
Antagonistic Regulation of Apoptosis and Differentiation by the Cut Transcription Factor Represents a Tumor-Suppressing Mechanism in
Apoptosis is essential to prevent oncogenic transformation by triggering self-destruction of harmful cells, including those unable to differentiate. However, the mechanisms linking impaired cell differentiation and apoptosis during development and disease are not well understood. Here we report that the Drosophila transcription factor Cut coordinately controls differentiation and repression of apoptosis via direct regulation of the pro-apoptotic gene reaper. We also demonstrate that this regulatory circuit acts in diverse cell lineages to remove uncommitted precursor cells in status nascendi and thereby interferes with their potential to develop into cancer cells. Consistent with the role of Cut homologues in controlling cell death in vertebrates, we find repression of apoptosis regulators by Cux1 in human cancer cells. Finally, we present evidence that suggests that other lineage-restricted specification factors employ a similar mechanism to put the brakes on the oncogenic process.
Published in the journal:
. PLoS Genet 8(3): e32767. doi:10.1371/journal.pgen.1002582
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002582
Summary
Apoptosis is essential to prevent oncogenic transformation by triggering self-destruction of harmful cells, including those unable to differentiate. However, the mechanisms linking impaired cell differentiation and apoptosis during development and disease are not well understood. Here we report that the Drosophila transcription factor Cut coordinately controls differentiation and repression of apoptosis via direct regulation of the pro-apoptotic gene reaper. We also demonstrate that this regulatory circuit acts in diverse cell lineages to remove uncommitted precursor cells in status nascendi and thereby interferes with their potential to develop into cancer cells. Consistent with the role of Cut homologues in controlling cell death in vertebrates, we find repression of apoptosis regulators by Cux1 in human cancer cells. Finally, we present evidence that suggests that other lineage-restricted specification factors employ a similar mechanism to put the brakes on the oncogenic process.
Introduction
It has been a long-standing paradigm that impaired cell fate commitment is a key initiator of cancer development [1], [2], since cancer cells display reduced differentiation properties compared to normal cells, while tumor formation can be suppressed by inducing the terminal cell fate in cancer cells [3]. The molecular basis of the interplay between cell differentiation and cancer has only recently been established. Bossuyt and colleagues (2009) demonstrated that loss of the proneural transcription factor Atonal not only leads to a loss of differentiated eye tissue but also promotes tumor formation and progression in this tissue context [4]. Thus, their work provided evidence that the maintenance of a differentiated state, which is critically controlled by a cell-type specification factor, is one crucial aspect to prevent the oncogenic process, whereas loss of this master regulator, together with other mutations creating a sensitized background, leads to the initiation of tumorigenesis. In order to evade tumor development, organisms have evolved potent mechanisms to protect themselves from the effects of mutations in their soma [5]. Programmed cell death, or apoptosis, plays a crucial role in removing abnormal cells, which could develop into tumors. This is supported by the observation that most types of cancers are associated with genetic alterations that deactivate this rescue pathway, most commonly via up-regulation of anti-apoptotic genes [6].
Since loss of terminal differentiation and the inability to activate apoptosis are crucial steps in cancer development, the existence of regulatory mechanisms preventing the accumulation of cells harboring mutations in both pathways seems essential for the survival of multi-cellular organisms. Consistently, mutations in differentiation genes very often result in the activation of the programmed cell death machinery [7], [8]. However, the mechanisms linking loss of differentiation and induction of apoptosis, which is crucial for the prevention of tumor formation, are still missing. Here we have used the Drosophila posterior spiracle (PS) as a model to analyze the interplay of differentiation and apoptosis at the mechanistic level. By studying the morphogenesis of this organ, we have identified a hard-wired program through which the cell-type specifying transcription factor Cut (Ct) controls in a subset of PS cells, the filzkörper cells, initiation of differentiation and simultaneous repression of apoptosis via the direct transcriptional regulation of the pro-apoptotic gene rpr. Using two well-established Drosophila in vivo eye cancer models, we demonstrate that this regulatory circuit instructed by the transcription factor Ct is a very potent mechanism to prevent and/or reduce tumor growth, as it allows the lineage-specific removal of abnormal cells at the time of their genesis. Moreover, our data show that a related regulatory wiring is used in vertebrates and that other cell-type specification factors might employ a similar mechanism for tumor suppression, thus suggesting that the coupling of differentiation and apoptosis by individual transcription factors is a widely used and evolutionary conserved cancer prevention module, which is hard-wired into the developmental program.
Results
Cut inhibits rpr expression and induction of apoptosis in the PS
The PS connects the Drosophila respiratory system to the environment and consists of an internal tube, the spiracular chamber with a refractile filter, the filzkörper, which is specified by the transcription factor Ct, and an external protrusion in which the spiracular chamber is located, the stigmatophore, which is under the control of the transcription factor Spalt (Sal) (Figure 1A; Figure S1A–S1D) [9]. In 1st instar ct mutant larvae filzkörper cells are not detectable (Figure 2A, 2D; Figure S5A, S5B), which a priori suggests that Ct is primarily required for the specification of the filzkörper cell fate. However, due to the fact that Ct has also been shown to regulate programmed cell death [7], we assumed that filzkörper cells in ct mutant embryos could be completely missing due to the induction of apoptosis. To test this hypothesis, we analyzed the expression of all Drosophila pro-apoptotic genes, which revealed the specific repression of reaper (rpr) (Figure 1B, 1C; Figure S1E, S1F; Figure S2A, S2B) but not of head involution defective (hid), grim and sickle (skl) (Figure S1I–S1N) transcription by Ct in embryonic filzkörper precursor cells. Strikingly, we only observed rpr de-repression in Ct-positive filzkörper, but never in Ct-neighboring, Sal-positive stigmatophore precursor cells (Figure 1B, 1C; Figure S1E, S1F), evidencing the cell-autonomous regulation of rpr by Ct. Rpr binds to Inhibitor of Apoptosis Protein (IAP), thereby releasing inhibition of caspases and promoting apoptosis [10]. Consistently we could demonstrate enhanced cell death in ct deficient filzkörper precursor cells of stage 11 embryos using the genetically-encoded caspase reporter Apoliner [11] as well as TUNEL and Acridine Orange (AO) stainings (Figure 1D, 1E, 1F, 1G; Figure S2G, S2H). Thus, rpr de-repression is followed by apoptosis induction in ct mutant embryos. To study the interplay between cell-type specification and cell death at the mechanistic level, we identified conserved Ct-dependent regulatory regions in the rpr intergenic regions using computational methods. Due to the principal requirement of the Hox transcription factor Abdominal-B (Abd-B) for PS development [9], we searched for clusters of binding sites for Abd-B and Ct and found a highly conserved 571 bp DNA element close to the rpr coding region, which we termed rpr-HRE-571 (Figure S4). Sequence-specific interaction of recombinant Ct protein with part of the enhancer module, the S2 sub-fragment, was detected by electrophoretic mobility shift assays (Figure 1K; Figure S1G). Immunostainings revealed rpr-HRE-571-GFP activity solely in stigmatophore (Figure 1I, 1L; Figure S3A, S3E, S3I, S3M) but not in Ct-positive filzkörper precursor cells (Figure 1L; Figure S3A, S3E, S3I, S3M), that do not express rpr (Figure 1B, 1H; Figure S1E). To validate the in vivo interaction of Ct with the identified enhancer module, we interfered with Ct-enhancer interaction in two ways: we mutated Ct binding sites within the rpr-HRE-571-S2 fragment (Figure 1J), a truncated module with identical activity as the rpr-HRE-571 enhancer (Figure 1M; Figure S3B, S3F, S3J, S3N), and eliminated all three sites in a small deletion version of the rpr-HRE-571 enhancer, termed rpr-HRE-571-S1 (Figure 1J). In both cases, GFP expression was ectopically activated in Ct-positive filzkörper precursor cells (Figure 1N, 1O; Figure S3C, S3D, S3G, S3H, S3K, S3L, S3O, S3P). These experiments demonstrated that Ct directly represses rpr transcription and thus apoptosis in the filzkörper precursor cells of the PS in a cell-autonomous manner by interacting with a small enhancer module located in the rpr intergenic region.


Repression of apoptosis by Ct is required for differentiation of filzkörper cells
Since our result showed that filzkörper cells are very efficiently eliminated by apoptosis in the absence of Ct function, we next asked whether Ct primarily acts as a repressor of programmed cell death or whether this factor is also required for the differentiation of filzkörper cells. To this end, we analyzed ct deficient cells, which were kept alive by expressing the caspase inhibitor p35 [12] in ct mutant embryos using the PS-specific driver ems-GAL4 [13]. In order to follow the cells normally under the control of Ct, these cells were GFP-labeled using the same driver, which is active only in a subset of Ct-expressing cells (Figure 2G, 2K). Our experiments revealed that Ct - and GFP-positive filzkörper cells found in the wild-type situation (Figure 2A, 2F, 2F′, 2B, 2G, 2G′, 2K; Figure S5A, S5D, S5G) are eliminated in ct mutant embryos (Figure 2D, 2I, 2I′, 2L; Figure S5B, S5E, S5H), whereas they remained viable when apoptosis is blocked (ct−; ems::p35) (Figure 2E, 2J, 2J). However, these ct deficient, undead cells had developmental defects, as they did not properly invaginate and did not acquire their terminal cell fate as indicated by reduced expression of the apical cell polarity marker Crumbs (Crb) and the cell adhesion molecule DE-Cadherin (Figure 2F′, 2J′; Figure S5D, S5F, S5G, S5I). Consistently, these cells never adopted a filzkörper cell fate (Figure 2E; Figure S5C). These defects were a consequence of blocking cell death in ct deficient, undifferentiated cells and were not due to a general response to the apoptosis inhibitor p35, as the filzkörper of ems::p35 control embryos (Figure 2B, 2G, 2G′) was indistinguishable form those of wild-type embryos (Figure 2A, 2F, 2F′). Local activation of apoptosis was sufficient to induce cell death in filzkörper cells, as expression of a rpr transgene resulted in their elimination (Figure 2C, 2H, 2H′). Taken together, our results revealed that Ct carries out two functions during PS morphogenesis: it allows the survival of uncommitted precursor cells by the transcriptional repression of the pro-apoptotic gene rpr and subsequently it drives these cells into a filzkörper-specific cell fate.
Simultaneous regulation of apoptosis and cell fate commitment is a general function of Cut
Ct is expressed in many different cell and tissue types [14], thus we tested the Ct switch function in diverse developmental contexts. Ct activity was eliminated in the Drosophila eye using RNAi (Figure 3C, 3G), resulting in an overall reduction of the eye size (Figure 4A, 4B), and a loss of interommatidial bristles (Figure 3A, 3E), which normally express Ct (Figure 3C). Consistently, expression of the bristle shaft progenitor marker DE-Cadherin [15] was lost in GMR::ctRNAi pupal retinas (Figure 3B, 3F). Expression of the apoptotic executor activated Caspase-3 was significantly increased in eye discs of ey::ctRNAi 3rd instar larvae (Figure 3D, 3H), and a significant induction of rpr RNA levels was observed using quantitative Real Time-PCR (qRT-PCR) (Figure 3K). Co-expression of either the apoptosis inhibitor p35, which rescues the eye size (Figure 4C), or of a rprRNAi construct along with the ctRNAi transgene resulted in a survival of ct deficient cells, as evidenced by the expression of the bristle shaft progenitor maker DE-Cadherin (Figure 3J). However, reminiscent to the phenotypes in the PS (Figure 2E, 2J′), these cells were unable to adopt their terminal fate due to the absence of the cell-specification factor Ct, and consequently fully differentiated interommatidial bristles were absent (Figure 3I). Similar results were obtained in other cell types specified by Ct (Figure S6), suggesting that the Ct-dependent switch between cell-type specification and programmed cell death is of general relevance.

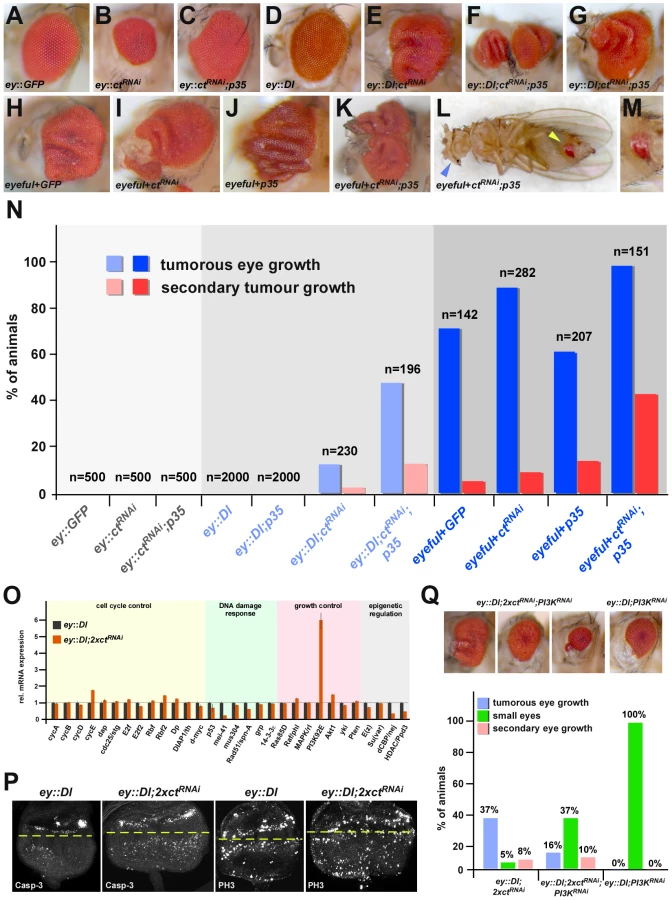
Simultaneous and antagonistic regulation of differentiation and apoptosis represents a cancer prevention mechanism
By analyzing the Ct-rpr interaction in two well-established in vivo Drosophila cancer models, we asked whether the combined transcriptional regulation of differentiation and apoptosis repression by Ct could represent a cancer prevention mechanism. In the oncogenic “eyeful” model [16], eye tumors occurred in 72.5% of control flies, with 4.9% of them showing macroscopically visible secondary tumor growths derived from the developing retina (Figure 4H, 4N) due to the eye-specific over-expression of the Notch ligand Delta (Dl) and the two epigenetic regulators longitudinals lacking (lola) and pipsqueak (psq) [16]. In contrast, pre-oncogenic ey::Dl flies over-expressing Dl exclusively in eye tissue [16] never displayed any eye tumors or invasive tumors but only mildly overgrown eyes (Figure 4D, 4N). Eye-specific inhibition of Ct activity alone only caused a small increase in primary and secondary tumor incidences in both sensitized backgrounds (Figure 4E, 4I, 4N), however, these numbers were dramatically increased when Ct function and the ability to activate apoptosis were simultaneously inhibited (Figure 4F, 4G, 4K, 4L, 4M, 4N). Consistently, increased numbers of apoptotic cells were found in tumorous tissue with reduced Ct levels (ey::Dl;2xctRNAi) (Figure 4P, 4Q), demonstrating that the coupled regulation of differentiation and apoptosis by a single transcription factor is an important mechanism to suppress cancer.
However, despite increased apoptosis activation in ey::Dl;2xctRNAi eye imaginal discs (Figure 4P), which should result in a reduction of tumor growth, tumor formation in these animals was increased (Figure 4N, 4Q). Using the proliferation marker Phosphorylated histone H3 (PH3), we could demonstrate that the tumor growth induced by differentiation loss is due to excessive cell proliferation (Figure 4P), which is in line with previous results [4]. What is the molecular basis for this phenotype? RT-PCR analysis of candidate genes involved in cell cycle and growth control using ey::Dl and ey::Dl;ctRNAi eye imaginal discs revealed a strong induction of phosphoinositide 3-kinase (PI3K) upon Ct depletion (Figure 4O). It has been shown before that PI3K overexpression in the ey::Dl pre-oncogenic background leads to tumor formation [17] and that PI3K is a limiting factor for RasV12 DlgRNAi induced tumor growth [18]. Thus we tested its contribution to tumor formation in Ct-induced oncogenic eyes by reducing its level in ey::Dl;2xctRNAi animals. Interestingly, we not only found a rescue of the tumorous eye growth, but also a dramatic increase in the occurrence of smaller eyes in ey::Dl;2xctRNAi;PI3KRNAi animals (Figure 4Q), which is similar to the apoptosis-induced “small eye” phenotype observed upon Ct depletion in the wild-type background (Figure 4B). Taken together, these results show that the Ct-dependent tumor growth is in part mediated by the up-regulation of the PI3K signaling pathway and that this pro-tumorigenic effect counteracts the anti-tumorigenic apoptosis effect of Ct.
Cell adhesive properties are critical for migratory behavior of tumor cells
We found cell clusters expressing the eye differentiation marker ELAV at abnormal, ectopic positions in undifferentiated tissue of 3rd instar eye-antennal discs (Figure S7), and it had been shown before that changes in the adhesive properties of cells are critical in inducing migratory behavior [19], [20]. Consistently, transcriptome profiling experiments revealed a reduction in the expression of cell adhesion genes in eye-imaginal discs of Ct depleted animals exhibiting primary and secondary tumor formation (ey::Dl;2xctRNAi) in comparison to control animals (ey::Dl) (Figure 5A). To test the significance of this finding, we interfered with the function of α-PS4 integrin, one of the genes identified as Ct responsive (Figure 5A), by reducing its expression and the expression of its heterodimeric interaction partner β-PS integrin (mys) [21] in the ey::Dl pre-oncogenic background. We observed an increase in primary and secondary tumor formation in both situations, while reducing the activity of a related but Ct-independent integrin, the α-PS2 integrin (if), did not have any effect (Figure 5B). Since decreasing the activity of another Ct responsive cell adhesion gene, namely Tissue inhibitor of metalloproteases (Timp), also induced an increase in secondary tumors (Figure 5B), we asked if restoration of cell adhesion would be able to rescue this phenotype in the Ct loss-of-function setting. To this end, we expressed one of the major adhesion genes regulated by Ct, DE-Cad (Figure 5C), in eye cells of eyeful+ctRNAi;p35 animals, which display high rates of invasive tumors (Figure 4N, Figure 5D), and observed a reduction of secondary tumor growth rate by more than 50% (Figure 5D). These results demonstrate that regulation of cell adhesiveness is one of the essential Ct-dependent mechanisms to suppress tumor spread. In vertebrates, invasive tumor growth requires the detachment of abnormal cells from tumor tissue and their circulation in the bloodstream [22]. To test if secondary tumor formation mediated by loss of Ct function is dependent on a similar mechanism, we analyzed the hemolymph, the insect “blood”, in our fly lines. Strikingly, we detected a significant increase in GFP-labeled eye-imaginal disc cells in the hemolymph of animals forming invasive tumors (eyeful+GFP;ctRNAi;p35) in comparison to control animals (ey::GFP) (Figure 5E, 5F), suggesting that tumor cells in flies indeed circulate through the bloodstream and invade ectopic locations. In sum, these results demonstrate that transcriptional coupling of differentiation and apoptosis is a cell-intrinsic mechanism to ensure normal development and to prevent tumor initiation, progression and invasion, which is at least in part achieved by fine-tuning the adhesive properties of cells required for tissue integrity.

Antagonistic coupling of cell fate commitment and apoptosis is a general and evolutionary conserved cancer prevention mechanism
We next explored whether the effective regulation of programmed cell death by Ct has been conserved during evolution. The vertebrate homologue of Cut, Cux1, has a well-documented function in cell differentiation during normal development as well as in tumor initiation and progression in specific cancer types [23]. In addition, several studies show that Cux1 represses apoptosis during normal vertebrate development [24], [25], [26], and just recently it has been demonstrated that Cux1 knock-down leads to activated apoptosis and to reduced growth of xenograft tumors in vivo [25], [27]. To further investigate the mechanistic basis of Cux1 function in mediating apoptosis repression in vertebrates, we suppressed Cux1 in Panc1 pancreatic cancer cells (Figure 6A) and determined the transcriptional response of human apoptosis genes. Strikingly, mRNA levels of the pro-apoptotic gene puma were consistently elevated, whereas the anti-apoptotic gene Bcl-2 was down-regulated upon Cux1 depletion (Figure 6A). Since BH3-only proteins, like Puma, and Bcl-2 are important for the release of the vertebrate functional equivalent of Rpr, Smac/DIABLO, from mitochondria [28] and since Cux1 binding sites are present close to the puma coding region (Table S1), these results suggest that the regulatory wiring of differentiation and apoptosis, at the level of Cut, is functionally conserved in vertebrates.

Does this regulatory layout represent a general mechanism employed by other differentiation factors? This would require a whole suite of cell-type specifying transcription factors to repress cell death genes by interacting with distinct enhancer modules located in their regulatory regions. In addition, these modules should follow a similar functional logic to the rpr-HRE-571 enhancer, in that cell-type specific gene activation is counteracted by strong repressing inputs from linked cis-elements (Figure 1L–1O). In line with this, we found that a different conserved enhancer module on the Drosophila rpr regulatory region (rpr-HRE-707) drove expression in CNS midline cells of stage 14 embryos (Figure 6F), which never express rpr at this and subsequent developmental stages (Figure 6L) [29]. However, extending the enhancer to include additional cis-elements (rpr-HRE-707+156) (Figure 6D) resulted in loss of enhancer activity (Figure 6H). Using the JASPAR database [30], we found consensus binding sequences for POU-domain containing transcription factors on the extended enhancer module, and one of these factors, Ventral veins lacking (Vvl), is known to function in midline glial cells and to repress apoptosis [31], [32]. Our analysis revealed a partial overlap of Vvl and reporter gene expression in rpr-HRE-707 embryos (Figure 6J), and consistently ectopic rpr transcripts were detected in several midline cells of vvl mutants (Figure 6L, 6M). Due to the existence of GFP-positive cells not expressing Vvl (Figure 6J), we assume that not only Vvl but also other POU transcription factors interact with the rpr-HRE-707+156 enhancer to repress rpr transcription in midline cells. Revisiting the rpr-HRE-571 enhancer module revealed that extension of the enhancer also led to a complete loss of reporter activity (compare Figure 1L, Figure 6E, 6G). Thus, complete repression of rpr transcription in the PS requires two inhibitory inputs: one active in filzkörper cells, which we had identified to be mediated by Ct, and one so-far unknown repressor functional in stigmatophore cells. Importantly, the functional analogy of Vvl and Ct also extended to the tumor suppression activity, since, like in the case of Ct (Figure 4N), primary and secondary tumor frequencies were increased when the ability to activate apoptosis and Vvl function was impaired at the same time (Figure 6K). Furthermore, we identified two unrelated cell-type specifying transcription factors in addition to Ct and Vvl, which showed similar behavior with regards to tumor suppression (Figure S8). Together with the fact that the regulatory sequences flanking the Drosophila rpr coding region show significantly less sequence divergence than expected and a high occurrence of conserved transcription factor binding motifs (Figure 6B, 6C), these findings lead us to propose that coupling of differentiation and cell death repression via a single transcription factor represents a general cancer prevention mechanism (Figure 7), which could be employed by a large number of developmental regulators in diverse organisms.

Discussion
Programmed cell death is an integral aspect of animal development [33]. Genetic studies in C. elegans, Drosophila and mouse have shown that apoptosis is used to sculpt tissues and to remove excessive and unwanted cells, thus defining the morphology required for diverse physiological functions [34]. In this context, apoptosis is usually regulated by cell signaling pathways [33], [35], [36]. In addition to its role in tissue morphogenesis, apoptosis is also required to eliminate potentially deleterious cells, which in most cases involves complex multi-step control mechanisms [33], [37]. One such situation generating harmful cells is the inability to differentiate or adopt the appropriate cell fate, which very often results in uncontrolled cell proliferation and cancer development, and thus requires the immediate killing of these cells. However, even though it is established that apoptosis is a protective mechanism against tumorigenesis in cases of aberrant cell differentiation [1], [34], [38], the interplay of the two processes at the mechanistic level has remained unclear. In our study, we show that the simultaneous and antagonistic regulation of differentiation and apoptosis is a hard-wired developmental program and carried out by individual transcription factors, such as Cut. Our results demonstrate that impairment of differentiation in the cell lineage specified by Cut instantaneously triggers locally restricted apoptosis by releasing transcriptional repression of the pro-apoptotic gene rpr in these cells. Due to its immediate effect, the coupling of differentiation and apoptosis on the transcriptional level represents one of the fastest and most direct mechanisms to eliminate abnormal cells in status nascendi and thereby immediately interferes with their potential to develop into harmful cells.
Interestingly, apoptosis induction as a consequence of aberrant cell-type specification is not only mediated by the cell death promoting gene rpr but also by hid [8]. However, despite the same trigger, which is the inability to properly differentiate, the transcriptional basis for inducing the expression of one of these two apoptosis genes seems to be quite different: in Drosophila early developmental mutants only the expression of the pro-apoptotic gene hid is up-regulated [8], whereas our study shows that exclusively the transcription of rpr is induced when a factor specifying a distinct cell type is lost. Although it is currently unknown how hid expression is regulated at the transcriptional level, this raises the possibility that the apoptosis gene hid acts a safeguard when broad positional information at the onset of embryogenesis is absent, whereas rpr might take over this function later in development when individual and specific cell types are defined by transcription factors restricting cell fate choices.
Given the well-known role of the vertebrate homologue of Cut, Cux1, in tumor initiation and progression in specific cancer types [23], we addressed whether the switch function of the cell specification factor Cut is also relevant in a pathological context. We found that simultaneous inhibition of Cut function and apoptosis within a sensitized background increases tumor formation and metastasis to secondary sites in the animal. In contrast, down-regulation of Cut and inhibition of apoptosis in a normal developmental context, such as in the Drosophila PS or the developing eye, only results in the survival of the Cut deprived cells, but not in tumor development. These results demonstrate that cells, which are unable to undergo the cell lineage-specific differentiation program, have to be eliminated, since they have the potential to develop into cancerous cells when other genetic or micro-environmental changes accumulate [19], [27], [39]. But why do differentiation-deprived cells form tumors in a cancer-prone tissue environment despite the ability to activate the apoptotic rescue pathway? This is due to the fact that the transcription factor Cut, as part of its selector gene function, coordinately regulates multiple cellular processes, including differentiation, apoptosis, cell adhesion, but also proliferation, which are all required for proper cell fate specification and the maintenance of a differentiated state (thereby preventing tumor formation). If, however, Cut activity is abolished, all its downstream functions are affected, leading not only to the activation of apoptosis, but also to reduced differentiation and adhesion properties and the activation of cell proliferation, which is, in the case of Cut, mediated (at least in part) by the PI3K signaling pathway. Thus, loss of Cut function stimulates tumor growth in a sensitized background, since the pro-tumorigenic effects of deregulated proliferation and cell adhesiveness out-compete the anti-tumorigenic apoptosis effects at work. However, when the anti-tumorigenic effect is eliminated in the differentiation-compromised cancer tissue, tumorigenesis is strongly enhanced, which resembles a prevalent situation in aggressive human cancers characterized by the loss of differentiation, the resistance to apoptosis activation and the mis-regulation of adhesion properties [1], [40], [41].
Several lines of evidence suggest that the dual role of Cut in differentiation and apoptosis for cancer prevention is conserved in evolution. First of all, the two vertebrate homologues of Cut, Cux1 and Cux2, code for homeobox-containing transcription factors, which are crucially involved in cell-type specific terminal differentiation [14], [23], [42]. Both, Cux1 and Cux2, have similar binding specificities to Drosophila Cut [43], they also operate as transcriptional repressors and activators of genes in multi-lineage differentiation pathways [26] and, like Drosophila Cut, they act as downstream effectors of the Notch signaling pathway [44], [45]. In addition to their well-established role in development and differentiation, there are also several examples linking the vertebrate Cut homologue Cux1 to apoptosis and cancer. First, inhibition or partial disruption of Cux1 function in mice leads to increased apoptosis rates in vivo [24], [26]. Second, Cux1 regulates normal hematopoiesis, in part by modulating the levels of survival and apoptosis factors [26]. Third, Cux1 plays a prominent role in cancer progression [23]. And fourth, induced down-regulation of Cux1 in subcutaneous xenograft tumors leads to activation of apoptosis and to reduced tumor growth [25]. Our results now show that the Cut-Rpr regulatory wiring of apoptosis and differentiation is conserved in vertebrates. In mammalian cells, the Rpr functional homologue, Smac/DIABLO, which is normally compartmentalized within mitochondria, has to be released to execute its pro-apoptotic function by binding to and inactivating Inhibitors of Apoptosis (IAPs) [46]. This process requires the permeabilization of the outer mitochondrial membrane (MOMP), which is achieved by the interaction of pro-apoptotic proteins like Puma with anti-apoptotic proteins like Bcl-2, which normally inhibit MOMP [47]. We now show that down-regulation of Cux1 in pancreatic cancer cell lines leads specifically to the transcriptional induction of the pro-apoptotic gene puma and the down-regulation of the anti-apoptotic gene Bcl-2. Thus, two crucial regulators for Smac/DIABLO release are controlled by Cux1 on the transcriptional level, showing that the basic design principle of the Cut-Rpr regulatory wiring is conserved but has been adapted to the system requirements in evolution. In future, it will be intriguing to study this mechanism in diverse cellular backgrounds, including stem cells, which neither die nor differentiate.
Materials and Methods
Bioinformatics
To identify Abd-B binding sites we used the method of Wasserman and Sandelin (2004) [48] with the Abd-B Position Frequency Matrix [49] (http://jaspar.genereg.net/) and 90% cut-off. Abd-B binding site clusters were identified if at least three Abd-B sites were present in a 400 bp window. Conserved enhancers were identified using PhastCon score [50]. Within conserved regions, Ct binding sites were identified using published sequence data [49]. Vvl and Cux1 binding sites were identified using the Vvl and Cux1 Position Frequency Matrices available at the JASPAR database [49] (http://jaspar.genereg.net/).
Genetics
Drosophila melanogaster strain Oregon R was used as wild type. Amorphic allele ctdb7/FM7 [51], ems-Gal4 and ems-Gal4, UAS-GFP/TM6B [13] were obtained from J. Castelli-Gair Hombria, UAS-Dcr2; ey-Gal4 from B. Dickson, eq-Gal4/TM6B [52] from H. Pi, UAS-Apoliner5 [11] from J. P. Vincent, UAS-ctEHK2/CyO [53], UAS-ctRNAi; UAS-ctRNAi (Grueber and Jan, unpublished) from Y.N. Jan and ey-Gal4, UAS-Dl/CyO and eyeful flies (ey-Gal4, GS88A8, UAS-Dl/CyO) from M. Domiguez [16]. UAS-Dcr2; C96-Gal4 (BL-25757), UAS-CD8::GFP (BL-5130) from Bloomington stock center. GMR-Gal4, UAS-p35, UAS-Abd-B, arm-Gal4, UAS-rpr, UAS-lacZ were described elsewhere [54], [55], [56], [57]. Other UAS-RNAi lines were obtained either from BDSC, VDRC or TRiP: DE-Cad (v8024), rpr (v12045), vvl (JF02126), gro (v6316), H (v24466), αPS2 (if) (BL27544), αPS4 (v109783), βPS (mys) (HMS00043), PI3K (v107390) and Timp (v109427). Five independent transgenic lines were analyzed for each reporter construct.
Mammalian cell culture and lentivirus-mediated Cux1 knock-down
Panc-1 human pancreatic cancer cells (Department of Surgery, Medical Faculty, University of Heidelberg) and 293 T cells were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, non-essential amino acids, 100 U/ml penicillin, 100 U/ml streptomycin, and 0.25 µg/ml amphotericin B. shRNA directed against human Cux1 was generated using the following complementary oligonucleotides (forward and reverse):
Cux1_KDa:
5′CCGGAAGAAGAACACTCCAGAGGATCTCGAGATCCTCTGGAGTGTTCTTCTTTTTTTG3′ and
5′AATTCAAAAAAAGAAGAACACTCCAGAGGATCTCGAGATCCTCTGGAGTGTTCTTCTT3′;
Cux1_KDb:
5′CCGGAAGAATCTTCTCGTTTGAAACCTCGAGGTTTCAAACGAGAAGATTCTTTTTTTG3′ and
5′AATTCAAAAAAAGAATCTTCTCGTTTGAAACCTCGAGGTTTCAAACGAGAAGATTCTT3′;
shRNA control (C),
5′CCGGAATTGCCAGCTGGTTCCATCACTCGAGTGATGGAACCAGCTGGCAATTTTTTTG3′ and
5′CCGGAATTGCCAGCTGGTTCCATCACTCGAGTGATGGAACCAGCTGGCAATTTTTTTG3′.
pLKO lentiviral vectors containing shRNA were transfected into 293 T cells together with psPAX2 (packaging vector) and pMD2.G (VSV-G envelope protein expression vector) using the calcium-phosphate transfection kit (Sigma). Panc-1 cells were infected using lentivirus-containing 293 T cell supernatant and Cux1 protein levels were assessed by Western blotting using anti-Cux1 (Sigma-Aldrich) and anti-β-Actin (GeneTex) antibodies.
Plasmid constructs
All enhancer fragments were PCR amplified from genomic DNA and cloned in pHPelican-GFP [58] or pHPelican-GFP_DEST [59]. For binding site mutations, a two-step overlap PCR was performed. For mutating Ct binding sites within the rpr-HRE-571-S2 enhancer fragment, mutation introduced into the Cut consensus sequences were identical to the ones introduced into the EMSA probes (see below). Primer sequences are available upon request.
Real-time PCR
Real Time PCR was performed following standard protocols using SYBR green. Expression was normalized to GAPDH for mammalian cells and to endogenous actin5C mRNA for imaginal disc analysis. Relative expression levels are based on three biological replicates.
Histology and scanning electron microscopy
Drosophila embryos were fixed as described [56]. Eye discs or wing discs were dissected in PBS and fixed in 4% paraformaldehyde/PBS for 10 min for immunostaining. In situ hybridization and immunochemistry were performed as described [56]. Fluorescent mRNA/protein double labeling and fluorescent duplex in situ hybridizations were done as described previously [60]. Primary Antibodies used were: mouse anti-Ct 2B10 (1∶200, DSHB), mouse anti-Crb cq4 (1∶200, DSHB), rat anti-DE-Cad DCAD2 (1∶100, DSHB), mouse anti-ELAV (1∶200, DSHB), rat anti-Sal (1∶800 kind gift from R. Barrio), rabbit anti-PH3 (1∶200, Santa Cruz), rabbit anti-Cleaved Caspase-3 (1∶50 Cell Signalling), mouse anti-GFP (1∶1000, Roche), rabbit anti-GFP (1∶2000, Sigma), anti-DIG POD (1∶200, Roche), Streptavidin HRP (1∶200, PerkinElmer). Scanning electron microscopy, Acridine Orange staining and cuticle preparation were carried out as described in Lohmann et al. (2002) [55] and Zhai et al. (2010) [61]. TUNEL assay was performed with the In Situ Cell Death Detection Kit (TMR) from Roche according to the manufacturer's instruction.
Protein purfication and electrophoretic mobility shift assays (EMSA)
Cut CR3HD (4849–5412 of ctRA from Y.N. Jan) was cloned into pMAL2-c2x vector (NEB) and expressed as Maltose Binding Protein (MBP) fusion proteins. EMSAs were carried out as described in Stöbe et al. (2009) [56]. The following oligonucleotides (S2 subfragment) were used for analyzing the Cut binding sequence in EMSA (only forward strand is shown):
Wild-type. 5′GCACTTTTGCCTGCAGTTCAACTCGGTTCAGTTCGGTTGTGTCATAAAAAATC3′
Mutated. 5′GCACTTTTGCCTGCAGTGGAACTCGGTGGAGTGGGGTTGTGTCATAAAAAATC3′
Cut consensus sequences are underlined, exchanged nucleotides in the mutated versus the wild-type sequence are shown in bold.
Quantification of GFP labeled eye cells in hemolymph of 3rd instar larvae
ey::CD8-GFP or eyeful+CD8-GFP;ctRNAi;p35 3rd instar larvae were dissected by rupturing the larval cuticle at the posterior end with a pair of fine forceps, the hemolymph was collected in ice-cold Schneider's medium (Invitrogen GIBCO) containing 1× Complete protease inhibitor mixture (Roche). Hemolymph cells were analyzed via FACSAria to quantify GFP-labeled cells circulating within the hemolymph. In addition, GFP mRNA levels within the hemolymph were measured by qRT-PCR.
Microarray
Eye-antennal discs of ey::Dl or ey::Dl::2xctRNAi 3rd instar larvae were dissected in cold PBS, total RNA was extracted using standard procedures. Microarray analysis was conducted at the Genomics Core Facility, EMBL, Heidelberg, Germany. Microarray data analysis was performed using the R package as described previously [54].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HanahanDWeinbergRA 2000 The hallmarks of cancer. Cell 100 57 70
2. HarrisH 2005 A long view of fashions in cancer research. Bioessays 27 833 838
3. ShahNSukumarS 2010 The Hox genes and their roles in oncogenesis. Nat Rev Cancer 10 361 370
4. BossuytWDe GeestNAertsSLeenaertsIMarynenP 2009 The Atonal proneural transcription factor links differentiation and tumor formation in Drosophila. PLoS Biol 7 e40 doi:10.1371/journal.pbio.1000040
5. VidalMCaganRL 2006 Drosophila models for cancer research. Curr Opin Genet Dev 16 10 16
6. PorttLNormanGClappCGreenwoodMGreenwoodMT 2011 Anti-apoptosis and cell survival: A review. Biochim Biophys Acta
7. PitsouliCPerrimonN 2010 Embryonic multipotent progenitors remodel the Drosophila airways during metamorphosis. Development 137 3615 3624
8. WerzCLeeTVLeePLLackeyMBolducC 2005 Mis-specified cells die by an active gene-directed process, and inhibition of this death results in cell fate transformation in Drosophila. Development 132 5343 5352
9. HuNCastelli-GairJ 1999 Study of the posterior spiracles of Drosophila as a model to understand the genetic and cellular mechanisms controlling morphogenesis. Dev Biol 214 197 210
10. GoyalLMcCallKAgapiteJHartwiegEStellerH 2000 Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J 19 589 597
11. BardetPLKolahgarGMynettAMiguel-AliagaIBriscoeJ 2008 A fluorescent reporter of caspase activity for live imaging. Proc Natl Acad Sci U S A 105 13901 13905
12. HayBAWolffTRubinGM 1994 Expression of baculovirus P35 prevents cell death in Drosophila. Development 120 2121 2129
13. MerabetSCatalaFPradelJGrabaY 2002 A green fluorescent protein reporter genetic screen that identifies modifiers of Hox gene function in the Drosophila embryo. Genetics 162 189 202
14. NepveuA 2001 Role of the multifunctional CDP/Cut/Cux homeodomain transcription factor in regulating differentiation, cell growth and development. Gene 270 1 15
15. HilgersVBushatiNCohenSM Drosophila microRNAs 263a/b confer robustness during development by protecting nascent sense organs from apoptosis. PLoS Biol 8 e1000396 doi:10.1371/journal.pbio.1000396
16. Ferres-MarcoDGutierrez-GarciaIVallejoDMBolivarJGutierrez-AvinoFJ 2006 Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature 439 430 436
17. PalomeroTSulisMLCortinaMRealPJBarnesK 2007 Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 13 1203 1210
18. WilleckeMToggweilerJBaslerK Loss of PI3K blocks cell-cycle progression in a Drosophila tumor model. Oncogene 30 4067 4074
19. PagliariniRAXuT 2003 A genetic screen in Drosophila for metastatic behavior. Science 302 1227 1231
20. MohametLHawkinsKWardCM 2011 Loss of function of E-cadherin in embryonic stem cells and the relevance to models of tumorigenesis. J Oncol 2011 352616
21. BokelCBrownNH 2002 Integrins in development: moving on, responding to, and sticking to the extracellular matrix. Dev Cell 3 311 321
22. ChafferCLWeinbergRA 2011 A perspective on cancer cell metastasis. Science 331 1559 1564
23. SansregretLNepveuA 2008 The multiple roles of CUX1: insights from mouse models and cell-based assays. Gene 412 84 94
24. QuagginSEYegerHIgarashiP 1997 Antisense oligonucleotides to Cux-1, a Cut-related homeobox gene, cause increased apoptosis in mouse embryonic kidney cultures. J Clin Invest 99 718 724
25. RipkaSNeesseARiedelJBugEAignerA 2010 CUX1: target of Akt signalling and mediator of resistance to apoptosis in pancreatic cancer. Gut 59 1101 1110
26. SinclairAMLeeJAGoldsteinAXingDLiuS 2001 Lymphoid apoptosis and myeloid hyperplasia in CCAAT displacement protein mutant mice. Blood 98 3658 3667
27. WoodhouseECLiottaLA 2004 Drosophila invasive tumors: a model for understanding metastasis. Cell Cycle 3 38 40
28. YuJWangPMingLWoodMAZhangL 2007 SMAC/Diablo mediates the proapoptotic function of PUMA by regulating PUMA-induced mitochondrial events. Oncogene 26 4189 4198
29. ZhouLSchnitzlerAAgapiteJSchwartzLMStellerH 1997 Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proc Natl Acad Sci U S A 94 5131 5136
30. Portales-CasamarEThongjueaSKwonATArenillasDZhaoX 2010 JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res 38 D105 110
31. CertelKAndersonMGShrigleyRJJohnsonWA 1996 Distinct variant DNA-binding sites determine cell-specific autoregulated expression of the Drosophila POU domain transcription factor Drifter in midline glia or trachea. Mol Cell Biol 16 1813 1823
32. InbalALevanonDSalzbergA 2003 Multiple roles for u-turn/ventral veinless in the development of Drosophila PNS. Development 130 2467 2478
33. ConradtB 2009 Genetic control of programmed cell death during animal development. Annu Rev Genet 43 493 523
34. BaehreckeEH 2002 How death shapes life during development. Nat Rev Mol Cell Biol 3 779 787
35. IgakiT 2009 Correcting developmental errors by apoptosis: lessons from Drosophila JNK signaling. Apoptosis 14 1021 1028
36. McNeillHDownwardJ 1999 Apoptosis: Ras to the rescue in the fly eye. Curr Biol 9 R176 179
37. RichardsonHKumarS 2002 Death to flies: Drosophila as a model system to study programmed cell death. J Immunol Methods 265 21 38
38. MolchadskyARivlinNBroshRRotterVSarigR 2010 p53 is balancing development, differentiation and de-differentiation to assure cancer prevention. Carcinogenesis 31 1501 1508
39. SchmeichelKL 2004 A fly's eye view of tumor progression and metastasis. Breast Cancer Res 6 82 83
40. BalzerEMKonstantopoulosK 2011 Intercellular adhesion: mechanisms for growth and metastasis of epithelial cancers. Wiley Interdiscip Rev Syst Biol Med
41. WirtzDKonstantopoulosKSearsonPC 2011 The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer 11 512 522
42. MichlPDownwardJ 2006 CUTL1: a key mediator of TGFbeta-induced tumor invasion. Cell Cycle 5 132 134
43. HaradaRBerubeGTamplinOJDenis-LaroseCNepveuA 1995 DNA-binding specificity of the cut repeats from the human Cut-like protein. Mol Cell Biol 15 129 140
44. IulianellaASharmaMVanden HeuvelGBTrainorPA 2009 Cux2 functions downstream of Notch signaling to regulate dorsal interneuron formation in the spinal cord. Development 136 2329 2334
45. SharmaMFopmaABrantleyJGVanden HeuvelGB 2004 Coexpression of Cux-1 and Notch signaling pathway components during kidney development. Dev Dyn 231 828 838
46. VerhagenAMVauxDL 2002 Cell death regulation by the mammalian IAP antagonist Diablo/Smac. Apoptosis 7 163 166
47. ChipukJEGreenDR 2009 PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle 8 2692 2696
48. WassermanWWSandelinA 2004 Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet 5 276 287
49. NoyesMBChristensenRGWakabayashiAStormoGDBrodskyMH 2008 Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133 1277 1289
50. SiepelABejeranoGPedersenJSHinrichsASHouM 2005 Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15 1034 1050
51. LovegroveBSomoesSRivasMLSotillosSJohnsonK 2006 Co-ordinated control of cell adhesion, cell polarity and cytoskeleton underlies Hox induced organogenesis in Drosophila. Curr Biol 16 2206 2216
52. PiHWuHJChienCT 2001 A dual function of phyllopod in Drosophila external sensory organ development: cell fate specification of sensory organ precursor and its progeny. Development 128 2699 2710
53. GrueberWBJanLYJanYN 2003 Different levels of the homeodomain protein Cut regulate distinct dendrite branching patterns of Drosophila multidendritic neurons. Cell 112 805 818
54. HueberSDBezdanDHenzSRBlankMWuH 2007 Comparative analysis of Hox downstream genes in Drosophila. Development 134 381 392
55. LohmannIMcGinnisNBodmerMMcGinnisW 2002 The Drosophila Hox gene Deformed sculpts head morphology via direct regulation of the apoptosis activator reaper. Cell 110 457 466
56. StobePSteinSMHabring-MullerABezdanDFuchsAL 2009 Multifactorial regulation of a Hox target gene. PLoS Genet 5 e1000412 doi:10.1371/journal.pgen.1000412
57. ZhaiZSteinMALohmannI 2009 Expression of the apoptosis gene reaper in homeotic, segmentation and other mutants in Drosophila. Gene Expr Patterns 9 357 363
58. BaroloSCarverLAPosakonyJW 2000 GFP and β-galactosidase transformation vectors for promoter/enhancer analysis in Drosophila. Biotechniques 29 726 732
59. BoyALZhaiZHabring-MullerAKussler-SchneiderYKasparP 2010 Vectors for efficient and high-throughput construction of fluorescent Drosophila reporters using the PhiC31 site-specific integration system. Genesis 48 452 456
60. KosmanDMizutaniCMLemonsDCoxWGMcGinnisW 2004 Multiplex detection of RNA expression in Drosophila embryos. Science 305 846
61. ZhaiZFuchsALLohmannI 2010 Cellular analysis of newly identified Hox downstream genes in Drosophila. Eur J Cell Biol 89 273 278
62. StratigopoulosGLeDucCACremonaMLChungWKLeibelRL 2011 Cut-like homeobox 1 (CUX1) regulates expression of the fat mass and obesity-associated and retinitis pigmentosa GTPase regulator-interacting protein-1-like (RPGRIP1L) genes and coordinates leptin receptor signaling. J Biol Chem 286 2155 2170
63. BlochlingerKJanLYJanYN 1993 Postembryonic patterns of expression of cut, a locus regulating sensory organ identity in Drosophila. Development 117 441 450
64. JackJDeLottoY 1995 Structure and regulation of a complex locus: the cut gene of Drosophila. Genetics 139 1689 1700
65. KruppJJYaichLEWessellsRJBodmerR 2005 Identification of genetic loci that interact with cut during Drosophila wing-margin development. Genetics 170 1775 1795
66. MicchelliCARulifsonEJBlairSS 1997 The function and regulation of cut expression on the wing margin of Drosophila: Notch, Wingless and a dominant negative role for Delta and Serrate. Development 124 1485 1495
67. LudlowCChoyRBlochlingerK 1996 Functional analysis of Drosophila and mammalian Cut proteins in flies. Dev Biol 178 149 159
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- PIF4–Mediated Activation of Expression Integrates Temperature into the Auxin Pathway in Regulating Hypocotyl Growth
- Metabolic Profiling of a Mapping Population Exposes New Insights in the Regulation of Seed Metabolism and Seed, Fruit, and Plant Relations
- A Splice Site Variant in the Bovine Gene Compromises Growth and Regulation of the Inflammatory Response
- Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson's Disease Genetics: The PDGene Database
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy