
The Atypical Calpains: Evolutionary Analyses and Roles in Cellular Degeneration
The calpains are physiologically important Ca2+-activated regulatory proteases, which are divided into typical or atypical sub-families based on constituent domains. Both sub-families are present in mammals, but our understanding of calpain function is based primarily on typical sub-family members. Here, we take advantage of the model organism Caenorhabditis elegans, which expresses only atypical calpains, to extend our knowledge of the phylogenetic evolution and function of calpains. We provide evidence that a typical human calpain protein with a penta EF hand, detected using custom profile hidden Markov models, is conserved in ancient metazoans and a divergent clade. These analyses also provide evidence for the lineage-specific loss of typical calpain genes in C. elegans and Ciona, and they reveal that many calpain-like genes lack an intact catalytic triad. Given the association between the dysregulation of typical calpains and human degenerative pathologies, we explored the phenotypes, expression profiles, and consequences of inappropriate reduction or activation of C. elegans atypical calpains. These studies show that the atypical calpain gene, clp-1, contributes to muscle degeneration and reveal that clp-1 activity is sensitive to genetic manipulation of [Ca2+]i. We show that CLP-1 localizes to sarcomeric sub-structures, but is excluded from dense bodies (Z-disks). We find that the muscle degeneration observed in a C. elegans model of dystrophin-based muscular dystrophy can be suppressed by clp-1 inactivation and that nemadipine-A inhibition of the EGL-19 calcium channel reveals that Ca2+ dysfunction underlies the C. elegans MyoD model of myopathy. Taken together, our analyses highlight the roles of calcium dysregulation and CLP-1 in muscle myopathies and suggest that the atypical calpains could retain conserved roles in myofilament turnover.
Published in the journal:
. PLoS Genet 8(3): e32767. doi:10.1371/journal.pgen.1002602
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002602
Summary
The calpains are physiologically important Ca2+-activated regulatory proteases, which are divided into typical or atypical sub-families based on constituent domains. Both sub-families are present in mammals, but our understanding of calpain function is based primarily on typical sub-family members. Here, we take advantage of the model organism Caenorhabditis elegans, which expresses only atypical calpains, to extend our knowledge of the phylogenetic evolution and function of calpains. We provide evidence that a typical human calpain protein with a penta EF hand, detected using custom profile hidden Markov models, is conserved in ancient metazoans and a divergent clade. These analyses also provide evidence for the lineage-specific loss of typical calpain genes in C. elegans and Ciona, and they reveal that many calpain-like genes lack an intact catalytic triad. Given the association between the dysregulation of typical calpains and human degenerative pathologies, we explored the phenotypes, expression profiles, and consequences of inappropriate reduction or activation of C. elegans atypical calpains. These studies show that the atypical calpain gene, clp-1, contributes to muscle degeneration and reveal that clp-1 activity is sensitive to genetic manipulation of [Ca2+]i. We show that CLP-1 localizes to sarcomeric sub-structures, but is excluded from dense bodies (Z-disks). We find that the muscle degeneration observed in a C. elegans model of dystrophin-based muscular dystrophy can be suppressed by clp-1 inactivation and that nemadipine-A inhibition of the EGL-19 calcium channel reveals that Ca2+ dysfunction underlies the C. elegans MyoD model of myopathy. Taken together, our analyses highlight the roles of calcium dysregulation and CLP-1 in muscle myopathies and suggest that the atypical calpains could retain conserved roles in myofilament turnover.
Introduction
Calpains are Ca2+-regulated neutral thiol proteases that perform a limited digestion of target substrates, and are thus considered to be regulatory, as opposed to strictly degradative [1]. Members of the calpain family are variously composed of discrete modular domains, numbered DI–DVI (Figure S1) [2], [3]; DI–DIV are domains associated with the large calpain catalytic subunit and DV–DVI are domains found in a common small regulatory subunit, named CAPNS1 (not shown) [4], [5]. All calpain large subunits share a signature domain DII, which contains the core catalytic triad of cysteine, histidine and asparagine [6], [7]. DII is further divided into subdomains, IIa and IIb, which change conformation to align the catalytic cleft upon binding of Ca2+ [8]. DIII has a C2-like domain, which is present in almost all calpains, whereas DI is usually composed of a short non-conserved sequence that can undergo autolysis [6], [7]. As discussed below, calpains differ in their domain architecture; however, a key feature that distinguishes the typical calpains from the atypical calpains is the presence of DIV. The most extensively studied calpains are the typical CAPN1 and CAPN2, which have a classic DIV composed of five EF hand motifs, also referred to as a penta EF hand (PEF) [4], [5]. A PEF domain is also present in DVI of CAPNS1, which heterodimerizes with CAPN1 and CAPN2 through DIV [4], [5], although not all typical calpains require CAPNS1 for activity [9]–[11]. Mammalian genomes also encode the endogenous inhibitor calpastatin, which is specific for typical calpains [12], [13].
By contrast to typical calpains, there is no evidence indicating that atypical calpains lacking DIV and EF hand motifs form heterodimers. This absence of EF hands was also responsible for the initial belief that atypical calpains were insensitive to Ca2+ regulation [1]. Both typical and atypical calpains can also carry a variety of alternative domains, including an additional C2-like domain, zinc fingers, glycine rich regions and microtubule interacting and transport (MIT) domains (Figure S1).
Genes encoding predicted calpain proteins have been identified in many organisms ranging from single-celled yeasts to higher vertebrates through the sequencing of whole genomes. Mammalian genomes encode nine typical and five atypical calpains [1], [14], [15]. The Drosophila genome encodes only three typical and one atypical calpain (CALPD), also known as small optic lobes (SOL) [16]. By contrast, the genome of the nematode C. elegans encodes multiple atypical calpains, but no typical calpains. Given the paucity of typical calpains in non-mammals, phylogenetic arguments have been presented to suggest that the EF hand motifs of typical calpains were late evolutionary additions [14]. Homologs of CAPNS1 and calpastatin, proteins that interact with typical calpain subunits, are also absent in C. elegans and Drosophila [16], [17].
Mammalian calpains participate in many cellular processes, including, but not limited to, cytoskeletal remodelling [18], cell mobility [19], myofibril maintenance [20], signal transduction [21], cell cycle progression [22], regulation of gene expression [15], apoptosis [23] and long term potentiation [24]. Genetic association studies have implicated calpains in disease pathologies including limb-girdle muscular dystrophy type 2A (LGMD2A, CAPN3) [25], susceptibility to type II diabetes (CAPN10) [26] and gastric cancer (CAPN8/9) [27], [28].
Despite their involvement in cell maintenance and disease, it has been difficult to predict the targets of calpain proteolysis with any precision, because substrate specificity is only weakly determined by primary sequence [29], [30]. In addition, a large number of in vitro substrates have been identified, but many await in vivo validation. Chemical calpain inhibitors have also been developed, including active site peptidomimetics and domain IV and VI non-peptidyl inhibitors [31]. Unfortunately, active site inhibitors are generally not specific for calpains, as other cysteine proteases, such as members of the cysteine cathepsin family, are also subject to inhibition [32]; in addition, DIV and DVI inhibitors would fail to inhibit atypical calpains.
By contrast to the many studies focused on the activity of the typical calpains, it is clear that the specific involvement of atypical calpains in development and disease pathologies remains underexplored. We are using C. elegans as a model for exploring the function of atypical calpains. Previous work in C. elegans has highlighted the importance of the tra-3/clp-5 calpain gene in sex determination [33], [34]; other studies have shown that the C. elegans clp-1 and tra-3/clp-5 genes are involved in neurodegeneration and necrosis of a subset of vulval cells and in the intestine [35]–[37]. In this paper, we have examined the evolution of atypical and typical calpains in metazoa and analyzed the effects of reducing or enhancing the activities of atypical calpains on C. elegans development. To gain information about the potential site of action of a subset of atypical calpain genes, we have examined their in vivo expression patterns. Given the association between calpain activity and degenerative pathologies, we have investigated factors that influence the activity of clp-1 in a C. elegans model of muscular dystrophy and reveal the importance of both sustained calpain expression and intracellular Ca2+ levels ([Ca2+]i). Taken together, we hypothesize that the muscle degenerative phenotype caused by ectopic clp-1 stems from a conserved physiological role for calpain in the turnover of sarcomeric muscle proteins.
Results
The C. elegans atypical calpain family
We searched the C. elegans genome sequence using the typical human CAPN1 sequence and identified 14 atypical calpain-like sequences. Seven of these genes had previously been named clp-1 to clp-7, so we named the remaining seven genes clp-8 to clp-10 and clpr-1 to clpr-4 (for clp-related) for reasons explained below (Figure S1). An earlier analysis predicted the existence of 17 C. elegans calpain-like sequences [35]; however, three of these genes are not valid family members. F44F1.1 is now considered to be a pseudogene (Wormbase, release WS225), and both M04F3.4 and T21H3.3 lack a catalytic domain II, although they carry EF hand motifs. The domain architecture of the C. elegans atypical calpain proteins and the typical and atypical calpain proteins found in humans and Drosophila is shown in Figure S1 for comparison; a multisequence alignment is also provided to highlight the conservation of catalytic domain II and the divergence of DI and DIII among members of the calpain superfamily (Figure S2).
The genome of C. briggsae, a nematode closely related to C. elegans [38], only encodes nine predicted calpain sequences. To explain this difference, we compared the sequences of calpain proteins from C. elegans, C. briggsae, Drosophila and also human, and generated a cladogram (Figure S3). By comparison to C. briggsae, it appears that a recent gene expansion specific to C. elegans created two paralogous gene clusters. The first cluster consists of the C. elegans CLP-9 (T11A5.6) and CLP-10 (W05G11.4) proteins, which are homologous to C. briggsae Cbr-G19393; the proteins in this cluster are each predicted to have a SolH domain. The second cluster includes clp-8 (F44F1.3) and four predicted clpr genes, which are missing a critical cysteine residue of the catalytic triad; together these genes are paralogous to Cbr-G04776 and Cbr-G00485, which encode proteins with an intact catalytic triad. The existence of clpr genes raises the possibility that these predicted calpains are inactive, or that they have possibly gained novel non-proteolytic activities (Figure S1).
The cladogram in Figure S3 highlights the phylogenetic relationships between the Caenorhabditis, Drosophila and human calpain proteins. Although typical calpains are absent in C. elegans, the CLP-2, TRA-3/CLP-5 and the paralogous CLP-9 and CLP-10 proteins share extensive homology (E-values<1e-94) with human CAPN7, CAPN5/CAPN6 and CAPN15, respectively (Figure S3). Little is known about the biological functions of these human atypical calpains; hence, an analysis of their C. elegans homologs would provide potential insights into their function.
Ancient metazoan origins of typical and atypical calpains
We examined the evolutionary history of the typical calpains in more depth to seek evidence that their absence in C. elegans might be due to a lineage specific loss. We restricted our analyses to genes in metazoan phyla, as an earlier study had shown that calpain sequences containing C-terminal EF hand motifs are absent in plants and fungi [14]. We were further aided by the availability of whole genome sequences, particularly those representative of more ancient phyla, such as the placozoan Trichoplax adhaerens, the cnidarians Nematostella vectensis and Hydra magnipapillata [39]–[41], and also the sponge Amphimedon queenslandica, which diverged from early metazoans over 600 million years ago [42]. We found that homologs of typical calpains with EF-hand domains were likely to be present not only in early metazoa, but also in sponge (Figure 1). We also noted that typical calpain genes were absent from the genomes of other nematodes, including P. pacificus, B. malayi and C. remanei. Surprisingly, typical calpain genes were also absent in the tunicate C. intestinalis, a primitive branching clade of chordates [43], further supporting the notion of lineage-specific loss of typical calpains. Genes encoding atypical calpain proteins, including those carrying SolH or PBH domains, were also found in all metazoan phyla examined and in sponge (Figure 1).

A profile hidden Markov model detects penta EF hand domains
When analysing the phylogeny of the typical calpains, we found that it was difficult to count EF-hand motifs in order to identify penta EF-hand (PEF) domains using existing models [44]; for example, PROSITE and pfam counted only four EF-hand motifs and thus failed to identify PEF domains in CAPN1, 2 and 3 and sorcin. The number of EF hand motifs is likely to affect the ability of these proteins to dimerize, so we developed 5 separate custom profile hidden Markov models (profile HMMs) based on the individual EF hand motifs present within a penta EF hand domain to improve the ability to count EF hand motifs [45] (Text S1, see Materials and Methods). These profiles were tested by showing that together they detected the presence of penta EF hand domains in human CAPN1, CAPN2, CAPNS1 and sorcin proteins (true positives), and their absence in human CAPN10 or the C. elegans atypical calpains (true negatives). Application of these profiles to other typical calpain proteins showed that the calpain from Trichoplax is predicted to have a penta EF hand whereas the sponge has only four predicted motifs (Figure 1).
An abundance of calpain sequences with an incomplete catalytic triad
As indicated above, many C. elegans calpain-like proteins are predicted to lack proteolytic activity because the catalytic triad has not been conserved. Such inactive calpains have also been identified in protozoa [14]. In humans, the atypical CAPN6 promotes microtubule stability despite lacking a key residue within the catalytic triad [46]. To examine the prevalence of calpain-like proteins, we selected proteins that were missing at least one catalytic residue by using Fast Statistical Alignment (FSA) to align 1234 proteins from the Uniprot database, which carry the signature calpain catalytic domain SAAS022684_004_001783 [47], [48]. After removing protein fragments and splice variants from this list, a total of 344 calpain-like proteins remained (Table S1). As might be predicted, this analysis successfully identified the C. elegans, CLP-3 and CLPR-1 to CLPR-4, Drosophila CALPC and mammalian CAPN6 proteins, which have incomplete catalytic triads. Surprisingly, putative inactive calpain proteins were conserved across the plant, animal and fungus kingdoms (Figure S4A), and were also detected in protists (Figure S4B). Thus, taken together, the abundance and retention of catalytically inactive calpains combined with the finding that CAPN6 is functionally active might argue that these proteins might have hitherto undiscovered functional roles.
Characterisation of C. elegans calpain mutants
Human pathologies, such as LGMD2A, an inherited autosomal-recessive pathology caused by mutations in the typical calpain CAPN3 gene [25], are associated with calpain dysregulation. To determine whether the atypical calpain proteases participate in physiological and degenerative processes similar to those attributed to typical calpains, we characterized C. elegans homozygous mutants carrying deletions within the calpain genes clp-1, -4, -6, -7, -8, -9, -10 and clpr-1; most of the deletions are predicted to disrupt the catalytic domain and to reduce relative mRNA steady-state levels by at least 70% (Table S2). In addition, we examined a clp-2 mutant carrying a Tc1 transposon inserted within an exon, and performed clp-3 (RNAi) [49]. Phenotypic analysis revealed that brood sizes were not significantly different from wild type for any of the calpain mutants or clp-3 (RNAi) treated animals, except for clp-10 (ok2713) mutants in which the average brood size was reduced by approximately 50% (144±8; n = 4) without a corresponding increase in embryonic lethality. Embryonic lethality was slightly elevated in mutants carrying deletions in calpain genes, but gross developmental, mobility or morphological defects were not observed. For reasons discussed below, we also stained the clp-1, -4, -6 and -7 deletion mutants with phalloidin, but failed to detect disruptions to the sarcomeric structure of adult body wall muscle (Figure S5). These results indicate that most calpain genes, except for clp-10 and the previously characterized tra-3/clp-5 sex determining gene [50], play non-essential roles in otherwise wild type animals, although we have not addressed whether these genes could be functionally redundant (Table S2).
Transcriptional expression profiles of clp-1 to clp-7
The typical CAPN1 and CAPN2 calpains are ubiquitously expressed in cells (for review, [1]); however, a temporal elevation in mouse CAPN2 mRNA levels was detected during an essential period of embryonic development [51], [52]. Other typical calpains are capable of displaying more restricted patterns of expression; for example, transcripts corresponding to CAPN3, the gene affected in LGMD2A muscular dystrophy, are detected only in skeletal muscle [25]. To gain insights into the potential roles of the C. elegans atypical calpain genes based on their expression patterns, we examined transgenic C. elegans carrying nuclear-localized mRFP transcriptional reporters driven from promoter regions corresponding to clp-1 to -7. A nuclear localization signal was included to facilitate tissue-specific localization. mRFP was expressed from all of the reporters, except clp-3 and clp-6 (Figure S6); only the clp-2 reporter showed limited expression confined to the intestine (Figure S6). These expression patterns remained unchanged over the course of larval development through to adulthood (Figure S7).
To aid in the identification of tissues displaying calpain gene expression, clp(p)::nls::mRFP transcriptional reporters were co-expressed in animals carrying one of five different tissue-specific GFP reporters (for details, see Materials and Methods). We found that the clp-1, -4 and -7 reporters were active in neurons and co-localized with both the pan-neural unc-119::gfp and the GABAergic unc-47::gfp reporters, whereas the tra-3/clp-5 reporter was only detected in non-GABAergic neurons (Figure 2 and Figure S8). In addition, the clp-1 and clp-4 reporters were expressed in cells of the ventral and dorsal nerve cords, whereas the tra-3/clp-5 and clp-7 reporters were only expressed in cells of the ventral nerve cord (Figure 2 and Figure S8). Given the association between human CAPN3 and LGMD2A [25], we next co-expressed the clp reporters with the body wall muscle marker myo-3p::gfp::nls and found that only the clp-1 and clp-4 promoters were active in muscle (Figure 3A, 3B).
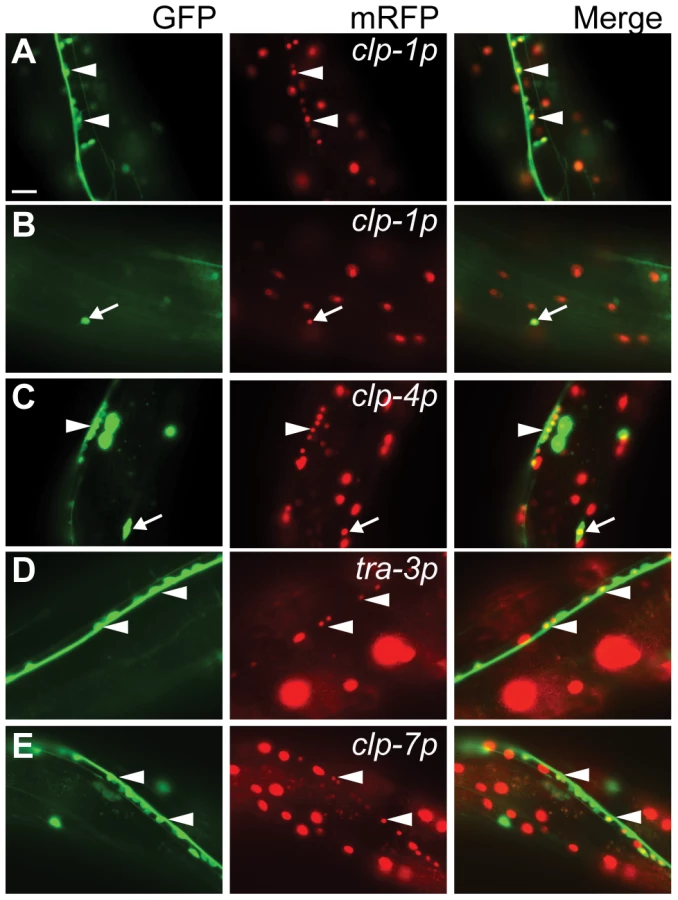

mRFP expressed from the clp-4, tra-3/clp-5 and clp-7 gene promoters also co-localized with a pes-6::gfp reporter, a marker for the excretory cell, which serves as the renal system of the worm (Figure 3C–3E). In addition, only the clp-7p::nls::mrfp transcriptional reporter co-localized with the seam cell reporter scm::gfp (Figure 3F). Calpain transcriptional reporters were also detected in other tissues, including the intestine (clp-1, -2, -7 and tra-3/clp-5), vulva (clp-1, tra-3/clp-5 and clp-7) and hypodermis (tra-3/clp-6 and clp-7) (Figure S9). The expression profiles of the clp-1 to clp-7 mRFP transcriptional reporters are summarized in Table 1.

Ectopic expression of CLP-1 in muscle causes paralysis
Increased calpain activity is associated with degenerative pathologies, such as cataract, neuronal degeneration and muscular dystrophy [25], [53]–[55]. Since a reduction in calpain activity failed to produce readily observable degenerative phenotypes in C. elegans, we next investigated the consequences of increasing calpain activity. This was first achieved by ectopically expressing full-length clp-1, -2, -4, -7 and tra-3/clp-5 cDNAs under the control of the inducible hsp-16.41 promoter, which is activated by heat shock in almost all tissues, except the germ line [56]. At least three independent transgenic strains were generated and tested for each construct; however, despite subjecting transgenic animals to daily doses of heat-shock driven calpain expression, we failed to detect abnormal phenotypes in any of the transgenic lines (n>1000 for each strain).
We considered the possibility that the transient nature of heat shock induced gene expression was insufficient for the purpose of eliciting ectopic phenotypes. To test this hypothesis, the constitutively active unc-54 promoter was used to drive ectopic expression of calpain cDNAs in body wall muscle [57]. Strikingly, we observed that the unc-54p::clp-1::myc transgene crEx325 caused 1.5%±0.4% of adult animals to develop paralysis (n>1000); similar results were obtained for two other independent transgenic lines (data not shown). Affected animals displayed an uncoordinated (Unc) phenotype at the L4/early adult stage, which progressed to paralysis and finally to premature death as animals matured to day 2 adults (Figure S10). This effect was not observed when the other muscle-associated gene, clp-4, was constitutively expressed from the unc-54 promoter, nor when clp-2, -7 or tra-3/clp-5 cDNAs were similarly expressed.
CLP-1–induced paralysis is dependent on expression levels and an intact catalytic triad
To understand why the hsp-16.41p::clp-1 transgene failed to cause paralysis, we compared the levels of CLP-1::MYC expressed from either the muscle-constitutive crEx325 [unc-54p::clp-1::myc] or the heat shock activated crEx329 [hsp-16.41p::clp-1::myc] transgene by western blot analysis (Figure S11A). We also observed that the level of CLP-1::MYC expressed from crEx329 peaked between 4 to 12 hours post-heat shock before declining (Figure S11B). By comparison, the level of CLP-1::MYC expressed from the crEx325 transgene was four-fold higher than that observed during the peak of crEx329 heat shock driven expression. These results indicate that sustained levels of elevated CLP-1 protein promote the development of paralysis.
To establish that an intact catalytic triad was required for CLP-1 to cause paralysis, four independent lines were established that were predicted to express a catalytically inactive CLP-1(C371A) from an unc-54p::clp-1(C371A) transgene [58], but none showed mobility defects or paralysis (n>1200). Additional lines expressing mRFP tagged CLP-1 proteins were generated to facilitate western blot analysis. In these lines, paralysis was not detected in crEx336 [unc-54p::clp-1(C371A)::mrfp] animals (n>1200), whereas paralysis developed in 1.9%±0.6% (n>300) of crEx335 [unc-54p::clp-1::mrfp] animals. We also observed that the level of CLP-1::mRFP expressed from crEx336, as measured by western blot analysis, was not reduced when compared to crEx335, and so could not account for the difference in their activities (Figure S11C). Attempts were also made to demonstrate CLP-1 proteolytic activity using casein zymography and the calpain-GLO protease™ assay (Promega), which are used to measure typical calpain activity [59]. Unfortunately, neither casein nor suc-LLVY-aminoluciferin was found to be a suitable substrate for CLP-1, although a similarly prepared recombinant rat CAPN2 was active in both assays.
Ectopic expression of clp-1 in neurons fails to elicit neurodegenerative or neuromuscular defects
We next examined clp-1 activity in neurons. Previous studies have shown that RNAi inactivation of clp-1 partially suppressed the degeneration of touch receptor neurons in animals carrying gain-of-function mutations in the mec-4 or deg-1 Na+ channel subunits [35], although degeneration was not observed when clp-1 was overexpressed in touch receptor neurons [35]. We also tested and failed to observe neurodegenerative phenotypes when either the unc-119p::clp-1 or unc-47::clp-1 reporter was expressed in other neurons.
Ectopic expression of clp-1 causes muscle cell abnormalities
In N2 wild type animals, body wall muscle cells form striated diamond shaped bundles that are arranged into four quadrants running down the length of the animal [60]. We hypothesized that constitutively elevated expression of CLP-1 in body wall muscle cells was causing extensive myofibrillar damage, which in turn was leading to paralysis. Before examining muscle morphology, we chromosomally integrated the crEx325 [unc-54p::clp-1] array to generate crIs4, in order to circumvent potential problems associated with mosaic expression [61]. We found that the integrated crIs4 array retained the same phenotypic characteristics as crEx325; 1.69±0.34 (n = 352) of crIs4 animals displayed paralysis. We next synchronized the growth of crIs4 worms and separated crIs4 adults into three distinct classes: 1) phenotypically wildtype with normal sinusoidal movement 2) Unc and 3) paralyzed, and examined the integrity of body wall muscles by staining actin thin filaments with phalloidin. We found that phenotypically wildtype crIs4 animals exhibited only occasional muscle cell abnormalities (Figure 4A, Table 2), whereas Unc crIs4 animals displayed disorganized bundles of actin fibers and were also missing body wall muscle cells (Figure 4B, Table 2). These abnormalities were even more extensive in paralyzed crIs4 animals (Figure 4C, Table 2). Thus, the paralysis observed in crIs4 animals can be attributed to the loss of sarcomere integrity.
![Ectopic <i>crIs4</i> [<i>unc-54p::clp-1</i>] expression disrupts body wall muscle.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/c46058d2de3126e58ef16060a5e9bfc9.png)

CLP-1 localizes to muscle M-lines and adhesion plaques
Given that CLP-1 promotes the degeneration of body wall muscle, we examined the intracellular localisation of CLP-1::mRFP in qyIs43 animals co-expressing a beta-integrin subunit [pat-3::gfp] (Figure 5) [62]; in muscle cells, PAT-3 is found at the base of thick filament M-lines, dense bodies (Z-disks) and in adhesion plaques that are formed between adjacent cells (Figure 5A). We found that CLP-1::mRFP was excluded from the nucleus (not shown) and dense bodies, but was present at structures immediately adjacent to dense bodies (Figure 5B). More specifically, CLP-1::mRFP co-localized with PAT-3::GFP at M-lines extending over the H-zone and at adhesion plaques (Figure 5C). We also generated a native clp-1::gfp translational reporter, which confirmed that CLP-1::GFP displayed the same pattern of sarcomeric localization, as CLP-1::mRFP driven from the unc-54 promoter (Figure 5D and Figure S12); CLP-1::GFP was also detected in other non-muscle tissues. Aggregates of CLP-1::mRFP were also observed, which might contribute to muscle degeneration and paralysis, although we cannot exclude the possibility that aggregation is an artifact of overexpression (Figure 5B). Thus, we speculate that sarcomeric proteins enriched at the sites of CLP-1 localization are potential targets for degradation.

Reduction of CLP-1 activity suppresses muscle degeneration in a C. elegans model of muscular dystrophy
In mammals, the absence of the large structural muscle protein dystrophin underlies the muscle degenerative disorder Duchenne muscular dystrophy (DMD) [63]. Similarly, a C. elegans DMD model is based on the progressive muscle degeneration observed in dys-1(cx18); hlh-1(cc561ts) animals [60]. In C. elegans, a null mutation in the only dystrophin-like protein gene, dys-1(cx18), caused only occasional muscle degeneration [64]. However, inclusion of a temperature sensitive allele of the MyoD transcription factor homolog, hlh-1(cc561ts), sensitized dys-1(cx18); hlh-1(cc561ts) mutants to become uncoordinated (Unc), but not paralyzed, and to display increased muscle degeneration [60].
We investigated whether CLP-1 contributed to the muscle degeneration associated with C. elegans DMD by constructing the dys-1(cx18); hlh-1(cc561ts); clp-1(tm690) strain. The clp-1(tm690) mutation deletes a 624 bp region of the clp-1 gene and introduces a translational frame-shift that is predicted to produce a 493 amino acid truncated protein, which lacks two of the three critical catalytic residues required for proteolytic activity. clp-1 (tm690) mutants are phenotypically wild-type and do not display any obvious defects in their muscle structure (Table 2, Figure S5). When the number of abnormal muscle cells present in dys-1(cx18); hlh-1(cc561ts); clp-1(tm690) was compared to that found in dys-1(cx18); hlh-1(cc561ts) animals after phalloidin staining, we found that the absence of clp-1 reduced the number of abnormal muscle cells by almost 50% (p<0.001). A body wall muscle cell was scored as abnormal when: 1) the classic striated pattern of actin filaments was disrupted 2) actin bundles were visible as puncta or 3) muscle cells were missing due to cell death. Thus, clp-1 is normally active in muscle and contributes to the muscle degeneration observed in dys-1(cx18); hlh-1(cc561ts) animals (Table 2).
Genetic manipulation of [Ca2+]i levels exacerbates clp-1–induced paralysis
Although ectopic expression of CLP-1 led to a degradative muscle pathology, we were surprised that the penetrance of the effect was not higher. To ask what other factors might modulate clp-1 activity in muscle, we first took a genetic approach to investigate the effect of intracellular calcium [Ca2+]i. Although calpains are referred to as Ca2+-activated proteases, little is known about the effect of physiological calcium levels on the activity of atypical calpains. In C. elegans, four allelic modifiers have been identified that are understood to increase [Ca2+]i: egl-19(ad695gf), itr-1(sy290gf), slo-1(js379) and unc-24(e138) (for descriptions of mutants, see Materials and Methods). We generated strains carrying each of these mutations in combination with crIs4 and scored adults for paralysis.
We found that the number of animals displaying paralysis was significantly increased when crIs4 was combined with mutations in egl-19(ad695), unc-24(e138) or slo-1(js379) (Figure 6A). Inclusion of the egl-19(ad695gf) mutation produced the most dramatic increase in paralysis (p<0.001), whereas the unc-24(e138) mutation caused a mild, but significant increase (p = 0.043) (Figure 6A). Although the slo-1(js379) mutation also increased the level of crIs4 paralysis, this effect could be attributed to either its role in Ca2+ regulation or to its participation in the Dystrophin Associated Protein Complex (DAPC), discussed below [65]. The itr-1(sy290gf) mutation did not significantly affect the number of paralyzed crIs4 animals; however, ITR-1 expression has not been detected in muscle [66].
![Genetic enhancers of <i>crIs4</i> [<i>unc-54p::clp-1</i>] induced paralysis.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/daa94d035cc9595618fe378d5e565f5e.png)
Genetic defects in the Dystrophin Associated Protein Complex (DAPC) do not enhance clp-1–induced paralysis
We next constructed a set of double mutants between crIs4 and members of a second group of allelic modifiers that form the Dystrophin Associated Protein Complex (DAPC): dys-1(cx18), snf-6(ok720), dyb-1(cx36) and stn-1(ok292), which encode a dystrophin-like protein, acetylcholine transporter, dystrobrevin and syntrophin, respectively [64], [67]–[69]. Constituents of the DAPC provide structural support to muscle by linking the cytoskeleton to the sarcolemma and extracellular matrix [70]. We hypothesized that mutations capable of destabilizing the integrity of the DAPC could sensitize muscle cells to CLP-1 induced damage. The four mutants listed above and slo-1(js379) share a similar phenotype marked by hyperactivity and exaggerated head bending [64], [65], [67]–[69]. An hlh-1(cc561ts) temperature sensitive allele of a MyoD homolog was also examined because this mutation sensitizes dystrophin dys-1(cx18) mutants to muscle damage [60].
We found that disrupting the structural proteins of the DAPC complex did not significantly sensitize animals to the effects of ectopic clp-1 provided by crIs4 (Figure 6B). By contrast, inclusion of the hlh-1(cc561ts) allele substantially increased the proportion of paralyzed crIs4 animals. We next tested whether the effects of crIs4 could be further enhanced in a dys-1(cx18); egl-19(ad695gf) genetic background by increasing [Ca2+]i. Adult dys-1(cx18); egl-19(ad695gf) double mutants in the absence of crIs4 are Unc, but not paralyzed, and exhibit moderate muscle degeneration [71]. We found that the percentage of dys-1(cx18); egl-19(ad695gf); crIs4 adults developing paralysis was vastly elevated (56.3±6.4%) when compared to animals expressing crIs4 in combination with a mutation in either dys-1(cx18) or egl-19(ad695gf) alone (Figure 6B). Moreover, the brood size of dys-1(cx18); egl-19(ad695gf); crIs4 animals was dramatically reduced and embryonic lethality elevated (Table S3).
The heightened sensitivity of dys-1(cx18); egl-19(ad695gf) mutants to crIs4 induced paralysis led us to investigate whether dys-1(cx18); egl-19(ad695gf) mutants might also be sensitized to the effects of ectopic clp-2, -4, -7 or tra-3/clp-5 expression in muscle under control of the unc-54 promoter. We examined over 1000 animals from at least three independent transgenic lines for each calpain, however, we failed to detect any exacerbated defects in movement aside from those already reported for dys-1(cx18); egl-19(ad695gf) mutants [71]. Hence, of the calpains tested, only clp-1 was found to induce paralysis when overexpressed in muscle under conditions of elevated [Ca2+]i.
Inhibition of the egl-19 channel reduces clp-1–induced paralysis
The EGL-19 L-type voltage gated Ca2+ channel is located along the basal membrane of muscle. To demonstrate that egl-19(gf) was exerting an effect on clp-1 activity specifically by altering [Ca2+]I, we asked if the small molecule antagonist, nemadipine-A, could suppress CLP-1 mediated muscle degeneration; nemadipine-A has been shown to be a specific and highly effective inhibitor of EGL-19 [72]. We found that a dose of 5 µM nemadipine-A was sufficient to abolish the enhanced level of paralysis observed in egl-19(ad695); crIs4 mutants (Figure 7A). We next treated hlh-1(cc561ts); crIs4 mutants with nemadipine-A and found that paralysis was also reduced by over 50% compared to untreated animals (Figure 7A). This result indicates that the hlh-1(cc561ts) mutation is likely to sensitize muscle cells to the effects of crIs4 indirectly by causing an increase in [Ca2+]I, which can be suppressed by inhibiting the activity of the EGL-19 Ca2+ channel.
![Small molecule and genetic inhibition of <i>crIs4</i> [<i>unc-54p::clp-1</i>] induced paralysis.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/55612e9202404135e22386c1d4fae282.png)
RNAi inactivation of aspartyl proteases suppresses clp-1–induced paralysis
The aspartyl proteases, asp-1, asp-3 and asp-4, have previously been shown to be constituents of a neural degenerative pathway involving the calpains clp-1 and tra-3/clp-5 [35]. To ask if the asp genes also promote muscle degeneration, we performed RNAi with the genes asp-1 to asp-6 on hlh-1(cc561ts); crIs4 animals. The effectiveness of RNAi knockdown was measured by quantitative PCR (qPCR) (Table S4). We found that asp-3(RNAi) reduced paralysis levels by over 40% and that asp-2(RNAi) and asp-4(RNAi) each reduced paralysis by ∼15%, whereas asp-1(RNAi) had no measurable effect in muscle. We further showed that clp-1(RNAi) almost completely abolished crIs4 induced paralysis in hlh-1(cc561ts); crIs4 animals, as might be expected (Figure 7B).
Discussion
Typical and atypical calpains are both detected in ancient metazoan phyla
CAPN1 and CAPN2 are still referred to as the main or major calpains despite the rich variety and abundance of genes encoding atypical calpain proteins (Figure 1). However, phylogenetic analyses performed here and by others indicate that the C-terminal EF hand motifs in typical calpains are unlikely to represent embellishments acquired late in metazoan evolution, as might be inferred by the greater importance attached to typical calpains [1], [14]. We detected typical calpain homologs in basal metazoan phyla, such as Nematostella, Trichoplax and Hydra [39]–[41], as well as in sponge, a representative of an early divergent metazoan clade [42], and found that the EF hand domain in early metazoan typical calpain proteins was already composed of 5 motifs (PEF) (Figure 1). The absence of typical calpain genes in C. elegans and the presence of both typical and atypical calpain genes in Schistosomes and Drosophila further suggests that C. elegans is likely to have undergone a lineage-specific loss of typical calpain genes; it is noteworthy that Drosophila and C. elegans have both been assigned to the clade Ecdysozoa [73]. A similar line of reasoning could also be used to explain the absence of typical calpain genes in the genome of the ascidian Ciona [43].
Studies directed toward understanding the function and regulation of atypical calpains have lagged, or have possibly been confounded by the presence of typical calpains. C. elegans presents an ideal model in which to examine the function of atypical calpains, because their genome encodes a range of variants that are representative of those found across phyla, including ancient metazoan lineages, and because of the availability of mutant alleles (Figure S1). Our analyses of these mutants have shown that clp-10(ok2713) mutants have a reduced brood size, but that the remaining mutants, with the exception of tra-3/clp-5, do not display obvious phenotypes affecting viability, motility or fertility [50]. Seven of the nine characterized mutants are represented by null or strong loss-of-function alleles that have eliminated or disrupted the catalytic triad; qPCR also shows that levels of affected transcripts were reduced in mutants by at least 70% (Table S2). Despite our inability to detect single mutant phenotypes, our results demonstrated that deletion of clp-1 suppressed muscle degeneration in dys-1(cx18); hlh-1(cc561ts) mutants (Table 2). In neurons, it was similarly shown that RNAi knockdown of either clp-1 or tra-3/clp-5 suppressed touch cell degeneration in mutants carrying a dominant-gain-of-function mec-4 allele [35]. These results indicate that the ability to detect phenotypes in atypical calpains might be dependent on the genetic background or physiological state of the animal. It also remains possible that the expansion in the family of C. elegans atypical calpain genes has made it difficult to detect single mutant phenotypes because of functional redundancy.
Regulation and specificity of atypical calpain activity
To gain insights into the physiological roles of atypical calpain proteins, we examined the consequences of calpain overexpression, but were unable to detect any obvious phenotypic changes resulting from heat-shock driven overexpression. CLP-1 protein expressed under these conditions peaked 4–8 hours post heat-shock before declining; by contrast, the level of CLP-1 protein resulting from unc-54 promoter activity was shown to be comparable to that detected during the peak period of expression after heat shock (Figure S11). The near-absence of phenotypes resulting from calpain overexpression could also indicate that enhanced atypical calpain activity is not necessarily detrimental in a healthy cell. One could speculate that if a given atypical calpain were constitutively active, then overexpression might have limited effect. For example, although heat shock driven expression of a wildtype tra-3/clp-5 transgene was clearly sufficient to prevent the sexual transformation of a tra-3/clp-5 null mutant, rescued animals did not display any degenerative phenotypes [34].
The substrate specificity of typical calpains is determined not only by primary sequence, but also by higher order structural features [30]. Very little is known about the substrate requirements for atypical calpains, but in our hands, CLP-1 and CLP-7 failed to cleave standard typical calpain substrates in vitro (data not shown). Our experiments also revealed that of all the clp genes tested only clp-1 driven from the unc-54 promoter led to muscle degeneration and paralysis (Figure S10). The inability of the other atypical calpains to cause paralysis would indicate that sarcomeric proteins are either not general substrates for atypical calpains or that they are inaccessible unless damaged. A third possibility is that an intracellular inhibitor regulates the activities of atypical calpains. In mammals, calpastatin directly inhibits CAPN1 and CAPN2 under pathological conditions [74]. The C. elegans genome lacks a calpastatin gene, but an intracellular serpin protease inhibitor, SRP-6, has been hypothesized to inhibit TRA-3/CLP-5 and CLP-10 in the intestine in response to hypo-osmotic shock [37]. A family of srp genes has been identified, so interactions with other clp genes might remain to be uncovered.
Calcium activation of atypical calpains
Typical calpain proteases require Ca2+ for activity, but under conditions of reduced [Ca2+]i, they can be stimulated through the interaction of the C2 domain with membrane phospholipids and through the autolysis of DI [6], [7], [15]. So, how does calcium influence atypical calpain activity? We previously showed that TRA-3/CLP-5 undergoes calcium-dependent autolysis [34], but it remains unknown if any of the other atypical calpains can undergo this process, or if autolysis enhances proteolytic activity. Structural analyses of a mini-calpain identified two Ca2+ binding sites within IIa and IIb of CAPN2 DII, which help to align the catalytic triad when Ca2+ is bound [8]. Extrapolating from this model, the conservation of key residues in DII would predict that atypical calpains would display a similar Ca2+ dependency (Figure S2). The potential influence of phospholipids on the activity of atypical calpains has yet to be examined, although DIII forms a C2 fold that could interact with Ca2+ and phospholipids [75].
It is clear that the physiological rise in [Ca2+]i achieved through the use of genetic mutants profoundly affected CLP-1 activity (Figure 6A). However, it is important to emphasize that it was the synergistic increase of both CLP-1 and [Ca2+]i levels that contributed to paralysis. None of the Ca2+ channel mutants examined developed paralysis, indicating that an increase in [Ca2+]i by itself is unable to pathologically activate clp-1. Nonetheless, removal of clp-1 substantially suppressed the muscle degeneration associated with C. elegans dys-1; hlh-1(cc561ts) DMD, which is based on a mouse model of myopathy involving the combined mutation of MyoD and dystrophin (Table 2) [60], [76]. In muscle degenerative conditions, such as DMD, a rise in [Ca2+]i is also hypothesized to activate calpain proteolysis [77]–[79].
We have also shown that the C. elegans hlh-1(cc561ts) allele of a MyoD homolog, sensitizes C. elegans to the effects of crIs4, as measured by the increase in paralysis, and that treatment of these worms with nemadine-A suppressed paralysis (Figure 7A). As nemadipine-A is a specific inhibitor of the EGL-19 L-type Ca2+ channel [72], it was not surprising to find that this drug also suppressed the paralysis associated with egl-19(gf); crIs4 mutants. Based on these results, we propose that the hlh-1(cc561ts) mutation creates a sensitized background for calpain activity by indirectly elevating [Ca2+]I, although it remains unclear if EGL-19 activity is disrupted. Following this logic, we speculate that inappropriate CLP-1 activity in a sensitized background could generate a positive feedback loop whereby calpain disrupts Ca2+ channel activity, leading to increased [Ca2+]i, and further activation of CLP-1. In support of this model, several studies have shown that typical calpains can cleave Ca2+ channels and disrupt their activities [80]–[82].
Physiological regulation of calpains in muscle
In mammals, inappropriate elevation of either CAPN1 or CAPN2 is associated with muscle degeneration [54]. Indirect evidence has also accumulated pointing to the involvement of these calpains and the skeletal muscle specific CAPN3 isoform in myofibrillar protein turnover, which promotes the replacement of damaged sarcomeric components in order to maintain efficient muscle contraction [7], [25], [83]–[88]. What roles do atypical calpains normally play in muscle? In our study and in reports by others, only the clp-1 and clp-4 genes appear to be expressed in muscle (Figure 3) [35], [89], and it was further found that a rescuing tra-3/clp-5::gfp translational fusion was not expressed in muscle [90]. Surprisingly, it has recently been reported that chronic RNAi knockdown of clp-1, clp-4, tra-3/clp-5, clp-6 or clp-7 was responsible for causing myofilament disruption [91], suggesting that calpains are involved in myofilament maintenance. By contrast, we failed to detect sarcomeric abnormalities in similarly staged adults when phalloidin was used to examine the body wall muscle of animals carrying deletion alleles in clp-1, clp-4, clp-6 or clp-7 (Figure S5). We are unable to account for these differences, although it has been reported elsewhere that a GFP-tagged myosin heavy chain reporter [myo-3::gfp] can independently cause an age-dependent sarcopenia in adults [92]. Nonetheless, our study independently supports a role for CLP-1 in the maintenance of muscle adhesion complexes and turnover of myofibrillar proteins.
We have obtained evidence that CLP-1 has the potential to disrupt sarcomeric integrity, indicating that CLP-1 is likely to target components of muscle adhesion complexes for destruction (Figure 4 and Figure 6). The localization of CLP-1 to M-lines and to structures immediately adjacent to Z-disks shows that CLP-1 is well positioned to participate in myofibrillar turnover or possibly remodelling of integrin-based muscle attachment assemblies (Figure 5); however, it is interesting that CLP-1 is excluded from dense bodies (Z-disks). CLP-1 could also regulate muscle cell-muscle cell interactions through its localization to adhesion plaques. In C. elegans, studies based on the use of FRAP show that C. elegans sarcomeric proteins undergo dynamic exchange suggestive of protein turnover [93]. Taken together, these observations suggest that CLP-1 might normally promote myofibrillar protein turnover and help to maintain the ordered alignment of adhesion complexes, or to accommodate changes to the sarcomere due to growth or cell damage. Evidence has also been obtained from mammalian systems indicating that the typical CAPN1 and CAPN2 calpains are able to regulate the dynamics of cell adhesion complexes [18], which are similar in composition to those found in C. elegans muscle [19].
Our data did not support the expectation that disruptions to the DAPC, an important complex that maintains muscle structural integrity in humans, would synergize with CLP-1 overexpression and lead to increased sarcomeric damage and paralysis in worms (Figure 6B) [70]. However, it has been reported that the C. elegans DAPC promotes, but is not essential for muscle integrity [64], [69], [94], [95], so destabilisation of the complex might not be sufficient to induce damage and heightened myofibril turnover, despite the increased availability of CLP-1.
A conserved calpain–cathepsin pathway of muscle degeneration
We propose that the death of muscle cells observed in paralyzed crIs4 animals is caused by activation of a pathway promoting necrosis. In mammalian neurons, a model has been proposed whereby inappropriate increases in [Ca2+]i caused by insults such as ischemia/reperfusion injury contribute to the activation of calpains and lead to the permeabilization of lysosomes. In turn, lysosomal rupture allows cathepsin proteases to leak into the cell and cause widespread degradation and necrotic cell death [55]. In C. elegans, support for this model was obtained when it was shown that clp-1, the aspartyl proteases asp-3 and asp-4 and to a lesser extent asp-1 were required for the necrotic death of neuronal touch receptors and vulval uv1 cells [35], [36]. Similarly, our data also revealed the importance of asp-3 in promoting clp-1 mediated paralysis in muscle (Figure 7B). By contrast to studies in neurons, we found that asp-4 has a reduced role in muscle degeneration and that asp-1 doesn't appear to be active in muscle, although we obtained evidence that asp-2 participates in this process. We, therefore, propose that a calpain-cathepsin pathway of degeneration has been conserved in C. elegans muscle.
Finally, the ability of nemadipine-A to reduce the level of paralysis in hlh-1; crIs4 worms shows that physiological conditions that lead to an indirect increase in [Ca2+]i can have a destructive and possibly necrotic outcome in muscle cells by activating atypical calpain activity (Figure 7A) [96]. Therefore, it will be of further interest to identify physiological conditions that could lead to elevated calpain expression or [Ca2+]i as causative factors in sarcopenia, a loss in muscle contractility observed in ageing cells [97], [98]. We propose that the identification of atypical calpain inhibitors would represent a fruitful area for pharmacological investigation. It should be possible to take advantage of the paralytic phenotype displayed by hlh-1(cc561ts); crIs4 mutants and to perform a chemical screen to identify compounds that ameliorate muscle damage. Such a screen could potentially identify calcium channel blockers or specific inhibitors of atypical calpains. Given that atypical calpain genes are also present in mammals, our results emphasize the importance of investigating the contribution of atypical calpains to degenerative disorders, especially under conditions associated with inappropriately elevated [Ca]I, such as muscular dystrophies, neurodegeneration and cataract.
Materials and Methods
Blast analyses and protein phylogeny
See Protocol S1 for sequence accession numbers and details about sequence similarity searches and protein alignments.
A custom profile hidden Markov model was created for each of the five EF-hand motifs of the penta EF hand, using the following proteins as a training set: HsCAPN1 (AAH75862.1), HsCAPN2 (NP_001739.2), HsCAPN3/p94 (AAI46650.1), HsCAPN8 (NP_001137434.1), HsCAPN9 (NP_006606.1), HsCAPN11 (EAX04252.1), DmCALPA (NP_001097378.1), DmCALPB (NP_524016.4), DmCALPC (AAF48591.2) and HsGRAN (P28676) [45]. These full-length proteins were aligned using FSA [48], and each of the EF hand motifs was sliced out of the alignment, based on the coordinates provided for the EF hand motifs of HsCAPN1, to generate five multiple sequence alignment files (MSF) [99]. The hmmbuild program in the HMMER software package was then used to create a profile HMM for each of the five EF-hand motifs [45]; pfam HMM files are available as Text S1. The EF-hand motif model was validated using true positive penta-EF hand domain containing proteins CAPNS1 (AAH64998.1) and Sorcin (AAA92155.1); HsCAPN10 and the C. elegans atypical calpains were shown to be true negatives. An E-value cutoff level of 0.01 was used.
C. elegans methods
Worms were grown and maintained at 20°C as described [100], except strains containing hlh-1(cc561ts) II, which were grown at 15°C [101]. The following strains were used: wild-type Bristol N2, HC46: ccIs4215 [myo-3::gfp::nls] I, LS292: dys-1(cx18) I, clp-8 [F44F1.3] (ok1878) I, clp-9 [T11A5.6] (ok1866) I, clpr-1 [W04A4.4] (ok2601) I, LS505: dyb-1(cx36) I, LS721: stn-1(ok292) I, LS587: dys-1(cx18) I; hlh-1(cc561ts) II, LS706: dys-1(cx18) I; egl-19(ad695gf) IV, NL2099: rrf-3(pk1426) II, PD4605: hlh-1(cc561ts) II, clp-1(tm690) III, clp-2 (pk323) III, clp-4 (ok2808) III, clp-10 [W05G11.4] (ok2713) III, JR667: unc-119 (e2498::Tc1) III, VC591: okIs53 snf-6(ok720) III, dpy-20(e1282) IV, DA695: egl-19(ad695gf) IV, clp-6 (ok1779) IV, clp-7 (ok2750) IV, itr-1(sy290) dpy-20(e1282) IV, unc-24(e138) IV, NM1968: slo-1(js379) V, EG1285: lin-15(n765); oxIs12 [unc-47::gfp+lin-15(+)] X, IM19: urIs13 [unc-119::gfp (IM#175; rol-6(su1006)], NK358: unc-119(ed4) III; qyIs43, UG756: bgIs312 (pes-6::gfp), wIs51 [scm::gfp (seam cell)+unc-119(+)]. The clp-1(tm690) III allele (kindly provided by Shohei Mitani) was sequenced using primers GGATGAGCTCTTCTATCGTG and GTCTGACCATGGTCCATTCC to confirm the presence of a 624 bp deletion, which is predicted to create a frame-shift at A(460), and produce a truncated protein of 493 amino acids.
egl-19 encodes an L-type voltage gated Ca2+ channel; an egl-19(ad695gf) gain-of-function mutation delays the inactivation of the EGL-19 L-type voltage gated Ca2+ channel, and hence leads to increased [Ca2+]i [102]. The itr-1 gene encodes an inositol 1,4,5-triphosphate receptor (IP3R) homolog that resides on the endoplasmic reticulum (ER) and releases Ca2+ into the cytoplasm in response to IP3 signalling [103]; the itr-1(sy290gf) allele is a gain-of-function mutation that increases [Ca2+]i. Because the itr-1(sy290gf) mutant carries a point mutation and does not display an obvious phenotype, the dpy-20(e1282) mutation, which is closely linked to the itr-1 gene, was included to facilitate the identification of itr-1 homozygotes. To account for potential marker effects, paralysis was also scored in a dpy-20(e1282); crIs4 genetic background. slo-1(js379) is a loss-of-function allele of a gene that encodes a Ca2+ activated potassium BK channel [104]. The SLO-1 channel is normally activated by a rise in [Ca2+]i, which leads to plasma membrane hyperpolarization. In turn, [Ca2+]i is reduced by the inactivation of L-type Ca2+ channels, such as EGL-19 [65], [104], [105]. unc-24(e138) is a loss-of-function allele of a gene encoding a protein containing stomatin-like and lipid transfer domains that indirectly regulates Na+ channels, and hence, similar to slo-1, unc-24 affects [Ca2+]i through plasma membrane hyperpolarisation [106]–[109].
Molecular biology and gene cloning
Primer sequences are available in Protocol S2. Protocol S3 provides details about gene cloning and reporter construction.
RNAi was performed as described by cloning PCR amplified products into the L4440 RNAi feeding vector [110]. RNAi constructs for asp-1 to asp-6 were obtained from a library [111].
qPCR
Animals were harvested and RNA was extracted as described by Hope (1999). cDNA was amplified from 1.5 µg of DNAse treated mRNA using Taqman Reverse Transcription Reagents (Applied Biosystems), as directed by the manufacturer. qPCR was performed in triplicate in 20 µl reactions using Fast SYBR Green Master Mix (Applied Biosystems) and analysed using Fast System SDS software (Applied Biosystems); PCR products were verified by agarose gel electrophoresis. Gene expression data were analysed using the ΔΔCT method and normalized using ama-1 as an endogenous reference gene relative to N2 wildtype animals [112]. Primers were designed to span exon-exon boundaries and when applicable, mRNA was amplified upstream of mutant deletion/insertion sites. The primers used are listed as follows: ama-1 (PK1084/PK1085); clp-1 (PK1062/PK1063); clp-2 (PK1113/PK1114); clp-3 (PK1066/PK1067); clp-4 (clp-4_f2/clp-4_r2); clp-6 (clp-6_f2/clp-6_r2); clp-7 (PK1074/PK1087); clp-8 (clp-8_f2/clp-8_r2); clp-9 (clp-9_f2/clp-9_r2); clp-10 (clp-10_f2/clp-10_r2); clpr-1 (clpr1_f2/clpr1_r2); asp-1 (PK1095/PK1096); asp-2 (PK1097/PK1098); asp-3 (PK1099/PK1100); asp-4 (PK1101/PK1102); asp-5 (PK1103/PK1104); asp-6 (PK1105/PK1106).
Generation of transgenic animals
Worms were transformed by germ line microinjection [113]. Microinjection solutions were composed of the plasmid of interest at 10 µg/ml and a co-transformation marker, either 80 µg/ml of pRF4 rol-6 (su1006) or 50 µg/ml of TG96 sur-5p::gfp. At least three independent transgenic strains were generated and examined for each construct, but only those displayed in this manuscript are listed, as they are representative of the patterns observed for a given construct. The following extrachromosomal arrays were generated with the pRF4 co-transformation marker: crEx65 (clp-1p::nls::mrfp), crEx70 (clp-2p::nls::mrfp), crEx72 (clp-3p::nls::mrfp), crEx74 (clp-4::nls::mrfp), crEx78 (tra-3p::nls::mrfp), crEx83 (clp-6p::nls::mrfp), crEx79 (clp-7::nls::mrfp), crEx202 (clp-1p::gfp), crEx110 (hsp-16.64p::clp-1), crEx86 (hsp-16.64p::clp-2), crEx88 (hsp-16.64p::clp-4), crEx8 (hsp-16.64p::tra-3), crEx96 (hsp-16.64p::clp-7) and crEx333 (unc-54p::clp-1::mrfp). The following extrachromosomal arrays were generated with the TG96 co-transformation marker: crEx325 (unc-54p::clp-1), crEx141 (unc-54p::clp-2), crEx147 (unc-54p::clp-4), crEx263 (unc-54p::tra-3), crEx258 (unc-54p::clp-7), crEx241 (unc-47p::clp-1), crEx190 (unc-119p::clp-1), crEx319 (unc-54p::clp-1(C371A)), crEx335 (unc-54p::clp-1::mrfp) and crEx336 (unc-54p::clp-1(C371A)::mrfp).
The unc-54p::clp-1(crEx325) extrachromosomal array, crEx325 was chromosomally integrated by gamma irradiation using a 137Cs source (RX30/50, Gravatom Industries). The integrated strain, crIs4 (unc-54p::clp-1) was outcrossed five times with N2 prior to study.
Phenotypic analyses
Tissue-specific GFP reporters used to identify tissues expressing calpain reporters include: unc-119::gfp, a pan-neuronal marker [114]; unc-47::gfp, which is specifically expressed in GABAergic neurons of the ventral nerve cord [115]; myo-3::gfp::nls, which is expressed in all muscle cells except those of the pharynx [116]; scm::gfp, a seam cell marker [117]; and pes-6::gfp, a excretory cell GFP reporter.
Brood size and embryonic lethality was scored by placing individual L4 staged hermaphrodites on NGM plates and transferring them daily to a fresh plate until egg laying had ceased. The number of dead eggs (embryonic lethality) and live animals (brood size) were scored two days after transfer of the mother.
The percentage of animals displaying paralysis was scored on day 2 of adulthood after first obtaining synchronized populations of animals of the specified genotype by bleaching gravid adults with alkaline hypochlorite [118]. Phenotypic scoring was performed by gently prodding animals with a platinum pick and registering their response: Unc animals retained the ability to move, but with impaired mobility; paralyzed animals failed to migrate.
Phalloidin staining
Animals were stained with Alexa Fluor 594 phalloidin (Invitrogen). Briefly, day 2 adults displaying wildtype, Unc or paralyzed phenotypes were lyophilized in an Automatic Environmental Speedvac (Savant) prior to fixation in ice-cold acetone. Animals were resuspended in 20 µl S-Mix (0.2 M Na phosphate (pH 7.5), 1 mM MgCl2, 0.004% (w/v) SDS) containing 2 U Alexa Fluor 594 phalloidin, incubated for 1 hour in the dark and washed twice with PBS-Tween 20 (0.5%) (Sigma) before viewing.
Western blotting
Eighty day 2 adult animals were placed in 20 µl of SDS protein sample buffer, electrophoresed on 10% or 4–12% gradient SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Blots were incubated with primary antibodies at the following concentrations: anti-mRFP/GFP primary antibody (Invitrogen), 1∶1000; anti-myc 9E10, 1∶1000; anti-α tubulin (Abcam), 1∶4000; anti-actin (Sigma), 1∶1000. Protein was visualized using anti-mouse or anti-rabbit horseradish peroxidase (HRP) linked antibodies (Amersham) at 1∶5000 dilution and Western Lightning™ chemiluminescent substrate (Perkin Elmer). Nitrocellulose membranes were stripped using Restore Plus stripping buffer (Thermo Scientific). Protein levels were quantified using ChemiDoc-It imaging system (UVP).
Nemadipine-A treatment
Worms were grown in wells of 24-well plates containing 1 ml of MYOB agar [119], including 5 µM nemadipine-A (kindly provided by Peter Roy) or 0.01% DMSO (control), as described [72]. Plates were incubated at 20°C, except those carrying hlh-1(cc561ts) mutants, which were raised at 15°C. Phenotypic scoring is described above.
Microscopy
Animals were immobilized using 10 mM sodium azide. Differential interference contrast (DIC) and fluorescent images were captured with a Zeiss Axioskop 2 fitted with an ORCA-ER (Hamamatsu) digital camera driven by Openlab 4 software (Improvision), or a Zeiss LSM 710 confocal driven by Zeiss Zen software.
Statistical analysis
Statistical analysis was performed using Student's T-test.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. SorimachiHHataSOnoY 2010 Expanding members and roles of the calpain superfamily and their genetically modified animals. Exp Anim 59 549 566
2. StroblSFernandez-CatalanCBraunMHuberRMasumotoH 2000 The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proc Natl Acad Sci U S A 97 588 592
3. HosfieldCMElceJSDaviesPLJiaZ 1999 Crystal structure of calpain reveals the structural basis for Ca(2+)-dependent protease activity and a novel mode of enzyme activation. EMBO J 18 6880 6889
4. LinGDChattopadhyayDMakiMWangKKCarsonM 1997 Crystal structure of calcium bound domain VI of calpain at 1.9 A resolution and its role in enzyme assembly, regulation, and inhibitor binding. Nat Struct Biol 4 539 547
5. BlanchardHGrochulskiPLiYArthurJSDaviesPL 1997 Structure of a calpain Ca(2+)-binding domain reveals a novel EF-hand and Ca(2+)-induced conformational changes. Nat Struct Biol 4 532 538
6. SorimachiHSuzukiK 2001 The structure of calpain. J Biochem 129 653 664
7. GollDEThompsonVFLiHWeiWCongJ 2003 The calpain system. Physiol Rev 83 731 801
8. MoldoveanuTHosfieldCMLimDElceJSJiaZ 2002 A Ca(2+) switch aligns the active site of calpain. Cell 108 649 660
9. HataSDoiNKitamuraFSorimachiH 2007 Stomach-specific calpain, nCL-2/calpain 8, is active without calpain regulatory subunit and oligomerizes through C2-like domains. J Biol Chem 282 27847 27856
10. KinbaraKIshiuraSTomiokaSSorimachiHJeongSY 1998 Purification of native p94, a muscle-specific calpain, and characterization of its autolysis. Biochem J 335 Pt 3 589 596
11. RavulapalliRDiazBGCampbellRLDaviesPL 2005 Homodimerization of calpain 3 penta-EF-hand domain. Biochem J 388 585 591
12. NishimuraTGollDE 1991 Binding of calpain fragments to calpastatin. J Biol Chem 266 11842 11850
13. OtsukaYGollDE 1987 Purification of the Ca2+-dependent proteinase inhibitor from bovine cardiac muscle and its interaction with the millimolar Ca2+-dependent proteinase. J Biol Chem 262 5839 5851
14. CroallDEErsfeldK 2007 The calpains: modular designs and functional diversity. Genome Biol 8 218
15. StorrSJCarragherNOFrameMCParrTMartinSG 2011 The calpain system and cancer. Nat Rev Cancer 11 364 374
16. FriedrichPTompaPFarkasA 2004 The calpain-system of Drosophila melanogaster: coming of age. Bioessays 26 1088 1096
17. Consortium 1998 Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282 2012 2018
18. LebartMCBenyaminY 2006 Calpain involvement in the remodeling of cytoskeletal anchorage complexes. FEBS J 273 3415 3426
19. FrancoSJHuttenlocherA 2005 Regulating cell migration: calpains make the cut. J Cell Sci 118 3829 3838
20. GollDENetiGMaresSWThompsonVF 2008 Myofibrillar protein turnover: the proteasome and the calpains. J Anim Sci 86 E19 35
21. EvansJSTurnerMD 2007 Emerging functions of the calpain superfamily of cysteine proteases in neuroendocrine secretory pathways. J Neurochem 103 849 859
22. JanossyJUbezioPApatiAMagocsiMTompaP 2004 Calpain as a multi-site regulator of cell cycle. Biochem Pharmacol 67 1513 1521
23. JohnsonJDHanZOtaniKYeHZhangY 2004 RyR2 and calpain-10 delineate a novel apoptosis pathway in pancreatic islets. J Biol Chem 279 24794 24802
24. TomimatsuYIdemotoSMoriguchiSWatanabeSNakanishiH 2002 Proteases involved in long-term potentiation. Life Sci 72 355 361
25. RichardIBrouxOAllamandVFougerousseFChiannilkulchaiN 1995 Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 81 27 40
26. HorikawaYOdaNCoxNJLiXOrho-MelanderM 2000 Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26 163 175
27. YoshikawaYMukaiHHinoFAsadaKKatoI 2000 Isolation of two novel genes, down-regulated in gastric cancer. Jpn J Cancer Res 91 459 463
28. HataSAbeMSuzukiHKitamuraFToyama-SorimachiN 2010 Calpain 8/nCL-2 and calpain 9/nCL-4 constitute an active protease complex, G-calpain, involved in gastric mucosal defense. PLoS Genet 6 e1001040 doi:10.1371/journal.pgen.1001040
29. TompaPBuzder-LantosPTantosAFarkasASzilagyiA 2004 On the sequential determinants of calpain cleavage. J Biol Chem 279 20775 20785
30. CuerrierDMoldoveanuTDaviesPL 2005 Determination of peptide substrate specificity for mu-calpain by a peptide library-based approach: the importance of primed side interactions. J Biol Chem 280 40632 40641
31. CarragherNO 2006 Calpain inhibition: a therapeutic strategy targeting multiple disease states. Curr Pharm Des 12 615 638
32. TurkVTurkBGuncarGTurkDKosJ 2002 Lysosomal cathepsins: structure, role in antigen processing and presentation, and cancer. Adv Enzyme Regul 42 285 303
33. BarnesTMHodgkinJ 1996 The tra-3 sex determination gene of Caenorhabditis elegans encodes a member of the calpain regulatory protease family. EMBO J 15 4477 4484
34. SokolSBKuwabaraPE 2000 Proteolysis in Caenorhabditis elegans sex determination: cleavage of TRA-2A by TRA-3. Genes Dev 14 901 906
35. SyntichakiPXuKDriscollMTavernarakisN 2002 Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 419 939 944
36. HuangLHanna-RoseW 2006 EGF signaling overcomes a uterine cell death associated with temporal mis-coordination of organogenesis within the C. elegans egg-laying apparatus. Dev Biol 300 599 611
37. LukeCJPakSCAskewYSNavigliaTLAskewDJ 2007 An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell 130 1108 1119
38. SteinLDBaoZBlasiarDBlumenthalTBrentMR 2003 The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol 1 e45 doi:10.1371/journal.pbio.0000045
39. SrivastavaMBegovicEChapmanJPutnamNHHellstenU 2008 The Trichoplax genome and the nature of placozoans. Nature 454 955 960
40. PutnamNHSrivastavaMHellstenUDirksBChapmanJ 2007 Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317 86 94
41. ChapmanJAKirknessEFSimakovOHampsonSEMitrosT 2010 The dynamic genome of Hydra. Nature 464 592 596
42. SrivastavaMSimakovOChapmanJFaheyBGauthierME 2010 The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466 720 726
43. DehalPSatouYCampbellRKChapmanJDegnanB 2002 The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 298 2157 2167
44. BatemanACoinLDurbinRFinnRDHollichV 2004 The Pfam protein families database. Nucleic Acids Res 32 D138 141
45. EddySR 1998 Profile hidden Markov models. Bioinformatics 14 755 763
46. TonamiKKuriharaYAburataniHUchijimaYAsanoT 2007 Calpain 6 is involved in microtubule stabilization and cytoskeletal organization. Mol Cell Biol 27 2548 2561
47. ConsortiumU 2011 Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res 39 D214 219
48. BradleyRKRobertsASmootMJuvekarSDoJ 2009 Fast statistical alignment. PLoS Comput Biol 5 e1000392 doi:10.1371/journal.pcbi.1000392
49. SimmerFMoormanCvan der LindenAMKuijkEvan den BerghePV 2003 Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol 1 e12 doi:10.1371/journal.pbio.0000012
50. HodgkinJ 1986 Sex determination in the nematode C. elegans: analysis of tra-3 suppressors and characterization of fem genes. Genetics 114 15 52
51. DuttPCroallDEArthurJSVeyraTDWilliamsK 2006 m-Calpain is required for preimplantation embryonic development in mice. BMC Dev Biol 6 3
52. RaynaudFMarcilhacAChebliKBenyaminYRosselM 2008 Calpain 2 expression pattern and sub-cellular localization during mouse embryogenesis. Int J Dev Biol 52 383 388
53. BiswasSHarrisFPhoenixDA 2001 Treatment of cataracts - vision for the future. Biologist (London) 48 273 277
54. SpencerMJMellgrenRL 2002 Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Hum Mol Genet 11 2645 2655
55. YamashimaT 2000 Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol 62 273 295
56. StringhamEGDixonDKJonesDCandidoEP 1992 Temporal and spatial expression patterns of the small heat shock (hsp16) genes in transgenic Caenorhabditis elegans. Mol Biol Cell 3 221 233
57. MillerDMStockdaleFEKarnJ 1986 Immunological identification of the genes encoding the four myosin heavy chain isoforms of Caenorhabditis elegans. Proc Natl Acad Sci U S A 83 2305 2309
58. ArthurJSGauthierSElceJS 1995 Active site residues in m-calpain: identification by site-directed mutagenesis. FEBS Lett 368 397 400
59. RaserKJPosnerAWangKK 1995 Casein zymography: a method to study mu-calpain, m-calpain, and their inhibitory agents. Arch Biochem Biophys 319 211 216
60. GieselerKGrisoniKSegalatL 2000 Genetic suppression of phenotypes arising from mutations in dystrophin-related genes in Caenorhabditis elegans. Curr Biol 10 1092 1097
61. KellyWGXuSMontgomeryMKFireA 1997 Distinct requirements for somatic and germline expression of a generally expressed Caernorhabditis elegans gene. Genetics 146 227 238
62. HagedornEJYashiroHZielJWIharaSWangZ 2009 Integrin acts upstream of netrin signaling to regulate formation of the anchor cell's invasive membrane in C. elegans. Dev Cell 17 187 198
63. KoenigMHoffmanEPBertelsonCJMonacoAPFeenerC 1987 Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50 509 517
64. BessouCGiugiaJBFranksCJHolden-DyeLSegalatL 1998 Mutations in the Caenorhabditis elegans dystrophin-like gene dys-1 lead to hyperactivity and suggest a link with cholinergic transmission. Neurogenetics 2 61 72
65. Carre-PierratMGrisoniKGieselerKMariolMCMartinE 2006 The SLO-1 BK channel of Caenorhabditis elegans is critical for muscle function and is involved in dystrophin-dependent muscle dystrophy. J Mol Biol 358 387 395
66. BaylisHAFuruichiTYoshikawaFMikoshibaKSattelleDB 1999 Inositol 1,4,5-trisphosphate receptors are strongly expressed in the nervous system, pharynx, intestine, gonad and excretory cell of Caenorhabditis elegans and are encoded by a single gene (itr-1). J Mol Biol 294 467 476
67. KimHRogersMJRichmondJEMcIntireSL 2004 SNF-6 is an acetylcholine transporter interacting with the dystrophin complex in Caenorhabditis elegans. Nature 430 891 896
68. GieselerKBessouCSegalatL 1999 Dystrobrevin - and dystrophin-like mutants display similar phenotypes in the nematode Caenorhabditis elegans. Neurogenetics 2 87 90
69. GrisoniKGieselerKMariolMCMartinECarre-PierratM 2003 The stn-1 syntrophin gene of C.elegans is functionally related to dystrophin and dystrobrevin. J Mol Biol 332 1037 1046
70. LapidosKAKakkarRMcNallyEM 2004 The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res 94 1023 1031
71. MariolMCSegalatL 2001 Muscular degeneration in the absence of dystrophin is a calcium-dependent process. Curr Biol 11 1691 1694
72. KwokTCRickerNFraserRChanAWBurnsA 2006 A small-molecule screen in C. elegans yields a new calcium channel antagonist. Nature 441 91 95
73. AguinaldoAMTurbevilleJMLinfordLSRiveraMCGareyJR 1997 Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387 489 493
74. TakanoJTomiokaMTsubukiSHiguchiMIwataN 2005 Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem 280 16175 16184
75. RizoJSudhofTC 1998 C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem 273 15879 15882
76. MegeneyLAKablarBGarrettKAndersonJERudnickiMA 1996 MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev 10 1173 1183
77. GaillyPDe BackerFVan SchoorMGillisJM 2007 In situ measurements of calpain activity in isolated muscle fibres from normal and dystrophin-lacking mdx mice. J Physiol 582 1261 1275
78. HopfFWTurnerPRSteinhardtRA 2007 Calcium misregulation and the pathogenesis of muscular dystrophy. Subcell Biochem 45 429 464
79. AllenDGZhangBTWhiteheadNP 2010 Stretch-induced membrane damage in muscle: comparison of wild-type and mdx mice. Adv Exp Med Biol 682 297 313
80. BanoDYoungKWGuerinCJLefeuvreRRothwellNJ 2005 Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120 275 285
81. KopilCMVaisHCheungKHSiebertAPMakDO Calpain-cleaved type 1 inositol 1,4,5-trisphosphate receptor (InsP(3)R1) has InsP(3)-independent gating and disrupts intracellular Ca(2+) homeostasis. J Biol Chem 286 35998 36010
82. Shoshan-BarmatzVWeilSMeyerHVarsanyiMHeilmeyerLM 1994 Endogenous, Ca(2+)-dependent cysteine-protease cleaves specifically the ryanodine receptor/Ca2+ release channel in skeletal muscle. J Membr Biol 142 281 288
83. DaytonWR 1982 Comparison of low - and high-calcium-requiring forms of the calcium-activated protease with their autocatalytic breakdown products. Biochim Biophys Acta 709 166 172
84. KumamotoTKleeseWCCongJYGollDEPiercePR 1992 Localization of the Ca(2+)-dependent proteinases and their inhibitor in normal, fasted, and denervated rat skeletal muscle. Anat Rec 232 60 77
85. Di LisaFDe TullioRSalaminoFBarbatoRMelloniE 1995 Specific degradation of troponin T and I by mu-calpain and its modulation by substrate phosphorylation. Biochem J 308 Pt 1 57 61
86. RaynaudFBonnalCFernandezEBremaudLCeruttiM 2003 The calpain 1-alpha-actinin interaction. Resting complex between the calcium-dependent protease and its target in cytoskeleton. Eur J Biochem 270 4662 4670
87. GalvezASDiwanAOdleyAMHahnHSOsinskaH 2007 Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ Res 100 1071 1078
88. NetiGNovakSMThompsonVFGollDE 2009 Properties of easily releasable myofilaments: are they the first step in myofibrillar protein turnover? Am J Physiol Cell Physiol 296 C1383 1390
89. FoxRMWatsonJDVon StetinaSEMcDermottJBrodiganTM 2007 The embryonic muscle transcriptome of Caenorhabditis elegans. Genome Biol 8 R188
90. SokolSB 2000 Regulatory proteolysis in C. elegans sex determination: Molecular, genetic and biochemical analysis of the calpain protease TRA-3 Cambridge Cambridge
91. EtheridgeTOczypokEALehmannSFieldsBDShephardF Calpains Mediate Integrin Attachment Complex Maintenance of Adult Muscle in Caenorhabditis elegans. PLoS Genet 8 e1002471 doi:10.1371/journal.pgen.1002471
92. MeissnerBWarnerAWongKDubeNLorchA 2009 An integrated strategy to study muscle development and myofilament structure in Caenorhabditis elegans. PLoS Genet 5 e1000537 doi:10.1371/journal.pgen.1000537
93. GhoshSRHopeIA 2010 Determination of the mobility of novel and established Caenorhabditis elegans sarcomeric proteins in vivo. Eur J Cell Biol 89 437 448
94. GrisoniKMartinEGieselerKMariolMCSegalatL 2002 Genetic evidence for a dystrophin-glycoprotein complex (DGC) in Caenorhabditis elegans. Gene 294 77 86
95. LecroiseyCMartinEMariolMCGrangerLSchwabY 2008 DYC-1, a protein functionally linked to dystrophin in Caenorhabditis elegans is associated with the dense body, where it interacts with the muscle LIM domain protein ZYX-1. Mol Biol Cell 19 785 796
96. DargelosEPoussardSBruleCDauryLCottinP 2008 Calcium-dependent proteolytic system and muscle dysfunctions: a possible role of calpains in sarcopenia. Biochimie 90 359 368
97. FraysseBDesaphyJFRollandJFPiernoSLiantonioA 2006 Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and restoration by growth hormone. Neurobiol Dis 21 372 380
98. AnderssonDCBetzenhauserMJReikenSMeliACUmanskayaA 2011 Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 14 196 207
99. MakiMNarayanaSVHitomiK 1997 A growing family of the Ca2+-binding proteins with five EF-hand motifs. Biochem J 328 Pt 2 718 720
100. BrennerS 1974 The genetics of Caenorhabditis elegans. Genetics 77 71 94
101. HarfeBDBrandaCSKrauseMSternMJFireA 1998 MyoD and the specification of muscle and non-muscle fates during postembryonic development of the C. elegans mesoderm. Development 125 2479 2488
102. LeeRYLobelLHengartnerMHorvitzHRAveryL 1997 Mutations in the alpha1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. EMBO J 16 6066 6076
103. ClandininTRDeModenaJASternbergPW 1998 Inositol trisphosphate mediates a RAS-independent response to LET-23 receptor tyrosine kinase activation in C. elegans. Cell 92 523 533
104. DaviesAGPierce-ShimomuraJTKimHVanHovenMKThieleTR 2003 A central role of the BK potassium channel in behavioral responses to ethanol in C. elegans. Cell 115 655 666
105. ZhaoGZhaoYPanBLiuJHuangX 2007 Hypersensitivity of BKCa to Ca2+ sparks underlies hyporeactivity of arterial smooth muscle in shock. Circ Res 101 493 502
106. BarnesTMJinYHorvitzHRRuvkunGHekimiS 1996 The Caenorhabditis elegans behavioral gene unc-24 encodes a novel bipartite protein similar to both erythrocyte band 7.2 (stomatin) and nonspecific lipid transfer protein. J Neurochem 67 46 57
107. PriceMPThompsonRJEshcolJOWemmieJABensonCJ 2004 Stomatin modulates gating of acid-sensing ion channels. J Biol Chem 279 53886 53891
108. SedenskyMMSiefkerJMKohJYMillerDM3rdMorganPG 2004 A stomatin and a degenerin interact in lipid rafts of the nervous system of Caenorhabditis elegans. Am J Physiol Cell Physiol 287 C468 474
109. ZhangSArnadottirJKellerCCaldwellGAYaoCA 2004 MEC-2 is recruited to the putative mechanosensory complex in C. elegans touch receptor neurons through its stomatin-like domain. Curr Biol 14 1888 1896
110. TimmonsLCourtDLFireA 2001 Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263 103 112
111. KamathRSAhringerJ 2003 Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30 313 321
112. LivakKJSchmittgenTD 2001 Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25 402 408
113. MelloCFireA 1995 DNA transformation. Methods Cell Biol 48 451 482
114. LimYSMallapurSKaoGRenXCWadsworthWG 1999 Netrin UNC-6 and the regulation of branching and extension of motoneuron axons from the ventral nerve cord of Caenorhabditis elegans. J Neurosci 19 7048 7056
115. McIntireSLReimerRJSchuskeKEdwardsRHJorgensenEM 1997 Identification and characterization of the vesicular GABA transporter. Nature 389 870 876
116. FireAXuSMontgomeryMKKostasSADriverSE 1998 Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391 806 811
117. TernsRMKroll-ConnerPZhuJChungSRothmanJH 1997 A deficiency screen for zygotic loci required for establishment and patterning of the epidermis in Caenorhabditis elegans. Genetics 146 185 206
118. SulstonJEHodgkinJ 1988 Methods. WoodWB The Nematode Caenorhabditis elegans Cold Spring Harbor Cold Spring Harbor University Press 587 606
119. ChurchDLGuanKLLambieEJ 1995 Three genes of the MAP kinase cascade, mek-2, mpk-1/sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development 121 2525 2535
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- PIF4–Mediated Activation of Expression Integrates Temperature into the Auxin Pathway in Regulating Hypocotyl Growth
- Metabolic Profiling of a Mapping Population Exposes New Insights in the Regulation of Seed Metabolism and Seed, Fruit, and Plant Relations
- A Splice Site Variant in the Bovine Gene Compromises Growth and Regulation of the Inflammatory Response
- Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson's Disease Genetics: The PDGene Database
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy