
Comparative Genomics Yields Insights into Niche Adaptation of Plant Vascular Wilt Pathogens
The vascular wilt fungi Verticillium dahliae and V. albo-atrum infect over 200 plant species, causing billions of dollars in annual crop losses. The characteristic wilt symptoms are a result of colonization and proliferation of the pathogens in the xylem vessels, which undergo fluctuations in osmolarity. To gain insights into the mechanisms that confer the organisms' pathogenicity and enable them to proliferate in the unique ecological niche of the plant vascular system, we sequenced the genomes of V. dahliae and V. albo-atrum and compared them to each other, and to the genome of Fusarium oxysporum, another fungal wilt pathogen. Our analyses identified a set of proteins that are shared among all three wilt pathogens, and present in few other fungal species. One of these is a homolog of a bacterial glucosyltransferase that synthesizes virulence-related osmoregulated periplasmic glucans in bacteria. Pathogenicity tests of the corresponding V. dahliae glucosyltransferase gene deletion mutants indicate that the gene is required for full virulence in the Australian tobacco species Nicotiana benthamiana. Compared to other fungi, the two sequenced Verticillium genomes encode more pectin-degrading enzymes and other carbohydrate-active enzymes, suggesting an extraordinary capacity to degrade plant pectin barricades. The high level of synteny between the two Verticillium assemblies highlighted four flexible genomic islands in V. dahliae that are enriched for transposable elements, and contain duplicated genes and genes that are important in signaling/transcriptional regulation and iron/lipid metabolism. Coupled with an enhanced capacity to degrade plant materials, these genomic islands may contribute to the expanded genetic diversity and virulence of V. dahliae, the primary causal agent of Verticillium wilts. Significantly, our study reveals insights into the genetic mechanisms of niche adaptation of fungal wilt pathogens, advances our understanding of the evolution and development of their pathogenesis, and sheds light on potential avenues for the development of novel disease management strategies to combat destructive wilt diseases.
Published in the journal:
. PLoS Pathog 7(7): e32767. doi:10.1371/journal.ppat.1002137
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002137
Summary
The vascular wilt fungi Verticillium dahliae and V. albo-atrum infect over 200 plant species, causing billions of dollars in annual crop losses. The characteristic wilt symptoms are a result of colonization and proliferation of the pathogens in the xylem vessels, which undergo fluctuations in osmolarity. To gain insights into the mechanisms that confer the organisms' pathogenicity and enable them to proliferate in the unique ecological niche of the plant vascular system, we sequenced the genomes of V. dahliae and V. albo-atrum and compared them to each other, and to the genome of Fusarium oxysporum, another fungal wilt pathogen. Our analyses identified a set of proteins that are shared among all three wilt pathogens, and present in few other fungal species. One of these is a homolog of a bacterial glucosyltransferase that synthesizes virulence-related osmoregulated periplasmic glucans in bacteria. Pathogenicity tests of the corresponding V. dahliae glucosyltransferase gene deletion mutants indicate that the gene is required for full virulence in the Australian tobacco species Nicotiana benthamiana. Compared to other fungi, the two sequenced Verticillium genomes encode more pectin-degrading enzymes and other carbohydrate-active enzymes, suggesting an extraordinary capacity to degrade plant pectin barricades. The high level of synteny between the two Verticillium assemblies highlighted four flexible genomic islands in V. dahliae that are enriched for transposable elements, and contain duplicated genes and genes that are important in signaling/transcriptional regulation and iron/lipid metabolism. Coupled with an enhanced capacity to degrade plant materials, these genomic islands may contribute to the expanded genetic diversity and virulence of V. dahliae, the primary causal agent of Verticillium wilts. Significantly, our study reveals insights into the genetic mechanisms of niche adaptation of fungal wilt pathogens, advances our understanding of the evolution and development of their pathogenesis, and sheds light on potential avenues for the development of novel disease management strategies to combat destructive wilt diseases.
Introduction
Vascular wilts caused by fungal pathogens are widespread and very destructive plant diseases, causing enormous economic losses. The survival structures produced by wilt pathogens may remain viable in the soil for more than 20 years [1], making them a major constraint on agricultural production. Control of wilt diseases is also complicated by the scarcity of sources of disease resistant host germplasm, and the soil and vascular habitats of the pathogens. Wilts caused by Verticillium species are among the most devastating of these types of diseases. The primary causal agent, V. dahliae (Vd), can cause diseases on over 200 plant species, including numerous economically important food crops, ornamental flowers, trees, and shrubs [2]–[4]. The list of hosts for V. dahliae is continually expanding, as new hosts in diverse ecological niches succumb to the pathogen [5].
Like other vascular pathogens, Vd enters and colonizes the plant vascular (xylem) system, disrupting water transport, and causing the characteristic symptoms of wilting, and often vascular discoloration (Fig. 1C, D and E), and death of aerial tissues [2]–[4]. A diverse arsenal of carbohydrate active enzymes, including cellulases and pectin degrading enzymes, may be important for each major phase of the infection pathway. These enzymes may be necessary during penetration of the plant roots to gain access to the plant xylem and to breach the plant defense structures (tyloses and pectin gels) released into xylem vessels in response to infection [6]–[8], and finally at the end of colonization, for the production of large numbers of survival structures in the plant tissue. Additionally, colonization of the xylem vessels requires the wilt pathogens to be adapted so that they may thrive in the xylem fluid, which undergoes diurnal fluctuations in osmolarity, and contains only low amounts of sugars, organic and amino acids, and inorganic ions [9].

We have sequenced the genomes of two closely related species of Verticillium, Vd and V. albo-atrum (Vaa) (Fig. 2). Their shared features include the formation of small, hyaline asexual spores for dispersal, absence of a sexual state, hemibiotrophic life style, and induction of wilting symptoms in a variety of different plants. More importantly, despite the phylogenetic relatedness, these two wilt pathogens differ significantly in host range, and the types of melanized survival structures they form to allow them to persist in the soil. Vd forms microsclerotia (long-lived survival structures) that are small clusters of melanized, thick-walled cells (Fig. 1 A and F), whereas Vaa produces melanized hyphae that are referred to as dark resting mycelia (Fig. 1 B). The microsclerotia produced by Verticillium dahliae can survive in the soil in the absence of a susceptible host plant, and under inhospitable conditions for more than 20 years [1], which may have conferred it a competitive edge relative to Vaa by enabling it to disperse and persist in regions inhospitable to Vaa. In addition, both pathogens are generally not host-specific, but individual strains of Vd or Vaa may be differentially virulent on different plant species [5] or show cultivar specificity within a single plant species [10], [11]. However, Vaa is limited to the more narrow range of hosts in temperate climates, while Vd is well known to have a very broad host range, and to infect over 200 plant hosts from temperate to subtropical climates [2].

Fusarium oxysporum (Fo) is another economically important wilt pathogen that infects over 100 plant species in diverse ecological niches worldwide [12], [13]. Both Verticillium and Fo belong to the subclass Hypocreomycetideae of ascomycete fungi, but are in different phylogenetic lineages (Fig. 2). Fo shares with Vd and Vaa the ecological niche of the plant vascular system and causes nearly identical disease symptoms, yet differs significantly as it produces wilt symptoms much more quickly, and individual strains of Fo exhibit a high degree of host specialization. Within the Fo species complex over 120 specialized forms (formae speciales; f.spp.) have been described based on their specificity to various host species [14], [15]. The recent Fusarium comparative genomics study revealed that Fo's lineage-specific chromosomes contribute to this strict host-specificity [12].
The comparative study presented here exposes the unique genomic profile of the Verticillium species, characterized by an enhanced capacity for degrading plant pectins. The comparison of the two Verticillium wilt pathogen genomes with that of Fo, the only other fungal wilt pathogen for which the complete genomic sequence is available [12], also reveals a conserved set of proteins that potentially sustain niche adaptation. Our study also uncovers genomic regions (genomic islands) in Vd that are repeat-rich, and may confer enhanced genetic diversity to this primary causal agent of Verticillium wilt. Taken together, this study provides key insights into niche adaptation of wilt pathogens, lays out a foundation for future functional studies, and sheds light into potential directions for development of novel management strategies for controlling wilt diseases.
Results/Discussion
High quality genome assemblies of V. dahliae and V. albo-atrum
The whole genome shotgun assemblies of Vd strain VdLs.17 (7.5×) and Vaa strain VaMs.102 (4×) were generated using Sanger sequencing technology, and assembled using Arachne [16] (Table S1 in Supporting Information and Methods). The current genome assembly of VdLs.17 comprises 52 sequence scaffolds with a total length of 33.8 Mb, and an N50 scaffold length of 1.27 Mb (that is, 50% of all bases are contained in scaffolds of at least 1.27 Mb). More than 95% of the sequence had quality scores of at least 40 (1 error/104 bases) (Table 1). An optical map of the Vd chromosomes was constructed using the restriction enzyme AflII. The resulting ∼300× physical coverage map consists of 8 linkage groups, with an estimated genome size of 35 Mb. More than 99.7% of the assembled scaffolds aligned to the optical map (Table S2 in Supporting Information), confirming the completeness and accuracy of the genome assembly. The map data enabled anchoring of the genome assembly to the linkage groups, and further allowed analyses of structural variation in the genome. Only 89% of the Vd reads were placed in the current assembly. Interestingly, when all the Vd reads were BLASTed against the assembled genome of Vd, over 97% of the non-ribosomal reads could be mapped onto the assembly.

Even though only 4× sequence coverage was generated for Vaa VaMs.102 (Table 1 and Table S1), we were able to deliver an assembly of 30.3 Mb in 26 sequence scaffolds, with an N50 scaffold length of 2.31 Mb (Table 1). The long continuity in this low coverage assembly was achieved through increasing the coverage with sequence from fosmid clones, and using Arachne-assisted assembly [16]. The assembly also benefited from the low repeat content of the Vaa genome, as less than 200 kb of the Vaa genome can be classified as repeats, whereas over 1.68 Mb of the Vd assembly were repetitive sequences (See Methods). Even at low coverage, the current Vaa assembly captures almost all of the genome, as more than 95.5% of all sequence reads could be assembled, which is much higher compared to the assembled Vd reads.
Proteome analysis of Verticillium species
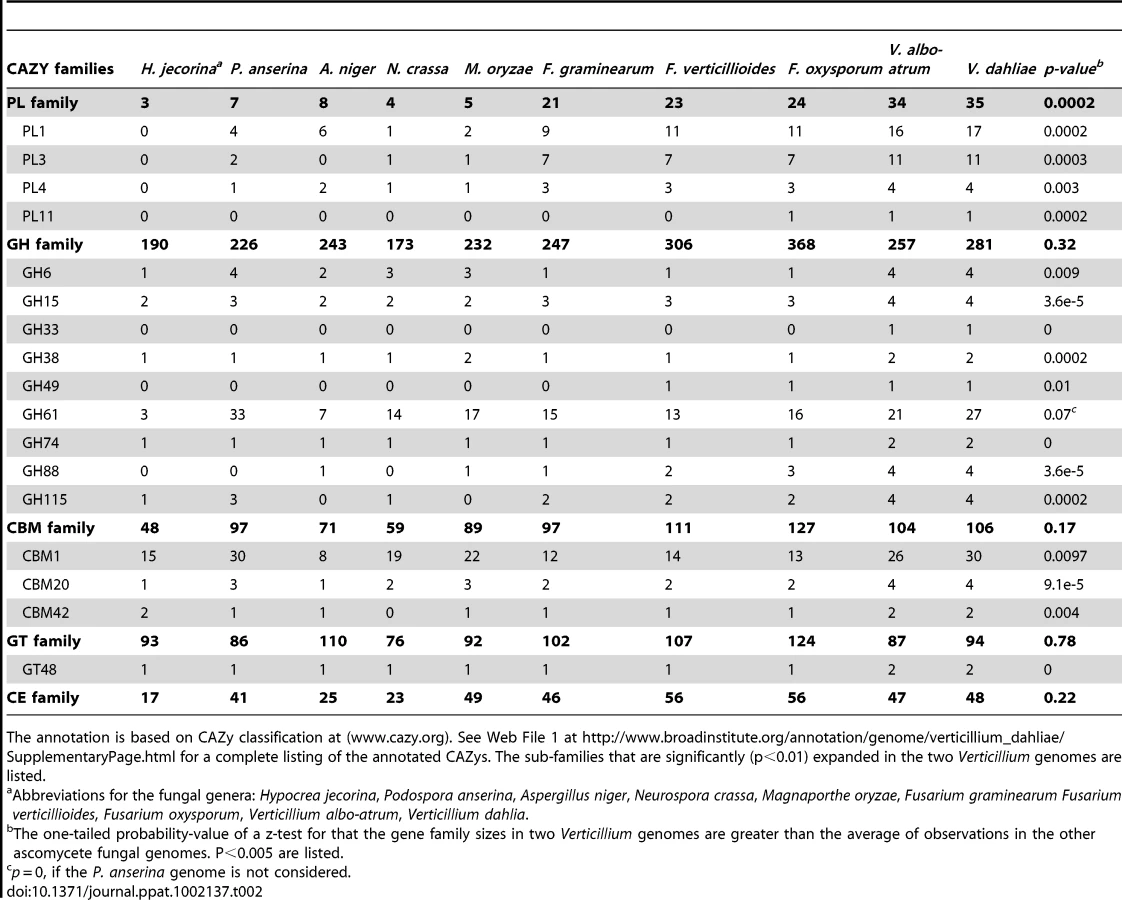
The genome of Vd strain VdLs.17 contains 10,535 predicted protein-encoding genes (Table 1), covering approximately 44% of the genome. Strain VaMs.102 of Vaa contains 10,221 predicted protein-encoding genes, covering approximately 41% of the genome. Among the annotated genes, 8699 of the proteins share 1∶1 orthologs between these two genomes, while 1357 and 1102 are specific to Vd and Vaa genomes respectively (Web File 1 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html, and Fig. S1). The two Verticillium genomes encode numerous carbohydrate-active enzymes, secreted proteins and transcription factors similar to those of other plant pathogenic fungi such as Fusarium spp. and Magnaporthe grisea [17] (Web Files 2 and 3 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html, Table S3, and Fig. S2). However, certain gene families, including those among the carbohydrate-active enzymes and secreted protein families, were significantly expanded in the two Verticillium genomes (Table 2). Such expansion provides a unique genomic signature of these two plant pathogens that live on a wide range of plant material and have an endophytic-like growth phase, living within the host plant for a long time before the disease state becomes evident.
Potential effectors and other secreted virulence factors
The arsenal of potentially secreted proteins (i.e. the secretome) of plant pathogens includes key pathogenicity molecules that are generally referred to as effectors. These effectors are molecules that are secreted by pathogens during host colonization and that modulate host biochemistry and physiology, including defense responses, to facilitate host colonization [18], [19], [20]. A combination of web-based software tools for the prediction of subcellular localization [21] and signal peptide motifs [22] revealed similar numbers of potentially secreted proteins encoded in each of the Verticillium genomes (Fig. S2; Web File 3 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html), namely 780 and 759 for Vd and Vaa, respectively. These numbers are comparable to those predicted in other fungi [17]. While 574 of these genes are conserved between the two species, 206 genes are specific to Vd and 185 are specific to Vaa.
Since many fungal effectors are small cysteine-rich proteins [23]–[25], all hypothetical proteins in the Vd and Vaa secretomes were classified based on their size and number of cysteine residues (Fig. S3). In total, 246 (conserved) hypothetical proteins can be designated as small (<400 amino acids), cysteine-rich (≥4 cysteine residues) proteins; 127 for Vd and 119 for Vaa, respectively. More than 60% of these predicted effectors are between 150 and 300 amino acids in size with 4–12 cysteines, typical for fungal effector proteins (Fig. S3). However, in neither Verticillium genome did we identify orthologs of well-characterized effectors reported in Fo [26], Phytophthora infestans [27], [28], or Cladosporium fulvum [24], [29], with the exception of homologs of the C. fulvum LysM effector Ecp6 [30], [31] and the C. fulvum virulence factor Ecp2 [32]. Under the selection conferred by a constant arms race between pathogens and their hosts, secreted proteins — especially effector proteins — are very diverse in pathogenic fungi. The elucidation of the roles of these potentially secreted proteins in Verticillium species therefore represents a challenging but potentially fertile ground for future functional studies.
We observed the expansion of some families of relatively conserved, secreted proteins known to play significant roles in pathogenesis, including LysM effectors [25], [31], [33], and the necrosis and ethylene-inducing-like protein (NLP) genes [34]–[36]. In contrast to C. fulvum and other Mycosphaerellaceae fungi that contain three LysM effector genes, the genomes of Vd and Vaa contain 7 and 6 LysM effector genes. Furthermore, while most fungal genomes contain two to three NLP genes, Vd and Vaa have eight and seven NLP gene homologs, respectively. It has previously been shown that NLPs display cytotoxic activity towards dicotyledonous and not towards monocotyledonous plant cells [35], [36] The expansion of the NLP family, also reported in the Fo genome, may therefore contribute to the broad host range among dicotyledonous plant hosts. Alternatively, some NLP family members may have diverged to exert completely different functions. Furthermore, both Vd and Vaa genomes encode four copies of a gene encoding a cysteine-rich, fungal-specific extracellular EGF-like (CFEM) domain, and some proteins containing this domain are proposed to play an important role in virulence and as effectors [37], [38].
Enhanced pectinolytic machinery in Verticillium spp.
The primary cell wall of dicotyledonous plants consists mainly of cellulose microfibrils embedded in a matrix of hemicelluloses, pectic polysaccharides, and glycoproteins [39]. Degradation of structurally complex pectin molecules requires numerous sugar-cleaving enzymes [40], [41]. For comparison of the carbohydrate-active enzymes from Verticillium species with those of other fungi (Table 2, Web File 2 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html), the boundaries of the carbohydrate active modules and associated carbohydrate-binding modules of the proteins encoded by each fungus in the comparison were determined, and classified using tools available at the Carbohydrate-Active-EnZymes database [42]. These comparisons revealed that despite the overall similar representation of Vd and Vaa carbohydrate active enzymes to those of other ascomycetes, polysaccharide lyase (PL) gene families that directly degrade pectin constituents are particularly expanded in Vd and Vaa (Table 2, Web File2, http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html). Among all sequenced fungal genomes, the Verticillium genomes encode the highest number and most diverse types of polysaccharide lyases to cleave different forms of pectins, including pectate lyases in the PL1, PL3, PL9 families, and rhamnogalacturonan lyases in the PL4 and PL11 families (Fig. 3). Interestingly, the PL11 family is present only in the wilt pathogens Vd, Vaa and Fo (Table 2, and Web File 2 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html). In addition to the significant expansion of polysaccharide lyase families (Table 2) many other enzymes, such as d-4,5 unsaturated α-glucuronyl hydrolase GH88 and GH105 families of enzymes (Fig. 3) that degrade the products generated by polysaccharide lyases [43], are also expanded in Verticillium. Such enhanced pectinolytic machinery illustrates the enhanced capacity of these species to degrade plant cell walls. Additionally, as pectins are released into the xylem vessels by infected plants and may form a barrier to prevent pathogen movement [7], [8], the pectin-degrading enzymes may contribute directly to the advancement of the Verticillium wilt pathogens within plant xylem vessels.

Expansion of carbohydrate-binding module 1 (CBM1) containing protein family
The conserved carbohydrate-binding module 1 (CBM1), generally referred to as a fungal-type cellulose binding domain, is usually appended to a diverse group of fungal enzymes. Through the conserved cysteine motif Cx10Cx5Cx9C, CBM1 anchors the enzyme's catalytic region to insoluble cellulose [44], enabling attachment to plant cell walls, and likely increasing enzyme efficiency. There are 30 CBM1-appended proteins in Vd (Table 2, Web File 2 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html, and Fig. S4). The majority (26) of these have orthologs in Vaa syntenic regions, reflecting their shared functional importance. Twenty-eight of the 30 Vd CBM1 proteins contain a signal peptide (Fig. S5), indicating that their enzymatic functions are extracellularly localized.
The Verticillium genomes as well as two saprobes, the white rot fungus Phanerochaete chrysosporium and the dung fungus Podospora anserina, encode the highest number of CBM1-containing proteins among the reported fungal genomes (Table 2) [45]. The putative enzymatic functions shared between the Vd and P. anserina CBM1-containing proteins are also remarkably similar, judging by the catalytic domains to which the CBM1s are appended (Fig. S5). For instance, in Vd there are a total of 18 CBM1-containing proteins encoding glycoside hydrolases, similar to 19 enzymes encoded in the P. anserina genome, and double the number (9) found in the Fo genome. P. anserina is an efficient saprobe. The shared profile of the CBM1-appended enzymes in P. anserina and the Verticillium species may indicate that the Verticillium species are also highly effective at utilizing diverse substrates for nutritional purposes. The enzymes may even contribute to saprophytic growth of Verticillium species after their emergence from the plant vascular system, and consequently to resting structure production.
There is one notable difference between the CBM1-appended proteins from wilt pathogens and those of the saprophytic fungi, namely that the wilt pathogens uniquely encode CBM1-containing polysaccharide lyases (from polysaccharide lyase families PL1 and PL3). There are three CBM1-containing polysaccharide lyases in Vd, Vaa and one (from polysaccharide lyase family PL1) in Fo, but none in P. anserina (Fig. S5). Interestingly, P. chrysosporium, which also possesses 30 CBM1-containing proteins, also lacks representative enzymes from polysaccharide lyase families PL1 and PL3 [45]. Therefore, conservation (for PL1) of the CBM1-appended polysaccharide lyase proteins among the wilt pathogens may indicate an important adaptation for the utilization of pectin from the cell walls of live plants, or from gels released into the xylem during infection.
Aside from their association with enzymes that degrade plant polysaccharides, an important role of CBM1 domains as elicitors of plant defense responses has been demonstrated experimentally in Phytophthora parasitica [46], and in root colonization by Trichoderma reesei [47]. Through our comparative study we have identified four such candidates among the Vd CBM1-containing proteins (Fig. S5) for future functional characterization.
Vd repeat-rich genomic islands confer genetic flexibility
The two sequenced Verticillium genomes are highly similar. On average, more than 90% of the sequence in any given 10 kb window can be unambiguously aligned to the other genome with an average 92% nucleotide sequence identity. This level of relatedness enabled the generation of large-scale alignments between Vd and Vaa genomes (Fig. 4, columns a and b, respectively), and the determination of synteny with high confidence (Methods, Supporting Information). However, the genome size of Vd is 2.6 Mb larger than that of Vaa assembly (Table 1). The colinearity of the syntenic maps revealed four regions of about 300 kb each in the genome of Vd, on chromosomes 3 and 4, that have no synteny with the Vaa genome (Fig. 4, circled in red), and contribute to the larger genome size in Vd. These four regions are hereafter referred to as Vd lineage-specific (LS) regions 1 to 4 for their unique presence in the Vd genome. Nucleic acid hybridizations using probes from four different genes (one from each of the four LS regions) revealed substantial genetic variation among the Vd strains tested (Fig. S6).

Structural flexibility of the LS regions
The four LS regions are repeat-rich (Fig. 4 column c), and the enriched repetitive sequences include DNA transposons, and LINE-like and long terminal repeat (LTR) retroelements based on manual curation (SG Amyotte et al, manuscript in preparation). Over 50% of all of the identifiable transposable elements in the Vd genome are found in the LS regions, contributing to an increased repetitive DNA content in the Vd genome (8-fold increase compared to that of the Vaa genome assembly). The skewed distribution of transposable elements in the LS regions is evident in the distribution of Pfam domains characteristic of the DNA transposon DDE superfamily endonucleases, and the retrotransposon RVE integrases (Fig. S7). Among the transposable elements in the LS regions are five different LTR (VdLTRE1–5), and we observed full-length, and actively transcribed copies of these elements in the Vd genome. Homologous sequences similar to elements VdLTRE1–4 were also found in Vaa. However, no significant matches to VdLTRE5 were detected in the Vaa genome assembly or Vaa unassembled sequence reads. In addition, within the Vd genome VdLTRE5 was present only in LS region 3, suggestive of its recent invasion into the genome.
Localized genomic dynamics is also reflected by the presence of genes that were duplicated either singly or in clusters within the four LS regions, with the cluster of seven genes in LS region 1 (VDAG_02357.1 to VDAG_02363.1) defined as ancestral based on structure and sequence conservation (Figs. S8 A and B). For example, the nucleotide sequences of VDAG_04863.1, VDAG_09199.1, VDAG_09219.1 are divergent from that of VDAG_02359.1 in the cluster VDAG_02363.1-VDAG_02357.1 (Fig. S8B). Moreover, while the complete cluster of the seven adjacent genes (VDAG_02363.1-VDAG_02357.1) is present in LS region 1, a cluster of four or more highly similar genes appears once in each of the other LS regions (Figs. S8A). The duplication could be the result of the enrichment of repetitive DNA (Fig. 4 column c) in these regions that provide localized sequence homology for intra - and interchromosomal recombination [48], [49]. The presence of such genomic islands and their contribution to genome innovation through duplication, diversification and differential gene loss were also reported in Aspergillus fumigatus [50].
Interestingly, the LS regions are flanked by extensive (1 to 5 kb) AT-rich sequences (Fig. 4 column c), a characteristic of sequences which may have undergone Repeat-Induced Point (RIP)-like mutation. RIP has been regarded as a genome defense mechanism in which duplicated DNA sequences are irreversibly altered by G∶C to A∶T transitions, and most notably has been observed following meiosis [51]. Single homologs of the gene encoding the DNA methyltranferase (DMT) RID, identified as part of the RIP machinery in N. crassa [52], were present in Vd (VDAG_01783.1) and Vaa (VDBG_01766.1). RIPCAL analyses [53] detected RIP-like mutations among copies of VdLTREs, 2, 3, and 4 (Fig. S9, and data not shown) but not in VdLTRE5, further confirming a different evolutionary history for these elements.
The conservation of VdLTREs 1–4 in the two Verticillium genomes, as well as the detectable signatures of RIPed sequences among the elements suggest that VdLTREs 1–4 elements were present in the ancestral species from which Vd and Vaa evolved, and that sexual reproduction existed in the pathogens' history (SG Amyotte et al, manuscript in preparation). However, VdLTRE5 would appear to have integrated into the Vd genome after the divergence of these species, and when sexual reproduction was no longer functional in the Vd lineage. Interestingly, single but different mating type loci, the MAT1-1 and MAT1-2 idiomorphs, were identified in the Vaa and Vd genomes respectively (Fig. S10), and although both mating type loci (MAT1-2 and MAT1-1) have been observed in Vd isolates tested [54], [55], a sexual phase has never been reported for either Vd or Vaa.
Functional diversity of the LS regions
The genetic flexibility achieved through the LS regions may provide capacity for Vd to rapidly adapt to different host niches. For instance, among the LS encoding genes, we identified two homologs (VDAG_04894.1 and VDAG_04836.1) of the vdt1 gene (GenBank Accession AB045985), associated with host range specificity in Vd [56]. Overall, the four LS regions contain 354 predicted protein-encoding genes. Rather than essential (“housekeeping”) gene functions, the genes encoded in LS regions are known to play roles in iron and lipid metabolism, environmental stress responses, and potentially secondary metabolism (Web File 4 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html), as well as pathotype specificity (VDAG_04894.1 and VDAG_04836.1). When compared to the core sequences of the VdLs.17 genome, gene families including those of bZIP transcription factors, ferric reductases, and phospholipases, are significantly enriched in the LS region (P<0.05) (odds ratio analyses [57]; Table S4 in Supporting Information). Of the 354 predicted proteins, 25 (7%) were predicted as secreted (Web File 4 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html), a number that is not significantly different from the overall representation of secreted proteins in Vd (7.4%) or Vaa (7.4%).
To further validate the potential functional importance of the genes encoded in these LS regions, we analyzed EST sequences generated from three different experimental conditions, and found evidence for expression of 1,372 genes. Among those, 23% of the genes encoded in the LS regions were transcribed under the tested conditions, significantly higher (P = 4e-6) than the 12.2% for genes located outside of the LS regions (Fig. 4, and Methods, Web File 5 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html). Even though the EST data only provide evidence for the functional importance of a small proportion (12%) of the genes in the genome, the randomness of the sampling reinforces the idea that the LS regions observed in this study do not simply serve as the sink of “junk DNA”, but instead encode genes that may be functionally important.
Among the expanded gene families, ferric reductase transmembrane proteins have important roles in cell differentiation through production of reactive oxygen species (ROS) [58], [59], and may influence pathogenic or symbiotic relationships between fungi and their host plants [60], [61]. In addition to the orthologous NADPH oxidase subfamilies (NoxA to NoxC) that are present in both Verticillium genomes, and are shared among fungi, the Vd genome possesses four additional copies of ferric reductase-like proteins that form a distinctive clade (Fig. S11A), suggesting a potentially important role of iron metabolism similar to those suggested for other plant host-pathogen interactions [62]. As a further indication of the importance of iron metabolism in Vd, an iron-binding ferritin (VDAG_02389.1) with a potential role in iron sequestration [63] was uniquely present within LS region 1. Among all four sequenced Fusarium genomes, homologs of this protein are present only in F. oxysporum (FOXG_16665, FOXG_16728) [12], and both are located on Fol chromosome 15, one of the four horizontally acquired chromosomes that are required for pathogenicity on tomato [12].
Members of the expanded basic-leucine zipper (bZIP) transcription factor family contain leucine zipper regions that mediate sequence-specific DNA-binding, and are predicted to have a nuclear localization (Web File 4 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html). Phylogenetically, four of the six bZIP TFs encoded in the Vd LS regions form a distinct clade when compared to those encoded in other regions of the genome (Fig. S11B). With the exception of the gene VDAG_09148.1, which was under positive evolutionary selection, purifying selection for retention of gene function is operating on the other bZIP TFs encoded in the LS regions (Fig. S12).
Apart from the bZIP factors, Vd carries an expanded family of phospholipases which includes a homolog of a patatin-like phospholipase (PLP; VDAG_02397.1; Fig. S11C) that catalyzes the nonspecific hydrolysis of various lipids, including phospholipids, glycolipids, sulfolipids, and mono - and diacylglycerols [64]. In addition to supplying energy for pathogen growth, lipid metabolism also produces signaling molecules that play crucial roles in intra - and inter-cellular signaling [65], [66], [67]. The expansion of the above regulatory factors, both TFs and phospholipases, may contribute/regulate pathogenic traits required for Verticillium wilt development [68], [69], [70].
LS region 1 encodes a sequence homologous to the high-osmolarity-glycerol response protein (Hog1p), a well known kinase involved in osmoregulation in Saccharomyces cerevisiae [71]. In yeast this protein is nuclear-localized, and mediates the up-regulation of nearly 600 genes [72]. Almost all ascomycete genomes have a single HOG1 homolog. However, in addition to the HOG1 ortholog (VDAG_08982.1 and VDBG_04396.1 in the core genomes of Vd and Vaa, respectively), Vd encodes an extra HOG1 sequence (VDAG_02354.1) nestled in LS region 1 between LINE-like retroelement sequences. The functional importance of this extra HOG1 homolog is suggested by its expression in both the nutritionally rich complete medium, and during nitrogen-starvation, with a 2.5-fold increase in expression level during growth under the nitrogen-starved conditions (Web file 5 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html, and Methods). The two Vd HOG1 homologs have different intron-exon structures, and the phylogenetic analysis of HOG1 from representative ascomycete fungi suggests that VDAG_02354.1 is not a duplicate of VDAG_08982.1 (Fig. S13).
Overall, the LS regions provide some genetic flexibility, and genes encoded in the LS regions play important roles in signaling/transcriptional regulation and iron/lipid metabolism, processes that are important in host-fungal interactions and pathogenesis. Coupled with the enhanced arsenal of plant cell wall-degrading enzymes in Verticillium genomes, these genomic islands may contribute to the increased genetic diversity of Vd.
Comparative analyses reveal wilt pathogen-specific proteins
As specific colonizers of plant xylem vessels, the major water transport system, the wilt pathogens must develop auxiliary osmoregulatory mechanisms to maintain osmotic stability and adapt to this unique ecological niche. Among the broad diversity of the fungal kingdom, fungal species from only four genera are reported to be able to colonize this particular ecological niche and induce wilts. These wilts, all notoriously destructive, include Fusarium wilt caused by members of the F. oxysporum species complex, Verticillium wilt caused by Verticillium spp., wilt of oak trees caused by Ceratocystis fagacearum, and Dutch elm disease caused by Ophiostoma ulmi and O. novo-ulmi [1]. With a specific interest in identifying potential wilt pathogenicity-related genes, or those that may confer the ability to colonize the plant xylem, BLASTp searches were conducted using BlastMatrix [73] to identify proteins that were common to three sequenced fungal wilt pathogens (Vd, Vaa, and Fo), but absent from the proteomes of F. solani, F. graminearum, and F. verticilloides. We identified 14 such candidates (Table 3).

Extraordinarily, one of the genes identified in the search for wilt-specific proteins encodes a glucan glucosyltransferase closely related to bacterial enzymes involved in production of osmoregulated periplasmic glucans. When exposed to low-osmolarity conditions, Gram-negative bacteria use osmoregulated periplasmic glucans to adjust the osmolarity of their periplasmic space to prevent swelling and rupturing of the cytoplasmic membrane [74]. One of these related bacterial proteins includes the Erwinia chrysanthemi opgH protein, which is required for the production of osmoregulated periplasmic glucans and pathogenicity [75]. Although homologs of the glucan glucosyltransferase gene are widely distributed and well conserved among proteobacteria [74], [75], the only eukaryotic counterparts we have identified are those in the sequenced wilt pathogens (VDAG_02071, VDBG_03162 and FOXG_02706), and in a fungal pathogen of insects, Metarhizium anisopliae, strain ARSEF 23 [76]. Searches of the NCBI database using BLAST searches (See Methods) revealed no homologs in any other fungal genomes or eukaryotic sequences.
Phylogenetic analysis of the four fungal glucosyltransferases and representative bacterial OPGH sequences showed that the fungal glucosyltransferases clustered together with 100% bootstrap support, and are most closely related to those of proteobacteria in the order Rhizobiales (bootstrap value 65%; Fig. 5), supporting a model of horizontal gene transfer. In support of a potential mechanism for horizontal transfer, genetic transformation of Vaa can occur when Vaa and the Rhizobiales bacterium Agrobacterium tumefaciens are co-cultivated at plant wound sites [77]. Interestingly, Metarhizium anisopliae is known to colonize plant roots [76], and the shared ecological niche and evolutionary lineage of plant pathogenic or endophytic fungi and Metarhizium spp. could potentially have been enabling factors in the acquisition of the glucosyltransferase in these fungal genera.

In the alignments of Vd and Vaa genome assemblies, the two Verticillium glucosyltransferases are located in a highly conserved syntenic block (scaffold 3 on chromosome 3 of the Vd genome). If horizontal gene transfer had occurred, it must have happened before the divergence of these two Verticillium species. As for the Fo glucosyltransferase, that gene is located in the core part of the Fo genome (scaffold 3 on chromosome 8 of Fo assembly), in an approx 7 kb region where the synteny breaks down between Fo and its closely related sister species F. verticillioides. The absence of this gene from F. verticillioides and other sequenced Fusarium genomes (F. graminearum, and F. solani) suggests that a horizontal gene transfer event may have occurred only in the F. oxysporum lineage, and independently from transfer of the gene homolog into Verticillium. In further support of a model for independent transfer into F. oxysporum, analysis of the 20 kb sequences flanking the predicted F. oxysporum glucosyltransferase (open reading frame FOXG_02706) did not reveal any conservation with sequences flanking the Verticillium spp. glucosyltransferases.
To assess the role in V. dahliae of the glucosyltransferase homolog (VDAG_02071), knock-out transformants were generated in the wild-type strain VdLs.17 (Fig. S14). No aberrant phenotype was observed during axenic growth (See Methods), and no significant difference in pathogenicity between either of the knockout strains of Vd, and the wild type VdLs.17 was observed on lettuce (Lactuca sativa, plant introduction 251246; P>0.05), under soilless pathogenicity assay test conditions (Table S5). However, a clear difference between the knockout and wild-type strains was observed during pathogenicity tests on Nicotiana benthamiana. At about ten days post-inoculation the first symptoms were observed on plants inoculated with the wild-type strain, and after 12 days unmistakable wilting was observed and the disease rapidly progressed. Upon inoculation with the knock-out transformants disease occurred more slowly, with plants showing less stunting and wilting than those inoculated with the wild-type strain (Fig. 6; Fig. S15). Thus, the gene is clearly a virulence factor that determines fungal aggressiveness in this host species.

Conclusion
As the first sequenced Verticillium species, the analysis of the V. dahliae and V. albo-atrum assemblies provides a genomic profile of the genus, characterized by an impressive arsenal of proteins with pectinolytic activity, and enzymes containing plant cell wall attachment modules. The significant expansion of polysaccharide lyases in the Vd and Vaa genomes is especially revealing considering that pectin gels are usually released by the hosts into the xylem in response to the wilt infections [7], [78]. Despite these obstructions in the xylem and the fact that pit membranes can effectively prevent the passage of large molecules to adjacent vessels [79], Vd hyphae are still able to systemically colonize the plant xylem within 1–4 days following inoculation [2], [80]. Such rapid establishment may rely on the presence of a diverse set of polysaccharide lyases that are able to rapidly breach the barriers around the pit membranes [81], and the pectin gels around tyloses [7]. Indeed, pectin degrading enzymes have long been suggested to contribute to virulence in Verticillium spp.-host interactions [2], and although disruption of single pectinase genes in the vascular wilt fungus Fo did not perturb virulence [78], this lack of effect is undoubtedly due to the functional redundancy of these genes.
Both Vd and Fo are able to attack a very broad range of plant species, but different mechanisms are employed to accomplish this. As a species complex, Fo causes wilts of over 120 plant species [15]. However, individual formae speciales of the fungus generally have host ranges restricted to a single family, or even genus of plant [82]. The recent comparative analysis of Fusarium genomes has clearly illustrated that such host specificity is conferred by a few lineage-specific chromosomes which encode genes conferring host specificity in the F. oxysporum species complex, and can be transmitted horizontally [12]. In contrast to such strict host-specificity, Vd is well known for its ability to rapidly adapt to new hosts, and the numbers of plant hosts reported to be susceptible to Vd continues to expand worldwide [2], [4]. While the machinery that enables Vd and Vaa to interact with the live plant or decaying plant material does not itself appear to contribute to major differences in pathogenicity between the two species, one of the key differences between these two genomes is the existence of more than 1 Mb of structurally flexible sequences within the Vd genome. These flexible “genomic islands” encode important regulatory genes and may enable Vd to rapidly adapt to new niches, as illustrated by the spread of Verticillium wilt on lettuce in California in the 1990s [83]. Overall, the comparative genomic study reported here provides a strong foundation for future studies, such as functional investigations of polysaccharide lyases, and genes encoded in the LS regions.
Methods
Phylogenetic comparison of fungal species
To highlight the phylogenetic relationships of the vascular wilt pathogens Verticillium dahliae, V. albo-atrum and Fusarium oxysporum and other fungi relevant to this study a neighbor joining tree was constructed using nuclear ribosomal large subunit (28S) DNA sequences. The sequences were retrieved for each species, and a 531 character alignment was analyzed using neighbor joining as implemented in PAUP v.4.0b [84]. Sequences used in the alignments included the following GenBank accessions: Aspergillus niger (EF661191), Fusarium oxysporum f. sp. lycopersici (EU214564), F. verticillioides (AB363766), F. graminearum (FJ755253), Hypocrea jecorina (AF510497), Magnaporthe grisea (AB026819), Neurospora crassa (FJ360521), V. albo-atrum (EF543839), and V. dahliae (DQ470945). The Podospora anserina 28S sequence was retrieved from the P. anserina genome sequencing project (http://podospora.igmors.u-psud.fr/). The phylogenetic topology obtained was consistent with the one based on larger studies comprising a representative sample of the Sordariomycetes [85].
Fungal strains and growth conditions
The fungal strains VdLs.17 (ATCC accession MYA-4575) and VaMS.102 (ATCC accession MYA-4576) were isolated from lettuce in California (CA), USA and alfalfa in Pennsylvania (PA), USA [5], [86], respectively. Other strains used in this study include VdLs.16 (lettuce isolate; CA, 1996); VdBob.70 (cauliflower isolate; CA, 1990); VdLe.88, (tomato isolate; CA, 1996); VaaMs.107 (alfalfa isolate; PA, 1986); VdLe.112 (tomato isolate; CA, 1997); VdSm.113 (eggplant isolate; CA, 1997); VdLs.439 (lettuce isolate; CA, 2001); VdLs.446 (lettuce isolate; CA, 2001) [86]; VdSo.925 (spinach isolate, the Netherlands, 2003); VdSo.936 (spinach isolate, Washington State, 2003); VdLe.1087 (tomato isolate, CA, 1970). Unless specified otherwise, cultures of these fungi were maintained on potato dextrose agar (PDA) or potato dextrose broth (PDB) media at 25°C prior to use. Cultures were maintained long term in closed vials on PDA, or as −80°C stocks in 20% glycerol.
Fungal DNA and EST library preparation
Protoplasts of strains VdLs.17 and VaMS.102 were produced by overnight incubation in a 5% (w/v) Glucanex (Sigma) enzyme mixture with buffer (0.8 M sorbitol, 1 M sodium citrate, and 10 mM EDTA), pH 5.8. An Omniprep kit (GBiosciences) was used to extract DNA from protoplasts derived from strain VdLs.17 conidia harvested from PDA plates, and mycelia of strain VaMs.102 from PDB shake cultures. The PDA or PDB was supplemented with streptomycin (50 µg/ml), kanamycin (50 µg/ml) and tetracycline (50 µg/ml) for the culture of these fungi.
Three EST libraries were produced from strain VdLs.17 cultured in complete medium, root extract medium, or low nitrogen medium. Complete and low nitrogen media were prepared as described previously [87], [88]. Root extract medium was prepared by the addition, to 100 ml basal medium [88], of 5 ml supernatant from a mixture of water and ground root tissue (4.5 g of ground root tissue per 10 ml of water) of lettuce cultivar Salinas (Pybas Seeds, Salinas, CA). Three shake (150 RPM) cultures of 100 ml of CM were each inoculated with 1×107 conidia/ml of strain VdLs.17, and maintained at 25°C in the dark. At 24 hrs, each of the cultures was centrifuged, washed with water, and resuspended in 100 ml of complete, low nitrogen or root extract medium. After an additional incubation period of 24 hrs, total RNA was extracted from each fungal culture with Trizol reagent (Invitrogen). The cDNA populations were prepared using a SMART cDNA Library Construction Kit (Clontech), normalized using a Trimmer kit (Evrogen) according to the manufacturer's instructions, and cloned into pCR2.1 (Invitrogen).
Genome sequencing, assembling and mapping
Whole genome shotgun assemblies of V. dahliae strain VdLs.17 (7.5×) and V. albo-atrum strain VaMs.102 (4×) were generated with Sanger technology at the Broad Institute using the approach outlined in Table S2, and assembled using Arachne [16]. To compensate for the lack of genetic mapping information, an optical map [89] of VdLs.17 was constructed (Genome Center of Wisconsin, Madison, WI). Optical mapping is a single-molecule approach for the construction of ordered restriction maps. It uses large (250–3,000 kb), randomly sheared genomic DNA molecules as the substrate for restriction map construction. By determining the presence of sequence-specific restriction enzyme cut sites and the distances between them, restriction maps of large DNA fragments can be created. Such maps provide a useful backbone for the alignment and verification of sequence data. The VdLs.17 optical map was constructed using the restriction enzyme AflII and aligned with in silico restriction maps of the genome assembly. The correspondence of the restriction enzyme cutting sites and the predicted fragment lengths have been used to order and orient the scaffolds to the optical map.
The Vd optical map corresponds to ∼300× physical coverage and consists of 8 linkage maps with an estimated genome size of 35 Mb. Alignments were made between optical maps and the in silico maps of the sequence scaffolds using map aligner software developed at the Broad Institute. The assembled sequence scaffolds were ordered and oriented, and gaps were estimated (Table S2). The optical linkage group maps for V. dahliae strain VdLs.17 can be accessed at http://www.broad.mit.edu/annotation/genome/verticillium_dahliae/maps/Index.html.
Gene annotation and gene families
Protein-encoding genes were annotated using a combination of manually curated genes, in addition to EST BLAST alignments, and ab initio gene predictions made by FGENESH, FGENESH+ (http://linux1.softberry.com), and GENEID (http://genome.crg.es/software/geneid). Additionally, protein-encoding genes were predicted based on BLASTs of known genes available in public databases. BLAST matches with E values<1e-10 were considered to be usable BLAST evidence. HMMER [90] searches were also performed using the Pfam library to find Pfam domains on six-frame translations of the genomic sequences.
V. dahliae and V. albo-atrum secretomes and annotation
Initially, subcellular localizations for all Vd and Vaa proteins were predicted using the WoLF PSORT software (http://wolfpsort.org; [21]), resulting in identification of 1383 putative extracellular Vd proteins and 1,310 putative extracellular Vaa proteins. Only proteins containing a signal peptide and a signal peptide cleavage site, but lacking transmembrane domains, were selected. To this end, signal peptides and signal peptide cleavage sites were predicted in the set of putative extracellular proteins using the SignalP3.0 program [22], where a final SignalP D-Score cut-off of 0.500 was used to increase specificity while retaining sensitivity. Subsequently, all proteins with signal peptides (1040 and 966 for Vd and Vaa respectively) were analyzed for the presence of transmembrane domains using the web servers Phobius [91] and TMHMM (version 2.0; [92]). Both servers identified differential, partially overlapping, sets of proteins with putative transmembrane domains. On average Phobius detected 22% more proteins with transmembrane domains than did TMHMM, and about 75% of the predictions were shared between the servers. For further analyses, all proteins with putative transmembrane domains as predicted by either of the two servers were removed from the dataset.
For functional classification of the secretomes of both Verticillium species we used a number of resources, including Broad Institute automatic annotations, and Psi-BlastP [93] hits to proteins in the nr database, the Uniprot knowledge database uniref90, and the Swissprot classified protein database. Furthermore, domain-calling analyses were performed using the Pfam database (release version 23) and HMMER [90]. Subsequently, all results were parsed through BioPerl (version 1.5). All proteins lacking significant BLAST hits (E-value<1e-10) in any of the databases and for which no significant Pfam domain was called by HMMER (E-value<1E-01) were annotated as hypothetical proteins, as were proteins for which an orthologous non-informative hit was found in the genome of the other Verticillium species. Proteins with no significant BLAST hit, but for which a particular Pfam domain was called by HMMER, were annotated as Pfam domain 1-containing proteins. All proteins with significant, though non-informative, hits in any of the BLAST analyses, and no Pfam domain call by HMMER were classified as conserved hypothetical proteins (hits were considered non-informative whenever their function could not be deduced from the hits, e.g. hits were to proteins with unknown function). Finally, all proteins with informative hits with or without recognized Pfam domain were annotated manually, and classified according to their potential function.
Analyses of carbohydrate-active enzymes and CBM1-containing proteins
The carbohydrate-active enzyme catalogs of VdLs.17 and VaMs.102 were compared with the corresponding catalogs from Aspergillus niger CBS 513.88, Neurospora crassa OR74A, Magnaporthe grisea 70-15, Gibberella zeae PH-1 (ana. Fusarium graminearum), Gibberella moniliformis 7600 (Fusarium verticillioides), Fusarium oxysporum f. sp. lycopersici 4286, Podospora anserina DSM 980, and Hypocrea jecorina (Trichoderma reesei). The boundaries of the carbohydrate-active modules and associated carbohydrate-binding modules of the proteins encoded by each fungus in the comparison were determined using the BLAST and HMM-based routines of the Carbohydrate-Active-EnZymes database ([42]; http://www.cazy.org/). The display of the modular structure of the proteins was subsequently done using Flymod (Lombard, Coutinho and Henrissat, unpublished).
For CBM1 identification, a total of 37 CBM1 domains were initially identified for Vd using a 31 amino acid sequence of the VDAG_07210.1 CBM1 in low stringency (E-value = 10) tBLASTn searches of the Verticillium group database. Thirty of the CBM1 domains resided in a predicted gene model. Further gene annotation corrections were performed manually, and cataloged for VDAG_07289.1, VDAG_01694.1, and VDAG_08156.1, incorporating the CBM1 in the revised gene prediction models (Verticillium Group, Broad Institute). Then the presence of the CBM1 module in each of the 30 predicted proteins was confirmed by searching with the protein sequences against a Pfam library (http://motif.genome.jp/), with cut-off E-values of ≤1e-6. The program WoLF PSORT (http://wolfpsort.org/; [21]) was used to predict subcellular locations of the CBM1-containing proteins. Sequence alignment of amino acids in the CBM1 domains of Vd was performed using DNASIS MAX v2.9 (MiraiBio, Hitachi Software).
Comparative searches of CBM1-containing proteins in Fo and Vaa were conducted using BLASTp with each of the identified predicted CBM1-containing proteins from Vd as a query. Only those searches having an E-value cut-off <1e-12 and >50% alignment were recorded as hits, and only the first hit was selected for comparison. For additional analysis, the set of 30 P. anserina CBM1-containing predicted proteins, and 13 Fo CBM1-containing protein sequences were downloaded from the carbohydrate-active enzyme database (http://www.cazy.org/geno/geno_eukarya.html) and the Fusarium group database (Broad Institute), respectively, and verified in motif searches with an E value cutoff of ≤1e-6 (http://motif.genome.jp/).
Repetitive sequences and transposable elements
Repeat sequences were detected using Cross_match [94] which searches the genome sequence against itself, filtering for alignments longer than 200 bp with greater than 60% sequence similarity. Full-length transposable elements were annotated using a combination of computational predictions based on BLAST analysis for transposase genes, and manual inspection using the DNASTAR-based GENEQUEST program (http://www.dnastar.com) to identify class I element open reading frames and terminal repeats. The genome distribution of repeated sequences was characterized using the sensitive mode of RepeatMasker version open-3.0.8, with Cross_Match version 0.990329, RepBase Update 9.04, RM database version 20040702. For the analyses of repeat-induced point mutations (RIP) in VdLTREs 2, 3, 4 and 5, sequences of at least 500 bp in length corresponding to each type of element, were identified in BLASTn searches of the Vd genome (E value cutoff <1e-5) with the type elements. Then the RIPCAL software tool for the automated analysis of RIP [53] was used for all retrieved sequences. For Vaa, BLASTn searches of the genome with the Vd type elements revealed a total of only 45 hits. AT-rich DNA sequences at the junctions of the lineage-specific (LS) regions of VdLs.17 were identified by manual inspections of these regions. The AT-rich sequences included: supercontig 4 : 311311–316103 (LS region 1, 4797 bp); two sequences in LS region 2 in supercontig 8 : 3028301–3029894 (1,593 bp) and 3412477–3413645 (1,169 bp) separated by approximately 383 kb; Supercontig 23 : 219657–220756 (LS region 3, 1100 bp); Supercontig 25 : 19431–22306 (LS region 4, 2875 bp).
Phylogenetic analyses of ferric reductase, and phospholipase sequences
The ferric reductase transmembrane domain-containing proteins were identified in feature searches of the Fusarium and Verticillium group databases (Broad Institute). Additional ferric reductase proteins and those of the NOX classes from other fungi were from Aguirre et al. [58], and GenBank sequences for each of the accessions were used for phylogenetic analyses. These sequences included: Saccharomyces cerevisiae, NP_014458; Candida albicans, EAK96678; N. crassa, XP_329210; Fusarium graminearum, XP_391371; M. grisea, EAA57330; and M. grisea, EAA56588. The Claviceps purpurea, CAP12327 and M. grisea, XP_368494 transmembrane domains were added to the analysis since these NADPH oxidases are virulence factors in the respective pathogens [60], [61]. Vd and Vaa ferric reductase proteins not included in the tree (Fig. S11A) were VDAG_06992, VDAG_07588, VDBG_05342, VDBG_05649, and VDBG_06458.
The protein sequences of patatin-like phospholipases were obtained from Vd, Vaa, and Fo databases by querying the databases using BLASTp with phospholipases predicted by the Broad annotation pipeline. Inspections of the protein alignments of patatin-like phospholipases from the Verticillium group database (Broad Institute) revealed major differences in the length and composition of these proteins. Therefore, domains common to patatin-like phospholipases were identified in the Vd and Vaa sequences using motif searches (http://motif.genome.jp). The identified domains of the oxyanion hole and the G-x-S-x-G motif (including noncanonical) from each protein were used for the phylogenetic analyses. The phylogenetic analyses included homologous sequences of plants and fungi that were obtained from literature searches, and identified in the tree (Fig. S11C) by GenBank accession: F. graminearum, FG06645.1; Aspergillus clavatus, XP_001268427; M. grisea, A4QVZ8; M. grisea, ABG79933; S cerevisiae, NP_014044, and sequences from five plant species (shown in purple in Supplementary Figure S10 C): Gossypium hirsutum, AAX99411; Nicotiana tabacum, AAF98368; Vitis vinifera, CAO61313; Oryza sativa, AAT77905, Arabidopsis thaliana, NP_180224.
Sequence and statistical analyses of lineage-specific regions
DNA alignments of duplicated lineage-specific (LS) sequences (as shown in Supplementary Fig. S7B) were performed using DNASIS MAX v2.9 (MiraiBio, Hitachi Software). The 354 predicted proteins from the Vd LS regions were downloaded from the Verticillium group database (Broad Institute). The program WoLF PSORT was used to predict subcellular location as described above, and BlastMatrix analyses were performed to identify putative orthologous sequences in other fungal species. BlastMatrix is a modified BLAST program that supports the simultaneous identification in multiple species of genes homologous to a query [73]. The fungal genome dataset archived in the web-based, comparative fungal genomics platform (CFGP; http://cfgp.snu.ac.kr) was queried, as were selected stramenopile, plant, protist and animal genomes.
The w statistics for odds ratio analyses [57] were calculated to compare the frequency of specific genes of interest within the LS regions, versus the their frequency in the remainder of the genome. Values significantly greater than 1 indicate the preferential (non-random) distribution of these genes within the LS region versus the non-LS regions. Transformation was done using the natural log (ln) of w, and the error and 95% CI were calculated for lnw. The odds ratios were based on 10,535 total predicted proteins encoded in the genome, and 354 total predicted proteins encoded in the LS regions of strain VdLs.17.
To elucidate potential evolutionary relationships among the bZIP TFs located in the Vd LS regions, a dN/dS analysis was employed. This analysis was done using the Phylogenetic Analysis by Maximum Likelihood (PAML) package [95] which estimates synonymous and non-synonymous substitution rates of nucleotide sequences using the pairwise codeml algorithm, assuming realistic evolutionary models. Prior to the dN/dS analysis, codons were reconstructed using Pal2Nal software ([96]; http://www.bork.embl.de/pal2nal).
Nucleic acid hybridizations
Each of the DNA sequences used as hybridization probes was amplified from genomic DNA of VdLs.17 by PCR, cloned into vector pCR4-TOPO and sequenced (MCLAB, San Francisco, CA) to ensure the correct probe sequence. PCR products were DIG-labeled using the random labeling method (Roche). The DNA probe for sequence VDAG_04871.1 was generated with primers 4871F (5′-TTTGGCCATCTCAAAAGATGG-3′) and 4871R (5′-TACTCATCTTGACCTTCTGTCC-3′). The probe for VDAG_05180.1 was generated with primers 5180F (5′-ACAATGCGGCCCGACGTTTTCG-3′) and 5180R (5′-AGCTGCACGGCACAACGATGTC-3′). Similarly, primers 9197F (5′-AGAGACTGTCCGACACAGGAAG-3′) and 9197R (5′ - CATCAGCTCGCGCAAACAATGG-3′) and 09220F (5′-GTGCATACTGATACGCAGTTGC-3′) and 09220R (5′-TGAGTTCCCAGAAAGAGCGGTGC-3′) were used for probes VDAG_09197.1 and VDAG_09220.1, respectively.
For DNA blot hybridizations, the DNA was extracted from the conidia of each of the strains using a bead beater protocol, and RNA was removed with RNAse A (Promega, Madison, WI). Five micrograms of DNA from each strain was digested overnight at 37°C with either HindIII or PstI enzyme (10 U/reaction, Promega). The entire reaction mix containing digested DNA was loaded onto a 0.8% agarose gel for electrophoresis. DNA was transferred to a Zeta Probe Membrane (Bio-Rad Laboratories, Hercules, CA) overnight by capillary transfer, using 20× saturated sodium citrate (SSC). Blots were fixed by cross-linking with a UVC500 cross-linker (Hoefer, San Francisco CA), rinsed with sterilized water (Millipore, Billerica, MA), and air-dried at room temperature. Pre-hybridization was done for 4 to 5 hours at 42°C, in 50% formamide, 5× SSC, 49 mM Na2H2PO4, 2.94% SDS and 0.2× blocking buffer (Roche, Mannheim, Germany). Hybridization was done overnight (16–18 hours), using 25 ng/ml DIG-labeled probe in 5 ml pre-hybridization buffer. Membranes were washed twice at 42°C for 30 minutes, with 125 ml 1 mM EDTA, 40 mM Na2H2PO4, 5% sodium dodecyl sulftate (SDS), and twice for 30 minutes at 55°C with 125 ml 1 mM EDTA, 40 mM Na2H2PO4, 1% SDS. Membranes were rinsed in 1× washing buffer (Roche) two times and once in 1× detection buffer (Roche), and hybrids detected with anti-digoxigenin-AP conjugate Fab fragments (Roche), according to the manufacturer's instructions, and exposure to BioMax film (Eastman Kodak Company, Rochester, NY).
Fungal vascular wilt pathogen-specific proteins
To identify potential wilt pathogen-specific proteins, the total protein set from Vd was used in comparative BlastMatrix [73] searches against sequences from the vascular wilt fungi Vaa and Fo, and predicted protein sets from other fungi, including Fusarium solani, F. graminearum, and F. verticilliodes. The same comparison was conducted with the other five genomes (15 genome comparisons in total were made via BLAST searches), resulting in the classification of the Vd genes into 32 classes (Web File 6 at http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/SupplementaryPage.html). For example, class 1 included proteins that did not display any significant matches of Vd proteins to any from the other five genomes, and can potentially be considered as Vd-specific. The class containing those predicted proteins present in Vd, Vaa and Fo but not present among the other protein sets included 28 candidate proteins (BLASTp E value cutoff <1e-6). Additional manual screening of these candidate genes was performed by BLASTp analysis (Verticillium and Fusarium group databases, Broad Institute), and by comparison of the protein alignment lengths (alignment lengths <50% were excluded). The manual screening limited the number of potential wilt-specific proteins to 14. Motif searches (http://motif.genome.jp/) were performed for each of the 14 proteins against the Pfam library, using the program Hmmpfam [90]. tBLASTn and BLASTp searches of the NCBI nr database were performed to identify similar proteins from other organisms (BLASTp E value cutoff <1e-6), and WoLF PSORT [21] was used to infer subcellular localization of the predicted proteins, as described above. Additional tBLASTn and BLASTp searches of the NCBI (nr and WGS) databases were performed using the glucosyltransferase gene ORFs of VDAG_02071, VDBG_03162 and FOXG_02706. Twenty kb windows flanking either side of the open reading frames for VDAG_02071, VDBG_03162 and FOXG_02706 were examined by BLASTn analyses to determine if these sequences were of fungal origin. The maximum-likelihood tree including four glucosyltransferase proteins was constructed employing a maximum likelihood-based package, PhyML [97]. Branch lengths in substitutions per site were calculated using the WAG evolutionary model [98].
Deletion of the glusosyltransferase gene in V. dahliae
The deletion construct for the knockout of gene VDAG_02071, the glusosyltransferase in Vd, was prepared by Paz et al [99] and used in Agrobacterium tumefaciens-mediated transformation of VdLs.17 to obtain independent mutant strains, ΔGT-A and ΔGT-B. The knockout of the glucosyltransferase gene in each was confirmed by nucleic acid hybridization (Fig. S14). The 1643 bp probe, DIG-labeled (Roche) as described above (See Nucleic acid hybridizations), was amplified using primers OSC-F 5′-CGCCAATATATCCTGTCAAACACT-3′ and Hyg-F, 5′-AGAGCTTGGTTGACGGCAATTTCG-3′. Five micrograms of DNA was obtained from VdLs.17 and the respective mutant strains, and digested with BamH1 enzyme (10 U/reaction, Promega) overnight at 37°C. DNA transfer for the blot, probe hybridization, and DIG detection was carried out as described above (See Nucleic acid hybridizations). Light microscopy analyses of the ΔGT-A and ΔGT-B strains, in comparison to strain VdLs.17, was performed to assess microsclerotia and conidia production, and morphology of conidiophores.
RT-PCR detection of VDAG_02071 expression
For reverse transcription-PCR detection of the VDAG_02071 transcripts, RNA was extracted from VdLs.17 and the mutant strains using the RNeasy Kit (Qiagen, La Jolla, CA) with an on-column DNAse digestion. Reverse transcription reactions included 100 ng RNA template and 0.5 ug oligo-dT15, were incubated at 70°C, cooled on ice and then added to 1× GoScript Buffer (Promega), 0.5 mM dNTPs, 7.5 mM MgCl2, 40 U RNAsin (Promega), and 1 ul GoScript Reverse Transcriptase (Promega) to a final volume of 20 ul. The reaction was incubated at 25°C for 5 min, 55°C for 45 min, and 70°C for 15 min. The amplification reactions included 5 µl of cDNA template, 1× Promega GoTaq Flexi Buffer, 3 mM MgCl2,0.2 mM dNTP's, 0.15 pmol ORF1F primer (5′-ATGAGCAACAACATCCTTACACC-3′), 0.15 pmol ORFR primer (5′-CTCTAGGGTTGAGCCGATG-3′), and 1.25 U GoTaq Flexi Polymerase. The thermocycling program included denaturation at 94°C for 3 min, followed by 40 cycles of 94°C for 30 sec, 55°C for 30 sec and 72°C 1 min 30 sec with a final extension of 72°C 5 min. For the β-tubulin control reaction, 2 µl of cDNA was used as template in a total reaction volume of 20 µl, containing 1× GoTaq Green Master Mix (Promega), 200 nM each of primers VertBtF and VertBtR (Atallah et al. 2007), and 1.25 U GoTaq Flexi polymerase. The thermocycling program consisted of a denaturation at 95°C for 3 min followed by 35 cycles of 95°C 15 sec, 63°C 35 sec, and 72°C 30 sec with a final extension of 72°C for 3 min.
Pathogenicity experiments
Two independent glucosyltransferase (VDAG_02071) knockout strains and the wild type VdLs.17 strain were subcultured on PDA for 10 days at 22°C. Inoculum was prepared by harvesting conidiospores and adjusting the concentration to 106 spores/ml in water. For each experiment six two-week-old Nicotiana benthamiana plants were inoculated with each of the Vd genotypes by dipping the roots for 5 min in inoculum, and transferring the plants into soil. Plants were scored at two weeks post-inoculation for the display of symptoms. The experiment was performed three times with similar results. ImageJ (http://rsb.info.nih.gov/ij/) was used to measure plant height, and data were analyzed using a T-test.
Pathogenicity tests on lettuce PI 251246 were conducted using a soilless assay as previously described [100], except that the inoculum was adjusted to 2×107 spores/ml and each treatment consisted of 15 plants inoculated with water or one of knockout or wild-type genotypes as described above. Dead plants were discarded from the data set for analyses. Data were analyzed using analysis of variance (ANOVA) statistics of ranked data using the PROC Mixed procedure of SAS (Version 9.1, SAS Institute, Cary, NC), with the LD_CI macro to generate relative effects (RME) for each treatment, and confidence intervals for detection of statistical differences between treatments [101], [102]. Leaf symptom data were expressed as the proportion of symptomatic leaves per treatment, and the root vascular discoloration data was expressed as the proportion of discolored roots per treatment. The data from the water control was not included in the analysis, but is summarized in Table S5. For the one-way ANOVA, “isolate” was treated as a fixed effect, and three independent experiments were combined into a single analysis with “experiment” treated as a random variable. The median and maximum percentage of symptomatic leaves and the percentage of plants with root vascular discoloration were calculated for each strain.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AgriosG 2005 Plant Pathology Burlington Elsevier Academic Press
2. PeggGFBradyBL 2002 Verticillium Wilts Wallingford, UK CABI Publishing
3. FradinEFThommaBPHJ 2006 Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol Plant Pathol 7 71 86
4. KlostermanSJAtallahZKValladGESubbaraoKV 2009 Diversity, pathogenicity, and management of Verticillium species. Ann Rev Phytopathol 47 39 62
5. BhatRGSubbaraoKV 1999 Host range specificity in Verticillium dahliae. Phytopathology 89 1218 1225
6. BishopCDCooperRM 1984 Ultrastructure of vascular colonization by fungal wilt pathogens. II. Invasion of resistant cultivars. Physiol Plant Pathol 24 277 289
7. ClerivetADeonVAlamiILopezFGeigerJ-P 2000 Tyloses and gels associated with cellulose accumulation in vessels are responses of plane tree seedlings (Platanus×acerifolia) to the vascular fungus Ceratocystis fimbriata f. sp platani. Trees (Berl West) 15 25 31
8. RiouxDNicoleMSimardMOuelletteGB 1998 Immunocytochemical evidence that secretion of pectin occurs during gel (Gum) and tylosis formation in trees. Phytopathology 88 494 505
9. AndersenPCBrodbeckBV 1989 Diurnal and temporal changes in the chemical profile of xylem exudate from Vitis rotundifolia. Physiologia Plantarum 75 63 70
10. HayesRJValladGEQinQ-MGrubeRCSubbaraoKV 2007 Variation for resistance to Verticillium wilt in lettuce (Lactuca sativa L.). Plant Disease 91 439 445
11. FradinEFZhangZAyalaJCJCastroverdeCDMNazarRN 2009 Genetic dissection of Verticillium wilt resistance mediated by tomato ve1. Plant Physiol 150 320 332
12. MaL-Jvan der DoesHCBorkovichKAColemanJJDaboussiM-J 2010 Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464 367 373
13. GordonTMartynR 1997 The evolutionary biology of Fusarium oxysporum. Ann Rev Phytopathol 35 111 128
14. ArmstrongGMArmstrongJK 1981 Formae speciales and races of Fusarium oxysporum causing wilt diseases. University Park, The Pennsylvania State University Press 392 399
15. MichielseCBRepM 2009 Pathogen profile update: Fusarium oxysporum. Mol Plant Pathol 10 311 324
16. JaffeDBButlerJGnerreSMauceliELindblad-TohK 2003 Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res 13 91 96
17. SoanesDMAlamICornellMWrongHMHederlerC 2008 Comparative genome analysis of filamentous fungi reveals gene family expansions associated with fungal pathogenesis. PLoS ONE 3 e2300
18. van der DoesHCRepM 2007 Virulence genes and the evolution of host specificity in plant-pathogenic fungi. Mol Plant Microbe Interac 20 1175 1182
19. de JongeRBoltonMDThommaBPHJ 2011 How filamentous pathogens co-opt plants: the ins and outs of fungal effectors. Curr Opin Plant Biol E-pub ahead of print. doi:10.1016/j.pbi.2011.03.005
20. OlivaRWinJRaffaeleSBoutemyLBozkurtTO 2010 Recent developments in effector biology of filamentous plant pathogens. Cell Microbiol 12 705 715
21. HortonPParkK-JObayashiTFujitaNHaradaH 2007 WoLF PSORT: protein localization predictor. Nucl Acids Res 35 W585 587
22. EmanuelssonOBrunakSvon HeijneGNielsenH 2007 Locating proteins in the cell using TargetP, SignalP and related tools. Nature Protoc 2 953 971
23. van EsseHPBoltonMDStergiopoulosIde WitPJGMThommaBPHJ 2007 The chitin-binding Cladosporium fulvum effector protein Avr4 is a virulence factor. Mol Plant Microbe Interac 20 1092 1101
24. van EsseHPVan't KloosterJWBoltonMDYadetaKAVan BaarlenP 2008 The Cladosporium fulvum virulence protein Avr2 inhibits host proteases required for basal defense. Plant Cell 20 1948 1963
25. BoltonMDvan EsseHPVossenJHde JongeRStergiopoulosI 2008 The novel Cladosporium fulvum lysin motif effector Ecp6 is a virulence factor with orthologues in other fungal species. Mol Microbiol 69 119 136
26. RepMvan der DoesHCMeijerMvan WijkRHoutermanPM 2004 A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol Microbiol 53 1373 1383
27. TianMWinJSongJvan der HoornRvan der KnaapE 2007 A Phytophthora infestans cystatin-like protein targets a novel tomato papain-like apoplastic protease. Plant Physiol 143 364 377
28. KamounS 2006 A catalogue of the effector secretome of plant pathogenic Oomycetes. Ann Rev Phytopathol 44 41 60
29. ThommaBPHJVan EsseHPCrousPWDe WitPJGM 2005 Cladosporium fulvum (syn. Passalora fulva), a highly specialized plant pathogen as a model for functional studies on plant pathogenic Mycosphaerellaceae. Mol Plant Pathol 6 379 393
30. de JongeRPeter van EsseHKombrinkAShinyaTDesakiY 2010 Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science 329 953 955
31. de JongeRThommaBPHJ 2009 Fungal LysM effectors: extinguishers of host immunity? Trends Microbiol 17 151 157
32. LaugeRJoostenMHAJVan den AckervekenGFJMVan den BroekHWJDe WitPJGM 1997 The in planta-produced extracellular proteins ECP1 and ECP2 of Cladosporium fulvum are virulence factors. Mol Plant Microbe Interac 10 725 734
33. de JongeRvan EsseHPKombrinkAShinyaTBoursR 2010 Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in tomato plants. Science 329 953 955
34. PembertonCLSalmondGPC 2004 The Nep1-like proteins - A growing family of microbial elicitors of plant necrosis. Mol Plant Pathol 5 353 359
35. QutobDKemmerlingBBrunnerFKufnerIEngelhardtS 2006 Phytotoxicity and innate immune responses induced by Nep1-like proteins. Plant Cell 18 3721 3744
36. GijzenMNürnbergerT 2006 Nep1-like proteins from plant pathogens: Recruitment and diversification of the NPP1 domain across taxa. Phytochemistry 67 1800 1807
37. ZhaoSQiX 2008 Signaling in plant disease resistance and symbiosis. J Integ Plant Biol 50 799 807
38. KulkarniRDKelkarHSDeanRA 2003 An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem Sci 28 118 121
39. CarpitaNCGibeautDM 1993 Structural models of primary cell walls in flowering plants: consistency of molecular structure with the physical properties of the walls during growth. Plant J 3 1 30
40. YadavPKSinghVKYadavSYadavKDSYadavD 2009 In silico analysis of pectin lyase and pectinase sequences. Biochemistry (Moscow) 74 1049 1055
41. MohnenD 2008 Pectin structure and biosynthesis. Curr Opin Plant Biol 11 266 277
42. CantarelBLCoutinhoPMRancurelCBernardTLombardV 2009 The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucl Acids Res 37 D233 238
43. GilkesNRHenrissatBKilburnDGMillerRCJrWarrenRA 1991 Domains in microbial beta-1, 4-glycanases: sequence conservation, function, and enzyme families. Microbiol Rev 55 303 315
44. ReceveurVCzjzekMSchuleinMPaninePHenrissatB 2002 Dimension, shape, and conformational flexibility of a two domain fungal cellulase in solution probed by small angle X-ray scattering. J Biol Chem 277 40887 40892
45. EspagneELespinetOMalagnacFDa SilvaCJaillonO 2008 The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol 9 R77
46. GaulinEDrameNLafitteCTorto-AlaliboTMartinezY 2006 Cellulose binding domains of a Phytophthora cell wall protein are novel pathogen-associated molecular patterns. Plant Cell 18 1766 1777
47. BrotmanYBriffEViterboAChetI 2008 Role of swollenin, an expansin-like protein from Trichoderma, in plant root colonization. Plant Physiol 147 779 789
48. DaboussiMJ 1996 Fungal transposable elements: Generators of diversity and genetic tools. J Genet 75 325 339
49. OliverKRGreeneWK 2009 Transposable elements: powerful facilitators of evolution. BioEssays 31 703 714
50. FedorovaNDKhaldiNJoardarVSMaitiRAmedeoP 2008 Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet 4 e1000046
51. CambareriEBSingerMJSelkerEU 1991 Recurrence of Repeat-Induced Point mutation (RIP) in Neurospora crassa. Genetics 127 699 710
52. FreitagMWilliamsRLKotheGOSelkerEU 2002 A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc Natl Acad Sci U S A 99 8802 8807
53. HaneJKOliverRP 2008 RIPCAL: a tool for alignment-based analysis of repeat-induced point mutations in fungal genomic sequences. BMC Bioinformatics 9 478
54. UsamiTItohMAmemiyaY 2009 Mating type gene MAT1-2-1 is common among Japanese isolates of Verticillium dahliae. Physiol Mol Plant Pathol 73 133 137
55. UsamiTItohMAmemiyaY 2009 Asexual fungus Verticillium dahliae is potentially heterothallic. J Gen Plant Pathol 75 422 427
56. UsamiTAbikoMShishidoMAmemiyaY 2002 Specific detection of tomato pathotype of Verticillium dahliae by PCR assays. J Gen Plant Pathol 68 134 140
57. SokalRRRohlfFJ 1995 Biometry, The Principles and Practice of Statistics in Biological Research, Analysis of Frequencies New York W.H. Freeman and Company
58. AguirreJRíos-MombergMHewittDHansbergW 2005 Reactive oxygen species and development in microbial eukaryotes. Trends Microbiol 13 111 118
59. Cano-DominguezNAlvarez-DelfinKHansbergWAguirreJ 2008 NADPH oxidases NOX-1 and NOX-2 require the regulatory subunit NOR-1 to control cell differentiation and growth in Neurospora crassa. Eukaryot Cell 7 1352 1361
60. EganMJWangZ-YJonesMASmirnoffNTalbotNJ 2007 Generation of reactive oxygen species by fungal NADPH oxidases is required for rice blast disease. Proc Natl Acad Sci U S A 104 11772 11777
61. GiesbertSSchurgTScheeleSTudzynskiP 2008 The NADPH oxidase Cpnox1 is required for full pathogenicity of the ergot fungus Claviceps purpurea. Mol Plant Pathol 9 317 327
62. ExpertDEnardCMasclauxC 1996 The role of iron in plant host-pathogen interactions. Trends Microbiol 4 232 237
63. LobreauxSMassenetOBriatJF 1992 Iron induces ferritin synthesis in maize plantlets. Plant Mol Biol 19 563 575
64. HirschbergHJHBSimonsJ-WFADekkerNEgmondMR 2001 Cloning, expression, purification and characterization of patatin, a novel phospholipase A. Eur J Biochem 268 5037 5044
65. La CameraSGeoffroyPSamahaHNdiayeARahimG 2005 A pathogen-inducible patatin-like lipid acyl hydrolase facilitates fungal and bacterial host colonization in Arabidopsis. Plant J 44 810 825
66. La CameraSBalagueCGobelCGeoffroyPLegrandM 2009 The Arabidopsis patatin-like protein 2 (PLP2) plays an essential role in cell death execution and differentially affects biosynthesis of oxylipins and resistance to pathogens. Mol Plant Microbe Interac 22 469 481
67. WangX 2004 Lipid signaling. Curr Opin Plant Biol 7 1 8
68. SheaJMDel PoetaM 2006 Lipid signaling in pathogenic fungi. Curr Opin Microbiol 9 352 358
69. LevSHadarRAmedeoPBakerSEYoderOC 2005 Activation of an AP1-like transcription factor of the maize pathogen Cochliobolus heterostrophus in response to oxidative stress and plant signals. Eukaryot Cell 4 443 454
70. BlancoFAJudelsonHS 2005 A bZIP transcription factor from Phytophthora interacts with a protein kinase and is required for zoospore motility and plant infection. Mol Microbiol 56 638 648
71. BrewsterJLde ValoirTDwyerNDWinterEGustinMC 1993 An osmosensing signal transduction pathway in yeast. Science 259 1760 1763
72. ProftMStruhlK 2004 MAP kinase-mediated stress relief that precedes and regulates the timing of transcriptional induction. Cell 118 351 361
73. ParkJParkBJungKJangSYuK 2008 CFGP: a web-based, comparative fungal genomics platform. Nucl Acids Res 36 D562 571
74. BohinJP 2000 Osmoregulated periplasmic glucans in Proteobacteria. FEMS Microbiol Lett 186 11 19
75. PageFAltabeSHugouvieux-Cotte-PattatNLacroixJMRobert-BaudouyJ 2001 Osmoregulated periplasmic glucan synthesis is required for Erwinia chrysanthemi pathogenicity. J Bacteriol 183 3134 3141
76. GaoQJinKYingS-HZhangYXiaoG 2011 Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet 7 e1001264
77. KnightCJBaileyAMFosterGD 2010 Investigating Agrobacterium-mediated transformation of Verticillium albo-atrum on plant surfaces. PLoS ONE 5 e13684
78. ReignaultPValette-ColletOBoccaraM 2008 The importance of fungal pectinolytic enzymes in plant invasion, host adaptability and symptom type. Eur J Plant Pathol 120 1 11
79. JansenSChoatBPletsersA 2009 Morphological variation of intervessel pit membranes and implications to xylem function in angiosperms. Am J Bot 96 409 419
80. HeinzRLeeSWSaparnoANazarRNRobbJ 1998 Cyclical systemic colonization in Verticillium-infected tomato. Physiol Mol Plant Pathol 52 385 396
81. ArendMMuningerMFrommJ 2008 Unique occurrence of pectin-like fibrillar cell wall deposits in xylem fibres of poplar. Plant Biol 10 763 770
82. KistlerHC 2001 Evolution of host specificity in Fusarium oxysporum. SummerellBALeslieJFBackhouseWLBurgessLW Fusarium: Paul E Nelson Memorial Symposium St. Paul APS Press 70 82
83. SubbaraoKVHubbardJCGreatheadASSpencerGA 1997 Verticillium wilt. DavisRMSubbaraoKVRaidRNKurtzEA Compendium of lettuce diseases St. Paul APS Press 26 27
84. SwoffordDL 2003 PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). 4 ed Sunderland, Massachusetts Sinauer Associates
85. ZhangNCastleburyLAMillerANHuhndorfSMSchochCL 2006 An overview of the systematics of the Sordariomycetes based on a four-gene phylogeny. Mycologia 98 1076 1087
86. QinQ-MValladGEWuBMSubbaraoKV 2006 Phylogenetic analyses of phytopathogenic isolates of Verticillium spp. Phytopathology 96 582 592
87. TrailFXuJ-RMiguelPSHalgrenRGKistlerHC 2003 Analysis of expressed sequence tags from Gibberella zeae (anamorph Fusarium graminearum). Fungal Genet Biol 38 187 197
88. CorrellJCKlittichCJRLeslieJF 1987 Nitrate nonutilizing mutants of Fusarium oxysporum and their use in vegetative compatibility test. Phytopathology 77 1649 1646
89. SchwartzDCLiXHernandezLIRamnarainSPHuffEJ 1993 Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science 262 110 114
90. EddyS 1998 Profile hidden Markov models. Bioinformatics 14 755 763
91. KallLKroghASonnhammerELL 2007 Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucl Acids Res 35 W429 432
92. KroghALarssonBvon HeijneGSonnhammerELL 2001 Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 305 567 580
93. AltschulSFKooninEV 1998 Iterated profile searches with PSI-BLAST–a tool for discovery in protein databases. Tr Biochem Sci 23 444 447
94. EwingBHillierLWendlMCGreenP 1998 Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res 8 175 185
95. YangZ 2007 PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 24 1586 1591
96. SuyamaMTorrentsDBorkP 2006 PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucl Acids Res 34 W609 612
97. GuindonSLethiecFDurouxPGascuelO 2005 PHYML Online – a web server for fast maximum likelihood-based phylogenetic inference. Nucl Acids Res 33 W557 559
98. WhelanSGoldmanN 2001 A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18 691 699
99. PazZGarcia-PedrajasMDAndrewsDLKlostermanSJBaeza-MontanezL 2011 One step construction of Agrobacterium-recombination-ready-plasmids (OSCAR), an efficient and robust tool for ATMT based gene deletion construction in fungi. Fungal Genet Biol 48 677 684
100. KlostermanSJHayesRJ 2009 A soilless Verticillium wilt assay using an early flowering lettuce. Plant Dis 93 691 698
101. BrunnerEDomhofSLangerF 2002 Nonparametric Analysis of Longitudinal Data New York John Wiley & Sons
102. ShahDAMaddenLV 2004 Nonparametric analysis of ordinal data in designed factorial experiments. Phytopathology 94 33 43
103. ValladGESubbaraoKV 2008 Colonization of resistant and susceptible lettuce cultivars by a green fluorescent protein-tagged isolate of Verticillium dahliae. Phytopathology 98 871 885
104. SaitouNNeiM 1987 The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4 406 425
105. FelsensteinJ 1995 PHYLIP: Phylogenetic inference programs. 3.572 ed Seattle Univ. Washington
106. ZuckerkandlEPaulingL 1965 Evolutionary divergence and convergence in proteins. BrysonVVogelHJ Evolving Genes and Proteins New York Academic Press 97 166
107. TamuraKDudleyJNeiMKumarS 2007 MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evolution msm092
108. AtallahZKBaeJJanskySHRouseDIStevensonWR 2007 Multiplex real-time quantitative PCR to detect and quantify Verticillium dahliae colonization in potato lines that differ in response to Verticillium wilt. Phytopathology 97 865 872
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Requires Glycerol for Maximum Fitness During The Tick Phase of the Enzootic Cycle
- Comparative Genomics Yields Insights into Niche Adaptation of Plant Vascular Wilt Pathogens
- The Role of IL-15 Deficiency in the Pathogenesis of Virus-Induced Asthma Exacerbations
- “Persisters”: Survival at the Cellular Level
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy