
Widespread Endogenization of Genome Sequences of Non-Retroviral RNA Viruses into Plant Genomes
Non-retroviral RNA virus sequences (NRVSs) have been found in the chromosomes of vertebrates and fungi, but not plants. Here we report similarly endogenized NRVSs derived from plus-, negative-, and double-stranded RNA viruses in plant chromosomes. These sequences were found by searching public genomic sequence databases, and, importantly, most NRVSs were subsequently detected by direct molecular analyses of plant DNAs. The most widespread NRVSs were related to the coat protein (CP) genes of the family Partitiviridae which have bisegmented dsRNA genomes, and included plant - and fungus-infecting members. The CP of a novel fungal virus (Rosellinia necatrix partitivirus 2, RnPV2) had the greatest sequence similarity to Arabidopsis thaliana ILR2, which is thought to regulate the activities of the phytohormone auxin, indole-3-acetic acid (IAA). Furthermore, partitivirus CP-like sequences much more closely related to plant partitiviruses than to RnPV2 were identified in a wide range of plant species. In addition, the nucleocapsid protein genes of cytorhabdoviruses and varicosaviruses were found in species of over 9 plant families, including Brassicaceae and Solanaceae. A replicase-like sequence of a betaflexivirus was identified in the cucumber genome. The pattern of occurrence of NRVSs and the phylogenetic analyses of NRVSs and related viruses indicate that multiple independent integrations into many plant lineages may have occurred. For example, one of the NRVSs was retained in Ar. thaliana but not in Ar. lyrata or other related Camelina species, whereas another NRVS displayed the reverse pattern. Our study has shown that single - and double-stranded RNA viral sequences are widespread in plant genomes, and shows the potential of genome integrated NRVSs to contribute to resolve unclear phylogenetic relationships of plant species.
Published in the journal:
. PLoS Pathog 7(7): e32767. doi:10.1371/journal.ppat.1002146
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002146
Summary
Non-retroviral RNA virus sequences (NRVSs) have been found in the chromosomes of vertebrates and fungi, but not plants. Here we report similarly endogenized NRVSs derived from plus-, negative-, and double-stranded RNA viruses in plant chromosomes. These sequences were found by searching public genomic sequence databases, and, importantly, most NRVSs were subsequently detected by direct molecular analyses of plant DNAs. The most widespread NRVSs were related to the coat protein (CP) genes of the family Partitiviridae which have bisegmented dsRNA genomes, and included plant - and fungus-infecting members. The CP of a novel fungal virus (Rosellinia necatrix partitivirus 2, RnPV2) had the greatest sequence similarity to Arabidopsis thaliana ILR2, which is thought to regulate the activities of the phytohormone auxin, indole-3-acetic acid (IAA). Furthermore, partitivirus CP-like sequences much more closely related to plant partitiviruses than to RnPV2 were identified in a wide range of plant species. In addition, the nucleocapsid protein genes of cytorhabdoviruses and varicosaviruses were found in species of over 9 plant families, including Brassicaceae and Solanaceae. A replicase-like sequence of a betaflexivirus was identified in the cucumber genome. The pattern of occurrence of NRVSs and the phylogenetic analyses of NRVSs and related viruses indicate that multiple independent integrations into many plant lineages may have occurred. For example, one of the NRVSs was retained in Ar. thaliana but not in Ar. lyrata or other related Camelina species, whereas another NRVS displayed the reverse pattern. Our study has shown that single - and double-stranded RNA viral sequences are widespread in plant genomes, and shows the potential of genome integrated NRVSs to contribute to resolve unclear phylogenetic relationships of plant species.
Introduction
Events of horizontal gene transfer (HGT) have been identified between various combinations of viruses and their eukaryotic hosts. HGT can occur during evolution in 2 inverse directions: “from host to virus” or “from virus to host.” In the host to virus direction, viral acquisition of host genes is observed as insertion of cellular genes for proteases (see [1] for review), ubiquitin [2], chloroplast protein [3] and heat-shock proteins [4], [5] into viral genomes. The virus to host direction involves endogenization of viral genes. Fossil sequences of viral origin, mostly from retroviruses, have been detected in many animal genomes. However, retrovirus sequences have not been identified in plants; instead, reverse-transcribing DNA viruses (pararetroviruses) have been identified. Although pararetroviral sequences have been found in some plant nuclear genomes [6], [7], [8], [9], only a limited number of integrated sequences are exogenized to launch virus infection; however, their cellular functions remain unclear in other examples.
In contrast, the sequences of non-retroviral RNA viruses were considered not to integrate into host chromosomes. However, recent reports identified endogenized genes of non-retroviral elements in mammals [10], [11], [12], [13]. Examples include the nucleocapsid protein (N) and nucleoprotein (NP) genes of bornaviruses and filoviruses, members of the negative-strand RNA virus group in the order Mononegavirales [11], [12], [14]. While some integrated N genes are expressed, their biological significance is unclear. Identification of these sequences contrasts with the lack of evidence for negative-strand RNA virus genome integration into plant genomes. Furthermore, RNA-dependent RNA polymerase (RdRp) and capsid protein (CP) coding domains from a group of monopartite dsRNA viruses have been identified in yeast chromosomes, and while some of these viruses appear to be expressed, their biological significance has not been explored [15], [16], [17].
The white root rot fungus Rosellinia necatrix is a soil-borne phytopathogenic ascomycetous fungus that causes damages to perennial crops. An extensive search of a large collection of field fungal isolates (over 1,000) was conducted to identify dsRNA (mycoviruses) that may serve as virocontrol (biological control) agents. Approximately 20% of field isolates were infected with known or unknown viral strains [18], [19], [20]. During molecular characterization of these viruses, we identified a novel partitivirus termed Rosellinia necatrix partitivirus 2 (RnPV2) in an ill-defined R. necatrix strain. The family Partitiviridae contains members with small bi-segmented dsRNA genomes [21] that infect plants, fungi or protozoa. They are thought to replicate using virion-associated RdRp in the host cytoplasm, which are phylogenetically related to those from the picorna-like superfamily [22]. Surprisingly, the RnPV2 CP showed the highest level of sequence identity to an Arabidopsis thaliana gene, IAA/LEU resistant 2 (ILR2), which was previously shown to regulate the activity of the phytohormone auxin [23]. Combined with information regarding integrated mononegaviral sequences in animals, this finding generated significant interest in searching currently available genome sequence data for not only dsRNA but also negative-strand viral sequences. In October 2010, Liu et al. [24] reported similar results based on an extensive search conducted in 2009. This group identified sequences in the chromosomes of diverse organisms that may have been acquired from monopartite (totiviruses and related unclassified viruses) and bipartite dsRNA viruses (partitiviruses).
We further examined plant genome sequences available as of December 10, 2010 for integrated sequences of not only partitivirus genomes but also negative-, and positive-strand RNA viruses (Table S1). Combining database searches and molecular analyses led to the identification of multiple endogenized sequences related to partitiviruses, cytorhabdoviruses, varicosaviruses and betaflexiviruses in the genomes of a variety of plants including those from the families Solanaceae and Brassicaceae. For example, while some partitivirus-related sequences are conserved on the orthologous locus across some genera, e.g., Arabidopsis, Capsella, Turritis, and Olimarabidopsis within the family Brassicaceae, others are retained in only a few species within a single genus, Arabidopsis. A similar integration pattern was observed for a rhabdovirus-related sequence in the family Solanaceae. These profiles of occurrence can potentially resolve unclear phylogenetic relationships between plants. Our study demonstrates widespread endogenization of non-retroviral RNA virus sequences (NRVSs) including sequences of plant positive - and negative-strand RNA viruses for the first time. We have proposed a model of viral gene transfer, in which NRVSs are suggested to be a factor constituting plant genomes.
Results
The CP sequence from a novel mycovirus shows the highest identity to a plant functional gene product, ILR2
We determined the complete nucleotide (nt) sequence of the genome segments (dsRNA1 and dsRNA2) of a novel partitivirus, RnPV2, from the white root rot fungus Rosellinia necatrix, a soil-borne phytopathogenic ascomycetous fungus. DsRNA2 was found to be 1828 nt long, encoding a polypeptide of 483 amino acids (aa) (CP, 54 kDa). Low-level sequence similarities among CPs from Partitiviridae family members were observed using a BLASTP search with RnPV2 CP against non-redundant sequences available in the NCBI database (http://www.ncbi.nlm.nih.gov/). Surprisingly, RnPV2 CP showed the highest degree of sequence similarity to ILR2 from Ar. thaliana. Notably, sequence similarities between RnPV2 CP and ILR2 were greater than those between the CP sequence from another mycovirus, Sclerotinia sclerotiorum partitivirus S (SsPV-S) and ILR2 noted previously [24]. ILR2 is known to regulate indole-3-acetic acid (IAA)-amino acid conjugate sensitivity and metal transport. An Ar. thaliana mutant with a single amino acid substitution in ILR2, known as ilr2-1, was shown to exhibit normal root elongation in the presence of a high concentration of exogenous IAA-leucine conjugates, which represses root elongation in wild-type lines [23].
RnPV2 CP-like sequences are conserved in some Brassicaceae spp. and Mimulus guttatus
Magidin et al. [23] identified 2 alleles of ILR2 in Ar. thaliana accessions (a long and a short allele) (Figure 1A). Although the authors confirmed ILR2 expression for only the WS ecotype (short allele), they determined that both short and long versions of ILR2 were functional. Given the similarity between ILR2 and RnPV2 CP sequences, we hypothesized that HGT occurred between the 2 organisms. Therefore, we assessed the extent to which ILR2 is conserved in plants. We used 3 approaches: BLAST search, genomic PCR, and Southern blot analyses. We first conducted an exhaustive BLAST (tblastn) search against genome sequence databases as described in the Materials and Methods. This search identified ILR2 homologs in Ar. lyrata and Mimulus guttatus (yellow monkey flower), which included both short and long versions of ILR2 homologs with modest levels of aa sequence identities (over 20%) to RnPV2 CP (Table S2, Figure 1A). Furthermore, a variety of partitivirus CP-related sequences with low-levels of aa sequence identities (approximately 20%) to RnPV2 CP were also detectable from genome sequences from other 17 plant species (Table 1). These sequences were classified into a total of 8 subgroups based on relatedness to best matched extant partitiviruses (Table 1). Their nomenclature is: AtPCLS1 (ILR2) is from Arabidopsis thaliana partitivirus CP-like sequence (PCLS) 1. Differently numbered PCLSs, referring to proteins potentially encoded by PCLSs, show the highest level of aa sequence identities to CPs encoded by different partitiviruses.


Genomic PCR analysis with primers corresponding to highly conserved 240-bp portions revealed that ILR2 homologs were retained in genera closely related to Arabidopsis, such as Capsella, Turritis, and Olimarabidopsis, but not in members of distantly-related genera, Brassica, Thellungiella, Crucihimalaya, Sisymbrium, and Thlaspi within the Brassicaceae family (Figure 1B). Genomic PCR fragments covering the entire ILR2-like domains of the plants shown in Table S4 were sequenced directly or after cloning into a plasmid. It should be noted that PCLS1s of closely related genera reside in an orthologous position [25], i.e., in a convergent configuration with the gene for the transmembrane Golgi matrix protein AtCASP, which shares a high degree of sequence similarity across kingdoms [26]. This notion was confirmed by genomic PCR in which a primer pair allowed detection of 0.75 - to 1-kb fragments spanning the CASP gene. Previous comparative genomics studies proposed a hypothesis that the Brassicaceae genomes consist of 24 (A to X) conserved genome blocks [27]. The ILR2 locus is on block F which is considered to be duplicated in B. rapa. A search against the Brassica database (BRAD) confirmed the absence of a PCLS1 on the 2 B. rapa loci that flank the CASP gene. Southern blotting with members of the Brassicaceae, Cucurbitaceae, Solanaceae, and Leguminosae families indicated that PCLS1 (ILR2) is present in Ar. thaliana and Cap. bursa-pastoris, but absent in the other plants (Figure 1C), consistent with BLAST results and genomic PCR analyses. Furthermore, the absence of ILR2 in Crucihimalaya lasiocarpa, Sisymbrium irio and B. rapa was confirmed by sequence analysis of genomic PCR fragments covering the entire ILR2 region and its flanking regions (Figure 1D).
Prevalence of partitivirus CP-like sequences (PCLS1 to PCLS8) in plant chromosomes
Genome sequences with low levels of similarities to RnPV2 CP included a number of PCLSs from various plants spanning more than 17 species from 8 families (Table 1). Most PCLSs confirmed to be present on their chromosomes of these organisms were identified by genomic PCR and/or Southern blotting and sequencing (Tables 1, S4). For instance, AtPCLS2 and Ar. lyrata PCLS3 (AlPCLS3) are retained on non-orthologous loci of ILR2s of Ar. thaliana and Ar. lyrata, respectively (Figure 2A). AtPCLS2 (At4g14104) resides between the genes for COP9 (constitutive photo-morphogenic-9, COP9) and an F-box protein, while AlPCLS3 is between 2 coding sequences for F-box domains corresponding to At4g02760 and At4g02740 [25]. AtPCLS2 and AlPCLS3 from 2 closely related plant species show the highest sequence identities to the CPs from 2 different partitiviruses: Raphanus sativus cryptic virus 2 (RSCV2) and Fragaria chiloensis cryptic virus (FCCV) (dsRNA2) [28]. The PCLS retention profile was revealed by genomic PCR using 2 primer sets. A primer set designed to amplify internal AtPCLS2 sequences provided DNA fragments of an expected size of 470 bp in Ar. thaliana accessions Col-0, Ler, and Shokei, but not in Ar. lyrata, Ar. Arenosa, or Cap. rubella (Figure 2B, top panel). A different primer set specific for AtPCLS2 and the F-box protein gene (At4g14103) gave the same amplification pattern (Figure 2B, second panel) as shown in the top panel. Using the same approach with 2 sets of primers, PCLS3 was detected by genomic PCR in Ar. lyrata and Ar. arenosa, while no such sequence was observed in Ar. thaliana ecotypes or Cap. rubella (Figure 2B, third and fourth panels). Although the COP9 and the F-box protein genes are conserved on the corresponding loci of Ar. lyrata, no counterpart of AtPCLS2 was identified between the genes (Phytozome). Similarly, no AlPCLS3 homolog was observed on the corresponding chromosomal position of Ar. thaliana [25].

PCLS4 and PCLS5 were found in the genome sequence databases of B. rapa (BrPCLS4 and 5), Solanum phureja (wild species of potato) (SpPCLS5) (Figure 3A, S2), and Nicotiana tabacum (NtPCLS5-1 and -2) (Figure S1A). These sequences commonly exhibited greater sequence similarity to CPs of previously reported plant partitiviruses than to RnPV2 CP (Tables 1). The 3 PCLS5s from the Solanaceae family were very similar to each other (approximately 60% aa sequence identity), and showed high sequence identity (over 45%) (Table S2) to CP of Raphanus sativus cryptic virus 1 (RSCV1, plant partitivirus) [29]. Two PCLSs, BrPCLS4 (Bra021820) and BrPCLS5 (Bra020160), which are detected on different scaffolds, were determined to not flank the CASP gene of B. rapa as AtPCLS1 (ILR2) does. BrPCLS4 and 5 show much greater aa sequence identities to CPs of RSCV1 and carrot cryptic virus 1 (CaCV1, plant partitivirus) [30] than it does to RnPV2 CP (Table S2).

Molecular analyses were performed to determine how widely these PCLS4 and PCLS5 are conserved. Genomic PCR using a primer set specific for BrPCLS4 detected related sequences in all Brassica species tested, but not in other plants including members of the family Solanaceae or genera other than Brassica in Brassicaceae, such as Ar. thaliana, Cru. lasiocarpa, Thellungiella parvula, Thl. arvense and Sis. irio, and Raphanus sativus (Figure 3B, top panel). For BrPCLS5, the primer set, PC5a-1 and PC5a-2 enabled detection of expected PCR fragments in all Brassica plants in addition to R. sativus, while no PCR fragments were amplified in the other plant species (Figure 3B, second panels). A different detection profile was obtained by genomic PCR with a primer set specific for SpPCLS5 in which PCLS5-related sequences were detectable only in Sol. tuberosum and Sol. lycopersicum (Figure 3B, third and fourth panels). We failed to yield amplification from all other tested plants in the families Brassicaceae and Solanaceae including Sol. melongena. Interestingly, PCLS5, but not PCLS4 fragments, were detected in R. sativus. Moreover, the presence or absence of PCLSs was confirmed by genomic Southern analysis. As expected from the genomic PCR results, hybridization signals were detected with a BrPCLS4 - or a BrPCLS5-specific probe in the Brassica species such as B. rapa and B. oleracea (Figure 3C, top and second panels); however, the numbers and signal positions differed between the 2 blots. The StPCLS4-specific probe allowed detection of 2 and 1 hybridization signals in Sol. tuberosum and Sol. lycopersicum, respectively, but not in any other plants examined in this study (Figure 3C, fourth panel).
In addition to PCLS1 to PCLS5, 2 other subgroups of PCLSs (PCLS6 and PCLS7) were observed in the GSS database of N. tabacum and showed an interesting detection pattern in Nicotiana species (Figure S1). NtPCLS6 and NtPCLS7 showed moderate aa sequence identities to CPs encoded by FCCV dsRNA3 (38%) [28] and RSCV3 dsRNA2 (30%) [29], respectively. Sequencing of genomic PCR fragments and Southern blotting (Figure S1B, E) suggested that NtPCLS5-1 and NtPCLS5-2 are retained only in N. tabacum, but not in other Nicotiana species examined, such as N. benthamiana and N. megalosiphon, whereas PCLS6 was detected in both N. tabacum and N. megalosiphon (Figure S1B). In contrast, PCLS7 is conserved in all 4 Nicotiana plants tested, although sequence divergence was observed among the PCLS7s. Other PCLSs from 2 legume plants, MtPCLS7 and LjPCLS8 were identified on their nuclear genomes by PCR (Figures S1A, C, D).
Phylogenetic analysis of the PCLSs
An expanded BLAST (tblastn) search against the EST sequence libraries (in NCBI) helped detect many related sequences of possible plant partitiviruses that shared moderate levels of sequence similarity. Some representative EST sequences, PCLSs and partitivirus CPs, whose entire sequences are available, were aligned using the MAFFT program. Three relatively well-conserved motifs are located on the N - terminal, central, and C-terminal regions of partitivirus CPs and PCLSs, and are represented by PGPLxxxF [31], F/WxGSxxL and GpfW domains (Figure S2). As expected from sequence similarities, phylogenetic analysis of partitivirus CPs and PCLSs identified in plant genomes clearly show that members of each PCLSs subgroup (PCLS1, 2, 4, 5, 7, 8) clusters together with the CP of the respective partitivirus that shows the highest sequence similarities (Figure 4, Table 1). For example, RnPV2 CP (in red), MgPCLS1, and ILR2 homologs (PCLS1s) from Arabidopsis-related genera (in green) constitute one group in the tree. The MgPCLS1 clade includes an assembled sequence in the EST database from meadow fescue (Festuca pratensis) (in purple) believed to be from a plant partitivirus. Another group includes PCLS5s from the families Brassicaceae and Solanaceae (in green), CPs of fungal (in red) and plant partitiviruses (in blue) are grouped together. Within this group, PCLSs from the families Brassicaceae (BrPCLS5, BoPCLS5, and BnPCLS5) and Solanaceae (StPCLS5, SpPCLS5, SlPCLS5, and NtPCLS5-1) comprised 2 subgroups that included CPs encoded by RSCV1 (CP) and RSCV1 dsRNA3 (Figure 4), respectively, which are considered to be from two different partitiviruses. PCLS4s from members of the genus Brassica clustered together with CPs of other plant partitiviruses including white clover cryptic virus 1 (WCCV1) [32], CaCV1, beet cryptic virus 1 (BCV1) [33], and vicia cryptic virus (VCV) [34].

The tree topology shown in Figure 4 was similar to that reported by Liu et al. [24]. The current study used more PCLSs detected in various plants but not partial PCLSs such as PCLS3 and NtPCLS5-2, 6 and 7 (Tobacco Contig-2, -3 and -4) analyzed phylogenetically by Liu et al. [24].
Detection of negative-strand RNA viral sequences in plant nuclear genomes
Because negative-strand RNA viral sequences are found in animal chromosomes, we searched for negative-strand RNA viral sequences (Table S1) in plant genomes as described in the Materials and Methods. This search identified sequences related to the N protein in members of the genus Cytorhabdovirus (Lettuce necrotic yellows virus, LNYV, Lettuce yellow mottle virus, LYMoV, and northern cereal mosaic virus, NCMV) and a CP of the genus Varicosavirus (Lettuce big-vein associated virus, LBVaV) in the genomes of a variety of plants such as Populus trichocarpa, N. tabacum, and B. rapa (Figures 5, S3, Table 2). While varicosaviruses have bipartite genomes replicated in the cytoplasm of infected plant cells, they are phylogenetically closely related to cytorhabdoviruses with monopartite genomes [35], [36]. Varicosavirus CP is phylogenetically and functionally equivalent to rhabdovirus N. Thus, these plant nuclear sequences were designated as rhabdovirus N-like sequences (RNLSs) and classified into 4 subgroups (RNLS1 to RNLS4) based on the sequences of presently existing viruses with the highest levels of sequence similarities (Table 2). Their potentially encoding proteins were designated as RNLSs as in the case for PCLSs.
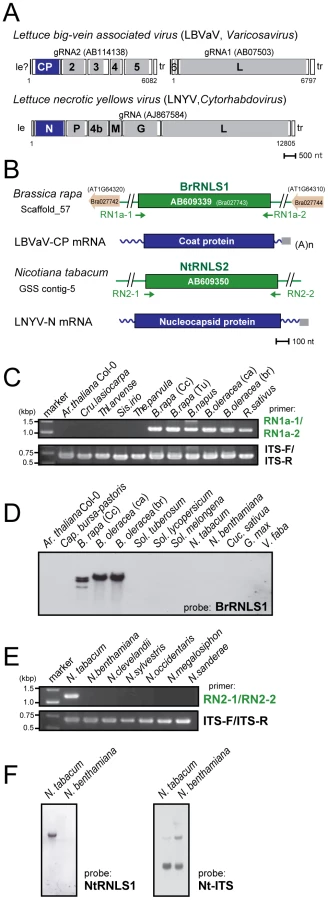

To confirm the presence of the RNLSs in plant chromosomes, we conducted genomic PCR and Southern blot analyses. Interestingly genomic PCR with primers specific for an RNLS1 from B. rapa (BrRNLS1) detected RNLS1s in R. sativus and all tested plants within the Brassica genus, but not in members in other genera (Figure 5C), in a pattern similar to that of PCLS5s from the family Brassicaceae (Figure 3B). Consistent with these results, Southern blotting detected hybridization signals in 3 Brassica plants (Figure 5D) with a probe specific for BrRNLS1.
The NtRNLS2 sequence was detected in N. tabacum, while no fragments were generated from other Nicotiana species using genomic PCR (Figure 5E). Southern blotting results supported this detection profile (Figure 5F); N. tabacum, but not N. benthamiana, was shown to carry an NtRNLS2-related sequence (Figure 5F, left panel).
All other RNLSs discovered through the similarity search of genome sequence databanks (Table 2), except for PtRNLS4 from Pop. trichocarpa and TcRNLS1 from Theobroma cacao, were shown to be retained on respective plant genomes by genomic PCR and subsequent sequencing (Figure S3). RNLS1s molecularly analyzed included those from Aquilegia flabellata (a close relative of Aq. coerulea) (AfRNLS1), Lotus japonicus (LjRNLS1), Malus x domestica (MdRNLS1) and Cucumis sativus (CsRNLS1) (Figure S3B–H). The AqfRNLS1 sequence defined in this article showed approximately 98% nt sequence identity to AcRNLS1 whose sequence is available in the database (Phytozome). LjRNLS1-1 from L. japonicus line B129 and CsRNLS1 from 3 cucumber varieties (Hokushin, Suyo, and ‘Borszcagowski’ line B10) were identical to the reported RNLS1 sequences for line MG-20 (Kazusa DNA Research Institute) and ‘Chinese long’ line 9930 [37], respectively. Approximately 97% nucleotide sequence identity was found between MdRNLS1s of cultivars ‘Sun-Fuji’ and ‘Golden Delicious.’ ‘Golden Delicious’ is currently used in the apple genome sequence project [38] (http://www.rosaceae.org/projects/apple_genome). These examined RNLS sequences are listed in Table S5.
Phylogenetic analysis of negative-strand RNA virus sequence in plant nuclear genomes
Several sequences found through searching plant EST databases (Table S6, Figure S4) were included in our phylogenetic analysis. Deduced amino acid sequences of plant RNLSs, the N (CP) proteins of negative-strand RNA viruses, and related EST entries were aligned using the MAFFT program (Figure S5). Pair-wise similarities between selected RNLSs and viral N (CP) sequences are shown in Table S7. Two amino acid segments, GmH and YaRifdxxxfxxLQtkxC are relatively well-conserved among these sequences. A dendrogram generated on the basis of alignment showed 4 major groups containing plant RNLSs (Figure 6). RNLS1s are separated into two major groups. The first group includes varicosavirus CPs and RNLS1s from apple, cucumber and Brassica plants (MdRNLS1, CsRNLS1, BoRNLS1, and BrRNLS1) in addition to a few ESTs. The second group accommodates RNLS1s from Aquilegia and Lotus (AqfRNLS1, AqcRNLS1, LjRNLS1), together with an RNLS2 from Mim. guttatus (MgRNLS2) and EST sequences from Cichorium intybus and B. oleracea. The placement of MgRNLS2 in this group may be explained by low-level sequence identity to its most closely related extant varicosavirus, LNYV (Table 2). NtRNLS3, PtRNLS4, and Ns of cytorhabdoviruses (LNYV, LYMoV, and NCMV) form the third group (Figure 6). A dichorhabdovirus (orchid fleck virus, OFV) and nucleorhabdoviruses (PYDV and SYNV), replicating in the nuclei of host plants, are placed into an independent clade.

Whether most of the analyzed ESTs originated from viruses or plant chromosomes is unknown. However, an EST from F. pratensis is presumed to originate from a plant virus in our preliminary experiment not only because the N (CP) - but also the L (RdRp)-derived ESTs were detected in the same EST library of F. pratensis. This suggests a presently existing virus more closely related to RNLSs of the genus Brassica than LBVaV, because both N - and L-related sequences are rarely found in a single plant genome (Table 2).
Database search for and molecular detection of plus-strand RNA viral sequences in plant genomes
Extensive searches of genome sequence databases for plant plus-strand RNA viral sequences were conducted using genome sequences of various plus-strand RNA viruses representing the major virus genera and families Potyviridae, Luteoviridae, Tombusviridae, and Bromoviridae (Table S1). Compared to searches for double - or negative-strand RNA viral sequences, the search for plus-strand RNA virus sequences yielded a much smaller number of hits. The Medicago truncatula database (HTGS) contains sequences of 320 and 475 nts with over 98% sequence identity to the capsid and movement protein genes of cucumber mosaic virus, a member of the family Bromoviridae. However, this sequence was not amplified in Med. truncatula line A17 used in the genome sequence project by genomic PCR with different sets of internal and external primers. A sequence similar to replication-related genes of citrus leaf blotch virus (CLBV) [39] belonging to the family Betaflexiviridae, is identified in the complete genome databases for the cucumber ‘Chinese long’ line 9930 [37] and termed Cucumis sativus flexivirus replicase-like sequence 1, CsFRLS1 (Figure 7A). The GSS database of cucumber ‘Borszczagowski’ line B10 also contains CsFRLS1 (http://csgenome.sggw.pl/), but its available sequence is fragmented (Figure 7A, dashed purple bar) and shorter than that in the complete genome sequence data base.
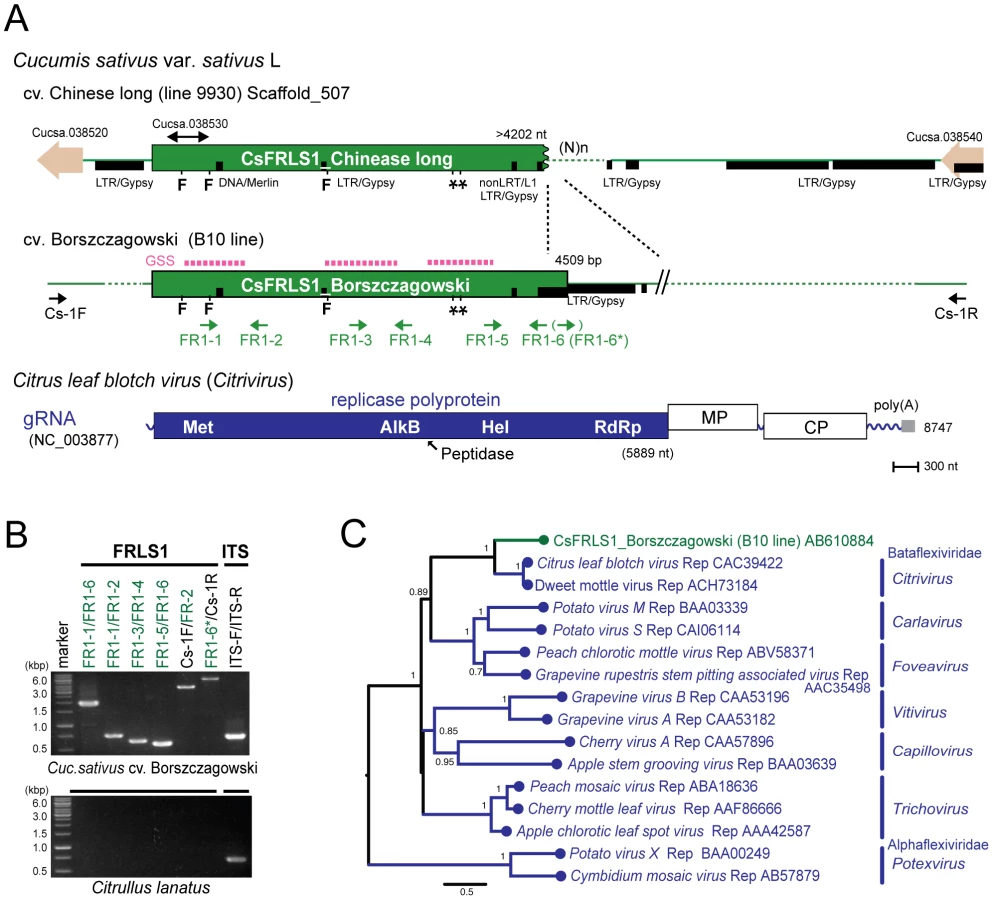
Two independent cucumber genome databases for 2 different lines strongly suggest the presence of CsFRLS1 in the cucumber chromosome. We confirmed this by genomic PCR using different sets of primers corresponding to methyltransferase (Met) and RNA helicase (Hel) domains, the inter-domain region (FR1-3 and FR1-4) and the entire CsFRLS1 region (Figure 7B). DNA fragments of expected sizes were amplified on genomic DNA from the ‘Borszczagowski’ line B10, but not from watermelon, Citrullus lanatus (Figure 7B). Furthermore, genomic PCR fragments covering FRLS1 and its flanking putative open reading frames (ORFs) were amplified, strongly suggesting that FRLS1 resides on the nuclear genome as shown in Figure 7A and B. The phylogenetic tree containing CsFRLS1 potentially encoded by CsFRLS1 and its counterparts from related viruses shows that CsFRLS1 is closely related to the genus Citrivirus within the family Betaflexiviridae (Figure 7C). The distance between CsFRLS1 and citriviruses are similar to intra-genus distances in the genera Carla-, Fovea-, Viti - and Potexviruses.
Discussion
The finding that the CP of a novel partitivirus, RnPV2 from a fungal phytopathogen matched a plant gene product, ILR2 from Ar. thaliana initiated a comprehensive search of the plant genomic sequence data available as of December 10, 2010 for non-retroviral RNA virus sequences (NRVSs) in plant genomes. While this study showed a variety of sequences related to the N (CP) genes of negative-stranded RNA viruses (cytorhabdoviruses and varicosaviruses) in members in the plant families including Solanaceae, Leguminosae, Brassicaceae and Phrymaceae, only one plus-sense RNA virus-related sequence (betaflexivirus replication-related gene) was found to be present in the cucumber genome. Furthermore, this survey detected sequences related to CP from dsRNA viruses (partitiviruses) (PCLSs) in various plants in addition to PCLSs reported by Liu et al. [24]. These authors performed a thorough search of eukaryotic genomic sequences available as of September 2009 for NRVSs and showed multiple dsRNA virus-related sequences not only in plants but also animals. Importantly, many of the NRVSs revealed by BLAST searches in this study were subsequently identified in plant genomes by Southern blotting, genomic PCR and sequence analyses (Figures 1–3, 5, 7, S1, S3). These findings provide interesting insights into plant nuclear genome evolution, plant phylogeny and virus/host interactions.
Horizontal gene transfer, HGT, can occur “from virus to plant” or “from plant to virus.” A retention profile of PCLS1 among plants strongly suggests that HGT may have involved the former direction. The family Brassicaceae of the order Brassicales includes the genus Arabidopsis, which is believed to have diverged after the split of the families Phrymaceae and Solanaceae, accommodates the genera Mimulus and Solanum and belong to different orders, Lamiales and Solanales, respectively (Figure 8). No PCLS1 homologs are found in Vitis vinifera or Carica papaya, and that this gene resides on non-orthologous chromosomal positions of Mim. guttatus (data not shown) and Arabidopsis-related species (Figure 1A). This strongly suggests that independent HGT events from virus to the Arabidopsis and Mim. guttatus lineages may have occurred (Figure 8). This observation is also true for other PCLSs. The families Solanaceae and Brassicaceae contain PCLS5s, while their counterparts are not found in other plants whose complete genome sequences are available (Figure 8). The observation that a relatively widely conserved gene PUX_4 is disrupted in Sol. phureja by SpPCLS5 (Figure 3A) provides additional evidence for its insertion into the PUX_4 locus. The HGT direction “from virus to plant” was further confirmed by phylogenetic analysis showing that plant PCLSs and partitivirus CPs are placed in a mixed way (Figure 4). Viral sequences are basal in each of the three major clades, supporting the direction of transfers from virus to plant.

The divergence time of plant lineages is estimated through a classical approach using fossils and mutations rates of some particular genes. Alternatively, if we assume that cellular genes evolve at a constant rate, their divergence time can be calculated from the genome-wide, spontaneous mutation rate determined on a generation basis in the laboratory [40]. Together with the patterns of occurrence of the non-retroviral integrated RNA virus sequences, these values allow us to estimate time of some, if not all, HGTs identified in this study. For example, the integration of PCLS1 (ILR2) may have post-dated the split of the lineages containing the genera Arabidopsis and Brassica (16.0–24.1 million years ago) and pre-dated the speciation of Arabidopsis spp., or more accurately the divergence of Arabidopsis and its closely related genera (Figure 8) (10–14 million years ago) [40], [41], [42]. The phylogenetic relation among PCLS1s from Arabidopsis and its close relatives within the tribe Camelina (Capsella, Olimarabidopsis, and Turritis) agrees with the phylogeny of the family Brassicaceae deduced from systematic analyses [43]. Moreover, assuming that the Ar. thaliana and Ar. arenosa separated 10 million years ago, the mutation rates calculated for PCLS1s between the 2 plants are estimated to be 6.8×10−9 base substitutions per site per year, a value close to the genome-wide base substitution rate, 7×10−9, reported for Ar. thaliana by Ossowski et al. [40]. These observations suggest that endogenized PCLS1s accumulated mutations in a manner similar to those of other nuclear sequences during the course of evolution after a single HGT event in an ancestral Arabidopsis plant.
The genome of B. rapa in the family Brassicaceae retained 2 PCLSs (BrPCLS4 and BrPCLS5) with low-level similarities to RnPV2 CP on chromosomal positions different from each other and from that of the PCLS1 (ILR2) homologs of Arabidopsis-related genera. No PCLS1 homolog was identified on the orthologous positions of the B. rapa genome, and no BrPCLS4 or BrPCLS5 homologs were found on the corresponding locus of the Ar. thaliana or Ar. lyrata genome. Therefore, BrPCLS4 and 5 may have been introduced into the B. rapa genome separately from each other and from PCLS1 (ILR2) after the divergence of the Brassica and Arabidopsis lineages (Figure 8). Similarly, the detection profile of AtPCLS2 and AlPCLS3 (Figure 2) shows that they may have been introduced into Ar. thaliana and Ar. lyrata chromosomes independently after the separation of 2 plant species (3.0–5.8 million years ago) (Figure 8); these are more recent HGT events than the PCLS1 integration into the Arabidopsis lineage. PCLS integrations into the Solanaceae lineage were slightly complex. Relatively high or moderate levels of aa sequence identities (47–68%) are shared within the PCLS5s from the family Solanaceae. However, a lack of information regarding genome sequences flanking the PCLS5s caused difficulty in determining whether a single event or multiple HGT events may have occurred within the lineage (Figure 8).
Gene sequences related to rhabdovirus or varicosavirus N (CP) genes (RNLSs) are detected in many genera including Brassica, Raphanus, Mimulus, Nicotiana, Lotus, Malus, Cucumis, Populus, Theobroma, and Aquilegia (Figures 5, 8, S3). Using similar rationale for the HGT of PCLSs, multiple integrations of RNLSs into plant chromosomes are thought to have occurred (Figure 8). RNLSs are distributed in an irregular manner in the plant lineage, while rhabdovirus N proteins show similar tree topology to that exhibited by corresponding RdRps. This is consistent with the hypothesis that HGT occurred “from virus to plants.” RNLS2 was detected in a very narrow range of plants, i.e., detectable only in N. tabacum but not other Nicotiana species (Figure 5). RNLS1 was detected in all tested Brassica species, R. sativus and Aq. coerulea, while it was not detected in the genomes of Ar. thaliana [25] or Ar. lyrata (Phytozome), which are much closely related to Brassica than Aq. coerulea to Brassica. If these sequences were of plant origin, homologous sequences are expected to be retained at least within some members of the families Brassicaceae and Solanaceae. However, Southern blotting and genomic PCR analyses with NtRNLS2 - and BrRNLS1-specific probes and primers failed to detect their related sequences in plants other than N. tabacum, and Brassica species and R. sativus, respectively (Figure 5C–F). A search using NtRNLS2 and BrRNL1 against the genome sequences of Ar. thaliana and Ar. lyrata did not yield any hits. This indicates that multiple HGTs of RNLSs occurred from “virus to plant.” While the BrRNLS1 integration may have postdated the split of the Arabidopsis and Brassica lineages (43.2–18.5 million years ago), NtRNLS2 and NtRNLS3 integration may have occurred after the divergence of N. tabacum (allotetraploid) and its maternal parent N. sylvestris (diploid) (0.2 million years ago) [44]. This hypothesis must be verified by sequence analysis of the corresponding regions of N. tabacum and other Nicotiana species.
The detection pattern of PCLSs within the family Brassicaceae provided an interesting insight into the phylogenetic relationship of some genera in the family. The family Brassicaceae is one of the largest families comprising over 300 genera and approximately 3,300 species that include an important plant biology model plant, Ar. thaliana, and agriculturally important Brassica species. Their phylogenetic relationships have been extensively studied and are occasionally controversial, because they rely on data sets and methods exploited for analyses. For example, placement of the genus Crucihimalaya is interesting to note in relation to this study. The genus is placed into a clade containing the genus Boechera, and is assumed to have separated from an ancestor common to the genus Capsella after the divergence of the Arabidopsis lineage based on phylogenetic analyses with a single nuclear gene (chalcone synthase gene) [45] or multiple data sets containing plastid and nuclear genes [45], [46], [47], [48]. However, utilization of different data sets shows different tree topologies, suggesting that the Crucihimalaya genus may have diverged before the split between Arabidopsis and Capsella [45], [49]. PCLS1s (ILR2 homologs) were detected in relatives of Arabidopsis but not in Cru. lasiocarpa (Figure 1B, D), strongly supporting the phylogenetic relation proposed by Lysak et al. [49]. The absence of the PCLS1 in a homologous position of the Cru. lasiocarpa chromosome was confirmed by sequencing of genomic PCR fragments generated with a specific primer set (Figure 1D). Therefore, these results clearly indicate that PCLSs have the potential to supplement phylogenetic estimates by serving as molecular markers. Furthermore, a similar insight into phylogenetic relations among Nicotiana species may be gained from data regarding 4 PCLSs identified in N. tabacum as more data in the genome and PCLS sequences of the genus Nicotiana become available.
Many examples of HGT from minus-sense RNA and dsRNA viruses, particularly from partitiviruses, have been found in plant nuclear genomes. Endogenization of NRVSs required 3 events to occur: (1) replication of the ancestral viral genome in the germ lines of host plants, (2) reverse transcription of genomic RNA, and (3) its subsequent integration into plant chromosomes. Many plant viruses are reported to be transmitted through pollens and seeds [50], while their transmission rates depended on virus/host combinations. Seed-transmitted viruses include positive-strand and negative-strand RNA viruses and partitiviruses with dsRNA genomes. The family Partitiviridae accommodates members that infect plants or fungi, and some plant and fungal partitiviruses are phylogenetically closely related ([21]; Figure 4). PCLS1 is most closely related to a novel fungal partitivirus, RnPV2, but the other PCLSs show the closest resemblance to plant partitiviruses (Table 1, Figure 4). Therefore, PCLS1 integration occurred when an ancestor of RnPV2 acquired the ability to infect an ancestral plant during endosymbiotic [51] or parasitic interactions between its host fungus and the plant, a host of the fungus, and to invade the plant germ cells. In support of this hypothesis, an assembled EST sequence is present in F. pratensis that is more closely related to PCLS1 than the RnPV2 CP gene and considered to have originated in a plant partitivirus (Figure 4). Such a virus may have been a direct source of plant PCLS1. Alternatively some fungal partitiviruses may be intrinsically able to infect plant cells. The expected capability of plant partitiviruses to replicate in host germ cells may be associated with their high rates (∼100%) of seed transmission via ovule and/or pollen [21], an uncommon phenomenon for plant viruses. Although germ lines are hypothesized to have the ability to eliminate virus infection, partitivirus may be able to overcome such a host mechanism. It is also likely that ancestral negative-strand RNA viruses may have invaded germ cells of host plants.
For the second required event, integration of NRVSs likely involved reverse transcription that may have been mediated by reverse transcriptase encoded by retrotransposons or pararetroviruses. However, the mechanism by which the viral RNA sequences were converted to DNA and introduced into plant genomes remains unknown. Interestingly LjPCLS8 harbors an unrelated sequence of 1.3-kb sequence in its central region (Figure S1A, D), suggesting a recombination event of during reverse transcription or a 2-step integration of 2 distinct molecules, PCLS8 and a sequence of an unknown origin. For the third event, as suggested by Liu et al. [24], transposon-mediated integration [52] and/or double-strand-break repair (non-homologous recombination) [53] may be involved. Flanking regions of some plant genome-integrated NRVSs (e.g., RNLS1s and CsFRLS1, see Figures 7, S3) carried transposable elements or multiple repeat sequences, supporting the first type of integration. Vertebrate cultured cells are useful for experimentally monitoring de novo integrations of negative-strand RNA viral sequences [11]; however, the agents that facilitate the reverse transcription and integration steps remain unknown.
In contrast to the nuclear integrations of partitivirus CP sequences and negative-strand RNA virus N sequences, plus-strand RNA virus endogenizations were observed much less frequently. A level of viral transcripts in germ cells may be one of factors governing the frequency of NRVSs. This is supported by the observation (data not shown) that, whereas we searched for integrated partitiviral RdRp sequences or other non-N sequences of rhabdoviruses, we could seldom detect them. Partitivirus CP and rhabdovirus N coding transcripts are highly likely to be produced in cells infected by the respective viruses more than other viral transcripts. Plus-strand RNA viruses, are believed to accumulate in infected plant cells much more than plant partitiviruses. However, plus-strand RNA viruses may generally be more able to be detected by a surveillance system of host germ cells and/or less competent to escape from their defense system. A smaller number of FRLS integrations observed in this study (Figure 7) may be associated with a lower ability of ancestral plus-strand RNA viruses to invade host germ cells, as predicted from the low seed transmissibility of CLBV [54]. Alternatively, plus-strand RNA virus sequences are disfavored by reverse transcriptase and agents that facilitate integration of their complementary DNA in the second and third events, respectively, although this possibility may be low.
Materials and Methods
Fungal strains and virus characterization
A virus-infected fungal strain of R. necatrix, W57, was isolated in the Iwate Prefecture, Japan. Molecular characterization of genomic dsRNAs were performed according to the methods described by Chiba et al. [55], unless otherwise mentioned.
Plant materials and gene characterization
Seeds for members of the Brassicaceae family, L. japonicus, Med. truncatula and Cuc. sativus cv. Borszczagowski B10 line were provided by the Arabidopsis Biological Resource Center of The Ohio State University, the Frontier Science Research Center, University of Miyazaki, and Drs. Kazuhiro Toyoda, Douglas Cook, and Grzegorz Bartoszewski, respectively. Seeds for members of the genus Nicotiana were originally obtained from Nihon Tabako, Inc (Tokyo, Japan) and maintained at Okayama University. Dr. Takashi Enomoto of Okayama University provided the remaining plants. Plant genomic DNA was isolated from seeds or fresh leaf materials and used in genomic PCR and Southern blot analyses as described by Miura et al. [56]. Sequences of ILR2 homologs (PCLS1s) from members of the family Brassicaceae, except for Ar. thaliana accessions Col-0 and WS, and Ar. lyrata, were obtained by sequencing genomic PCR fragments. Genomic PCR fragments or clones were used to determine the sequences of other selected PCLSs, RNLSs and FRLSs. Digoxigenin (DIG)-labeled DNA, prepared as described by Chiba et al. [55], was used as probes in Southern blotting analyses as described by Faruk et al. [57]. Table S3 includes sequences of primers used in this study.
Database search and phylogenetic analysis
BLAST (tblastn) searches [58] were conducted against genome sequence databases available from the NCBI (nucleotide collection, nr/nt; genome survey sequences, GSS; high-throughput genomic sequence, HTGS; whole-genome shotgun reads, WGS; non-human, non-mouse ESTs, est others) (http://www.ncbi.nlm.nih.gov/), Phytozome v6.0 (http://www.phytozome.net/), Brassica database (BRAD) (http://brassicadb.org/brad/), Potato Genome Sequencing Consortium (http://potatogenomics.plantbiology.msu.edu/), and Kazusa DNA Research Institute (http://www.kazusa.or.jp/e/index.html). The databanks covered the complete and partial genome sequences of 20 plant species. Transposable element sequences were identified using the Censor (http://www.girinst.org/censor/index.php) [59]. Obtained non-retroviral integrated sequences were translated to amino acid sequences and aligned with MAFFT version 6 under the default parameters [60] (http://mafft.cbrc.jp/alignment/server). For some non-retroviral integrated sequences with interrupted ORFs, frames were restored by adding Ns as unknown sequences to obtain continuous aa sequences (edited residues are shown as Xs). Alignments were edited by using MEGA version 4.02 software [61]. To obtain appropriate substitution models for the maximum likelihood (ML) analyses, each data set was subjected to the Akaike information criterion (AIC) calculated using ProtTest server [62] (http://darwin.uvigo.es/software/prottest_server.html). According to ProtTest results, WAG+I+G+F, LG+I+G, and LG+I+G+F were selected for PCLSs and partitiviruses, for RNLSs, plant rhabdoviruses and varicosaviruses, and for FRLS and flexiviruses, respectively. Phylogenetic trees were generated using the appropriate substitution model in PhyML 3.0 [63] (http://www.atgc-montpellier.fr/phyml/). In each analysis, four categories of rate variation were used. The starting tree was a BIONJ tree and the type of tree improvement was subtree pruning and regrafting (SPR) [64]. Branch support was calculated using the approximate likelihood ratio test (aLRT) with a Shimodaira–Hasegawa–like (SH-like) procedure [65]. The tree was midpoint-rooted using FigTree version 1.3.1 software (http://tree.bio.ed.ac.uk/software/).
Data deposition
Two mycoviral genome sequences and a total of 73 non-retroviral integrated RNA virus sequences were analyzed. Sequence data (1 of the 2 genome segments of RnPV2, 21 PCLSs, 12 RNLSs and 1 FRLS) used for phylogenetic analyses in this article have been deposited into the EMBL/GenBank/DDBJ Data Library under the following accession numbers: AB569998 (RnPV2 dsRNA2), AB576168–AB576175, AB609326–AB609329 (ILR2-like sequences: PCLS1s), AB609330–AB609338 (PCLS2–PCLS8), AB9339–AB609350 (RNLSs), and AB610884 (CsFRLS1) (Tables S4 and S5). Other non-retroviral integrated RNA virus elements whose sequences were partially determined and analyzed in this study are available upon request.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GorbalenyaAE 1992 Host-related sequences in RNA viral genomes. Semin Virol 3 359 371
2. MeyersGRumenapfTThielHJ 1989 Ubiquitin in a togavirus. Nature 341 491
3. MayoMAJollyCA 1991 The 5′-terminal sequence of potato leafroll virus RNA: evidence of recombination between virus and host RNA. J Gen Virol 72 2591 2595
4. AgranovskyAABoykoVPKarasevAVKooninEVDoljaVV 1991 Putative 65 kDa protein of beet yellows closterovirus is a homologue of HSP70 heat shock proteins. J Mol Biol 217 603 610
5. DoljaVVKreuzeJFValkonenJP 2006 Comparative and functional genomics of closteroviruses. Virus Res 117 38 51
6. BertschCBeuveMDoljaVVWirthMPelsyF 2009 Retention of the virus-derived sequences in the nuclear genome of grapevine as a potential pathway to virus resistance. Biol Direct 4 21
7. GayralPNoa-CarrazanaJCLescotMLheureuxFLockhartBE 2008 A single Banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J Virol 82 6697 6710
8. Richert-PoggelerKRNoreenFSchwarzacherTHarperGHohnT 2003 Induction of infectious petunia vein clearing (pararetro) virus from endogenous provirus in petunia. EMBO J 22 4836 4845
9. KuniiMKandaMNaganoHUyedaIKishimaY 2004 Reconstruction of putative DNA virus from endogenous rice tungro bacilliform virus-like sequences in the rice genome: implications for integration and evolution. BMC Genomics 5 80
10. GeukingMBWeberJDewannieuxMGorelikEHeidmannT 2009 Recombination of retrotransposon and exogenous RNA virus results in nonretroviral cDNA integration. Science 323 393 396
11. HorieMHondaTSuzukiYKobayashiYDaitoT 2010 Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature 463 84 87
12. BelyiVALevineAJSkalkaAM 2010 Unexpected inheritance: multiple integrations of ancient bornavirus and ebolavirus/marburgvirus sequences in vertebrate genomes. PLoS Pathog 6 e1001030
13. KatzourakisAGiffordRJ 2010 Endogenous viral elements in animal genomes. PLoS Genet 6 e1001191
14. TaylorDJLeachRWBruennJ 2010 Filoviruses are ancient and integrated into mammalian genomes. BMC Evol Biol 10 193
15. KooninEV 2010 Taming of the shrewd: novel eukaryotic genes from RNA viruses. BMC Biol 8 2
16. TaylorDJBruennJ 2009 The evolution of novel fungal genes from non-retroviral RNA viruses. BMC Biol 7 88
17. FrankACWolfeKH 2009 Evolutionary capture of viral and plasmid DNA by yeast nuclear chromosomes. Eukaryot Cell 8 1521 1531
18. ArakawaMNakamuraHUetakeYMatsumotoN 2002 Presence and distribution of double-stranded RNA elements in the white root rot fungus Rosellinia necatrix. Mycoscience 43 21 26
19. IkedaKNakamuraHArakawaMMatsumotoN 2004 Diversity and vertical transmission of double-stranded RNA elements in root rot pathogens of trees, Helicobasidium mompa and Rosellinia necatrix. Mycol Res 108 626 634
20. GhabrialSASuzukiN 2009 Viruses of plant pathogenic fungi. Annu Rev Phytopathol 47 353 384
21. GhabrialSAOchoaWFBakerTNiberML 2008 Partitiviruses: general features. MahyBWJVRM Encyclopedia of Virology 3rd edn Oxford Elsevier 68 75
22. KooninEVWolfYINagasakiKDoljaVV 2008 The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups. Nat Rev Microbiol 6 925 939
23. MagidinMPittmanJKHirschiKDBartelB 2003 ILR2, a novel gene regulating IAA conjugate sensitivity and metal transport in Arabidopsis thaliana. Plant J 35 523 534
24. LiuHFuYJiangDLiGXieJ 2010 Widespread horizontal gene transfer from double-stranded RNA viruses to eukaryotic nuclear genomes. J Virol 84 11876 11887
25. InitiativeTAG 2000 Analysis of the genome of the flowering plant Arabidopsis thaliana. Nature 408 796 815
26. RennaLHantonSLStefanoGBortolottiLMisraV 2005 Identification and characterization of AtCASP, a plant transmembrane Golgi matrix protein. Plant Mol Biol 58 109 122
27. SchranzMELysakMAMitchell-OldsT 2006 The ABC's of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends Plant Sci 11 535 542
28. TzanetakisIEPriceRMartinRR 2008 Nucleotide sequence of the tripartite Fragaria chiloensis cryptic virus and presence of the virus in the Americas. Virus Genes 36 267 272
29. ChenLChenJSZhangHChenSN 2006 Complete nucleotide sequences of three dsRNA segments from Raphanus sativus-root cv. Yidianhong [corrected] with leaf yellow edge symptoms. Arch Virol 151 2077 2083
30. WillenborgJMenzelWVettenHJMaissE 2009 Molecular characterization of two alphacryptovirus dsRNAs isolated from Daucus carota. Arch Virol 154 541 543
31. BlawidRSDMaissE 2008 Alphacryptovirus and Betacryptovirus. MahyBWJVRM Encyclopedia of Virology 3rd edn Oxford Elsevier 98 104
32. BoccardoGCandresseT 2005 Complete sequence of the RNA1 of an isolate of White clover cryptic virus 1, type species of the genus Alphacryptovirus. Arch Virol 150 399 402
33. SzegoAEnunluNDeshmukhSDVeliceasaDHunyadi-GulyasE 2010 The genome of Beet cryptic virus 1 shows high homology to certain cryptoviruses present in phylogenetically distant hosts. Virus Genes 40 267 276
34. BlawidRStephanDMaissE 2007 Molecular characterization and detection of Vicia cryptic virus in different Vicia faba cultivars. Arch Virol 152 1477 1488
35. SasayaTIshikawaKKoganezawaH 2002 The nucleotide sequence of RNA1 of Lettuce big-vein virus, genus Varicosavirus, reveals its relation to nonsegmented negative-strand RNA viruses. Virology 297 289 297
36. KondoHMaedaTShirakoYTamadaT 2006 Orchid fleck virus is a rhabdovirus with an unusual bipartite genome. J Gen Virol 87 2413 2421
37. HuangSLiRZhangZLiLGuX 2009 The genome of the cucumber, Cucumis sativus L. Nat Genet 41 1275 1281
38. VelascoRZharkikhAAffourtitJDhingraACestaroA 2010 The genome of the domesticated apple (Malus x domestica Borkh.). Nat Genet 42 833 839
39. VivesMCGalipiensoLNavarroLMorenoPGuerriJ 2001 The nucleotide sequence and genomic organization of Citrus leaf blotch virus: candidate type species for a new virus genus. Virology 287 225 233
40. OssowskiSSchneebergerKLucas-LledoJIWarthmannNClarkRM 2010 The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327 92 94
41. ClaussMJKochMA 2006 Poorly known relatives of Arabidopsis thaliana. Trends Plant Sci 11 449 459
42. BeilsteinMANagalingumNSClementsMDManchesterSRMathewsS 2010 Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc Natl Acad Sci U S A 107 18724 18728
43. OyamaRKClaussMJFormanováNKroymannJSchmidKJ 2008 The shrunken genome Arabidopsis thaliana. Plant Syst Evol 273 257 271
44. ClarksonJJLimKYKovarikAChaseMWKnappS 2005 Long-term genome diploidization in allopolyploid Nicotiana section Repandae (Solanaceae). New Phytol 168 241 252
45. KochMHauboldBMitchell-OldsT 2001 Molecular systematics of the Brassicaceae: evidence from coding plastidic matK and nuclear Chs sequences. Am J Bot 88 534 544
46. KochMADobesCMatschingerMBleekerWVogelJ 2005 Evolution of the trnF(GAA) gene in Arabidopsis relatives and the Brassicaceae family: monophyletic origin and subsequent diversification of a plastidic pseudogene. Mol Biol Evol 22 1032 1043
47. BaileyCDKochMAMayerMMummenhoffKO'KaneSLJr 2006 Toward a global phylogeny of the Brassicaceae. Mol Biol Evol 23 2142 2160
48. CouvreurTLFranzkeAAl-ShehbazIABakkerFTKochMA 2010 Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae). Mol Biol Evol 27 55 71
49. LysakMAKochMABeaulieuJMMeisterALeitchIJ 2009 The dynamic ups and downs of genome size evolution in Brassicaceae. Mol Biol Evol 26 85 98
50. MinkGI 1993 Pollen and seed-transmitted viruses and viroids. Annu Rev Phytopathol 31 375 402
51. RoossinckMJ 2003 Plant RNA virus evolution. Curr Opin Microbiol 6 406 409
52. MaoriETanneESelaI 2007 Reciprocal sequence exchange between non-retro viruses and hosts leading to the appearance of new host phenotypes. Virology 362 342 349
53. ManiRSChinnaiyanAM 2010 Triggers for genomic rearrangements: insights into genomic, cellular and environmental influences. Nat Rev Genet 11 819 829
54. GuerriJPinaJAVivesMCNavarroLMorenoP 2004 Seed Transmission of Citrus leaf botch virus: Implications in Quarantine and Certification Programs. Plant Dis 88 906
55. ChibaSSalaipethLLinYHSasakiAKanematsuS 2009 A novel bipartite double-stranded RNA mycovirus from the white root rot fungus Rosellinia necatrix: molecular and biological characterization, taxonomic considerations, and potential for biological control. J Virol 83 12801 12812
56. MiuraEKatoYMatsushimaRAlbrechtVLaalamiS 2007 The balance between protein synthesis and degradation in chloroplasts determines leaf variegation in Arabidopsis yellow variegated mutants. Plant Cell 19 1313 1328
57. FarukMIEusebio-CopeASuzukiN 2008 A host factor involved in hypovirus symptom expression in the chestnut blight fungus, Cryphonectria parasitica. J Virol 82 740 754
58. AltschulSFMaddenTLSchafferAAZhangJZhangZ 1997 Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 3389 3402
59. KohanyOGentlesAJHankusLJurkaJ 2006 Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7 474
60. KatohKTohH 2008 Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9 286 298
61. TamuraKDudleyJNeiMKumarS 2007 MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24 1596 1599
62. AbascalFZardoyaRPosadaD 2005 ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21 2104 2105
63. GuindonSDufayardJFLefortVAnisimovaMHordijkW 2010 New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59 307 321
64. HordijkWGascuelO 2005 Improving the efficiency of SPR moves in phylogenetic tree search methods based on maximum likelihood. Bioinformatics 21 4338 4347
65. AnisimovaMGascuelO 2006 Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst Biol 55 539 552
66. KochMBJMitchell-OldsT 1999 Molecular systematics and evolution of Arabidopsis and Arabis. Plant Biol 1 529 537
67. SasayaTKusabaSIshikawaKKoganezawaH 2004 Nucleotide sequence of RNA2 of Lettuce big-vein virus and evidence for a possible transcription termination/initiation strategy similar to that of rhabdoviruses. J Gen Virol 85 2709 2717
68. DietzgenRGCallaghanBWetzelTDaleJL 2006 Completion of the genome sequence of Lettuce necrotic yellows virus, type species of the genus Cytorhabdovirus. Virus Res 118 16 22
69. GroupAP 2003 An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APGII. Bot J Linn Soc 141 399 436
70. UdvardiMKTabataSParniskeMStougaardJ 2005 Lotus japonicus: legume research in the fast lane. Trends Plant Sci 10 222 228
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Requires Glycerol for Maximum Fitness During The Tick Phase of the Enzootic Cycle
- Comparative Genomics Yields Insights into Niche Adaptation of Plant Vascular Wilt Pathogens
- The Role of IL-15 Deficiency in the Pathogenesis of Virus-Induced Asthma Exacerbations
- “Persisters”: Survival at the Cellular Level
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy