
Nos2 Inactivation Promotes the Development of Medulloblastoma in Mice by Deregulation of Gap43–Dependent Granule Cell Precursor Migration
Medulloblastoma is the most common malignant brain tumor in children. A subset of medulloblastoma originates from granule cell precursors (GCPs) of the developing cerebellum and demonstrates aberrant hedgehog signaling, typically due to inactivating mutations in the receptor PTCH1, a pathomechanism recapitulated in Ptch1+/− mice. As nitric oxide may regulate GCP proliferation and differentiation, we crossed Ptch1+/− mice with mice lacking inducible nitric oxide synthase (Nos2) to investigate a possible influence on tumorigenesis. We observed a two-fold higher medulloblastoma rate in Ptch1+/− Nos2−/− mice compared to Ptch1+/− Nos2+/+ mice. To identify the molecular mechanisms underlying this finding, we performed gene expression profiling of medulloblastomas from both genotypes, as well as normal cerebellar tissue samples of different developmental stages and genotypes. Downregulation of hedgehog target genes was observed in postnatal cerebellum from Ptch1+/+ Nos2−/− mice but not from Ptch1+/− Nos2−/− mice. The most consistent effect of Nos2 deficiency was downregulation of growth-associated protein 43 (Gap43). Functional studies in neuronal progenitor cells demonstrated nitric oxide dependence of Gap43 expression and impaired migration upon Gap43 knock-down. Both effects were confirmed in situ by immunofluorescence analyses on tissue sections of the developing cerebellum. Finally, the number of proliferating GCPs at the cerebellar periphery was decreased in Ptch1+/+ Nos2−/− mice but increased in Ptch1+/− Nos2−/− mice relative to Ptch1+/− Nos2+/+ mice. Taken together, these results indicate that Nos2 deficiency promotes medulloblastoma development in Ptch1+/− mice through retention of proliferating GCPs in the external granular layer due to reduced Gap43 expression. This study illustrates a new role of nitric oxide signaling in cerebellar development and demonstrates that the localization of pre-neoplastic cells during morphogenesis is crucial for their malignant progression.
Published in the journal:
. PLoS Genet 8(3): e32767. doi:10.1371/journal.pgen.1002572
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002572
Summary
Medulloblastoma is the most common malignant brain tumor in children. A subset of medulloblastoma originates from granule cell precursors (GCPs) of the developing cerebellum and demonstrates aberrant hedgehog signaling, typically due to inactivating mutations in the receptor PTCH1, a pathomechanism recapitulated in Ptch1+/− mice. As nitric oxide may regulate GCP proliferation and differentiation, we crossed Ptch1+/− mice with mice lacking inducible nitric oxide synthase (Nos2) to investigate a possible influence on tumorigenesis. We observed a two-fold higher medulloblastoma rate in Ptch1+/− Nos2−/− mice compared to Ptch1+/− Nos2+/+ mice. To identify the molecular mechanisms underlying this finding, we performed gene expression profiling of medulloblastomas from both genotypes, as well as normal cerebellar tissue samples of different developmental stages and genotypes. Downregulation of hedgehog target genes was observed in postnatal cerebellum from Ptch1+/+ Nos2−/− mice but not from Ptch1+/− Nos2−/− mice. The most consistent effect of Nos2 deficiency was downregulation of growth-associated protein 43 (Gap43). Functional studies in neuronal progenitor cells demonstrated nitric oxide dependence of Gap43 expression and impaired migration upon Gap43 knock-down. Both effects were confirmed in situ by immunofluorescence analyses on tissue sections of the developing cerebellum. Finally, the number of proliferating GCPs at the cerebellar periphery was decreased in Ptch1+/+ Nos2−/− mice but increased in Ptch1+/− Nos2−/− mice relative to Ptch1+/− Nos2+/+ mice. Taken together, these results indicate that Nos2 deficiency promotes medulloblastoma development in Ptch1+/− mice through retention of proliferating GCPs in the external granular layer due to reduced Gap43 expression. This study illustrates a new role of nitric oxide signaling in cerebellar development and demonstrates that the localization of pre-neoplastic cells during morphogenesis is crucial for their malignant progression.
Introduction
Medulloblastoma (MB) is a highly malignant tumor of the cerebellum that preferentially develops in children and adolescents. Although the survival rate for standard risk MB is around 70% [1] surviving patients often suffer from neurodevelopmental and cognitive side effects of the aggressive therapy [2]. Therefore, improved understanding of the molecular pathomechanisms driving MB growth is necessary to develop less toxic and more effective treatments. Recent molecular profiling studies suggested at least four MB subtypes that are associated with distinct expression profiles, genomic aberrations and clinical features [3], [4]. One of these MB subtypes is characterized by aberrant activation of the hedgehog (Hh) pathway and typically corresponds to the desmoplastic (nodular) MB variant. This subtype is supposed to develop from granule cell precursors (GCPs) of the external granular layer (EGL) [5].
The EGL is a transient germinal zone at the subpial cerebellar surface consisting of rhombic lip-derived progenitor cells that have migrated tangentially to the emerging cerebellar cortex at late stages of embryonal brain development [6]. During the early postnatal period in mouse, the morphogenic factor sonic hedgehog (Shh) is secreted by subjacent Purkinje cells and binds to patched receptors (Ptch1 and Ptch2) expressed on the GCP surface [7]. Ligand binding to Ptch1 then leads to functional de-repression of Smoh (Drosophila smoothened homolog) and subsequent activation of Gli (Glioma-associated oncogene family zinc finger) transcription factors [8]. This launches a temporally concerted gene expression pattern causing a proliferation burst and massive expansion of the GCP population during the first two postnatal weeks [7]. In particular, the direct Gli-target N-myc [9], [10] and D type cyclins [11] were shown to be crucial for the growth and neoplastic transformation of GCPs [12]. In addition, the set of genes targeted by activated Gli transcription factors also include components of the canonical Hh pathway for feedback-loop regulation, such as the receptors Ptch1 and Ptch2 as well as the hedgehog-interacting protein (Hip) [10], [13]. After several rounds of cell division, GCPs normally exit cell cycle and accumulate at the inner site of the EGL [14], where they start to migrate through the molecular layer (ML) and the Purkinje cell layer to form the internal granular layer (IGL) [15]. The mechanisms underlying the attenuation of the mitotic response and eventually the stop of GCP proliferation are not well understood. The most evident concepts describe extrinsic cues in gradient-based models to trigger GCP differentiation with increasing distance to the region of the outer EGL [16]. Finally, the EGL disappears at about three weeks after birth in mice.
PTCH1 was identified as a frequent target of inactivating mutations or genomic loss in sporadic MBs [17]–[19] that belong to the molecular subtype hallmarked by an aberrant activity of hedgehog signaling. The monoallelic inactivation of the Ptch1 gene in mice and thus downstream activation of the Hh pathway leads to MB development at a frequency of about 10–15% [20]. This mouse model has provided substantial insights into the pathogenesis of Hh-dependent MBs and has been used in different cross-breeding experiments to investigate tumor suppressor gene functions in this particular context [21], [22].
Nitric oxide (NO) is a highly reactive gaseous molecule involved in various physiological processes ranging from vasculature modulation to neurotransmission [23], [24]. NO is produced by three distinct enzyme isoforms: i) neuronal nitric oxide synthase (nNos/Nos1), ii) inducible nitric oxide synthase (iNos/Nos2), and iii) endothelial nitric oxide synthase (eNos/Nos3). Though being constitutively expressed in their respective tissue, nNos and eNos activity strongly depends on calcium [25], whereas calcium-independent iNos is primarily regulated by transcriptional induction, e.g. by inflammatory cytokines and endotoxins [26], which permits higher quantities of NO generation. The role of NO in cancer initiation and progression is heterogeneous with opposing effects in different malignancies [27]. Considering effects of tumor stroma, increased angiogenesis was reported to be associated with elevated Nos activity [28] and some immune-related processes were found to be mediated by NO [29], including cytotoxicity of activated microglia [30]. Finally, NO released by vascular endothelial cells was reported to build a niche-like microenvironment for maintenance of glioma stem cells [31]. In the context of cerebellar development, Nos2 (inducible Nos) is initially expressed in early GCPs, whereas Nos1 (neuronal Nos) is hardly present before postnatal day 7 (Cerebellar Development Transcriptome Database [32]). Successively, Nos1 expression increases along with granule cell differentiation [33] and predominantly contributes to the common NO signaling that becomes apparent in the IGL as development proceeds [34]. Evidence has been provided that NO negatively acts on proliferation of neuronal precursors during adult neurogenesis [35]. Similarly, Ciani and colleagues demonstrated enhanced proliferation of cerebellar precursor cells upon inhibition of NO synthases [36].
Here, we report on the generation of Ptch1+/− Nos2−/− mice to investigate the impact of Nos2 on tumor development in Ptch1 hemizygous mutant mice. Interestingly, we observed an approximately two-fold increase in the incidence of spontaneous MB in Ptch1+/− Nos2−/− mice in comparison to Ptch1+/− Nos2+/+ mice. To characterize the molecular pathomechanism underlying the tumor-promoting effect of Nos2 deficiency in Ptch1+/− mice, we performed comprehensive expression and DNA copy number profiling of MB tumors (Ptch1+/− Nos2+/+ versus Ptch1+/− Nos2−/−) as well as expression profiling of normal cerebellar tissue samples from different developmental stages and various genotypes (Ptch1+/− Nos2+/+, Ptch1+/− Nos2−/−, Ptch1+/+ Nos2−/− and wild-type mice). Downregulation of the growth-associated protein 43 (Gap43) was the most striking feature in the cerebellum of Nos2-deficient mice when compared to Ptch1+/− Nos2+/+ and wild-type mice. Subsequent functional analyses and results from in situ studies of GCPs in postnatal cerebellum allowed us to formulate a model for the tumor promoting role of Nos2 deficiency in Ptch1 mutant mice via deregulation of Gap43-dependent migration of GCPs.
Results
Loss of Nos2 increases the rate of spontaneous MB in Ptch1+/− mice
Survival analyses of 315 wild-type mice, 412 Ptch1+/+ Nos2−/− mice, 215 Ptch1+/− Nos2+/+ mice and 221 Ptch1+/− Nos2−/− mice demonstrated a significantly higher MB incidence in the group of Ptch1+/− Nos2−/− mice relative to the group of Ptch1+/− Nos2+/+ mice (p = 0.0007, Logrank test, Figure 1A). In total, 11% of the Ptch1+/− Nos2+/+ mice (24/215) and 21% of the Ptch1+/− Nos2−/− mice (47/221) were sacrificed due to the development of cerebellar MB. None of the 315 wild-type and the 412 Ptch1+/+ Nos2−/− mice developed MBs. These observations indicate a MB-promoting role of Nos2 deficiency in Ptch1+/− mice.

MBs in Ptch1+/− Nos2+/+ and Ptch1+/− Nos2−/− mice show identical histological features
In humans, Hh-dependent MBs typically correspond to the desmoplastic subtype. MBs in Ptch1+/− mice, however, microscopically resemble the classic MB subtype [20]. Histological analysis of MBs in Ptch1+/− Nos2+/+ and Ptch1+/− Nos2−/− mice demonstrated similar morphological features (Figure 1B–1E). The tumors were composed of densely packed sheets of cells with hyperchromatic carrot-shaped nuclei and scant cytoplasm. There were no obvious histopathological differences between MBs of the two genotypes.
Molecular analyses of MBs in Ptch1+/− Nos2+/+ and Ptch1+/− Nos2−/− mice
For an initial assessment of the molecular tumor characteristics, gene expression of hedgehog signaling pathway components were measured in 21 MBs and 24 normal (adult) cerebellar tissue samples from both Ptch1+/− Nos2+/+ and Ptch1+/− Nos2−/− mice. Using quantitative real-time PCR (qRT-PCR), significant downregulation of the wild-type Ptch1 transcript and upregulation of the Shh target genes Gli1 and N-myc were generally observed in the tumor samples (Figure S1), indicating all examined MBs to be of the same Hh-dependent molecular subtype. However, there were no significant differences for these genes between MBs of the two genotypes. Furthermore, targeted genetic analyses showed a loss of the wild-type Ptch1 allele in 10 of the 21 MBs investigated, while none of the tumors demonstrated a Tp53 mutation or N-myc amplification. The Cdkn2a/p16INK4a locus was retained in all tumors while a single MB demonstrated a homozygous p19ARF deletion (see Table S1 and Text S1 for details).
In order to identify the molecular pathomechanism contributing to the increased MB rate in Nos2-deficient Ptch1 mutant mice, we performed array-based gene expression profiling of three Ptch1+/− Nos2+/+ versus six Ptch1+/− Nos2−/− and comparative genomic hybridization (array-CGH) analyses of five Ptch1+/− Nos2+/+ versus seven Ptch1+/− Nos2−/− MB tissue samples. All specimens investigated had tumor cell contents between 70% and 90% as determined on corresponding formalin-fixed and paraffin-embedded (FFPE) reference sections. Differential expression of selected candidate genes was validated by qRT-PCR on an expanded, partially overlapping tumor set of seven Ptch1+/− Nos2+/+ versus seven Ptch1+/− Nos2−/− MB samples.
The expression profiling analysis revealed a total of 87 differentially regulated genes between tumors of the two genotypes (Table S2) with the vast majority (87%) showing lower transcript levels in Ptch1+/− Nos2−/− when compared to Ptch1+/− Nos2+/+ mice. As expected from the initial targeted qRT-PCR measurements, there was no difference detectable concerning the activation of Hh pathway genes. Due to the important role of Nos2 during angiogenesis and cancer-associated immune response, including microglia, stromal effects need to be particularly considered in a systemic Nos2 knockout model. However, neither the set of significantly deregulated genes nor selective determination of marker expression for pericytes, vascular endothelial cells or microglia suggested any differences in the tumor stroma between the two genotypes (see Table S3 and Text S1 for details). According to the findings of Ciani and co-workers [37], reduction of NO enhances GCP proliferation through an increased expression of the proto-oncogene N-myc. Therefore, protein levels were particularly examined for differences between tumor samples from Ptch1+/− Nos2+/+ and Ptch1+/− Nos2−/− mice. The results shown in Figure S2, however, revealed similar expression of N-myc in all MBs.
Analyses of genomic copy number alterations revealed a trisomy of chromosome 6 in the majority of MBs from both groups (11/12, Figure 2A and 2B). Moreover, a small region on chromosome 13, approximately 1.5 Mb upstream of the Ptch1 gene, showed a hemizygous deletion in healthy cerebella of Ptch1-mutant mice (data not shown) but a homozygous deletion in most tumors (10/12). Similarly, a second small region 3.8 Mb downstream of the last Ptch1 exon exhibited a loss in 9 of 12 MBs. This suggests structural changes flanking the Ptch1 locus that likely contribute to inactivation of the wild-type allele. The frequencies of genomic aberrations showed a more heterogeneous karyotype with gross structural changes in Ptch1+/− Nos2+/+ MBs when compared to Ptch1+/− Nos2−/− MBs (see Figure 2A, 2B and Text S1 for details). However, a general difference in chromosomal instability was not obvious between both genotypes. Only a small region (205.6 kb) on chromosome 14 containing the Entpd4 (ectonucleoside triphosphate diphosphohydrolase 4) gene was more frequently gained in Ptch1+/− Nos2−/− MBs (7/7) than in Ptch1+/− Nos2+/+ MBs (1/5, Figure 2A). Accordingly, Entpd4 expression appeared to be specifically upregulated in expression profiles of Ptch1+/− Nos2−/− tumors. QRT-PCR validation confirmed an elevated mean expression in Ptch1+/− Nos2−/− compared to Ptch1+/− Nos2+/+ MBs in those samples that overlapped with the array-CGH analysis but revealed no significant difference across the expanded tumor set (Figure 2C).

Nos2 inactivation is associated with low expression of mitotic genes in postnatal cerebellum
As GCPs are considered the cells of origin for the Hh-dependent MB subtype, we examined the effect of Nos2 ablation in the context of cerebellar development. Therefore, gene expression profiles of normal cerebellar tissue samples from postnatal day 9 (P9), 6 weeks after birth (6W), and 1 year of age (1Y) were generated from wild-type, Ptch1+/− Nos2+/+, Ptch1+/− Nos2−/−, and Ptch1+/+ Nos2−/− animals. While specimens of mature cerebellum (6W and 1Y) were investigated separately in 3 biological replicates per genotype and developmental stage, samples of postnatal cerebellum consisted of pooled RNA from 4–5 individuals processed in technical replicates due to limited tissue amounts.
An unsupervised hierarchical cluster analysis of transcriptome data clearly separated developing cerebellum (P9) of wild-type mice and the two Ptch1-mutated genotypes from mature cerebellum. Interestingly, P9 cerebellum of Ptch1+/+ Nos2−/− mice displayed different properties highly similar to mature cerebellum (Figure 3A). Expression profiles of MBs formed a distinct cluster clearly separated from all healthy tissue samples.

A direct comparison between gene expression profiles from Ptch1+/+ Nos2−/− and wild-type P9 cerebellar tissue samples resulted in a total of 984 deregulated genes with 755 genes (76.7%) showing a decreased expression in Ptch1+/+ Nos2−/− mice (Table S4). P9 cerebellum from Ptch1+/− Nos2+/+ and Ptch1+/− Nos2−/− mice revealed only 5 and 32 deregulated genes relative to wild-type, respectively (Table S5 and Table S6). This large deviation of postnatal gene expression in the Ptch1+/+ Nos2−/− genotype included a set of downregulated genes that are essential for proliferation of GCPs (e.g. cyclin D1, cyclin D2 and N-myc, Figure 3B). As hedgehog signaling constitutes the main regulatory pathway for neonatal cell proliferation in GCPs of the EGL, the 984 deregulated genes were analyzed for enrichment of Gli transcription factor targets. Matching this list to a set of recently identified Gli-targets in GCPs [10] yielded a significant overrepresentation of Gli1-regulated genes (p = 0.005, chi-square test). Hence, the reduced transcript levels of these target genes suggests an attenuated hedgehog signaling in postnatal Ptch1+/+ Nos2−/− cerebellum compared to wild-type (or any other genotype).
Notably, the decreased expression of Gli1-targets and proliferation-associated genes observed in Nos2-deficient cerebellar tissue was abolished upon additional inactivation of the hedgehog receptor Ptch1 (in Ptch1+/− Nos2−/− mice). Therefore, we examined the transcript levels of patched receptors themselves in more detail. While neither Ptch1 nor Ptch2 expression was changed between wild-type and Ptch1+/− Nos2+/+ P9 cerebellum, a significant increase of Ptch1 and a minor increase of Ptch2 expression were observed in Ptch1+/+ Nos2−/− mice relative to wild-type mice (Figure 3C). Notably, in Ptch1+/− Nos2−/− cerebellar tissue samples, Ptch2 expression was more elevated than Ptch1. However, since Ptch2 is not capable of inhibiting smoothened (Smoh), it probably failed to take over the attenuating effect on Gli activity [38]. MB specimens from Ptch1+/− Nos2+/+ versus Ptch1+/− Nos2−/− mice showed no significant difference in expression levels of either patched receptor, with Ptch2 being substantially increased over Ptch1 in both groups (Figure 3C). These findings indicate that Nos2 deficiency leads to an upregulation of Ptch1 in GCPs, which results in a downregulation of mitotic genes and Gli-targets only in a Ptch1-wild-type background.
Decreased expression of Gap43 is the most common effect of Nos2 inactivation
So far, Nos2 inactivation was shown to counteract proliferation and antagonize hedgehog signaling in developing cerebella. To identify those Nos2-dependent effects promoting MB induction, we determined the features that were common to Ptch1+/+ Nos2−/− and Ptch1+/− Nos2−/− genotypes and persisted in the tumor tissues. Accordingly, the overlap of differential gene expression from three comparisons was built: i) Ptch1+/+ Nos2−/− versus wild-type P9 cerebellum, ii) Ptch1+/− Nos2−/− versus Ptch1+/− Nos2+/+ P9 cerebellum; and iii) Ptch1+/− Nos2−/− versus Ptch1+/− Nos2+/+ MB. As a result, only 2 genes were observed to be deregulated in a Nos2-dependent manner during cerebellar development and in MBs (Figure 4A). Although Stmn1 (stathmin 1) appeared to be upregulated in Ptch1+/− Nos2−/− MBs relative to Ptch1+/− Nos2+/+ MBs, this could not be confirmed by qRT-PCR (Figure S3). Gene expression of Gap43 was consistently reduced in Nos2-deficient cerebellar tissue samples and downregulation in Ptch1+/− Nos2−/− tumors relative to Ptch1+/− Nos2+/+ tumors was also significant in the expanded validation set (Figure 4C and 4D). To further assess the immediacy of Nos2 inactivation and Gap43 deregulation, Gap43 transcript levels were determined in expression profiles of healthy cerebella from all developmental stages (P9, 6W and 1Y). Groups for comparison were built according to presence or absence of Nos2, irrespective of the Ptch1 status. The results clearly demonstrated a close association of altered Gap43 transcript levels and Nos2 status (Figure 4B), and indicated downregulation of Gap43 to be the most common effect of Nos2 deficiency in the cerebellum.
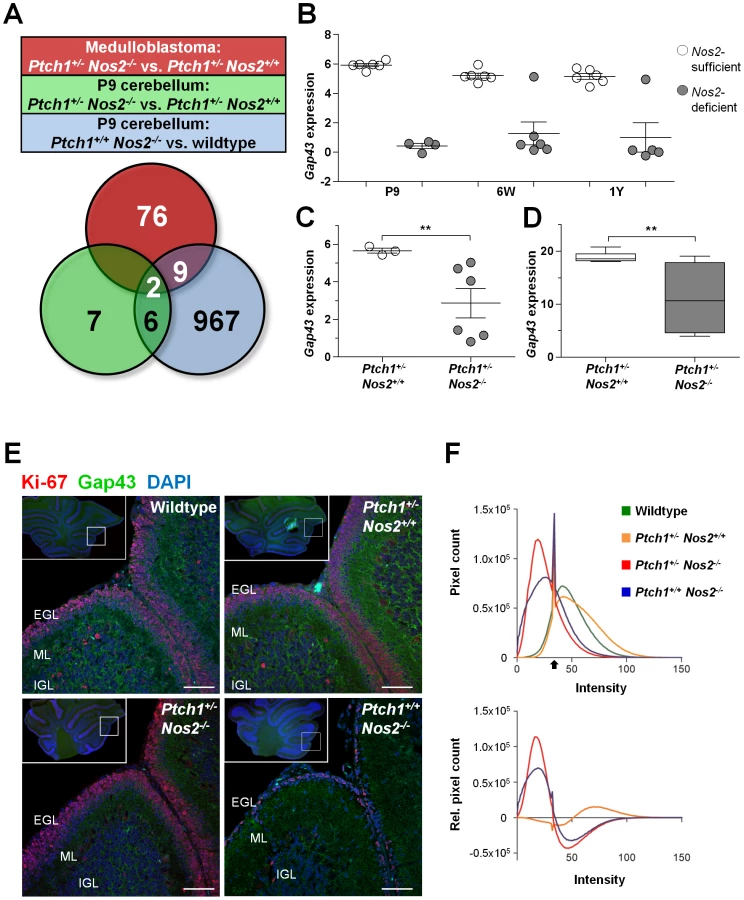
To investigate differences in Gap43 expression on protein level in situ we performed immunofluorescent double stainings of Gap43 and the proliferation marker Ki-67 on FFPE sections of P9 cerebella from wild-type, Ptch1+/− Nos2+/+, Ptch+/+ Nos2−/− and Ptch1+/− Nos2−/− mice. As illustrated in Figure 4E, Gap43 immunofluorescence was particularly prominent in the outer region of the molecular layer (ML) that is connected to and partially comprised of radial GCP process extensions. Image quantification further indicate a quantitative difference of Gap43 expression in this region with sections from wild-type and Ptch1+/− Nos2+/+ mice showing a more intense staining than sections from Ptch1+/− Nos2−/− and Ptch1+/+ Nos2−/− mice (Figure 4F).
Impaired NO signaling reduces Gap43 transcript levels
The association of Nos2 inactivation and decreased Gap43 expression suggests a gene-regulatory function of NO signaling. In order to investigate this possible link in vitro, we used the murine cerebellar precursor cell line c17.2 and the human MB cell line D458 (see Text S2 for details). Both cell lines were treated either with the Nos inhibitor L-NAME (Nω-nitro-L-arginine methyl ester) to reduce NO levels or solvent control (Figure S4). Relative expression of Gap43 was assessed every 24 hours by qRT-PCR. In c17.2 cells, Gap43 transcript abundance was generally low and increased with culture duration. We observed a slightly decreased expression of Gap43 upon L-NAME treatment that reached significance (p = 0.023) after 120 hours (Figure 5A). NOS inhibition in D458 human MB cells resulted in a significant reduction of Gap43 transcript levels starting already after 72 hours with further decrease after 96 hours and 120 hours (Figure 5B). FACS analyses of apoptosis and cell cycle excluded these observations to be attributed to secondary effects of changing cell conditions (Figure S5). These results suggest Gap43 downregulation as a direct consequence of reduced NO levels in murine neuronal precursors and human MB cells.

Increase of Ptch1 expression and impairment of GCP migration upon knockdown of Gap43
The dependency of Gap43 expression on NO signaling suggests this gene as key mediator of the effects observed in Nos2-deficient P9 cerebellum and Ptch1+/− Nos2−/− MB, in particular, the upregulation of functional Ptch1 in Ptch1+/+ Nos2−/− mice. Mishra et al. recently reported a central role of Gap43 in the polarization of developing GCPs by regulating centrosome positioning and thus defining correct orientation towards the IGL [39]. Since this is a prerequisite for directed migration, reduced levels of Gap43 in P9 cerebellar tissue may lead to retention of GCPs in the EGL. To test these hypotheses, shRNA-mediated knockdown of Gap43 was performed in c17.2 cells (see Text S1 for details). Upon knockdown of Gap43 we observed a strong inverse behavior of Ptch1 and Gap43 transcript levels (Figure 5C). Changes in migration characteristics were assayed in a Boyden chamber using recombinant SDF-1α (CXCL12) as chemoattractant, which was reported to participate in guiding migration of embryonal GCPs in vivo [40]. Downregulation of Gap43 yielded a significant decrease in cell migration between 14% (p = 0.013) and 20% (p = 0.007) (Figure 5D; Figure S6C). A pseudo-effect of the knockdown due to altered proliferation of the v-myc-immortalized c17.2 cells was excluded by FACS-based cell cycle analysis (Figure S7).
GCPs of the external granular layer show Gap43–associated phenotypes
Transcriptome and functional analyses suggest that a decreased Gap43 expression accounts for Ptch1 upregulation and impairment of directed neuronal precursor migration in vitro. Accordingly, Ptch1+/+ Nos2−/− P9 cerebella are supposed to increasingly retain GCPs with reduced mitotic activity in the EGL compared to wild-type and Ptch1+/− Nos2+/+ mice. Moreover, the Ptch1+/− Nos2−/− genotype is also expected to exhibit retention of GCPs, but not to show any cell cycle arrest. To further verify this hypothesis in situ we performed immunofluorescent double staining of proliferating (Ki-67+) and post-mitotic GCPs on FFPE sections of postnatal cerebellum (Figure 6A). Here, post-mitotic cells were delineated by the neuronal marker NeuN (neuronal nuclear antigen A60) [41]. At least three different regions of each mouse cerebellum were analyzed in three to four animals per genotype using confocal laser scanning microscopy. In accordance with the microarray data, averaged cell counts of wild-type and Ptch1+/− Nos2+/+ mice did not show significant difference. In contrast, an increase of post-mitotic GCPs (NeuN+, Ki-67−) was detectable in the EGL of Ptch1+/− Nos2−/− and Ptch1+/+ Nos2−/− mice (Figure 6C). Concurrently, the ratio of dividing to non-dividing GCPs was similar in Ptch1+/− Nos2−/−, wild-type and Ptch1+/− Nos2+/+ P9 cerebella but markedly decreased in Ptch1+/+ Nos2−/− mice. This recapitulated the downregulation of mitotic genes observed in the expression profiles. However, the total amount of proliferating GCPs per EGL section was significantly higher in Ptch1+/− Nos2−/− mice compared to any other genotype (Figure 6C). These results demonstrate a tissue phenotype that corresponds to the effects of reduced Gap43 in developing cerebellar neuronal precursors (in vitro). The increased accumulation of proliferating GCPs in the EGL observed in the Ptch1+/− Nos2−/− genotype supposedly leads to a larger pool of cells susceptible to neoplastic transformation and is therefore likely to promote medulloblastoma development.

Discussion
The Ptch1+/− MB mouse model has been intensively studied and has greatly contributed to our understanding of Hh-dependent MB tumorigenesis in the context of cerebellar development. The data presented here indicate a role of Nos2 and hence NO signaling in Hh-dependent MB by demonstrating a significantly increased MB rate in Ptch1+/− Nos2−/− mice compared to Ptch1+/− Nos2+/+ mice. The global genome-wide screens performed in the present study did not reveal obvious molecular differences between MBs in Ptch1+/− Nos2+/+ versus Ptch1+/− Nos2−/− animals. Assessment of genomic alterations using array-CGH identified trisomy of chromosome 6 as a recurrent feature in tumors of both genotypes. This corresponds to a recent report on MBs of the same molecular subtype with inactivated double-strand break repair proteins targeted to neuronal progenitors of p53−/− mice [42]. The most common loss identified in our analyses affected two small regions on chromosome 13 encompassing the Ptch1 gene and possibly indicate acquired homozygosity for the mutant allele or somatic rearrangements rather than a broad deletion of the locus. Targeted duplex PCR further confirmed loss of the functional wild-type allele to be a frequent event in these MBs. Notably, tumors of the Ptch1+/− Nos2−/− genotype showed a higher frequency of a small gain on chromosome 14. The affected Entpd4 gene encodes for an apyrase located at the internal membrane of lysosomal vacuoles and the Golgi apparatus. It preferentially catalyzes the hydrolysis of UDP to UMP [43] and thereby facilitates the inverse directed import of UDP-GlcNAc [44]. This in turn was reported to increase glycosylation of surface receptors (e.g. EGFR and PDGFR) and foster cell growth [45]. According to the microarray and qRT-PCR expression data, Entpd4 transcript levels were indeed increased in tumors with this chromosomal gain. However, this effect did not turn out to be Nos2-dependent in an expanded sample set. Consequently, Entpd4 likely plays a role in MB pathogenesis but is not directly linked to loss of Nos2.
The examination of tumor-relevant changes in developing cerebellum as a consequence of impaired Nos2 activity and hence NO signaling surprisingly revealed a decreased proliferation of GCPs in the cerebellum of Ptch1+/+ Nos2−/− mice. The concurrent upregulation of Ptch1 and the significant enrichment of downregulated Gli1-target genes strongly suggest that this effect is a consequence of reduced hedgehog signaling. Moreover, this phenotype was completely abrogated by a concomitant Ptch1 mutation. The slight increase of Ptch2 in Ptch1+/− Nos2−/− cells points to a compensatory effect and further supports the notion of an inhibitory function of Nos2 loss on the hedgehog pathway in postnatal cerebellum. Since neither a Smoh-regulating domain [38] nor a function for cell cycle arrest through seizing cyclin B1 [46] were reported for Ptch2, its upregulation may be insufficient for preventing MB induction. In contrast to these observations, Ciani et al. demonstrated that proliferation of cultured GCPs increased upon withdrawal of NO and that this effect was mediated by augmented N-myc levels [37]. However, N-myc was not differentially expressed between Ptch1+/− Nos2−/− and Ptch1+/− Nos2+/+ MBs of our series. A possible explanation for this discrepancy might be an unrecognized heterogeneity in the isolated cerebellar cell population used in the Ciani study. Since eNos and nNos are known to attenuate the mitotic activity of subventricular neuronal stem cells [47], [48] Nos inhibitor treatment possibly resulted in a selective growth advantage over GCPs. Downregulation of Gap43 was the only feature observed in Nos2-deficient versus Nos2-proficient postnatal cerebella irrespective of the Ptch1 status. This difference was also conserved between Ptch1+/− Nos2−/− and Ptch1+/− Nos2+/+ MBs, and particularly visible in outer regions of the molecular layer, where maturating GCPs of the EGL develop contact forming projections prior to radial migration. Other studies already suggested a link between Gap43 mRNA levels and NO signaling due to co-induction of nNos and Gap43 during axon regeneration and reactive synaptogenesis following injury of spinal motoneurons [49] and sensory neurons [50]. Furthermore, a downregulation of Gap43 was reported after silencing of soluble guanylate cyclase subunits, the central elements of cGMP-mediated NO signaling [51]. Finally, the present study demonstrates Gap43 downregulation to be a consequence of NO withdrawal in neuronal progenitors and MB cells. A possible mechanism for this regulation refers to decreased protein levels of the poly(U)-binding and degradation factor AUF1 upon NO-dependent cGMP production [52]. AUF-proteins generally bind to AU-rich elements of the 3′UTR (untranslated region) of coding transcripts and associate with proteins of the ELAV-like family to control gene expression via mRNA decay [53]. Tsai et al. demonstrated that Gap43 mRNA levels are post-transcriptionally regulated during neuronal differentiation and that elements of the 3′UTR confer transcript instability, which is abolished upon TPA treatment (inter alia inducing NOS2) [54]. At the same time, Chung et al. demonstrated that indeed ELAV-like family member HuD was binding to 3′UTR regions of GAP43 [55]. Taken together, NO accumulation possibly decreases cellular levels of mRNA-destabilizing AUF1 protein and thus might contribute to a high transcript abundance of Gap43.
Gap43 is a membrane-anchored protein at the cytoplasmic side of neuronal cell projections and found to be highly expressed during development of the CNS [56]. It is particularly localized in axonal growth cones and participates in the coordination of extrinsic stimuli and intrinsic cell remodeling [57] by regulating cytoskeleton dynamics [58]. Granule cell (GC) migration follows a sequence of tangential and radial movements controlled by successive formation of leading projections [59]. As maturating GCPs exit cell cycle, positioning of the centrosome determines the site of axon growth cone emergence and thus neuronal polarity including localization of such projections [60]. This defines the structural orientation of GCPs in terms of directing its dendrite to descend across the molecular and Purkinje cell layers to populate the IGL. However, centrosome positioning and therefore accurate polarization of GCPs require phosphorylated Gap43 to bind to the centrosome-associated microtubule-organizing center [61]. Hence, inaccurate GCP migration was observed in Gap43−/− animals [39], a finding that is in full agreement with our data from the functional Gap43 knockdown assays. Downregulation of Gap43 in Nos2-deficient P9 cerebellum therefore likely mediates the retention of GCPs observed in FFPE sections. Accordingly, NO/cGMP signaling was demonstrated to be crucial for accurate migration of the neuronal precursor cell line NT2 [62]. Furthermore, slice culture experiments of neonatal cerebella (P9) exhibited a substantial reduction of proliferation and migration of maturating granule cells to the IGL upon application of NO synthases inhibitors [63]. The elevation of Ptch1 levels upon Gap43 reduction in vitro fits to the data by Shen et al. who reported an upregulation of Ptch1 gene expression in inner EGL regions of Gap43−/− mice compared to wild-type animals. Moreover, cultured Gap43-deficient GCPs show decreased proliferation in response to administered recombinant Shh protein [64]. A possible regulatory link was recently provided as the activation of the hedgehog signaling component Smoh was found to depend on PI4P (phosphatidylinositol 4-phosphate) levels that immediately increase when Shh binds to Ptch1 or when functional Ptch1 is absent [65]. The authors further showed that imbalanced conversion of the precursor molecule PI into PI4P influences hedgehog pathway activity. Alternatively, the production of PI4P can also result from a specific dephosphorylation of PI(4,5)P2 [66]. In this context, Gap43 protein was recently demonstrated to build oligomeric structures in the plasma membrane which sequester specifically PI(4,5)P2 [67]. A similar finding has been reported earlier showing that GAP43 participates in the accumulation of plasmalemma rafts, which promoted retention of PI(4,5)P2 [68]. The amount of Gap43 associated with the plasma membrane therefore possibly modulates the utilization of PI(4,5)P2, including its conversion into PI4P, which in turn directly affects hedgehog signaling through Smoh activation. However, the effective impact on downstream Gli-targets would still be difficult to conclude regarding the multitude of responses to Shh, including negative feedback regulation [13]. Further studies applying depletion and enrichment of specific phosphatidyl derivatives and selective silencing of hedgehog pathway elements will be necessary to elucidate the molecular nature of this proposed signaling axis.
The increased accumulation of mitotic granule cells at the EGL seen in the combined Ptch1+/− Nos2−/− genotype supposedly gives a special clue to MB induction. In contrast to the classical view of neonatal EGL organization, which describes radial migration of granule cells to follow a proliferation stop, more and more evidence arises showing that cell cycle arrest is not a prerequisite for migration but rather occurs during a temporally coordinated interplay of gene expression patterns [69]. This corroborates our data shown in Figure 6 (white arrows), where proliferation is still maintained in migrating granule cells and even in cells of the IGL of wild-type cerebellum. Regulation of such expression patterns is largely dependent on Shh stimuli being most intensive in the EGL [70], as well as gradients of other soluble factors such as Bmps, which were reported to account for a regulatory environment along the transition through the cerebellar layers [71]. Further evidence for a niche-like-concept was provided by Choi et al. in Bdnf−/− mice that displayed a severe retardation of GCP migration [16]. The authors could demonstrate that mitotic activity of maturating GCPs was significantly enhanced when cells were retained in the EGL and declined with increasing distance from outer EGL regions. Therefore, the accumulation of GCPs in the EGL in combination with the insensitivity to Ptch1-mediated cell cycle arrest in Ptch1+/− Nos2−/− mice provide a growth advantage and increase the number of putative transformation targets over Ptch1+/− Nos2+/+ mice (Figure 7C).

In conclusion, the following picture emerged from our data: Homozygous deletion of Nos2 leads to a reduction of basic NO levels in immature GCPs of the EGL during postnatal development of the cerebellum. This reduction causes a downregulation of Gap43 expression, which results in an increased expression of Ptch1 and impaired directed migration of maturating GCPs. As a consequence, undifferentiated granule cell precursors exit cell cycle and are retained at the EGL (Figure 7B). In case of an additional heterozygous Ptch1 mutation, upregulation of this receptor does not suffice to exert the anti-proliferative stimulus following Gap43 decrease, which results in an increased fraction of continuously dividing cells in the EGL (Figure 7C). As reduced migration towards the IGL further leads to a withdrawal of growth-limiting signals, expansion of the GCP population is additionally supported. Finally, this advances medulloblastoma development in Ptch1+/− Nos2−/− mice compared to Ptch1+/− Nos2+/+ mice. The mechanism described here illustrates a new tumor-promoting concept in MB showing that the localization of pre-neoplastic cells within the developing cerebellum is important for pathogenesis.
Materials and Methods
Generation of Ptch1+− Nos2−/− mice
Ptch1+/− mice (B6; 129P2-Ptch1tm1Mps/Ptch+; [20]) and Nos2−/− mice (B6;129P2-Nos2tm1Lau; [72]) were obtained from the Jackson Laboratory (Bar Harbor, Maine, USA) and crossbred to generate double heterozygous mice (Ptch1+/− Nos2+/−). The F1 hybrids were backcrossed with Ptch1+/+ Nos2−/− mice to generate Ptch1+/− Nos2−/− mice. Later on, Ptch1+/− Nos2−/− mice were directly mated. For details on housing and genotyping see Text S2 and Table S9. All animal experiments were approved by the responsible federal authorities (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, Recklinghausen, Germany, Az. 50.05-230-17/06).
Microarray-based genomic and expression profiling
Total RNA of tumor specimens and normal cerebellar tissue samples was isolated via CsCl density gradient centrifugation [73] and assessed for integrity using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA). For gene expression microarrays, linear amplification of mRNA and labeling of cDNA were conducted on samples and Mouse Universal Reference RNA (Stratagene, La Jolla, USA) according to the TAcKLE protocol [74]. Both were combined for two-color hybridizations with each sample being performed as two replicates of inverse dye orientation. Global gene expression profiling was performed on self-printed oligonucleotide microarrays. Further details of microarray production and hybridization are described in Text S2. Genomic DNA of tumor specimens was isolated from the interphase of the CsCl gradient by ethanol precipitation, proteinase K digest, and phenol/chloroform extraction. DNA samples were monitored for purity and adequate fragment size using spectrophotometric measurements and gel electrophoresis. Array-based comparative genomic hybridization (array-CGH, matrix-CGH, [75]) was performed on Mouse Genome CGH 244 k Microarrays (Agilent). Cy5-labeled tumor DNA was combined with corresponding Cy3-labeled reference (wild-type genomic DNA) to receive either sex-matched sample pairs or pairs of different gender for internal negative or positive control. Sample preparation, microarray hybridization, and washing procedures were carried out as described in the manufacturer's protocol. Microarray data are available in GEO (http://www-ncbi.nlm.nih.gov/geo), under accession number GSE29201.
qRT–PCR analyses
Total RNA isolated from cell culture samples using the RNeasy Mini Kit (Qiagen, Hilden, Germany) or RNA from tissue specimens was subjected to oligo(dT)-primed reverse transcription. QRT-PCR measurements were conducted in an ABI PRISM 7900HT thermal cycler (Applied Biosystems, Foster City, USA) using the SYBR green reaction and detection system (ABgene, Epsom, UK). For relative quantification mean ratios were calculated between genes of interest and a set of five housekeeping genes (Table S7) according to the Pfaffl method [76].
The expression levels of Ptch1, Gli1, N-myc, and Nos2 were determined by real-time reverse transcription PCR analysis using the ABI PRISM 5700 system (Applied Biosystems) as reported before [73]. For these experiments, the mRNA expression level of mitochondrial ribosomal protein L32 (Mrpl32) served as housekeeping reference. All primer sequences are depicted in Table S8.
Cell culture experiments
Inhibition of NO synthases was performed in c17.2 and D458 cells which were seeded at densities of 2×105 and 4×105 cells per well in 12-well plates, respectively. Cells were daily treated with either 1 mM of the inhibitor L-NAME or 1× PBS as solvent control. For knockdown experiments of Gap43, c17.2 cells were grown in a 12-well plate to 80% confluency and transfected with 2 µg of pLKO.1-puro vector that contained either shRNA constructs targeting Gap43, shRNA against GFP, or non-target shRNA as a control (Sigma-Aldrich, St. Louis, USA) using 9 µl FuGene HD reagent (Roche, Basel, Switzerland). Transfection was repeated 2 times each after 8 hours and subjected to selection conditions (1 µg/ml puromycin) for 24 hours. Subsequently, cells were trypsinized, adjusted to 4×105 cells/ml and seeded into the inserts of a Costar Polycarbonate Membrane Transwell plate (8 µm pores, Corning, USA). After 24 hours cells were either harvested for gene expression and protein analyses or 0.1 µg/µl recombinant SDF-1α was applied to the lower compartment for migration assays. Following 12 hours of incubation, cells at the bottom of the insert membrane were methanol-fixed, hematoxylin-stained, and counted.
Immunofluorescence analyses
FFPE sections of postnatal cerebella were pre-processed as described in Text S2. For immunofluorescence co-staining, Gap43 (Sigma-Aldrich, clone GAP-7b10) or NeuN (Millipore, clone A60) first primary antibodies were diluted 1∶1000 or 1∶200, respectively and applied using the Dako REAL Detection System (Dako, Glostrup, Denmark). Following over night incubation at 4°C, washing in TBS, and blocking of residual biotin/streptavidin, sections were subsequently incubated with biotinylated anti-mouse secondary antibody (Dako) and stained with 20 ng/µl FITC-conjugated streptavidin (Invitrogen, Carlsbad, USA). The second primary antibody against Ki-67 (Novocastra, Wetzlar, Germany) was diluted 1∶1000 and accordingly applied using biotinylated anti-rabbit secondary antibody (Dako) and 20 ng/µl Cy5-conjugated streptavidin (Invitrogen). Co-stained sections were then covered with DAPI-containing VECTASHIELD Mounting Medium (Vector, Burlingame, USA) and subjected to confocal laser scanning microscopy.
Quantification of Gap43 staining was performed for areas of interest using Image J software (NIH). Numbers of dividing and non-dividing cells in the EGL of postnatal cerebellar tissue sections were counted manually and normalized to the corresponding length of the EGL edge. Cell counts for each region were averaged across three sections, each with 10–20 µm distance in z-axis, and per individual.
Statistical analyses
Kaplan-Meier survival plots were calculated for a total of 1167 mice, including 315 wild-type mice, 412 Ptch1+/+ Nos2−/− mice, 215 Ptch1+/− Nos2+/+ mice and 221 Ptch1+/− Nos2−/− mice. MB-free survival was plotted using the GraphPad Prism 5 software (GraphPad, La Jolla, USA). The logrank test was applied to compare survival (tumor occurrence) of the different genotypes. For comparisons of differences of means between two groups of replicates, p-value calculations were performed using an unpaired, two-tailed t-test, unless indicated otherwise. Calculated error bars represent the standard error of the mean (SEM). For details on microarray statistics please refer to Text S2.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. EllisonDWKocakMDaltonJMegahedHLusherME 2011 Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol 29 1400 1407
2. MulhernRKMerchantTEGajjarAReddickWEKunLE 2004 Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol 5 399 408
3. NorthcottPAKorshunovAWittHHielscherTEberhartCG 2011 Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29 1408 1414
4. KoolMKosterJBuntJHasseltNELakemanA 2008 Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 3 e3088 doi:10.1371/journal.pone.0003088
5. SchullerUHeineVMMaoJKhoATDillonAK 2008 Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14 123 134
6. MillenKJGleesonJG 2008 Cerebellar development and disease. Curr Opin Neurobiol 18 12 19
7. Wechsler-ReyaRJScottMP 1999 Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22 103 114
8. CohenMMJr 2003 The hedgehog signaling network. Am J Med Genet A 123A 5 28
9. KenneyAMColeMDRowitchDH 2003 Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 130 15 28
10. LeeEYJiHOuyangZZhouBMaW 2010 Hedgehog pathway-regulated gene networks in cerebellum development and tumorigenesis. Proc Natl Acad Sci U S A 107 9736 9741
11. PogorilerJMillenKUtsetMDuW 2006 Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development 133 3929 3937
12. KesslerJDHasegawaHBrunSNEmmeneggerBAYangZJ 2009 N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes Dev 23 157 170
13. LumLBeachyPA 2004 The Hedgehog response network: sensors, switches, and routers. Science 304 1755 1759
14. EspinosaJSLuoL 2008 Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. J Neurosci 28 2301 2312
15. ten DonkelaarHJLammensMWesselingPThijssenHORenierWO 2003 Development and developmental disorders of the human cerebellum. J Neurol 250 1025 1036
16. ChoiYBorghesaniPRChanJASegalRA 2005 Migration from a mitogenic niche promotes cell-cycle exit. J Neurosci 25 10437 10445
17. PietschTWahaAKochAKrausJAlbrechtS 1997 Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res 57 2085 2088
18. WolterMReifenbergerJSommerCRuzickaTReifenbergerG 1997 Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res 57 2581 2585
19. JohnsonRLRothmanALXieJGoodrichLVBareJW 1996 Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272 1668 1671
20. GoodrichLVMilenkovicLHigginsKMScottMP 1997 Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277 1109 1113
21. BriggsKJCorcoran-SchwartzIMZhangWHarckeTDevereuxWL 2008 Cooperation between the Hic1 and Ptch1 tumor suppressors in medulloblastoma. Genes Dev 22 770 785
22. UzielTZindyFXieSLeeYForgetA 2005 The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev 19 2656 2667
23. MoncadaS 1997 Nitric oxide in the vasculature: physiology and pathophysiology. Ann N Y Acad Sci 811 60 67; discussion 67–69
24. ArancioOKieblerMLeeCJLev-RamVTsienRY 1996 Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell 87 1025 1035
25. NathanCXieQW 1994 Nitric oxide synthases: roles, tolls, and controls. Cell 78 915 918
26. KleinertHSchwarzPMForstermannU 2003 Regulation of the expression of inducible nitric oxide synthase. Biol Chem 384 1343 1364
27. FukumuraDKashiwagiSJainRK 2006 The role of nitric oxide in tumour progression. Nat Rev Cancer 6 521 534
28. YamaguchiKSaitoHOroSTatebeSIkeguchiM 2005 Expression of inducible nitric oxide synthase is significantly correlated with expression of vascular endothelial growth factor and dendritic cell infiltration in patients with advanced gastric carcinoma. Oncology 68 471 478
29. HussainSPTriversGEHofsethLJHePShaikhI 2004 Nitric oxide, a mediator of inflammation, suppresses tumorigenesis. Cancer Res 64 6849 6853
30. ManderPBorutaiteVMoncadaSBrownGC 2005 Nitric oxide from inflammatory-activated glia synergizes with hypoxia to induce neuronal death. J Neurosci Res 79 208 215
31. CharlesNOzawaTSquatritoMBleauAMBrennanCW 2010 Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 6 141 152
32. SatoASekineYSarutaCNishibeHMoritaN 2008 Cerebellar development transcriptome database (CDT-DB): profiling of spatio-temporal gene expression during the postnatal development of mouse cerebellum. Neural Netw 21 1056 1069
33. JuradoSSanchez-PrietoJTorresM 2004 Elements of the nitric oxide/cGMP pathway expressed in cerebellar granule cells: biochemical and functional characterisation. Neurochem Int 45 833 843
34. JeskoHChalimoniukMStrosznajderJB 2003 Activation of constitutive nitric oxide synthase(s) and absence of inducible isoform in aged rat brain. Neurochem Int 42 315 322
35. PackerMAStasivYBenraissAChmielnickiEGrinbergA 2003 Nitric oxide negatively regulates mammalian adult neurogenesis. Proc Natl Acad Sci U S A 100 9566 9571
36. CianiECalvaneseVCrochemoreCBartesaghiRContestabileA 2006 Proliferation of cerebellar precursor cells is negatively regulated by nitric oxide in newborn rat. J Cell Sci 119 3161 3170
37. CianiESeveriSContestabileABartesaghiR 2004 Nitric oxide negatively regulates proliferation and promotes neuronal differentiation through N-Myc downregulation. J Cell Sci 117 4727 4737
38. RahnamaFToftgardRZaphiropoulosPG 2004 Distinct roles of PTCH2 splice variants in Hedgehog signalling. Biochem J 378 325 334
39. MishraRGuptaSKMeiriKFFongMThostrupP 2008 GAP-43 is key to mitotic spindle control and centrosome-based polarization in neurons. Cell Cycle 7 348 357
40. ZhuYYuTZhangXCNagasawaTWuJY 2002 Role of the chemokine SDF-1 as the meningeal attractant for embryonic cerebellar neurons. Nat Neurosci 5 719 720
41. WeyerASchillingK 2003 Developmental and cell type-specific expression of the neuronal marker NeuN in the murine cerebellum. J Neurosci Res 73 400 409
42. FrappartPOLeeYRussellHRChalhoubNWangYD 2009 Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc Natl Acad Sci U S A 106 1880 1885
43. BiederbickARosserRStorreJElsasserHP 2004 The VSFASSQQ motif confers calcium sensitivity to the intracellular apyrase LALP70. BMC Biochem 5 8
44. FangMShenZHuangSZhaoLChenS 2010 The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 143 711 724
45. TaniguchiN 2007 A sugar-coated switch for cellular growth and arrest. Nat Chem Biol 3 307 309
46. BarnesEAKongMOllendorffVDonoghueDJ 2001 Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J 20 2214 2223
47. TorroglosaAMurillo-CarreteroMRomero-GrimaldiCMatarredonaERCampos-CaroA 2007 Nitric oxide decreases subventricular zone stem cell proliferation by inhibition of epidermal growth factor receptor and phosphoinositide-3-kinase/Akt pathway. Stem Cells 25 88 97
48. MatarredonaERMurillo-CarreteroMMoreno-LopezBEstradaC 2004 Nitric oxide synthesis inhibition increases proliferation of neural precursors isolated from the postnatal mouse subventricular zone. Brain Res 995 274 284
49. YuanQHuBChuTHSuHZhangW 2010 Co-expression of GAP-43 and nNOS in avulsed motoneurons and their potential role for motoneuron regeneration. Nitric Oxide 23 258 263
50. ChenTJHuangCWWangDCChenSS 2004 Co-induction of growth-associated protein GAP-43 and neuronal nitric oxide synthase in the cochlear nucleus following cochleotomy. Exp Brain Res 158 151 162
51. Lopez-JimenezMEBartolome-MartinDSanchez-PrietoJTorresM 2009 Suppression of guanylyl cyclase (beta1 subunit) expression impairs neurite outgrowth and synapse maturation in cultured cerebellar granule cells. Cell Death Differ 16 1266 1278
52. JuradoSRodriguez-PascualFSanchez-PrietoJReimundeFMLamasS 2006 NMDA induces post-transcriptional regulation of alpha2-guanylyl-cyclase-subunit expression in cerebellar granule cells. J Cell Sci 119 1622 1631
53. DeMariaCTBrewerG 1996 AUF1 binding affinity to A+U-rich elements correlates with rapid mRNA degradation. J Biol Chem 271 12179 12184
54. TsaiKCCansinoVVKohnDTNeveRLPerrone-BizzozeroNI 1997 Post-transcriptional regulation of the GAP-43 gene by specific sequences in the 3′ untranslated region of the mRNA. J Neurosci 17 1950 1958
55. ChungSEckrichMPerrone-BizzozeroNKohnDTFurneauxH 1997 The Elav-like proteins bind to a conserved regulatory element in the 3′-untranslated region of GAP-43 mRNA. J Biol Chem 272 6593 6598
56. GorgelsTGVan Lookeren CampagneMOestreicherABGribnauAAGispenWH 1989 B-50/GAP43 is localized at the cytoplasmic side of the plasma membrane in developing and adult rat pyramidal tract. J Neurosci 9 3861 3869
57. MeiriKFSaffellJLWalshFSDohertyP 1998 Neurite outgrowth stimulated by neural cell adhesion molecules requires growth-associated protein-43 (GAP-43) function and is associated with GAP-43 phosphorylation in growth cones. J Neurosci 18 10429 10437
58. ShenYManiSDonovanSLSchwobJEMeiriKF 2002 Growth-associated protein-43 is required for commissural axon guidance in the developing vertebrate nervous system. J Neurosci 22 239 247
59. ChedotalA 2010 Should I stay or should I go? Becoming a granule cell. Trends Neurosci 33 163 172
60. ZmudaJFRivasRJ 1998 The Golgi apparatus and the centrosome are localized to the sites of newly emerging axons in cerebellar granule neurons in vitro. Cell Motil Cytoskeleton 41 18 38
61. GuptaSKMeiriKFMahfoozKBhartiUManiS 2010 Coordination between extrinsic extracellular matrix cues and intrinsic responses to orient the centrosome in polarizing cerebellar granule neurons. J Neurosci 30 2755 2766
62. TegengeMABickerG 2009 Nitric oxide and cGMP signal transduction positively regulates the motility of human neuronal precursor (NT2) cells. J Neurochem 110 1828 1841
63. TanakaMYoshidaSYanoMHanaokaF 1994 Roles of endogenous nitric oxide in cerebellar cortical development in slice cultures. Neuroreport 5 2049 2052
64. ShenYMishraRManiSMeiriKF 2008 Both cell-autonomous and cell non-autonomous functions of GAP-43 are required for normal patterning of the cerebellum in vivo. Cerebellum 7 451 466
65. YavariANagarajROwusu-AnsahEFolickANgoK 2010 Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev Cell 19 54 65
66. SkwarekLCBoulianneGL 2009 Great expectations for PIP: phosphoinositides as regulators of signaling during development and disease. Dev Cell 16 12 20
67. ZakharovVVMosevitskyMI 2010 Oligomeric structure of brain abundant proteins GAP-43 and BASP1. J Struct Biol 170 470 483
68. LauxTFukamiKThelenMGolubTFreyD 2000 GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol 149 1455 1472
69. ArgentiBGalloRDi MarcotullioLFerrettiENapolitanoM 2005 Hedgehog antagonist REN(KCTD11) regulates proliferation and apoptosis of developing granule cell progenitors. J Neurosci 25 8338 8346
70. BlaessSGraus-PortaDBelvindrahRRadakovitsRPonsS 2004 Beta1-integrins are critical for cerebellar granule cell precursor proliferation. J Neurosci 24 3402 3412
71. GrimmerMRWeissWA 2008 BMPs oppose Math1 in cerebellar development and in medulloblastoma. Genes Dev 22 693 699
72. LaubachVESheselyEGSmithiesOShermanPA 1995 Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci U S A 92 10688 10692
73. van den BoomJWolterMKuickRMisekDEYoukilisAS 2003 Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol 163 1033 1043
74. SchlingemannJThuerigenOIttrichCToedtGKramerH 2005 Effective transcriptome amplification for expression profiling on sense-oriented oligonucleotide microarrays. Nucleic Acids Res 33 e29
75. Solinas-ToldoSLampelSStilgenbauerSNickolenkoJBennerA 1997 Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer 20 399 407
76. PfafflMW 2001 A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29 e45
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- PIF4–Mediated Activation of Expression Integrates Temperature into the Auxin Pathway in Regulating Hypocotyl Growth
- Metabolic Profiling of a Mapping Population Exposes New Insights in the Regulation of Seed Metabolism and Seed, Fruit, and Plant Relations
- A Splice Site Variant in the Bovine Gene Compromises Growth and Regulation of the Inflammatory Response
- Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson's Disease Genetics: The PDGene Database
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy