
Akirin Links Twist-Regulated Transcription with the Brahma Chromatin Remodeling Complex during Embryogenesis
The activities of developmentally critical transcription factors are regulated via interactions with cofactors. Such interactions influence transcription factor activity either directly through protein–protein interactions or indirectly by altering the local chromatin environment. Using a yeast double-interaction screen, we identified a highly conserved nuclear protein, Akirin, as a novel cofactor of the key Drosophila melanogaster mesoderm and muscle transcription factor Twist. We find that Akirin interacts genetically and physically with Twist to facilitate expression of some, but not all, Twist-regulated genes during embryonic myogenesis. akirin mutant embryos have muscle defects consistent with altered regulation of a subset of Twist-regulated genes. To regulate transcription, Akirin colocalizes and genetically interacts with subunits of the Brahma SWI/SNF-class chromatin remodeling complex. Our results suggest that, mechanistically, Akirin mediates a novel connection between Twist and a chromatin remodeling complex to facilitate changes in the chromatin environment, leading to the optimal expression of some Twist-regulated genes during Drosophila myogenesis. We propose that this Akirin-mediated link between transcription factors and the Brahma complex represents a novel paradigm for providing tissue and target specificity for transcription factor interactions with the chromatin remodeling machinery.
Published in the journal:
. PLoS Genet 8(3): e32767. doi:10.1371/journal.pgen.1002547
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002547
Summary
The activities of developmentally critical transcription factors are regulated via interactions with cofactors. Such interactions influence transcription factor activity either directly through protein–protein interactions or indirectly by altering the local chromatin environment. Using a yeast double-interaction screen, we identified a highly conserved nuclear protein, Akirin, as a novel cofactor of the key Drosophila melanogaster mesoderm and muscle transcription factor Twist. We find that Akirin interacts genetically and physically with Twist to facilitate expression of some, but not all, Twist-regulated genes during embryonic myogenesis. akirin mutant embryos have muscle defects consistent with altered regulation of a subset of Twist-regulated genes. To regulate transcription, Akirin colocalizes and genetically interacts with subunits of the Brahma SWI/SNF-class chromatin remodeling complex. Our results suggest that, mechanistically, Akirin mediates a novel connection between Twist and a chromatin remodeling complex to facilitate changes in the chromatin environment, leading to the optimal expression of some Twist-regulated genes during Drosophila myogenesis. We propose that this Akirin-mediated link between transcription factors and the Brahma complex represents a novel paradigm for providing tissue and target specificity for transcription factor interactions with the chromatin remodeling machinery.
Introduction
A fundamental question in embryonic development is how diverse cell lineages are specified, patterned and organized from a single common progenitor. These processes are governed by distinct gene expression programs administered by tightly regulated transcription factor activities during embryonic development. One mechanism whereby transcription factor activities are modulated is through direct interactions with cofactors. In addition, links between transcription factors and the general transcription machinery can be indirect, such as the actions of secondary factors that recruit chromatin remodeling complexes to modify the local chromatin environment and allow gene activation. The identification of these secondary effectors and their in vivo function is critical for understanding the regulation of transcription factor activity during embryonic development. Such interactions have significant ramifications for developmental disorders and diseases such as cancer.
The Twist transcription factor represents an ideal model for studying the regulation of transcription factor activity throughout development. Twist is a highly conserved transcription factor that is a key regulator of many developmental programs during embryogenesis as well as cancer metastasis [1]–[3]. In Drosophila melanogaster, Twist regulates multiple, discrete steps of mesoderm development, including gastrulation, segregation of populations of mesodermal cells, establishment and formation of the somatic musculature, and finally establishment of the adult musculature [4]–[7]. To achieve these diverse activities, Twist regulates a large number of target genes temporally within a discrete lineage resulting in a diverse array of outputs during mesodermal development [8]. A key question is how these diverse outputs of Twist activity during development are achieved.
The varied roles of Twist during different phases of embryonic development and the large number of Twist-regulated target genes suggest complex regulation of Twist activity [4], [8], [9]. Twist activity is often modulated by interactions with other transcription factors [10], [11]. Twist is a basic Helix-loop-Helix (bHLH) transcription factor that can homodimerize or heterodimerize, with distinct activities for each dimer pair. For example, Twist homodimers are responsible for activating target genes that direct cells to the somatic myogenic lineage [10]. By contrast, heterodimers between Twist and another bHLH protein, Daughterless, repress the somatic myogenic lineage [10], [11]. Twist activity is also regulated by interactions with other transcription regulators that bind closely linked DNA regulatory elements. One such interaction, with the NF-κB orthologue Dorsal, produces synergistic rather than cooperative activation of Dorsal targets during development [12]–[14]. Hence, Daughterless and Dorsal are examples of transcription regulators that physically interact with Twist to modify its output.
Other mechanisms that regulate Twist activity during development are less clear. One mechanism whereby transcriptional activity is indirectly regulated is through the action of chromatin modifying factors that generate a local chromatin environment that allows and/or favors the expression of a specific target gene. The structure of chromatin is modified by two predominant mechanisms: the marking of nucleosome tails with post-translational modifications such as acetylation, phosphorylation and methylation, and the remodeling of the local chromatin environment via factors that reposition nucleosomes in a local gene environment [15]. Nucleosome repositioning occurs via the activity of ATP-dependent chromatin remodeling complexes such as the Brahma-containing (BRM) complex, the Drosophila orthologue of the yeast SWI/SNF chromatin remodeling complex [15], [16]. Mutations in BRM complex subunits have revealed essential roles for BRM in such processes as homeotic gene expression, oogenesis, and cell cycle control [17]–[22]. Loss-of-function studies have determined that BRM-regulated chromatin remodeling activity is required for most RNA Polymerase II-regulated transcription [23]. Most eukaryotic organisms have two different compositions of the SWI/SNF complex; in Drosophila, these complexes are designated the BAP (Osa-containing) and PBAP (polybromo/Bap180, Bap170 and SAYP-containing) complexes [15], [16], [24]. Both BAP and PBAP are linked to gene activation and repression, are present in the same cells, and perform unique yet cooperative functions during development [19], [24]–[28]. Both the association of BRM complexes with transcription factors and BRM complex targeting and regulation during embryogenesis remain an area of considerable interest. Moreover, links between BRM complexes, chromatin remodeling and Twist have not yet been elucidated.
The nuclear protein Akirin was shown to regulate gene expression in several different transcription pathways, yet its mechanism of action remained unclear [29], [30]. Here we identify Akirin as a factor that facilitates an interaction between Twist and the BRM chromatin remodeling complex to promote gene expression. We find that Akirin interacts both physically and genetically with Twist at Twist-dependent enhancer regions and positively regulates expression of Dmef2, a Twist-regulated gene that is critical for somatic myogenesis during development. As would be predicted by this interaction, akirin mutant embryos show a range of somatic muscle phenotypes. Akirin is widely associated with regions of active transcription. We find that Akirin colocalizes with subunits of the BRM chromatin remodeling complex on polytene chromosomes and interacts genetically with core subunits of the BRM complex during myogenesis. Finally, we verify with chromatin immunoprecipitation that Twist, Akirin, and a core subunit of the BRM complex all occupy the Twist-dependent Dmef2 enhancer. Curiously, Akirin/Twist interactions are not required at all Twist-dependent enhancers, as evidenced by our chromatin immunoprecipitation experiments at the eve MHE. These results suggest that Akirin functions as a BRM accessory protein that links the BRM chromatin remodeling machinery to Twist transcription factor activity at a subset of Twist-regulated enhancers. These results provide a common mechanism by which Akirin links chromatin remodeling factors to spatiotemporal-specific gene activation.
Results
Akirin is a conserved Twist-interacting nuclear protein
To identify proteins that interact with Twist, we performed a yeast double interaction screen [31], [32]. Two Twist-regulated enhancers from the Dmef2 [33] and tinman [34] genes were cloned upstream of a HIS3 reporter. Yeast strains containing these enhancer/reporter constructs were first transformed with a Twist expression vector and then with a 0–6 hr embryonic cDNA library fused to a Gal4 activation domain. Transformants were positively scored for cDNAs that activated HIS3 expression at Dmef2, tinman, or both enhancers in a Twist-dependent manner. We identified 28 cDNAs that activated expression from both enhancers. From this group, we selected the then-unknown gene CG8580 for further study. CG8580 was found to require the presence of Twist for activation of the HIS3 reporter [32].
We initially named CG8580 as bhringi [32], but recent groups have re-designated this gene and its orthologues as akirin [29], [35]. For clarity, we have adopted this nomenclature. The Drosophila akirin gene encodes a highly conserved 201-residue protein with orthologues present in over 24 different metazoan genomes [35]. Aside from a predicted nuclear localization sequence, Drosophila Akirin has no known protein motifs [35]. We raised antibodies against a peptide sequence found in the N-terminal region of Drosophila Akirin (Figure S1). Immunofluorescence staining of embryos at various stages of development showed that Akirin is ubiquitously expressed and found in the nuclei of all cells of the developing embryo (Figure S2).
akirin mutants display a range of muscle phenotypes
The somatic muscles of Drosophila embryos are arranged in a stereotypic, repeated pattern of 30 muscles within each abdominal hemisegment (Figure 1) [36]. Because Akirin interacted with Twist, a transcription factor that is essential to the proper patterning of the somatic musculature, we examined the somatic musculature of akirin mutant embryos, using publicly available and newly generated akirin alleles (See Materials and Methods and Figure S3). akirin mutant embryos exhibit a range of somatic body wall muscle phenotypes. Three classes of defects were observed: missing muscles, attachment defects, and duplicated muscles (Figure 1). While these defects were seen easily in the 4 lateral transverse (LT) muscles, the same classes of defects were detected in several other muscles, particularly the DT1, DO3, DO4 and DA3 muscles. Misattached, missing or duplicated muscles were found in stage 16 (15 h after egg laying (AEL)) akirin2 homozygous mutant embryos (46.5%, n = 114 embryos), and embryos from crosses of akirin2 females with akirin3 males (67%, n = 52 embryos), or akirin3 females with akirin5 (44.7%, n = 38 embryos) males (Table 1). The low penetrance of muscle phenotypes seen in akirin mutants is likely due to the high degree of maternal loading of akirin RNA (Figure S3). We attempted to generate akirin maternal/zygotic mutants by making germline clones using akirin3 mutants [37]. However, no akirinMZ mutant embryos were recovered, suggesting that Akirin is important for oocyte formation (data not shown). Therefore, homozygous viable akirin2 females likely produce embryos that are more highly sensitized to low levels of akirin mRNA in the oocyte. Importantly, the missing muscle phenotype is not due to a failure to properly specify muscle founder cells, as immunohistochemistry of founder cell identity proteins, including Slouch, Even-skipped and Krüppel, did not show a decrease in the numbers of founder cells in akirin mutants (Figure S4). Also, in akirin mutant embryos there were no obvious signs of muscle degeneration or late differentiation defects [38] (data not shown). Epidermal development was normal, and the cuticle developed normally (data not shown).


Akirin interacts both genetically and physically with Twist
The muscle phenotypes in akirin mutant embryos were reminiscent of those observed in embryos in which twist expression is modified by RNAi in the during muscle fiber formation [11]. To determine if akirin interacts genetically in vivo with twist, we examined the patterning of the somatic body wall musculature in embryos that are heterozygous for both twist and akirin. While twi1/+ embryos [39], akirin2/+ and akirin3/+ embryos have a wild-type somatic muscle pattern (data not shown), stage 16 embryos that are heterozygous for both twist and akirin2 and akirin3 (twi1/+; akirin2/+, and twi1/+; akirin3/+) show a general disruption of the muscle pattern, with missing and misattached muscles (twiI1/+; akirin2/+ : 21.6% of 60 embryos examined and twi1/+; akirin3/+: 18.5% of 105 embryos examined, Figure 2A).

Additionally, in vitro-translated Akirin was successfully pulled down using a GST-Twist fusion protein indicating a physical interaction between Akirin and Twist (Figure 2B). Taken together, these data confirmed our initial results of the yeast double interaction screen and demonstrated both a physical and functional interaction between Akirin and Twist.
Dmef2 enhancer activity is reduced in akirin mutant embryos
Akirin was initially identified in our screen as a Twist interacting protein in the context of the Dmef2 enhancer. In addition, the muscle defects in akirin mutants are similar to the phenotypes in embryos with modified Dmef2 or twist expression [7], [11], [40]–[42]. These data suggested that Akirin and Twist interact to positively regulate transcription from Dmef2. To test this in vivo, we analyzed akirin mutant embryos that carry a lacZ transgene regulated by the same Dmef2 muscle enhancer [33] used in our initial double interaction screen. This particular enhancer requires Twist activity for early Dmef2 expression during somatic myogenesis. Regulation of Dmef2 by Twist is critical for the subsequent establishment of the somatic musculature [8], [33], [40], [41], [43]–[48]. Whole-mount antibody staining for β-galactosidase (β-gal) indicated that akirin mutants have reduced expression of β-gal compared to wild-type embryos with the same transgene at the same developmental stage (Figure 3A, 3B). Densitometric analysis of Western blotting supported the observation that total β-gal levels were changed in akirin mutant extracts relative to wild-type embryo extracts (Figure S5). To confirm that endogenous Dmef2 expression levels were affected, we performed RT-qPCR analysis on total mRNA prepared from wild-type and akirin2 mutant embryos at the same developmental age (4–6 h AEL). Accordingly, we observed a 2.75-fold reduction in Dmef2 transcripts in akirin2 mutant embryos compared to wild-type embryos at this time (Figure 3C).

For comparison, we examined a second Twist target gene, even-skipped. No reduction in protein levels of Even-skipped (Eve) or levels of expression from eveMHE-lacZ reporter constructs were observed in akirin mutant embryos at the same stage (Figure 3B and Figure S5). These data indicated that Twist and Akirin interact to positively regulate expression of the Dmef2 reporter. These data also suggested that not all Twist target genes and/or their associated enhancer elements require Akirin activity for their proper expression.
Akirin protein is detected at actively transcribed gene loci
In addition to our observed relationship with Twist, Akirin has been linked to other transcriptional regulators in various insect and mouse contexts [29], [30], [49]. However, in each of these contexts, the mechanism by which Akirin promotes gene expression remained unclear. Because Akirin does not have a consensus DNA-binding domain or predicted catalytic activity, we hypothesized that Akirin regulates gene expression through its interactions with transcriptional regulators and would therefore localize to regions of active transcription. We analyzed the distribution of Akirin on polytene chromosomes and found Akirin localization throughout the genome including puffed regions of polytene chromosomes (Figure 4A, 4B), which indicate active transcription [50]. For further confirmation, co-immunostaining for Akirin and Serine 10-phosophorylated histone H3, a histone modification that serves as a marker of actively transcribing loci in polytene chromosomes, was performed [51]–[53]. Akirin partially colocalized with Ser10-phosphohistone H3 staining (43.8% colocalization, see Figure 4C, 4E and Figure S6A). To strengthen these results, we performed additional co-immunostaining for Akirin and Ser7-phosophorylated RNA polymerase II [54], which also showed partial colocalization between these two stains (55.4%, see Figure 4D, 4F and Figure S6B). These data confirmed that Akirin is associated with some actively transcribing loci throughout the genome. In addition, because Twist is not expressed in wandering third instar salivary glands ([55] and data not shown), these data suggested that Akirin interacts with other transcriptional regulators for its function in this tissue. However, when we ectopically expressed Twist in the salivary glands using Sgs3-GAL4 [56], we found that Twist and Akirin partially colocalize on polytene chromosomes (54% colocalization, Figure 4G). Furthermore, under these conditions both Twist and Akirin colocalized at the Dmef2 locus (Figure 4G), and Dmef2, which is not normally expressed in salivary glands, is now expressed (Figure S7). We note that Akirin is not localized to the Dmef2 locus when Twist is not present (Figure 4H). Together, these data supported our earlier results indicating that Akirin acts with Twist to promote the expression of a Twist-regulated target. Finally, the colocalization of Akirin with regions of active gene expression in a tissue that does not normally contain Twist confirmed that Akirin functions as a general regulator of gene expression.

Akirin colocalizes with components of the Brahma chromatin remodeling complex
One mechanism whereby Akirin might function as a general cofactor for gene expression would be through interactions with chromatin remodeling complexes. A Drosophila whole-genome yeast 2-hybrid experiment [57] suggested that Akirin interacts with BAP60, a core subunit of the Drosophila SWI/SNF class Brahma (BRM) chromatin remodeling complex [58]. Immunostaining of polytene chromosomes with antibodies against the Brahma (Figure 5A) and Snr1 (Figure 5B) core subunits revealed that Akirin colocalized with BRM (67% and 59.1% colocalization respectively, Figure 5D and Figures S8, S9). Akirin also colocalized with Osa (65.3% colocalization), a subunit exclusive to the BAP complex (Figure 5C, 5D and Figure S9). These data therefore suggested that Akirin associates with BRM complex components but is not a core BRM complex subunit.

Akirin interacts with BRM complex subunits during embryonic myogenesis
Given the observed colocalization between Akirin and the Brahma, Snr1 and Osa BAP subunits on polytene chromosomes, we hypothesized that there is a functional association of these proteins during embryonic development. To test this, the patterning of the somatic muscles in stage 16 embryos that are double heterozygotes for both akirin and brahma was examined (Figure 6B). Both brmI21/+ and akirin2/+ (single heterozygous) embryos have a normal somatic muscle pattern (Figure S10), but the brmI21/+,akirin2/+ double heterozygous embryos (35%, n = 55) displayed disruptions in the somatic muscles, with both missing and improperly attached muscles (Figure 6B, Table 2). We further analyzed embryos that are double heterozygotes of both akirin and other BRM complex core subunits (Figure 6, Table 2). Although heterozygous embryos for other BRM complex subunits do not show a somatic muscle phenotype (Figure S10), muscle phenotypes were observed in akirin2/+,moira1/+ (40%, n = 48, Figure 6C), akirin2/+,Snr101319/+ (45%, n = 52, Figure 6D), and Bap601/+;akirin3/+ embryos (40%, n = 55, Figure 6E). Additionally, akirin2/+,bap180Δ86/+ (38%, n = 45, Figure 6F) and akirin2/+,osa2/+ (42%, n = 52, Figure 6G) embryos also had disrupted muscle patterning. To ensure that these defects were specific for subunits of the BRM complex and not due to a general interaction between akirin and other chromatin factors, double heterozygous combinations of akirin with alleles of Polycomb, Nurf-38, Iswi, and Su(var)3-9 were tested. Each of these allelic combinations showed muscle patterning defects in fewer than 5% of embryos. These results confirmed a functional interaction during muscle development between Akirin and core BRM subunits, as well as PBAP and BAP-specific subunits. Based on our data thus far, we hypothesized that Akirin mediates Twist-BRM interactions on a subset of Twist target genes. To begin to test this, we examined whether whether twi and brm genetically interact. We find that the somatic muscle pattern is disrupted in twiI1/+; brmI21/+ double heterozygous embryos (23%, n = 51, Figure 6H), supporting a functional interaction between twist and brahma.


To determine whether the Akirin protein associates with the BRM complex in vivo, we attempted co-immunoprecipitation experiments from embryonic lysates using tagged Akirin. While we did observe weak physical interaction between tagged Akirin and the Brahma core subunit (data not shown), these appear to be highly transient and not robust. As we had identified a functional interaction between twist and brahma (Figure 6H), we examined whether Brahma and Twist physically interact. Antibodies against Brahma successfully co-immunoprecipitated Twist from whole embryonic extracts (Figure S11). Together these data suggest that Twist and Akirin both interact functionally with the BRM complex during myogenesis.
Twist, Akirin, and a core BRM complex subunit are localized to Twist-dependent enhancers in vivo
To further support our interaction data, we next tested whether Twist, Akirin, and the BRM complex were localized to Twist-regulated enhancer elements in embryos when the muscle pattern is being established. Using chromatin immunoprecipitation and quantitative PCR, we examined the occupancy of the Dmef2 enhancer and the eve MHE element by Twist and Akirin (Figure 7). Both of these enhancer regions contain E-box elements that are bound by and regulated by Twist during development [8], [33], [46], [59]. In agreement with previously published data [8], antibodies against Twist and Akirin both successfully immunoprecipitated the Dmef2 enhancer in extracts prepared from 2–4 and 4–6 hour-old embryos (Figure 7A, 7B). These data suggested that Akirin localizes to the Dmef2 enhancer element with Twist during these time periods. However, while Twist occupancy at the eve MHE was detected as previously reported [8], Akirin protein was not significantly enriched at the eve MHE region in 2–4, 4–6, and 6–10 hour embryo extracts compared to preimmune antisera controls. We concurrently examined occupancy of these elements by Twist and Akirin in extracts prepared from akirin2 mutant embryos. As we would predict, Twist occupancy at both Dmef2 and eve MHE enhancers was not significantly affected by the absence of Akirin (Figure 7A).

We next tested the occupancy of Dmef2 and eve MHE enhancers by a BRM subunit. We found that the BRM core subunit Moira occupied the Dmef2 enhancer early, in extracts prepared from 2–4 hour old embryos (Figure 7C), with a similar occupancy profile as that observed with Twist at this time. However, the reduction in Akirin level appeared to affect Moira occupancy at the Dmef2 enhancer, as we observed a 64% reduction in Moira levels at this enhancer in 2–4 hour old akirin2 mutant extracts versus wild-type. We further found that Moira occupied the eve MHE, albeit at relatively low levels, at all time points examined. Further, this occupancy was not affected by the absence of Akirin, as the levels did not appear to change in akirin2 mutant extracts versus wild-type (Figure 7C). For comparison, we also examined occupancy of Twist, Akirin and Moira at the enhancer region of the oskar gene, which is not normally expressed in the embryo at this time [60]. We found no detectable occupancy of any of these factors at this region at the same timepoints (see Figure S12). In summary, we find that Twist, Akirin, and core subunits of the BRM complex all co-occupy the Dmef2 Twist-dependent enhancer together at a temporally critical time for establishing the muscle pattern during embryogenesis, and the occupancy of Moira at this element is dependent upon the presence of Akirin. In contrast, we find that Akirin is not present at a second Twist-regulated enhancer, the eve MHE, at the same period of embryonic development.
Discussion
The Twist transcription factor controls many key processes in the establishment and patterning of the mesoderm, including organogenesis of the somatic body wall musculature during Drosophila embryonic development [6]. As Twist activity regulates a large number of genes and processes over the course of mesodermal development [8], [9], specific Twist functions are likely conferred through secondary and tertiary proteins that interact with Twist [11]. In a screen for such interacting proteins, we identified the highly conserved nuclear protein Akirin. Akirin mutants show defects in myogenesis consistent with a role in regulating Twist activity, in particular at the Dmef2 gene. Akirin accomplishes this regulation through interactions with core subunits of the Brahma chromatin remodeling complex. Finally, we find Twist, Akirin and a BRM complex subunit all co-occupy the Dmef2 enhancer during early embryogenesis, and this occupancy of a core BRM subunit requires the presence of Akirin. These data suggest that Akirin is required for optimal expression of this Dmef2 enhancer element. By contrast, the Twist-regulated eve MHE regulatory element does not require Akirin for its expression. Together, these results suggest that Akirin is an accessory protein that links Twist and the BRM chromatin remodeling complex for activation of specific Twist-regulated enhancers during Drosophila embryonic development. We propose that this Akirin-mediated link between Twist and the Brahma complex represents a novel paradigm for providing tissue and target specificity for Twist transcription factor activity with chromatin remodeling machinery during embryogenesis.
Akirin and Twist interact during embryonic development
Establishment of the somatic musculature during embryogenesis requires precisely regulated Twist activity [4]. The muscle phenotype of akirin mutants and twist and akirin double heterozygotes indicates a functional interaction between Twist and Akirin during myogenesis. This interaction is direct, as Akirin and Twist physically bind. Additionally, both Twist and Akirin co-occupy the Dmef2 enhancer during embryogenesis, and their association is important for robust expression from the Dmef2 enhancer. Dmef2 is a critical regulator of myogenesis and is expressed throughout this process [33], [40], [45], [47], [48]. Dmef2 coordinates multiple processes necessary for proper somatic myogenesis and regulates, in combination with Twist, a subset of Twist-regulated genes in a feed-forward manner [8], [47]. The missing, misattached, or duplicated muscle phenotypes that we observe in akirin mutants are similar to the phenotypes of embryos in which Dmef2 or twist expression levels are modified [11], [40]–[42]. For example, missing and misattached muscles were reported in incompletely rescued Dmef2 mutant embryos [41], while muscle duplications have been reported as a result of RNAi-mediated knockdown of twist [11]. Therefore, akirin mutant muscle phenotypes likely reflect a perturbation in Twist activity, resulting in an early alteration in Dmef2 expression levels during embryogenesis. There are other elements in the Dmef2 enhancer that are bound and controlled by transcription factors such as Pannier, Medea and Tinman, that regulate Dmef2 expression during embryogenesis [8], [33], [46], [59]. It is unknown whether Akirin interacts with these factors at other Dmef2 control elements for their optimal expression or whether interaction with just one factor is sufficient. These questions are subjects for further study.
Whole genome ChIP-on-chip experiments have identified almost 500 cis-regulatory elements that are bound by Twist during mesodermal development [8]. Further, Twist co-regulates a number of targets together with Dorsal, Dmef2 and Tinman in a feed-forward manner. Interestingly, our data would argue that Twist does not require Akirin for optimal expression of all Twist-dependent enhancers. First, our analysis of polytene chromosomes indicates that Akirin colocalizes with Twist at only 54% of Twist binding sites. Second, our chromatin immunoprecipitation experiments indicate that Akirin is enriched at the Dmef2 enhancer during the first 10 hours of embryonic development, but not at the even-skipped MHE enhancer at the same time. Accordingly, we did not observe a noticeable decrease in Eve expression, a reduction in Eve-positive clusters, or defects in patterning of the DA1 muscle in akirin mutants (Figure 3, Figure S3, and data not shown). What could explain the different requirements for Akirin for optimal expression of Dmef2 versus eve? On one level, there are inherent differences in the complexities of these enhancers: While both the eve MHE and Dmef2 enhancers contain E-boxes bound and regulated by Twist, the eve MHE element also contains multiple DNA elements bound by a large array of factors from the Wingless, Decapentaplegic, and RTK-Ras-MAPK pathways [59]. This greater complexity of regulation may obviate the need for Akirin to achieve optimal expression. On another level, the difference in Akirin regulation of these enhancers could be reflected in the local chromatin environment of these enhancers, and whether the individual enhancer needs to be remodeled by SWI/SNF activity for optimal expression. Analysis of deposited Modencode data [61] suggests that the histone modification profile of the eve MHE remains largely static during the same temporal window as our chromatin immunoprecipitation experiment, while the environment surrounding the Dmef2 enhancer element changes during this same time (data not shown). Further studies are required to identify the basis of this different requirement for Akirin at these Twist targets. Our polytene analysis indicates that a large number (>200) of Akirin-positive bands colocalize with Twist in the genome (Figure 4). Identifying the complete list of Twist target genes that require Akirin will likely yield further clues as to the regulatory logic of Akirin together with chromatin remodeling during Twist target expression.
Akirin interactions with transcription factors other than Twist
Our data establishes Akirin as a Twist-interacting protein that promotes expression from a Twist-regulated enhancer; however, the results presented in this study also indicate that Akirin does not act solely with Twist.Analysis of salivary gland polytene chromosomes demonstrated that Akirin is associated with numerous actively transcribed gene loci. Twist is not normally expressed in salivary glands, therefore this result suggests that Akirin has roles in activation of non-Twist regulated genes. Moreover, the widespread expression of Akirin throughout the entire embryo suggests that specificity of Akirin function is determined not by restriction of Akirin expression, but rather by the associated transcription factor. Indeed, potential interactions between Akirin and other transcription factors have been described: Akirin misexpression enhances phenotypes resulting from mutations in the GATA-2 homologue pannier [62]. Additionally, whole genome yeast 2-hybrid analysis [57] suggests an interaction between Akirin and Charlatan, a zinc-finger transcription factor involved in development of the peripheral nervous system [63]. Finally, recent work has identified Akirin as a promyogenic factor and target for Myostatin regulation [30], as well as NF-κB target gene expression in the Drosophila innate immunity pathway [29]. Taken together, these interactions with transcription factors other than Twist, and roles in non-Twist-dependent pathways further support our model whereby Akirin functions as a general transcription cofactor. We propose that the regulatory mechanism involving Akirin and the Brahma chromatin remodeling complex at specific enhancers is applicable to these transcriptional regulators in these other contexts. Further experimentation is required to validate this model.
Akirin is likely a novel accessory of the Brahma chromatin remodeling complex
Our studies identify Akirin as a nuclear factor that genetically interacts with the BRM complex and is required for optimal expression of the Twist-dependent Dmef2 enhancer. This association between Akirin and BRM complexes is likely the mechanism whereby Akirin is linked to gene activation. The Brahma complex (BRM) promotes gene activation by remodeling the local chromatin environment allowing components of the general transcription machinery greater accessibility to the DNA [15]. BRM complexes are tightly associated with regions of transcriptionally active chromatin, and are associated with both promoter paused (initiating) and actively elongating RNA Polymerase II complexes throughout the Drosophila genome [23]. Although a strong physical association of BRM complexes with RNA Polymerase II has not been confirmed, loss of BRM function leads to a severe impairment in transcription by RNA polymerase II [23]. Based on our genetic and ChIP data, we conclude from our results that Akirin is not a core BRM subunit, but is rather an accessory protein that is capable of interacting with BRM complexes. We base this conclusion on the observation that the distribution of Akirin and BRM subunits on polytene chromosomes do not completely overlap, and because numerous biochemical analyses of BRM complex composition to date have failed to identify Akirin as a BRM subunit [24], [64]. Further, we did not observe a specific interaction of akirin with either BAP or PBAP complex-specific subunits during myogenesis (Figure 6). In Drosophila, both BAP and PBAP complexes have both been linked to gene activation and repression, are present in the same cells, and perform unique, yet cooperative, functions during development [15], [16], [19], [24]–[28]. Indeed, while we were able to observe weak physical interactions between Akirin and the Brahma core subunit in vivo (data not shown), these interactions were not overly robust. Moreover, it is unknown if Akirin needs to be post-translationally modified or to further associate with other factors to mediate a physical interaction with the Brahma subunit. Further, while we tested interactions with the core Brahma subunit, it remains to be determined whether Akirin may be interacting instead with other core BRM subunits. Nevertheless, our data strongly suggests a likely association as an accessory of the BRM complex. As an accessory protein, Akirin would likely confer tissue, target, and even temporal specificity on BRM complex activity by connecting BRM complexes with a particular transcription factor for promotion of gene expression (see below).
Interactions among Akirin, BRM complexes, and Twist
Our data suggest that Twist target genes have different requirements for the presence of chromatin remodeling factors during gene activation and imply that the chromatin environments at these genes are varied. This also would suggest that the local chromatin environment of a particular Twist target changes over developmental time. Further experiments will be required to validate this hypothesis. As an accessory protein, Akirin optimizes Twist transcription factor activity outputs. Akirin likely accomplishes this optimization by facilitating an interaction between Twist and BRM complexes and as such, we would predict, change the local chromatin environment to one more favorable for transcription. The exact nature of the interface between bHLH transcription factors such as Twist and chromatin remodeling complexes such as BRM has not been previously determined. Our data would suggest that Akirin would be a suitable candidate for mediating this relationship between Twist and chromatin remodeling complexes. Mammalian SWI/SNF complexes are positively associated with bHLH transcription factor activity; however, the precise role of their remodeling activity during expression of bHLH target genes remains unclear [65]. Whether a similar linkage via Akirin is at play with mammalian Twist during development or in a cancer context remains to be tested. Nevertheless, in keeping with our proposed model of Akirin function, our data suggest a relationship between Twist and BRM during development: twiI1/+;brm2/+ double heterozygous embryos show muscle patterning defects similar to twi1/+;akirin2/+ double heterozygotes (Figure 6). Also, forced expression of Twist in salivary glands and subsequent analysis of colocalization on polytene chromosomes indicated that Twist and Brahma colocalized 58% of the time (Figure S11), a frequency similar to that observed between Twist and Akirin (54%, Figure 4 and data not shown).
Our finding that early (i.e., 2–4 hours) occupancy of the Moira core subunit at the Dmef2 enhancer was decreased in akirin mutants would suggest that Akirin contributes to BRM complex localization. However, our co-immunoprecipitation experiments (data not shown) would suggest that any physical interaction between these two proteins would be either highly transient, or exquisitely sensitive to the presence of interfering factors such as protein tags. Therefore, the mechanism by which Akirin would increase Moira occupancy remains unclear. The result of such a recruitment or stabilization of BRM complexes by Akirin at Twist-target loci, would presumably result in remodeling of the local environment by BRM for optimal gene expression. Further experiments, aimed at understanding the nature of the Akirin/BRM complex association are currently underway. Together, this association between Twist, Akirin and the BRM complex would provide a novel mechanism linking chromatin remodeling factors to spatiotemporal-specific gene activation by the Twist transcription factor. Our work provides another venue to investigate how changes in the chromatin environment at specific targets leads to optimal gene expression and how these local changes impact the development of specific tissues.
Materials and Methods
Drosophila genetics
Flies were reared using standard conditions. Three different alleles of akirin were used: akirinKG01343 (referred to in this work as akirin2), akirinEY08097 (akirin1) and akirinEP(3)0906 (akirin3). akirin1 and akirin2 are viable P-element insertions into the first intron of akirin, while the homozygous lethal akirin3 allele is an insertion into the first exon (Figure 1A). We further generated two additional homozygous lethal akirin alleles by excision of the akirin1 (EY08097) P-element (akirin4 and akirin5). Mobilization of the EY08097 P-element resulted in the isolation of two different mutations: akirin5, which is an insertion of approximately 1 kb of DNA into the second akirin exon due to incomplete P-element excision, and akirin4, which is a deletion of 6 kb containing the first exon and intron of akirin, as well as a portion of the upstream SH3β open reading frame. The akirinEP(3)0906 mutant chromosome possessed a second unidentified lethal mutation in addition to the EP(3)0906 insertion. This second lethal mutation was removed by recombination with a rucuca-marked chromosome [66] to produce the akirin3 line used in this study. akirin3, akirin4, and akirin5 homozygous mutant embryos die shortly after stage 16 and do not hatch into larvae (data not shown). For analysis, akirin3, akirin4, and akirin5 were balanced over TM3, Dfd-lacZ or TTG [67]. For genetic interactions, the following alleles were used: moira1, Snr101319, brm2 and brmI21 (Bloomington stock collection). osa308 and bap180Δ86 (a gift of J. Treisman), Bap601 (a gift of G. Mardon), nurf38, Iswi1, and Su(var)3-91 (a gift of V. Corces), and twi1 [39]. Dmef2-lacZ [33] and eveMHE-lacZ (a gift of A. Michelson) were used for expression studies. GAL4 expression lines [68] used were twi-GAL4;Dmef2-GAL4 [69] and Sgs3-GAL4 [56]. OreR and yw flies were used as wild-type stocks where indicated. Germline clones [37] were generated by heat shock of hs-FLP; ovoD1,FRT2A/akirin3,FRT2A larvae. Germline clone females were then mated to akirin3/TM3,Dfd-lacZ at 20–22°C to create akirin3 maternal/zygotic embryos.
Production of UAS-HA–tagged Akirin flies
The HA-tag was fused in-frame at the Akirin C-terminus using PCR techniques (Table 3). PCR products were cloned into pUASt and injected into yw embryos as described [4]. Multiple independent transformant lines were generated and evaluated for Akirin-HA expression.
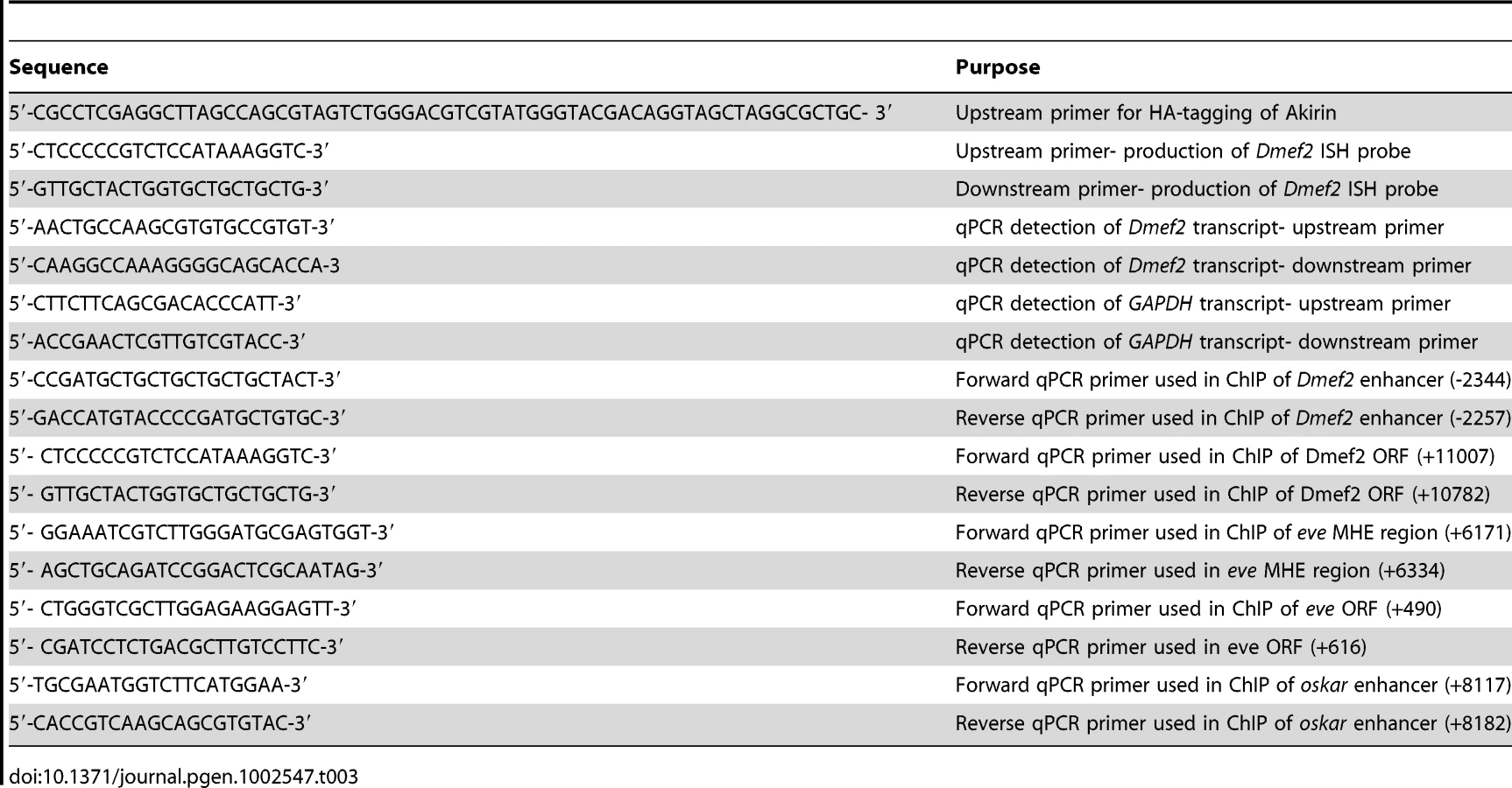
Double-interaction screen
Briefly, we adapted the method described by L. Pick and colleagues [31]. Enhancers tested were the 175-bp upstream enhancer of Dmef2 [33] and the 375-bp enhancer contained in the first intron of the tinman gene [34] cloned upstream of a HIS3 reporter. Reporter strains were created using these constructs. Screening for Twist-interacting proteins was performed by introducing a cDNA library prepared from 0–6 hour Drosophila embryos fused to the GAL4 activation domain [31]. Akirin was identified as a cDNA that expressed the HIS3 reporter in a Twist-dependent manner at both Dmef2 and tinman enhancers [32].
Immunohistochemistry and production of Akirin antibody
Antibodies and dilutions used: anti-tropomyosin (1∶1000, Abcam), anti-myosin monoclonal antibody (1∶500, a gift of S. Abmayr), anti-Twist (1∶100, a gift of M.Levine) anti-Osa (1∶50, DSHB), anti-Brm (1∶50, gift of C.P. Verrijzer), anti-HA 3F10 (1∶25, Roche), anti-Dmef2 (1∶1000, a gift of B. Patterson), anti-Snr1 (1∶100, gift of A. Dingwall), anti-Eve (1∶3000, a gift of M. Frasch), anti-phosphohistone H3 (mouse monoclonal, 1∶100, Millipore), anti-Serine-7-phosphorylated RNA polymerase II (1∶100, Millipore) and anti-β-galactosidase (1∶1000, Abcam). Whole-mount embryo immunohistochemistry was performed as described [70]. Comparison of β-galactosidase expression levels in wild-type and akirin mutant embryos was performed as per [11]. For production of polyclonal Akirin antibody, the peptide sequence of CESMIKERENQLR corresponding to residues 151–163 in full-length Drosophila Akirin was synthesized and injected into rabbits (Sigma). Production bleeds were tested for immunoreactivity using pre-immune serum as a negative control (See Figure S2).
Microscopy
Bright field and immunofluorescent images were obtained using a Zeiss Axiophot microscope. Images were processed using Adobe Photoshop. Confocal images were acquired using either a Zeiss LSM 510 confocal scanning system mounted on an Axiovert 100 M microscope equipped with a 63× 1.2 NA C-Apochromat water objective, or a Leica SP5 confocal microscope equipped with a 63× 1.4 NA HCX PL Apochromat oil objective. Pinholes were set to capture optical slices of 1.0 µm. All images were processed using Adobe Photoshop. Maximum intensity projections of confocal Z-stacks were rendered using Volocity Visualization (Improvision).
GST-pulldown assays
Twist was cloned into pGEX-2T in frame and fusion protein expressed via IPTG induction. Glutathione Sepharose 4B beads (Amersham Pharmacia Biotech) bound with GST or GST-Twist were suspended in ZTx buffer (25 mM HEPES, pH = 7.5, 12.5 mM MgCl2, 20% glycerol, 0.1% Triton-X 100, 150 mM KCl)+0.25 mg/ml BSA and 1 mM DTT and incubated for 10 mins with rotation. 35S-Methionine-labeled Akirin and Daughterless proteins were produced in vitro using the TnT coupled reticulocyte lysate system (Promega). Beads and translated proteins were then incubated at room temperature with rotation for 1 hour. Beads were spun and washed in NETTx buffer (20 mM Tris, 100 mM NaCl, 1 mM EDTA and 0.25% Triton-X 100). Proteins were eluted from beads by boiling in Laemmli sample buffer and resolved by SDS-PAGE. Gels were fixed in 50% Methanol, 10% Acetic Acid, incubated in Amplify solution (Amersham Pharmacia Biotech), and dried for autoradiography.
Western blotting
Whole-embryo extracts were prepared and resolved by SDS-PAGE as described [71]. Western detection was performed as described [72]. Antibodies and dilutions used: anti-Dmef2 (rabbit, 1∶1000) (a gift of B. Patterson), anti-alpha-tubulin (mouse, 1∶5000) (Sigma), anti-Brahma (1∶1000) (a gift of C.P.Verrijzer), anti-β-galactosidase (mouse, 1∶1000) (Promega).
Densitometry
Autoradiographs were scanned using a BioRad GS-800 Calibrated Densitometer to determine optical density of each band, which was then divided by the area examined. For analysis of β-galactosidase levels (see Figure S5), this value was normalized to similar results obtained from anti-α-tubulin loading controls. For analysis of co-immunoprecipitations (see Figure 6), this value was reported as a percentage of the input sample analyzed. For each condition, background (chosen from blank region of the film piece, with no sample present) was subtracted from all analyzed regions prior to analysis.
Polytene chromosome immunohistochemistry, in situ hybridization, and immunocolocalization analysis
Polytene squash preparations and immunostaining was performed as described [52]. For colocalization analysis, separate channel information was compared for signal bands in polytene squashes. Contrast settings were set to maximum for each channel, eliminating >80% of the background signal. A band was scored as “colocalized” when it appeared in both channels. Bands that appeared solely in either signal channel were scored accordingly. A minimum of 150 bands were counted from at least three different squash preparations. Data was plotted in percentage bar graphs using Microsoft Excel.
In situ probes were generated using the Roche DIG-labeled PCR kit, using oligonucleotides to amplify exon 8 of the Dmef2 open reading frame (Table 3). In situ probe purification and hybridization was performed as described [73]. Following hybridization, slides were washed in increasing stringency SSC solutions, and then incubated in polytene buffer. Antibody staining was carried out as described above, using 1∶100 rhodamine-conjugated anti-DIG antibodies (Roche) to detect DIG-labeled probes.
Production of cDNA and quantitative PCR
wild-type (yw) and akirin2 embryos were collected on 10 cm apple juice agar plates and allowed to grow to 4–6 hours of age after egg laying. Embryos were dechorionated in bleach and homogenized in RNA TriReagent (Sigma). Total mRNA was prepared according to manufacturer's instructions, and resuspended in RNAse-free water. Following purification, total mRNA was treated with DNAseI to degrade genomic DNA, and re-purified using phenol∶chloroform. 1 microgram of total mRNA was used to prepare first-strand cDNA using the RevertAid First Strand Synthesis Kit (Fermentas). First-strand product was diluted to produce a working concentration of 10 ng/µL. For quantitative PCR, first-strand cDNA product was diluted 1∶2 with Lightcycler 480 SYBR green master mix (Roche). qPCR was conducted using a Lightcycler 480 qPCR machine (Table 3). qPCR results were analyzed and relative ratios of Dmef2 transcripts (normalized to GAPDH levels) in yw and akirin2 mutants were determined using described methods [74].
Co-immunoprecipitations
Total nondenatured extracts were prepared from yw embryos collected on apple juice plates. To prepare total nondenatured extracts, embryos were dechorinated in 50% bleach, rinsed in distilled water, and immediately homogenized in one mL of extraction buffer (50 mM HEPES, pH 7.6, 385 mM NaCL, 0.1% Tween-20, 0.1 mM EGTA, 1.1 mM MgCl2 and 100 µg/mL PMSF, and 1 µg/mL each of Aprotinin, leupeptin, and pepstatin A), using a 1 mL Dounce homogenizer. Extracts were spun at 15,000 g at 4°C to pellet debris. Supernatants were removed and following the addition of glycerol to 10%, extracts were snap-frozen in liquid nitrogen before storing at −80°C for later use. For immunoprecipitations, extracts were thawed and quantitated using the BCA assay method (Pierce). Extracts were diluted in IP buffer (10 mM HEPES pH 8, 100 mM NaCl, 10% glycerol, 0.05% Tween-20, 100 µg/mL PMSF, and 1 µg/mL each of Aprotinin, leupeptin, and pepstatin A) to produce a final concentration of 3 mg/mL of total protein. Extracts were incubated with 10 µL of anti-Brahma antibody (a gift of P. Verrijzer) for 1 hour at 4°C. Immunocomplexes were pulled-down using 20 µL of a 50% protein A-agarose slurry. Beads were washed four times at 4°C, and immunocomplexes were released by boiling in Laemmli sample buffer.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitations were performed essentially as described [75] with several modifications. In brief, 2–4 hour, 4–6 hour, 6–10 hour yw Drosophila embryos were collected, dechorionated, and fixed in fixing solution (1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl, 2% formaldehyde (v/v), 50 mM HEPES, pH 8.0) with an equal volume of n-heptane by vigorous shaking for 25 min at room temperature. Fixed embryos were washed twice with methanol, once with storage buffer (50 mM Tris-HCl pH 8.0, 1 mM EDTA), and kept at −80°C. Approximately 100 µL of fixed embryos were washed with 1 ml of IP buffer (100 mM NaCl, 67 mM Tris pH 8.0, 0.33% SDS, 1.66% Triton X-100, 5 mM EDTA) for 10 min. The following procedures were all carried out at 4°C. Embryos were resuspended in IP buffer containing protease inhibitor cocktail, were then sonicated fourteen times for 12 sec by a Branson 250 sonicator at a power setting of 4 and 30% duty cycle. After sonication, the Whole Cell extract; (WCE) was obtained by centrifugation. 30 µL of WCE was diluted, reverse cross-linked, and treated with proteinase K. DNA was purified using QIAprep Spin Miniprep columns and recovered in 100 µL of elution buffer (Qiagen) to check genomic DNA fragments with an approximate bulk size of 300–800 bp. For each immunoprecipitation, WCE containing 75 µg chromatin DNA was treated with either 5 µL of nonimmune guinea pig serum, anti-Twist antiserum (guinea pig), anti-Akirin antiserum (rabbit), and anti-Moira antiserum (rabbit), together with 25 µL of protein A agarose coated with salmon sperm DNA (Upstate) for overnight incubation. Mock immunoprecipitations were performed using nonimmune guinea pig serum, preimmune rabbit serum, and nonimmune rabbit serum. Precipitated agarose beads were washed with mixed micelle buffer, buffer 500, LiCl/Detergent buffer, and TE, then treated with RNase A. The precipitated immune complexes were eluted by Bicarbonate/SDS buffer and cross-links were reversed in the presence of proteinase K overnight at 65°C. DNA was purified using QIAprep Spin Miniprep columns and recovered in 200 µL of elution buffer. Quantitative PCR was performed using the Applied Biosystems (ABI) Prism 7700 Real-Time qPCR instrument (AQ method). One of triplicates of reaction mixture were prepared by adding 4 ul 2× SYBR mix (usb 75762), 0.4 µL 2.5 uM Primers (Table 3), and 3.6 µL DNA template. To depict standard curves, duplicates of each 5, 50, and 500 folds diluted DNA from WCE as described above were used. %input and SD were calculated from triplicated scores of immunoprecipitations over that of input WCE.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BarnesRMFirulliAB 2009 A twist of insight - the role of Twist-family bHLH factors in development. The International Journal of Developmental Biology 53 909 924
2. YangJManiSADonaherJLRamaswamySItzyksonRA 2004 Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117 927 939
3. MiraouiHMariePJ 2010 Pivotal role of Twist in skeletal biology and pathology. Gene 468 1 7
4. BayliesMKBateM 1996 twist: a myogenic switch in Drosophila. Science 272 1481 1484
5. BernardFKrejciAHousdenBAdryanBBraySJ 2010 Specificity of Notch pathway activation: Twist controls the transcriptional output in adult muscle progenitors. Development
6. CastanonIBayliesMK 2002 A Twist in fate: evolutionary comparison of Twist structure and function. Gene 287 11 22
7. LeptinM 1991 twist and snail as positive and negative regulators during Drosophila mesoderm development. Genes & Development 5 1568 1576
8. SandmannTGirardotCBrehmeMTongprasitWStolcV 2007 A core transcriptional network for early mesoderm development in Drosophila melanogaster. Genes & Development 21 436 449
9. ZeitlingerJZinzenRPStarkAKellisMZhangH 2007 Whole-genome ChIP-chip analysis of Dorsal, Twist, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes & Development 21 385 390
10. CastanonIvon StetinaSKassJBayliesMK 2001 Dimerization partners determine the activity of the Twist bHLH protein during Drosophila mesoderm development. Development 128 3145 3159
11. WongM-CCastanonIBayliesMK 2008 Daughterless dictates Twist activity in a context-dependent manner during somatic myogenesis. Developmental Biology 317 417 429
12. González-CrespoSLevineM 1993 Interactions between dorsal and helix-loop-helix proteins initiate the differentiation of the embryonic mesoderm and neuroectoderm in Drosophila. Genes & Development 7 1703 1713
13. JiangJKosmanDIpYTLevineM 1991 The dorsal morphogen gradient regulates the mesoderm determinant twist in early Drosophila embryos. Genes & Development 5 1881 1891
14. ShirokawaJMCoureyAJ 1997 A direct contact between the dorsal rel homology domain and Twist may mediate transcriptional synergy. Molecular and Cellular Biology 17 3345 3355
15. ClapierCRCairnsBR 2009 The Biology of Chromatin Remodeling Complexes. Annual Review of Biochemistry 78 273 304
16. MohrmannLVerrijzerCP 2005 Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochimica et Biophysica Acta 1681 59 73
17. BrizuelaBJElfringLBallardJTamkunJWKennisonJA 1994 Genetic analysis of the brahma gene of Drosophila melanogaster and polytene chromosome subdivisions 72AB. Genetics 137 803 813
18. BrizuelaBJKennisonJA 1997 The Drosophila homeotic gene moira regulates expression of engrailed and HOM genes in imaginal tissues. Mechanisms of Development 65 209 220
19. CollinsRTFurukawaTTaneseNTreismanJE 1999 Osa associates with the Brahma chromatin remodeling complex and promotes the activation of some target genes. The EMBO journal 18 7029 7040
20. CrosbyMAMillerCAlonTWatsonKLVerrijzerCP 1999 The trithorax group gene moira encodes a brahma-associated putative chromatin-remodeling factor in Drosophila melanogaster. Molecular and Cellular Biology 19 1159 1170
21. ElfringLKDanielCPapoulasODeuringRSarteM 1998 Genetic analysis of brahma: the Drosophila homolog of the yeast chromatin remodeling factor SWI2/SNF2. Genetics 148 251 265
22. TreismanJELukARubinGMHeberleinU 1997 eyelid antagonizes wingless signaling during Drosophila development and has homology to the Bright family of DNA-binding proteins. Genes & Development 11 1949 1962
23. ArmstrongJAPapoulasODaubresseGSperlingASLisJT 2002 The Drosophila BRM complex facilitates global transcription by RNA polymerase II. The EMBO journal 21 5245 5254
24. ChalkleyGEMoshkinYMLangenbergKBezstarostiKBlastyakA 2008 The Transcriptional Coactivator SAYP Is a Trithorax Group Signature Subunit of the PBAP Chromatin Remodeling Complex. Molecular and Cellular Biology 28 2920 2929
25. CarreraIZavadilJTreismanJE 2008 Two subunits specific to the PBAP chromatin remodeling complex have distinct and redundant functions during drosophila development. Molecular and Cellular Biology 28 5238 5250
26. CollinsRTTreismanJE 2000 Osa-containing Brahma chromatin remodeling complexes are required for the repression of wingless target genes. Genes & Development 14 3140 3152
27. RendinaRStrangiAAvalloneBGiordanoE 2010 Bap170, a Subunit of the Drosophila PBAP Chromatin Remodeling Complex, Negatively Regulates the Egfr Signaling. Genetics
28. CarreraITreismanJE 2008 Message in a nucleus: signaling to the transcriptional machinery. Current Opinion in Genetics & Development 18 397 403
29. GotoAMatsushitaKGesellchenVel ChamyLKuttenkeulerD 2008 Akirins are highly conserved nuclear proteins required for NF-kappaB-dependent gene expression in drosophila and mice. Nature Immunology 9 97 104
30. MarshallASalernoMSThomasMDaviesTBerryC 2008 Mighty is a novel promyogenic factor in skeletal myogenesis. Experimental Cell Research 314 1013 1029
31. YuYYussaMSongJHirschJPickL 1999 A double interaction screen identifies positive and negative ftz gene regulators and ftz-interacting proteins. Mechanisms of Development 83 95 105
32. GonzalezKBayliesM 2005 bhringi: A novel Twist co-regulator. Program and Abstracts 46th Annual Drosophila Research Conference, San Diego, CA, 2005 320B
33. CrippsRMBlackBLZhaoBLienCLSchulzRA 1998 The myogenic regulatory gene Mef2 is a direct target for transcriptional activation by Twist during Drosophila myogenesis. Genes & Development 12 422 434
34. YinZXuXLFraschM 1997 Regulation of the twist target gene tinman by modular cis-regulatory elements during early mesoderm development. Development 124 4971 4982
35. MacqueenDJJohnstonIA 2009 Evolution of the multifaceted eukaryotic akirin gene family. BMC Evolutionary Biology 9 34
36. BateM 1990 The embryonic development of larval muscles in Drosophila. Development 110 791 804
37. ChouTBPerrimonN 1996 The autosomal FLP-DFS technique for generating germline mosaics in Drosophila melanogaster. Genetics 144 1673 1679
38. BulchandSMenonSDGeorgeSEChiaW 2010 Muscle wasted: a novel component of the Drosophila histone locus body required for muscle integrity. Journal of Cell Science 123 2697 2707
39. SimpsonP 1983 Maternal-Zygotic Gene Interactions during Formation of the Dorsoventral Pattern in Drosophila Embryos. Genetics 105 615 632
40. BourBAO'BrienMALockwoodWLGoldsteinESBodmerR 1995 Drosophila MEF2, a transcription factor that is essential for myogenesis. Genes & Development 9 730 741
41. GunthorpeDBeattyKETaylorMV 1999 Different levels, but not different isoforms, of the Drosophila transcription factor DMEF2 affect distinct aspects of muscle differentiation. Developmental Biology 215 130 145
42. RanganayakuluGZhaoBDokidisAMolkentinJDOlsonEN 1995 A series of mutations in the D-MEF2 transcription factor reveal multiple functions in larval and adult myogenesis in Drosophila. Developmental Biology 171 169 181
43. ElgarSJHanJTaylorMV 2008 mef2 activity levels differentially affect gene expression during Drosophila muscle development. Proceedings of the National Academy of Sciences of the United States of America 105 918 923
44. JunionGJaglaTDuplantSTapinRDa PonteJ-P 2005 Mapping Dmef2-binding regulatory modules by using a ChIP-enriched in silico targets approach. Proceedings of the National Academy of Sciences of the United States of America 102 18479 18484
45. LillyBGalewskySFirulliABSchulzRAOlsonEN 1994 D-MEF2: a MADS box transcription factor expressed in differentiating mesoderm and muscle cell lineages during Drosophila embryogenesis. Proceedings of the National Academy of Sciences of the United States of America 91 5662 5666
46. NguyenHTXuX 1998 Drosophila mef2 expression during mesoderm development is controlled by a complex array of cis-acting regulatory modules. Developmental Biology 204 550 566
47. SandmannTJensenLJJakobsenJSKarzynskiMMEichenlaubMP 2006 A temporal map of transcription factor activity: mef2 directly regulates target genes at all stages of muscle development. Developmental Cell 10 797 807
48. TaylorMVBeattyKEHunterHKBayliesMK 1995 Drosophila MEF2 is regulated by twist and is expressed in both the primordia and differentiated cells of the embryonic somatic, visceral and heart musculature. Mechanisms of Development 50 29 41
49. SalernoMSDyerKBracegirdleJPlattLThomasM 2009 Akirin1 (Mighty), a novel promyogenic factor regulates muscle regeneration and cell chemotaxis. Experimental Cell Research 315 2012 2021
50. SpradlingAPenmanSPardueML 1975 Analysis of drosophila mRNA by in situ hybridization: sequences transcribed in normal and heat shocked cultured cells. Cell 4 395 404
51. BuszczakMSpradlingAC 2006 The Drosophila P68 RNA helicase regulates transcriptional deactivation by promoting RNA release from chromatin. Genes & Development 20 977 989
52. KaramCSKellnerWATakenakaNClemmonsAWCorcesVG 2010 14-3-3 mediates histone cross-talk during transcription elongation in Drosophila. PLoS Genet 6 e1000975 doi:10.1371/journal.pgen.1000975
53. NowakSJCorcesVG 2000 Phosphorylation of histone H3 correlates with transcriptionally active loci. Genes & Development 14 3003 3013
54. ChapmanRDHeidemannMAlbertTKMailhammerRFlatleyA 2007 Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 318 1780 1782
55. ThisseCThisseB 1992 Dorsoventral development of the Drosophila embryo is controlled by a cascade of transcriptional regulators. Dev Suppl 173 181
56. ZhimulevIFBelyaevaESSemeshinVFShlomaVVMakuninIV 2003 Overexpression of the SuUR gene induces reversible modifications at pericentric, telomeric and intercalary heterochromatin of Drosophila melanogaster polytene chromosomes. Journal of Cell Science 116 169 176
57. GiotLBaderJSBrouwerCChaudhuriAKuangB 2003 A protein interaction map of Drosophila melanogaster. Science 302 1727 1736
58. MöllerAAvilaFWEricksonJWJäckleH 2005 Drosophila BAP60 is an essential component of the Brahma complex, required for gene activation and repression. Journal of Molecular Biology 352 329 337
59. HalfonMSCarmenaAGisselbrechtSSackersonCMJiménezF 2000 Ras pathway specificity is determined by the integration of multiple signal-activated and tissue-restricted transcription factors. Cell 103 63 74
60. Kim-HaJWebsterPJSmithJLMacdonaldPM 1993 Multiple RNA regulatory elements mediate distinct steps in localization of oskar mRNA. Development 119 169 178
61. CelnikerSEDillonLALGersteinMBGunsalusKCHenikoffS 2009 Unlocking the secrets of the genome. Nature 459 927 930
62. Peña-RangelMTRodriguezIRiesgo-EscovarJR 2002 A misexpression study examining dorsal thorax formation in Drosophila melanogaster. Genetics 160 1035 1050
63. EscuderoLMCamineroESchulzeKLBellenHJModolellJ 2005 Charlatan, a Zn-finger transcription factor, establishes a novel level of regulation of the proneural achaete/scute genes of Drosophila. Development 132 1211 1222
64. VorobyevaNESoshnikovaNVNikolenkoJVKuzminaJLNabirochkinaEN 2009 Transcription coactivator SAYP combines chromatin remodeler Brahma and transcription initiation factor TFIID into a single supercomplex. Proceedings of the National Academy of Sciences of the United States of America 106 11049 11054
65. OhkawaYYoshimuraSHigashiCMarfellaCGADacwagCS 2007 Myogenin and the SWI/SNF ATPase Brg1 maintain myogenic gene expression at different stages of skeletal myogenesis. The Journal of Biological Chemistry 282 6564 6570
66. MartinSGDobiKCSt JohnstonD 2001 A rapid method to map mutations in Drosophila. Genome Biology 2 RESEARCH0036
67. HalfonMSGisselbrechtSLuJEstradaBKeshishianH 2002 New fluorescent protein reporters for use with the Drosophila Gal4 expression system and for vital detection of balancer chromosomes. Genesis (New York, NY : 2000) 34 135 138
68. BrandAHPerrimonN 1993 Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118 401 415
69. CoxVTBayliesMK 2005 Specification of individual Slouch muscle progenitors in Drosophila requires sequential Wingless signaling. Development 132 713 724
70. RichardsonBEBeckettKNowakSJBayliesMK 2007 SCAR/WAVE and Arp2/3 are crucial for cytoskeletal remodeling at the site of myoblast fusion. Development 134 4357 4367
71. BeckettKBayliesMK 2006 Parcas, a regulator of non-receptor tyrosine kinase signaling, acts during anterior-posterior patterning and somatic muscle development in Drosophila melanogaster. Developmental Biology 299 176 192
72. NowakSJNahirneyPCHadjantonakisA-KBayliesMK 2009 Nap1-mediated actin remodeling is essential for mammalian myoblast fusion. Journal of Cell Science 122 3282 3293
73. NowakSJPaiC-YCorcesVG 2003 Protein phosphatase 2A activity affects histone H3 phosphorylation and transcription in Drosophila melanogaster. Molecular and Cellular Biology 23 6129 6138
74. PfafflMW 2001 A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29 e45
75. LiljaTAiharaHStabellMNibuYMannervikM 2007 The acetyltransferase activity of Drosophila CBP is dispensable for regulation of the Dpp pathway in the early embryo. Developmental Biology 305 650 658
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- PIF4–Mediated Activation of Expression Integrates Temperature into the Auxin Pathway in Regulating Hypocotyl Growth
- Metabolic Profiling of a Mapping Population Exposes New Insights in the Regulation of Seed Metabolism and Seed, Fruit, and Plant Relations
- A Splice Site Variant in the Bovine Gene Compromises Growth and Regulation of the Inflammatory Response
- Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson's Disease Genetics: The PDGene Database
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy