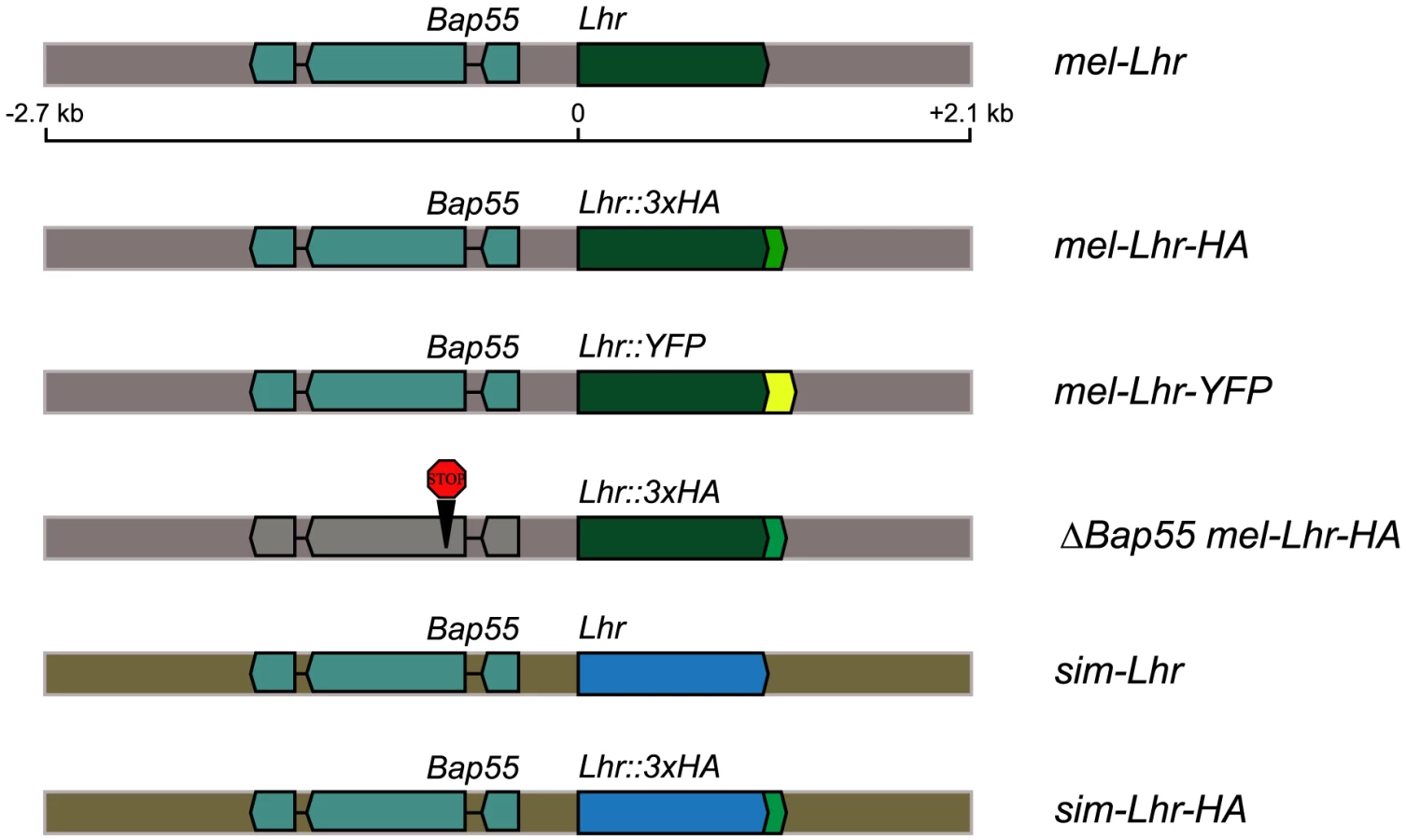
Complementation tests were performed by crossing D. melanogaster mothers heterozygous for an Lhr-HA transgene to D. simulans Lhr1 fathers. This cross generates two classes of hybrid sons: the control class that lacks the transgene and has white eyes, and the experimental class that inherits the transgene and has orange eyes. Complementation is detected as the lethality of orange-eyed sons. If hybrid lethal activity partitions discretely between Lhr orthologs, as expected from the functional divergence interpretation of genetic asymmetry, sons inheriting the φ{Dsim\Lhr-HA} transgene should be lethal, while those inheriting φ{Dmel\Lhr-HA} should be viable.
Unexpectedly, both transgenes fully complemented the D. simulans Lhr1 mutation (Table 1, crosses 1 thru 4), suggesting that both D. simulans and D. melanogaster Lhr orthologs have hybrid lethal activity. As this result was contrary to expectation we tested several possible causes of artifacts. First, the C-terminal HA-tag does not affect Lhr function because untagged versions of both mel-Lhr and sim-Lhr also complement Lhr1 (Table 1, crosses 5 and 6). Second, the adjacent gene Bap55 present in these constructs is not responsible for complementation because a modified mel-Lhr-HA transgene, φ{ΔBap55 Dmel\ Lhr-HA}, in which the Bap55 CDS is interrupted by two stop codons and a frameshift mutation, also complements Lhr1 (Table 1, cross 7). Third, the results are not caused by other unknown aspects of the strain background or by the attP2 site because the attP2 site itself without an integrated transgene does not complement Lhr1 (Table 1, cross 8). Furthermore mel-Lhr-HA integrated into a different site (attP86Fb) also complements Lhr1 (Table 1, cross 4). Fourth, these results are not due to an over-expression artifact because data presented below demonstrate that the mel-Lhr-HA transgene expresses Lhr at a level similar to the endogenous wild type locus (see section “cis-by-trans regulatory divergence causes functional divergence of D. melanogaster and D. simulans Lhr” below). These results clearly show that D. melanogaster Lhr has hybrid lethal activity even when expressed at its wild type level.

How can these results be reconciled with the original observation that only a mutation in D. simulans Lhr, and not the D. melanogaster ortholog, rescues hybrid sons? Those experiments were done in hybrid genotypes that had only a single dose of either mel-Lhr or sim-Lhr [16]. In contrast, the experiments here were performed by adding a transgenic copy of either mel-Lhr or sim-Lhr to hybrids that also carried the endogenous chromosomal copy of mel-Lhr. Increased dosage of mel-Lhr in the current experiments may therefore explain why we have not observed a difference between the mel-Lhr and sim-Lhr transgenes. This hypothesis raises the question of whether the hybrid lethal activity of the mel-Lhr-HA transgene would be eliminated in a background lacking the chromosomal copy of mel-Lhr. To test this we crossed D. melanogaster mothers that were doubly heterozygous for the mel-Lhr-HA transgene and an Lhr− deficiency to D. simulans Lhr1 fathers. If transgenic mel-Lhr behaves identically to the endogenous locus, then hybrid sons inheriting the Lhr− deficiency along with the mel-Lhr transgene should be equivalent in Lhr dosage to rescued +/Lhr1 hybrid males and thus be viable. However, hybrid sons from this cross were also inviable (Table S3). This result indicates that the mel-Lhr-HA transgene does not precisely phenocopy the native chromosomal mel-Lhr locus. In the Discussion we consider possible causes of this difference.
Interactions with Hmr reveal a difference in lethal activity of Lhr orthologs
Because the complementation tests did not reveal a difference in the hybrid lethal effects of Lhr orthologs we used a more sensitive genetic assay to test for functional divergence between mel-Lhr and sim-Lhr. We previously demonstrated that Lhr-dependent hybrid lethality requires the presence of its D-M partner, the D. melanogaster gene Hmr [16].
We reasoned that the hypomorphic allele Hmr1 might exhibit different sensitivities to the HI effects of the different Lhr alleles, but that the null allele Df(1)Hmr− would not. We therefore introduced each of our Lhr transgenes into these Hmr mutant backgrounds and tested the effect of the transgenes on hybrid male viability in crosses to D. mauritiana and D. simulans. Crosses with the sim-Lhr-HA transgene recapitulated our previous experiments: Hmr1 hybrid males carrying sim-Lhr-HA were essentially inviable at room temperature and showed strongly reduced viability at 18°C, while Df(1)Hmr− hybrid males were equally viable with and without the transgene (Table 2). We then performed similar crosses with mel-Lhr-HA. This transgene had little effect on viability of males with the null mutation Df(1)Hmr− and the results were in general not significantly different compared to the crosses with sim-Lhr-HA (Table 2, sets 1 & 2). In crosses with the hypomorphic mutation Hmr1, hybrids carrying mel-Lhr-HA had reduced viability compared to their non-transgene carrying siblings, particularly at room temperature. Strikingly, we found that in all four cross conditions the magnitude of the viability reduction was significantly less for mel-Lhr-HA compared to sim-Lhr-HA (Table 2, sets 3 & 4). These data demonstrate that sim-Lhr is more potent than mel-Lhr in creating the hybrid lethal interaction with Hmr, and that our Lhr transgenes thus do in fact reveal a significant degree of functional divergence.

A sensitive genetic screen reveals weak hybrid rescue by deletion of mel-Lhr
Having demonstrated that wild type mel-Lhr has hybrid lethal activity, we reinvestigated whether removal of mel-Lhr has any detectable hybrid rescue activity. We previously showed that deletion of mel-Lhr does not rescue hybrids with D. simulans [16]. We therefore looked for rescue in hybrids with D. mauritiana at 18°C, conditions that are maximally conducive for hybrid viability [17]. Unrescued hybrid males die as larvae [23]. We found that two D. melanogaster Lhr− deletions rescued 7–21% of males to the pharate adult stage (Table 3). This is clearly a modest rescuing effect and did not occur in one of the genetic backgrounds tested (Df(2R)BSC49 crossed to D. mauritiana W139), but it is significant because crosses with 45 other deletions across chromosome 2R gave no rescue. A third Lhr− deletion, Df(2R)BSC44, did not rescue hybrids, demonstrating that hybrid viability is sensitive to genetic background effects. The difference in magnitude of rescue for deletion of mel-Lhr versus sim-Lhr further supports our conclusion using transgenes that sim-Lhr has greater hybrid lethality activity than mel-Lhr.

LHR partially localizes to the dodeca satellite within heterochromatin in D. melanogaster
We next set out to determine why sim-Lhr is more potent than mel-Lhr in causing hybrid lethality. Coding sequence evolution leading to different protein localization patterns is one possible cause of Lhr functional divergence. In order to test this hypothesis we examined the cellular localization of LHR orthologs in their wild type backgrounds using our Lhr transgenes. In D. melanogaster LHR protein is most abundant during embryogenesis (Figure S1). We therefore analyzed the distribution of LHR during early embryogenesis and found a cyclical on-off pattern through the cell cycle, with localization to chromatin mainly during interphase (Figure S2). This pattern is identical to its interaction partner, Heterochromatin Protein 1 (HP1) [24]. Thus, we focused on interphase nuclei, and unless otherwise specified all images were taken at embryonic nuclear cycles 12–14, when heterochromatin is first observed. Consistent with previous results, LHR-HA colocalized with HP1 at DAPI-rich heterochromatic foci on the apical surface of the nuclei (Figure 2A). Unlike HP1, however, which is found throughout the nuclear compartment including euchromatin, LHR is restricted to heterochromatin. Consistent with being localized to a sub-domain of HP1, LHR strongly overlapped with Histone-3 lysine 9 dimethylation (H3K9me2), a histone modification specific to pericentric heterochromatin [25], but not with Cid, a histone variant specific to the centromere.

LHR was also observed in the embryonic germline precursors, the pole cells, and in the somatic and germline cells of the ovary (Figure S3A), where it again colocalized with H3K9me2 (Figure S3B). However, LHR was excluded from the nucleolus, a sub-compartment within heterochromatin consisting of rDNA repeats (Figure S3B). This observation suggested that LHR has a specific distribution within heterochromatin. We therefore used immuno-FISH to investigate the localization pattern of LHR relative to various pericentric satellites in D. melanogaster. We observed no overlap between LHR and the 359 bp satellite, a 4–5 Mb block on the X-chromosome [26], [27], nor between LHR and the highly abundant AATAT satellite, which is distributed across multiple chromosomes [28] (Figure 2C). In contrast, LHR consistently overlapped with dodeca, a G/C-rich pericentric satellite on the third chromosome [29], although a substantial amount of LHR is also found in other heterochromatic regions that we have not mapped. During metaphase, however, four discrete foci of LHR were visible along the metaphase plate. Noticeably, each LHR focus corresponded to the pericentric region of the third chromosome, as identified by overlapping dodeca signal (Figure 2D).
sim-LHR also associates with the dodeca satellite in D. simulans
We next tested whether LHR localization is conserved in D. simulans. We constructed transgenic lines of D. simulans using the sim-Lhr-HA construct described above. Like mel-LHR in D. melanogaster, sim-LHR in D. simulans also localized to apical heterochromatic foci, as marked by DAPI (Figure 3C). We were particularly interested to determine whether sim-LHR associated with the dodeca satellite, because the distribution of dodeca varies among melanogaster subgroup species [30]. In particular, dodeca satellite is present only in the pericentric region of the third chromosome in D. melanogaster, but is present in the pericentric heterochromatin of both the second and the third chromosomes in D. simulans [30]. We confirmed this difference and found that the dominant dodeca signal is on the D. simulans second chromosome in mitotic brain squashes (Figure 3A). We also noted significant differences in the interphase organization of dodeca between species. We quantified the number of dodeca foci per nucleus and the fraction of nuclear space occupied in interphase nuclei from wild type brains. The dodeca signal in D. simulans appeared fragmented into more foci and occupied a greater nuclear volume, indicating that dodeca-containing heterochromatin has evolved species-specific nuclear organization properties (Figure 3B).

Despite this divergence in both chromosomal location and structure of dodeca, immuno-FISH mapping in D. simulans showed that sim-LHR partially colocalized with dodeca in interphase nuclei (Figure 3C). As with mel-LHR, a substantial amount of sim-LHR localizes to other regions of heterochromatin which we have not mapped. However, our results show that its association with dodeca is conserved between species.
We were unable to detect sim-LHR on chromosomes during metaphase (data not shown). We note, however, that only a small fraction of mel-LHR appears to be on metaphase chromosomes in D. melanogaster (see Figure S2) and we have found that challenging to image. We are thus unable to determine whether the apparent absence of sim-LHR from metaphase chromosomes reflects a true difference between species or instead is due to technical limitations.
sim-LHR colocalizes with mel-LHR within D. melanogaster
It is unclear how LHR localizes to specific domains within heterochromatin, but it might require associations with other heterochromatin proteins, some of which are also rapidly evolving [21]. If LHR is co-evolving with other rapidly evolving proteins, then its heterochromatic localization might be altered when expressed in a foreign species.
To test this possibility we examined the localization of sim-LHR-HA in D. melanogaster. We found that sim-LHR-HA localized to the H3K9me2-enriched heterochromatic regions (Figure 3D), and colocalized with the dodeca satellite in a pattern identical to that seen for mel-LHR above (see Figure 2C). In order to directly compare the localization of LHR orthologs within the same nucleus, we generated a recombinant transgenic line that expressed both YFP-tagged mel-LHR and HA-tagged sim-LHR. The two LHR orthologs showed complete overlap, demonstrating that the heterochromatic localization properties of LHR orthologs are conserved (Figure 3D).
Incompatible hybrids have wild-type heterochromatin states and LHR localization
To determine whether heterochromatin states are perturbed in hybrids we examined HP1 and H3K9me2 localization. Although hybrid embryos were not sexed in this experiment, the staining appeared uniformly wild type in all embryos (Figure 4A). In order to specifically compare LHR and/or dodeca localization in hybrid males versus females, we developed a FISH probe that hybridized to the D. simulans Y-chromosome (Figure S4). We found that mel-LHR staining was enriched within apical heterochromatin in both sexes, and that it overlapped partially with dodeca (Figure 4C). Importantly, we detected no difference in dodeca organization and LHR localization between lethal hybrid males and viable hybrid females. Since heterochromatin defects might become more apparent later in development we then looked at heterochromatin states in hybrid larval neuroblasts. Consistent with the embryo staining, we saw no defects in the organization of either dodeca or the 2L3L satellite in either inviable male or viable female larvae (Figure 4D). Furthermore, despite differences in the pericentric heterochromatic sequences between homologous chromosomes, somatic pairing during interphase appeared unaffected in hybrid nuclei.

cis-by-trans regulatory divergence causes functional divergence of D. melanogaster and D. simulans Lhr
In spite of the adaptive protein sequence divergence between D. melanogaster and D. simulans orthologs of Lhr, our results surprisingly suggest only a limited degree of functional divergence of Lhr, with both orthologs having significant hybrid lethal activity and similar patterns of protein localization within heterochromatin. We therefore asked if gene regulatory divergence of Lhr between D. melanogaster and D. simulans might instead be responsible for the asymmetry of the lethal effects of Lhr in hybrids. We first surveyed Lhr transcript levels using qRT-PCR in three strains from each of the two species, and found no significant difference between the two species (Figure 5A). Consistent with this, we detected similar levels of LHR protein between the species (Figure 5B). Expression levels of mel-Lhr-HA and sim-Lhr-HA transgenes were each at a wild type level in their own species background, as total Lhr transcript level was approximately double in strains homozygous for the transgenes compared to wild type controls (Figure 5C). However, sim-Lhr was significantly overexpressed in D. melanogaster. The different expression levels of the sim-Lhr-HA and mel-Lhr-HA transgenes in the same D. melanogaster background indicate that cis-regulatory divergence has occurred at Lhr (Figure 5C). Furthermore, the fact that wild type levels of Lhr are not significantly different between the species (Figure 5A) despite these cis-regulatory differences suggests that trans acting factors that regulate Lhr have diverged. Taken together these data demonstrate that Lhr has undergone cis-by-trans compensatory regulation, such that cis-regulatory regions and trans-factors have co-evolved within each species to maintain a constant level of gene expression [31]. The uncoupling of such species-specific compensatory changes in a foreign genetic background would explain why sim-Lhr is hyper-expressed in D. melanogaster.

Given these results, we hypothesized that such a mechanism might cause asymmetric expression of Lhr orthologs in hybrids and by extension underlie the asymmetric rescue properties of Lhr orthologs. To test this hypothesis, we did allele-specific pyrosequencing to estimate the relative expression levels of the two Lhr orthologs in hybrids (Figure 6). We examined 3–5 day-old larvae because temperature shift experiments have shown that the L2/L3 stage is the critical phase of the lethality [17]. As expected Lhr transcript from the pure species parents was essentially 100% for their respective species-specific SNP. However, there was a significant overrepresentation of the D. simulans-specific SNP in both hybrid males and females, with ∼65% of Lhr transcripts deriving from the D. simulans ortholog in hybrid males and ∼60% in hybrid females. These data confirm our expectation that cis-by-trans divergence of Lhr regulation causes asymmetric expression in hybrids, and strongly suggests that a D. simulans mutation rescues hybrid sons because it removes a greater fraction of the total pool of Lhr, compared to a mutation in the D. melanogaster ortholog. We emphasize that this regulatory evolution leads to asymmetric expression of Lhr in hybrids but does not appear to cause an increase in total levels. The abundance of transgenic mel-LHR protein is not elevated in hybrids compared to pure species, as determined by Western blots (Figure 5D). Moreover, because protein levels of LHR orthologs appear equivalent in hybrids, we infer that levels of D. simulans LHR are also not visibly elevated in hybrids (Figure 5D). We therefore conclude that hybrid male lethality is not caused by Lhr over-expression. As we discuss below, lethality instead appears to result from hybrids becoming sensitive to Lhr activity due to its interaction with additional genes including Hmr.

Discussion
Lhr and Hmr are D-M interaction partners that cause hybrid lethality [16]. Population genetic analyses of Lhr, Hmr and other HI genes found their coding sequences to be evolving rapidly under positive selection [3], [4]. These results imply that selection-driven protein divergence is the molecular basis of incompatibility in hybrids. An experimental prediction then is that independently evolving orthologs of a D-M gene should be non-equivalent with respect to the HI phenotype. Our initial genetic data supported this expectation for Lhr, because a loss of function mutation in D. simulans Lhr rescues lethal hybrid sons, while a loss-of-function mutation in D. melanogaster Lhr does not [16]. These findings led to several hypotheses: 1) HI is due to divergence specific to the D. simulans lineage; 2) this divergence has caused significant changes in the heterochromatin association properties of LHR proteins from D. melanogaster and D. simulans; and 3) defects in heterochromatin states directly cause hybrid lethality.
Contrary to some of these expectations, a subsequent study found that both D. melanogaster and D. simulans Lhr could cause HI when overexpressed in hybrids and that both proteins localized to heterochromatin when ectopically expressed in salivary gland cells [21]. In order to further explore functional differences between sim-Lhr and mel-Lhr we developed a native-promoter-driven transgenic system and performed higher resolution mapping of LHR protein localization. We found that both Lhr orthologs suppress hybrid rescue by D. simulans Lhr1, supporting the inference that hybrid lethal activity is a shared ancestral function. However, using a more sensitive interaction assay with Hmr, we detected that the lethal interaction was greater with sim-Lhr (Table 2). This finding is consistent with the pattern of genetic asymmetry where a mutation in D. simulans Lhr rescues hybrid lethality, while a deficiency removing D. melanogaster Lhr does not [16]. Our further investigation here reveals that removing mel-Lhr does in fact provide a modest level of hybrid rescue (Table 3). The fact that this rescue only occurs to the pharate adult stage in a minority of male hybrids underscores our conclusion that mel-Lhr has weaker hybrid lethal activity than sim-Lhr. A major focus of this study then became to understand the cause of this difference.
Assessing transgene function
We attempted to create transgenic constructs of Lhr that were functionally identical to the wild type locus. To achieve this we generated Lhr transgenes that were driven by their native cis-regulatory sequences (Figure 1). Although the boundary of the regulatory regions included in the constructs was arbitrary we did quantitative RT-PCR assays on the transgenes to confirm that they expressed at wild type levels in both D. melanogaster and D. simulans (Figure 5C). Additionally, we infer from western blots that the abundance of transgenic LHR protein is similar in hybrids and pure species (Figure 5D), suggesting comparable expression levels in both backgrounds.
Nevertheless, we found that our mel-Lhr-HA transgene has greater activity than wild type Lhr when directly tested against an Lhr− deletion (Table S3). We consider two explanations: One possibility is that the construct has aberrant expression in a limited number of tissues or developmental stages that is beyond the resolution of detection in qRT-PCR assays of whole embryos or animals. Two, genetic assays for Lhr rescue are highly sensitive to genetic background effects; for example a large screen for suppression of Lhr rescue found a wide range of rescue even in the control balancer-chromosome classes [32]. We also observed here variable effects of D. melanogaster Lhr− deletions on hybrid viability (Table 3). Thus it is possible that this anomalous result results from an interaction with the multi-locus deficiency used and/or its genetic background.
While the result in Table S3 remains unexplained, we emphasize that the major conclusions of this study are not affected. The inference that mel-Lhr has hybrid lethal activity is independently shown by the rescue activity of the mel-Lhr deletion (Table 3). That result also demonstrates the asymmetric lethal activity of mel-Lhr and sim-Lhr, as does pyrosequencing of cDNA from hybrids (Figure 6). Likewise, the inference from transgenic assays that Lhr has undergone cis-by-trans compensatory evolution (Figure 5C) is fully consistent with the quantification of Lhr transcription by qRT-PCR in pure species (Figure 5A) coupled with the pyrosequencing result in hybrids.
Conserved heterochromatic localization of LHR orthologs
Our first hypothesis to explain the differential effects of mel-Lhr versus sim-Lhr on hybrid viability was that their respective proteins might have different localization patterns. Previous studies found the LHR localizes to heterochromatin in D. melanogaster, but did not determine whether it is a general heterochromatin factor or instead has a specific localization within heterochromatin [16], [20], [21]. The heterochromatic landscape is dramatically different in closely related species [33], which raises the question of whether rapid evolution of Lhr orthologs reflects functional divergence necessitated by its association with fast-evolving heterochromatic sequences.
We addressed this question by (1) mapping LHR localization within D. melanogaster pericentric heterochromatin, (2) comparing its localization in D. simulans, and (3) examining sim-LHR localization in a D. melanogaster background. Within both species LHR localized to heterochromatic foci but was not ubiquitous (Figure 2A). For example, mel-LHR does not overlap with the AATAT or the 359 bp satellites, two major components of D. melanogaster pericentric heterochromatin [28]. In contrast, a portion of LHR consistently colocalized with the dodeca satellite in both species during interphase. The conservation of this colocalization pattern was particularly striking, given that dodeca repeats are found only on chromosome III in D. melanogaster but on both chromosomes II and III in D. simulans (see Figure 3A and reference [30]). Thus, the chromosomal distribution of LHR between the two species is different.
However, despite this divergence in the genomic location of dodeca, sim-LHR when expressed in D. melanogaster colocalized perfectly with mel-LHR (Figure 3D), demonstrating full conservation of LHR's heterochromatic localization properties. For three reasons, it is highly unlikely that this conserved pattern is because LHR orthologs share a DNA-binding activity specific to the dodeca sequence. First, LHR contains no recognizable DNA-binding domain. Second, LHR localization to heterochromatin is dependent on HP1 binding [20], [21]. Finally, LHR signal is neither restricted to dodeca nor perfectly overlapping with it (Figure 2C and Figure 3C). Thus, it is unclear what features of DNA or chromatin are configuring this localization pattern of LHR.
No evidence for heterochromatic defects or satellite DNA-mediated genetic conflicts in incompatible hybrids
Neither the structure of the dodeca satellite nor LHR localization differed between pure species and hybrids, nor between lethal male and viable female hybrids (Figure 4C and 4D). These results set Lhr apart from two other well-characterized heterochromatin-associated HI genes. OdsH is a fast-evolving homeodomain protein that mislocalizes to the heterochromatic Y-chromosome in hybrids [5]. Zhr is a species-specific satellite DNA that causes hybrid lethality by improperly segregating during mitosis [6]. Such defects have been interpreted as support for the hypothesis that internal conflict with selfish heterochromatic elements is driving HI [3], [4], [7], [8]. We cannot rule out the possibility that there are defects in heterochromatin undetectable by our cytological analyses, or that Lhr may have other functions related to telomeric [16] or euchromatic [20] localization that have been affected by genetic conflicts. Nevertheless, the observations that heterochromatin appears normal in hybrids and that LHR localizes normally in both hybrids and when expressed in foreign species are not consistent with straightforward expectations of genetic conflict theories involving satellite DNAs [3]. Further work will be required to understand how Lhr causes lethal hybrids to have defects in cell proliferation and abnormally few larval cells entering mitosis [34], [35].
Regulatory divergence causes the asymmetric hybrid lethal effects of Lhr orthologs
Having found that LHR orthologs have not diverged in their heterochromatin localization, we tested whether the asymmetric effects of mutations in mel-Lhr versus sim-Lhr on hybrid lethality reflect a history of regulatory sequence divergence rather than protein sequence divergence. In particular, we hypothesized that asymmetric expression of Lhr orthologs in hybrids could explain the aforementioned genetic asymmetry. We tested this hypothesis by measuring allele-specific expression of Lhr orthologs in hybrid larvae. Our results strongly support this hypothesis: we found that approximately 66% of the total Lhr transcripts in lethal hybrid male larvae originates from the D. simulans allele (Figure 6). Thus a mutation in D. simulans Lhr creates hybrid sons with only 1/3rd the wild type level of Lhr transcript, while hybrid sons with a mutation in the D. melanogaster ortholog have twice that amount. We conclude that only a loss-of-function mutation in D. simulans Lhr produces viable hybrids because it removes a greater proportion of the total Lhr gene product.
The divergence leading to asymmetric expression does not, however, reflect species-specific divergence in expression levels, because Lhr expression is not significantly different between D. melanogaster and D. simulans (Figure 5A). Instead asymmetric Lhr expression in hybrids is likely caused by the uncoupling of species-specific compensatory changes between cis-regulatory sequences and trans-factors. Interestingly, studies comparing the evolution of transcriptional networks between species have found that this type of regulatory divergence is frequently associated with gene mis-expression in interspecific hybrids [14], [31]. Furthermore, Takahasi et al. recently found evidence that stabilization of expression levels within a species involves widespread cis - and trans-compensatory mutations that can be detected as incompatibilities between heterospecific regulatory elements in interspecific hybrids [36]. The authors also suggest that signatures of adaptive evolution might result from the rapid accumulation of compensatory changes, and thus reflect the maintenance of an existing function rather than the evolution of a novel one. To our knowledge Lhr is the first example of cis-by-trans compensatory evolution occurring at an adaptively evolving hybrid incompatibility gene. An intriguing possibility is that the rapid evolution of the protein coding region reflects compensatory changes required to maintain an existing regulatory function of Lhr, rather than to alter its protein function.
A Dobzhansky-Muller interaction between a derived and an ancestral allele
We emphasize that cis-by-trans regulatory divergence explains the asymmetric effect of Lhr mutations on hybrid viability, but is not the direct cause of Lhr having hybrid lethal activity. Instead our data argue that the hybrid male genotype has evolved an acute sensitivity to Lhr dosage. Our genetic assays further suggest that the activity of Lhr that causes hybrid lethality was likely present in the ancestral state because it is shared by both mel-Lhr and sim-Lhr. This hypothesis is further supported by the observation that GAL4-UAS driven expression of Lhr from D. yakuba, an outgroup species, also kills hybrid sons [21]. Unlike Lhr, however, transgenic assays with its D-M partner, Hmr, showed that only the D. melanogaster ortholog but not the D. simulans ortholog is capable of causing hybrid lethality [37]. That result is consistent with the HI effect of Hmr being derived during evolution in the D. melanogaster lineage.
HI involving ancestral gene function is compatible with the D-M model, and was first considered by Muller [9]. One model he proposed involves incompatibility between an ancestral and a derived allele, with loss of a suppressor allele being required to ‘release’ the incompatibility. Here, this would require a suppressor to evolve first and become fixed in the D. melanogaster lineage, before the incompatibility-causing substitutions evolved in Hmr (Figure 7A). In the hybrid background, the suppressor is diluted or inactivated, exposing the lethal interaction. Alternatively, incompatibility could result from a complex epistatic interaction involving three or more loci. In the simplest case, changes at a single D. simulans locus, Sen*, cause the hybrid background to become sensitive to the dosage of Lhr in the presence of Hmr from the D. melanogaster lineage (Figure 7B). We favor the latter model because in the first model over-expression of sim-Lhr in D. melanogaster might be expected to at least partially overcome the suppressor and create the incompatible interaction. However, GAL4-UAS over-expression of sim-Lhr has no effect in a D. melanogaster pure species background [16], [21].

Although we diagram only a single sensitizing locus, a polygenic model involving multiple genes is equally possible, because available data only establish that Hmr and Lhr are insufficient to cause hybrid lethality [16]. If many additional genes are involved, then the distinction between ancestral and derived alleles may become blurred. For example, interacting genes may co-evolve, and have high evolutionary rates that maintain interactions rather than alter molecular functions.
Other examples of ancestral-derived incompatibilities have been discovered, such as the inter-allelic incompatibility at the S5 locus in rice, and the bi-locus incompatibility between the derived S. cerevisiae splicing factor MRS1 and the ancestral COX1 mRNA [11], [12]. However, unlike the incompatible S5 alleles which differ by only two amino acid substitutions, and COX1 which retains the ancestral intron that causes HI, Lhr orthologs have diverged rapidly under selection [16]. It is therefore remarkable that despite extensive protein sequence divergence between the hybridizing species, hybrid lethality has evolved as sensitivity to the dosage of an ancestral function. The key mechanistic implication is that instead of searching for a process or function that differentiates Lhr orthologs as the source of hybrid lethality, we now know that the sensitivity to Lhr in hybrids is based on a function and/or interaction that is common to both orthologs.
Role of positive selection in the evolution of hybrid incompatibilities
There are least 6 HI genes known that are rapidly diverging under selection [3]. With the exception of OdsH and Prdm9, where the signature of selection is restricted to a single functional domain [38], [39], in the other HI genes peaks of nonsynonymous substitutions do not coincide with a specific functional domain within the protein coding sequence. In these cases, it has been assumed that changes derived under selection have led to functional divergence, in turn causing incompatibility. However, it remains to be tested if that is truly the case.
We have assayed the hybrid lethal activity of both Lhr orthologs and found that despite extensive selection-driven divergence of the protein sequence, hybrid lethal activity is a shared ancestral function. We do not rule out the possibility that protein divergence makes some minor difference in hybrid lethal activity. However, our results suggest that the asymmetric effect of Lhr in causing hybrid lethality is explained by regulatory divergence. This finding demonstrates the need to consider regulatory divergence when interpreting interspecies experiments. Our results also highlight the complexity of the interspecific background and emphasize that hybrids are far from being the stoichiometric sum of two parental genomes. We suggest that while positive selection of protein-coding sequences remains a characteristic of HI genes, the phenotypic target of selection and its connection to HI are in some cases much less direct than expected.
Materials and Methods
Drosophila crosses and stocks
All crosses were done at room temperature, or at 18°C where explicitly stated. At least 2 replicates were done for each cross. Each interspecific cross was initiated with ∼15–20 1-day-old D. melanogaster virgin females and ∼30–40 3–4-day-old sibling-species males. The nomenclature used for the transgenic lines and a complete description of the constructs used to generate them are included in Table S1. Genetic markers, deficiencies, and balancer chromosomes are described on FlyBase [40]. We previously showed that the D. melanogaster stock y1,w67c23; P{w+mC = lacW}l(2)k01209[k08901a]/CyO, used here in Table 3 and Table S3, is deleted for Lhr (see Fig. S4 in ref. [16]).
DNA constructs
To make a modified pCasper4 containing the attB site, we PCR amplified a 280 bp fragment using the pTA plasmid (gift from Michele Calos) as the template [22]. This PCR product, along with flanking SalI sites was cloned into the compatible XhoI site of pCasper4 to create the plasmid pCasper4\attB. In order to construct Lhr transgenes with Lhr under the control of its native regulatory sequences, we used a 4.8 kb genomic fragment that spans 2.7 kb upstream and 1 kb downstream of the Lhr CDS. This fragment includes the complete CDS of the adjacent gene Bap55 (Figure 1).
To generate the p{sim-Lhr} construct we amplified this fragment from D. simulans w501 genomic DNA, using primer pairs 691/664 (see Table S2 for primer sequences). This PCR product was gel purified and cloned into the pCR-BluntII TOPO vector (Invitrogen), according to manufacturer's directions. The insert was sequenced completely and subcloned into pCasper4\attB using NotI and KpnI restriction enzymes. Note that this transgene contains more upstream DNA than the sim-Lhr transgene used by Prigent et. al. [41], which was also functional.
The p{mel-Lhr} construct was generated similarly, a 4.8 kb fragment was PCR amplified from wild type D. melanogaster (strain Canton-S) genomic DNA using primer pairs 597/598, and TOPO cloned into pCR-BluntII vector. The forward primer contains a NotI site, allowing the insert to be released as a NotI fragment and cloned into the NotI site of pCasper4\attB. A clone was chosen with the same orientation as in p{sim-Lhr}.
To construct p{sim-Lhr-HA} a triple-HA tag was added in-frame to the C-terminus of the Lhr CDS using a two-piece fusion PCR strategy. The two overlapping PCR products were amplified using p{sim-Lhr} as the template, with primer pairs 691/728 and 729/664. These fragments were used as templates for the fusion PCR, and the gel-purified product was TOPO cloned into the pCRBluntII vector and sequenced completely. The insert was then subcloned into pCasper4\attB exactly as in p{sim-Lhr}. The construction of p{mel-Lhr-HA} followed the same logic, using the primer pairs 597/728 and 729/598. To synthesize the p{mel-Lhr-YFP} construct a three-piece fusion PCR strategy was used, the first and last PCR products, containing upstream and downstream genomic regions respectively, were amplified using p{mel-Lhr} as the template, with primer pairs 597/730 and 733/598. The central PCR product containing the YFP-tag was amplified from p{w+mC UAS-Lhr::Venus = UAS-Lhr::YFP} [16], with primer pair 731/732. The 3 overlapping PCR products were used as templates for the fusion PCR, and cloned into the pCR-BluntII vector and sequenced completely. The insert was subcloned into pCasper4\attB exactly as in p{mel-Lhr}.
The p{ΔBap55 mel-Lhr-HA} construct is identical to p{mel-Lhr-HA} except that the Bap55 CDS is interrupted by the insertion of “TAA TGA C”, i.e. two stop codons and a frame shift mutation after the second methionine at position 6. Two overlapping PCR products were amplified using p{mel-Lhr-HA} as template, with primer pairs 597/1171 and 1172/598. The products were stitched together using fusion PCR and cloned into pCasper4\attB exactly as done in p{mel-Lhr}.
Transgenic fly lines
φC31-mediated transformation of D. melanogaster was performed by Genetic Services Inc. The integration sites used were: i) P{CaryP}attP2 and ii) M{3xP3-RFP.attP}ZH-86Fb at cytological positions 68A4 and 86Fb, respectively [22], [42]. P{CaryP}attP2 carries the body color marker yellow+ (y+). Site specificity of integration was tested using the PCR assays of ref. [43]. We also developed attP docking-site specific PCR assays, primer pairs1086/1087 for attP2, and 949/1177 for ZH-86Fb. All D. melanogaster transformants were crossed into the strain w1118. P-element mediated integration was used to transform the D. simulans w501 strain with P{sim-Lhr-HA}.
Quantitative RT–PCR
Total RNA was isolated using the Trizol Reagent (Invitrogen), followed by DNaseI (Roche) treatment and purification using RNeasy columns (Qiagen). First strand cDNA was synthesized from 4 µg of total RNA using the SuperScriptIII first-strand synthesis system (Invitrogen) with the oligo(dT)20 primer in a 20 µl reaction according to the manufacturer's instructions. Quantitative real time PCR (qRT-PCR) was performed on a Biorad MyiQ cycler with SYBR detection using the 2× supermix from Biorad. Relative concentrations of Lhr transcripts were calculated against rpl32 as the reference gene with rpl32 primers from reference [44]. The rpl32 gene sequence is 99% identical between the species. For Lhr primer pair 1147/1148 was developed to recognize conserved sequences and to amplify both D. melanogaster and D. simulans Lhr with equal and high efficiency. For each sample real-time PCR on test and reference genes was done in technical triplicates, and the standard curve method was used to estimate transcript abundance. For each genotype RNA was isolated from between 3 and 4 independent 6–10 hr-old embryo collections. For all genotypes except D. simulans P{sim-Lhr-HA} cDNA was synthesized twice from each RNA isolate.
Pyrosequencing
RNA was extracted from 3–5 day-old larvae collected from non-crowded vials. In hybrid crosses the D. melanogaster mothers carried the X-linked mutation y− allowing the sex of larvae to be determined by using mouth hook coloration (daughters are y+ and sons y−). Total RNA and genomic DNA were simultaneously extracted from the same biological samples using the SV RNA system (Promega). For the pure species control, RNA and genomic DNA were extracted once from a single biological collection, followed by a single round of cDNA synthesis. For the hybrid samples, RNA and genomic DNA were extracted from four independent biological samples. cDNA was synthesized twice from each independent RNA isolate. Pyrosequencing measurements were performed in triplicate on each cDNA and in duplicate on each genomic DNA.
Western blotting
Whole cell extracts were obtained by grinding samples in ∼3 volumes of lysis buffer (50 mM Tris-HCl pH 7.5, 10 mM EDTA, 1.25% TritonX-100, 1× Roche protease inhibitor tablet). Extracts were cleared by centrifugation at 14,000 rpm for 10 min at 4°C. Total protein concentration of the cleared extracts was measured using Bradford assay (Biorad) and the samples were boiled in 0.5× volume of 4× SDS-Sample buffer. For most westerns 40 µg of total protein was loaded in each lane. Primary antibodies used were: rat anti-HA 3F10 (Roche; 1∶1000) and mouse anti-tubulin T5168 (Sigma; 1∶10,000). HRP conjugated goat anti-rat and goat anti-mouse secondary antibodies (Jackson; 1∶5,000) were used and detected with ECL Western blotting substrate (Pierce).
FISH and immuno-staining
Embryo FISH and immuno-FISH were performed as in reference [6] and immunostaining of ovarioles was performed as in reference [45] with the following antibodies: Rat anti-HA 3F10 (Roche; 1∶100), mouse anti-HP1 C1A9 (DSHB; 1∶100), rabbit anti-histone H3 lysine 9 dimethylation (Upstate 07-441; 1∶100), rabbit anti-Cid (a gift from S. Henikoff; 1∶1000), rabbit anti-GFP (Abcam ab6556; 1∶300), mouse anti-Fibrillarin (Cytoskeleton Inc. AFb01; 1∶400) and mouse anti-Hts 1B1 (DSHB; 1∶4). FISH probes are described in reference [6]. DNA was stained using TOPRO-3 iodide (Molecular Probes) or Vectashield containing DAPI (Vector Laboratories). All imaging was conducted at the Cornell University Core Life Sciences Microscopy and Imaging Facility, using either a Leica DM IRB confocal microscope or an Olympus BX50 epifluorescent microscope, except for embryo images with a DAPI channel which were taken in the Plant Cell Imaging Center at the Boyce Thompson Institute, with a Leica TCS SP5 confocal microscope. Images were processed using Photoshop (Adobe, version 7.0). Contrast and brightness changes, when used, were applied globally across images.
Quantification of dodeca signal in interphase larval brain tissue was done using ImageJ [46]. Watershed segmentation was applied on the DAPI-channel to generate a mask of nuclear territories. The Analyze Particle function was then used to identify individual nuclei as ROIs (regions of interest) and screened to exclude aberrant nuclear segmentations and non-nuclear entities. Each ROI was individually selected on the dodeca FISH channel of the same image and the FociPicker3D plug-in was used to identify regions of local maxima. We then calculated two measures to estimate the nuclear dispersion of dodeca satellite: (1) the total number of foci per nucleus and (2) the fraction of total nuclear area occupied by the dodeca signal.
Supporting Information
{kind=link}
Zdroje
1. DobzhanskyT 1937 Genetics and the origin of species New York Columbia Univ. Press
2. MullerHJ 1940 Bearing of the Drosophila work on systematics. HuxleyJS The new systematics Oxford Clarendon Press 185 268
3. MaheshwariSBarbashDA 2011 The genetics of hybrid incompatibilities. Annu Rev Genet 45 331 355
4. PresgravesDC 2010 The molecular evolutionary basis of species formation. Nat Rev Genet 11 175 180
5. BayesJJMalikHS 2009 Altered heterochromatin binding by a hybrid sterility protein in Drosophila sibling species. Science 326 1538 1541
6. FerreePMBarbashDA 2009 Species-specific heterochromatin prevents mitotic chromosome segregation to cause hybrid lethality in Drosophila. PLoS Biol 7 e1000234 doi:10.1371/journal.pbio.1000234
7. BrownJDO'NeillRJ 2010 Chromosomes, conflict, and epigenetics: chromosomal speciation revisited. Annu Rev Genomics Hum Genet 11 291 316
8. JohnsonNA 2010 Hybrid incompatibility genes: remnants of a genomic battlefield? Trends Genet 26 317 325
9. MullerHJ 1942 Isolating mechanisms, evolution and temperature. Biol Symp 6 71 125
10. CattaniMVPresgravesDC 2009 Genetics and lineage-specific evolution of a lethal hybrid incompatibility between Drosophila mauritiana and its sibling species. Genetics 181 1545 1555
11. ChenJDingJOuyangYDuHYangJ 2008 A triallelic system of S5 is a major regulator of the reproductive barrier and compatibility of indica-japonica hybrids in rice. Proc Natl Acad Sci U S A 105 11436 11441
12. ChouJYHungYSLinKHLeeHYLeuJY 2010 Multiple molecular mechanisms cause reproductive isolation between three yeast species. PLoS Biol 8 e1000432 doi:10.1371/journal.pbio.1000432
13. GrazeRMMcIntyreLMMainBJWayneMLNuzhdinSV 2009 Regulatory divergence in Drosophila melanogaster and D. simulans, a genomewide analysis of allele-specific expression. Genetics 183 547 561
14. McManusCJCoolonJDDuffMOEipper-MainsJGraveleyBRWittkoppPJ 2010 Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20 816 825
15. SturtevantAH 1920 Genetic studies on Drosphila simulans. I. Introduction. Hybrids with Drosophila melanogaster. Genetics 5 488 500
16. BrideauNJFloresHAWangJMaheshwariSWangXBarbashDA 2006 Two Dobzhansky-Muller genes interact to cause hybrid lethality in Drosophila. Science 314 1292 1295
17. HutterPAshburnerM 1987 Genetic rescue of inviable hybrids between Drosophila melanogaster and its sibling species. Nature 327 331 333
18. WatanabeTK 1979 A gene that rescues the lethal hybrids between Drosophila melanogaster and D. simulans. Jpn J Genet 54 325 331
19. BarbashDASiinoDFTaroneAMRooteJ 2003 A rapidly evolving MYB-related protein causes species isolation in Drosophila. Proc Natl Acad Sci U S A 100 5302 5307
20. GreilFde WitEBussemakerHJvan SteenselB 2007 HP1 controls genomic targeting of four novel heterochromatin proteins in Drosophila. EMBO J 26 741 751
21. BrideauNJBarbashDA 2011 Functional conservation of the Drosophila hybrid incompatibility gene Lhr. BMC Evol Biol 11 57
22. GrothACFishMNusseRCalosMP 2004 Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 166 1775 1782
23. SánchezLDübendorferA 1983 Development of imaginal discs from lethal hybrids between Drosophila melanogaster and Drosophila mauritiana. Roux's Arch Dev Biol 192 48 50
24. KellumRRaffJWAlbertsBM 1995 Heterochromatin protein 1 distribution during development and during the cell cycle in Drosophila embryos. J Cell Sci 108 1407 1418
25. EbertALeinSSchottaGReuterG 2006 Histone modification and the control of heterochromatic gene silencing in Drosophila. Chromosome Res 14 377 392
26. HsiehTBrutlagD 1979 Sequence and sequence variation within the 1.688 g/cm3 satellite DNA of Drosophila melanogaster. J Mol Biol 135 465 481
27. KäsELaemmliUK 1992 In vivo topoisomerase II cleavage of the Drosophila histone and satellite III repeats: DNA sequence and structural characteristics. EMBO J 11 705 716
28. LoheARHillikerAJRobertsPA 1993 Mapping simple repeated DNA sequences in heterochromatin of Drosophila melanogaster. Genetics 134 1149 1174
29. AbadJPCarmenaMBaarsSSaundersRDGloverDM 1992 Dodeca satellite: a conserved G+C-rich satellite from the centromeric heterochromatin of Drosophila melanogaster. Proc Natl Acad of Sci U S A 89 4663
30. CarmenaMAbadJPVillasanteAGonzalezC 1993 The Drosophila melanogaster dodeca satellite sequence is closely linked to the centromere and can form connections between sister chromatids during mitosis. J Cell Sci 105 41 50
31. LandryCRWittkoppPJTaubesCHRanzJMClarkAGHartlDL 2005 Compensatory cis-trans evolution and the dysregulation of gene expression in interspecific hybrids of Drosophila. Genetics 171 1813 1822
32. PresgravesDC 2003 A fine-scale genetic analysis of hybrid incompatibilities in Drosophila. Genetics 163 955 972
33. LoheARobertsP 1988 Evolution of satellite DNA sequences in Drosophila. VermaRS Heterochromatin, Molecular and Structural Aspects Cambridge Cambridge Univ. Press 148 186
34. OrrHAMaddenLDCoyneJAGoodwinRHawleyRS 1997 The developmental genetics of hybrid inviability: a mitotic defect in Drosophila hybrids. Genetics 145 1031 1040
35. BolkanBJBookerRGoldbergMLBarbashDA 2007 Developmental and cell cycle progression defects in Drosophila hybrid males. Genetics 177 2233 2241
36. TakahasiKRMatsuoTTakano-Shimizu-KounoT 2011 Two types of cis-trans compensation in the evolution of transcriptional regulation. Proc Natl Acad Sci U S A 108 15276 15281
37. BarbashDAAwadallaPTaroneAM 2004 Functional divergence caused by ancient positive selection of a Drosophila hybrid incompatibility locus. PLoS Biol 2 e142 doi:10.1371/journal.pbio.0020142
38. OliverPLGoodstadtLBayesJJBirtleZRoachKC 2009 Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet 5 e1000753 doi:10.1371/journal.pgen.1000753
39. TingCTTsaurSCWuMLWuCI 1998 A rapidly evolving homeobox at the site of a hybrid sterility gene. Science 282 1501 1504
40. TweedieSAshburnerMFallsKLeylandPMcQuiltonP 2009 FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res 37 D555 D559
41. PrigentSRMatsubayashiHYamamotoMT 2009 Transgenic Drosophila simulans strains prove the identity of the speciation gene Lethal hybrid rescue. Genes Genet Syst 84 353 360
42. BischofJMaedaRKHedigerMKarchFBaslerK 2007 An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 104 3312 3317
43. VenkenKJHeYHoskinsRABellenHJ 2006 P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314 1747 1751
44. FiumeraACDumontBLClarkAG 2005 Sperm competitive ability in Drosophila melanogaster associated with variation in male reproductive proteins. Genetics 169 243 257
45. ArunaSFloresHABarbashDA 2009 Reduced fertility of Drosophila melanogaster Hybrid male rescue (Hmr) mutant females is partially complemented by Hmr orthologs from sibling species. Genetics 181 1437 1450
46. AbràmoffMDMagalhãesPJRamSJ 2004 Image processing with ImageJ. Biophotonics international 11 36 42
47. BonaccorsiSLoheA 1991 Fine mapping of satellite DNA sequences along the Y chromosome of Drosophila melanogaster: relationships between satellite sequences and fertility factors. Genetics 129 177 189
48. StolcVGauharZMasonCHalaszGvan BatenburgMF 2004 A gene expression map for the euchromatic genome of Drosophila melanogaster. Science 306 655 660
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- PIF4–Mediated Activation of Expression Integrates Temperature into the Auxin Pathway in Regulating Hypocotyl Growth
- Metabolic Profiling of a Mapping Population Exposes New Insights in the Regulation of Seed Metabolism and Seed, Fruit, and Plant Relations
- A Splice Site Variant in the Bovine Gene Compromises Growth and Regulation of the Inflammatory Response
- Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson's Disease Genetics: The PDGene Database
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy