
E6 and E7 from Beta Hpv38 Cooperate with Ultraviolet Light in the Development of Actinic Keratosis-Like Lesions and Squamous Cell Carcinoma in Mice
Cutaneous beta human papillomavirus (HPV) types appear to be involved in the development of non-melanoma skin cancer (NMSC); however, it is not entirely clear whether they play a direct role. We have previously shown that E6 and E7 oncoproteins from the beta HPV type 38 display transforming activities in several experimental models. To evaluate the possible contribution of HPV38 in a proliferative tissue compartment during carcinogenesis, we generated a new transgenic mouse model (Tg) where HPV38 E6 and E7 are expressed in the undifferentiated basal layer of epithelia under the control of the Keratin 14 (K14) promoter. Viral oncogene expression led to increased cellular proliferation in the epidermis of the Tg animals in comparison to the wild-type littermates. Although no spontaneous formation of tumours was observed during the lifespan of the K14 HPV38 E6/E7-Tg mice, they were highly susceptible to 7,12-dimethylbenz(a)anthracene (DMBA)/12-0-tetradecanoylphorbol-13-acetate (TPA) two-stage chemical carcinogenesis. In addition, when animals were exposed to ultraviolet light (UV) irradiation, we observed that accumulation of p21WAF1 and cell-cycle arrest were significantly alleviated in the skin of Tg mice as compared to wild-type controls. Most importantly, chronic UV irradiation of Tg mice induced the development of actinic keratosis-like lesions, which are considered in humans as precursors of squamous cell carcinomas (SCC), and subsequently of SCC in a significant proportion of the animals. In contrast, wild-type animals subjected to identical treatments did not develop any type of skin lesions. Thus, the oncoproteins E6 and E7 from beta HPV38 significantly contribute to SCC development in the skin rendering keratinocytes more susceptible to UV-induced carcinogenesis.
Published in the journal:
. PLoS Pathog 7(7): e32767. doi:10.1371/journal.ppat.1002125
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002125
Summary
Cutaneous beta human papillomavirus (HPV) types appear to be involved in the development of non-melanoma skin cancer (NMSC); however, it is not entirely clear whether they play a direct role. We have previously shown that E6 and E7 oncoproteins from the beta HPV type 38 display transforming activities in several experimental models. To evaluate the possible contribution of HPV38 in a proliferative tissue compartment during carcinogenesis, we generated a new transgenic mouse model (Tg) where HPV38 E6 and E7 are expressed in the undifferentiated basal layer of epithelia under the control of the Keratin 14 (K14) promoter. Viral oncogene expression led to increased cellular proliferation in the epidermis of the Tg animals in comparison to the wild-type littermates. Although no spontaneous formation of tumours was observed during the lifespan of the K14 HPV38 E6/E7-Tg mice, they were highly susceptible to 7,12-dimethylbenz(a)anthracene (DMBA)/12-0-tetradecanoylphorbol-13-acetate (TPA) two-stage chemical carcinogenesis. In addition, when animals were exposed to ultraviolet light (UV) irradiation, we observed that accumulation of p21WAF1 and cell-cycle arrest were significantly alleviated in the skin of Tg mice as compared to wild-type controls. Most importantly, chronic UV irradiation of Tg mice induced the development of actinic keratosis-like lesions, which are considered in humans as precursors of squamous cell carcinomas (SCC), and subsequently of SCC in a significant proportion of the animals. In contrast, wild-type animals subjected to identical treatments did not develop any type of skin lesions. Thus, the oncoproteins E6 and E7 from beta HPV38 significantly contribute to SCC development in the skin rendering keratinocytes more susceptible to UV-induced carcinogenesis.
Introduction
Non-melanoma skin cancer (NMSC) is the most common cancer in adult fair-skinned populations [1]. Ultraviolet light (UV) is a key risk factor for the NMSC [2]–[4]. In addition, it appears that infectious agent(s) may favor skin carcinogenesis. This is suggested by the fact that immuno-compromised organ transplant recipients (OTRs) have a 50–100-fold higher risk of developing NMSC compared to the general population [5], [6] [6] [7] [8]. A sub-group of cutaneous human papillomavirus (HPV) types, belonging to the genus beta of the HPV phylogenetic tree, are putative etiological factors of NMSC [9] [10]. These HPV types were first isolated in individuals with an autosomal recessive disorder, termed epidermodysplasia verruciformis (EV). EV individuals are susceptible to infection by beta HPV types and have a propensity to develop confluent flat warts, which, in approximately 30% of the cases, progress to squamous cell carcinomas (SCC) on sun-exposed areas [9] [10]. Accordingly, DNA from several beta HPV types was found in a high percentage of precursor lesions, actinic keratoses, and SCC from OTRs [11] [12] [13]. More recent studies indicate that beta HPV types are also involved in skin carcinogenesis in the normal population. Detection of antibodies against the major capsid protein L1 showed an increased seroreactivity to beta HPV types in patients with cutaneous SCC in comparison to healthy individuals [14] [15] [16] [17] [18] [19].
Functional studies have provided further evidence of an association of beta HPV types with NMSC. Since previous studies demonstrated the key role in cellular transformation of E6 and E7 oncoproteins from cervical cancer-associated mucosal high-risk (HR) HPV types, functional investigations on beta HPV types focused on the characterization of E6 and E7 biological properties. These studies showed that E6 and E7 from beta HPV types also displayed transforming capability in in vitro and in vivo experimental models [10]. E6 from beta HPV types associates with the pro-apoptotic protein Bak, a member of the Bcl-2 family, promoting its proteasomal degradation and preventing apoptosis in response to genomic stress [20] [21]. Studies from our group have shown that E6 and E7 from beta HPV38 are able to immortalize primary human keratinocytes [22] [23], similarly to E6 and E7 from the mucosal HR HPV types. Accordingly, we observed that HPV38 E6 and E7 expression in these cells leads to the accumulation of ΔNp73α, which in turn alters the p53 transcriptional functions [24].
Tg mouse lines expressing the entire early region of beta HPV8 (E6, E7, E1 E2 and E4 genes) or the E6 gene alone driven by the K14 promoter, spontaneously developed multifocal skin tumours and, in approximately 6% of the cases, SCC [25] [26]. In addition, a single dose of UV rapidly promoted papillomas and SCC formation [26]. In another study, Tg mouse models for the beta HPV20 and the benign cutaneous HPV27 were generated, in which E6 and E7 oncoproteins were expressed as single polycistronic transcript under the control of the K10 promoter that is active in the supra-basal differentiated layer of the skin epidermis [27]. Both Tg models developed skin lesions, including SCC after exposure to UV irradiation. However, no significant difference in the skin tumour incidence was observed between HPV 20 and 27 Tg animals [27]. Based on our in vitro data [22] [23] [24], we generated K10 HPV38 E6/E7 Tg mice [28]. These animals displayed hyperplastic and dysplastic patches in the skin epidermis, but no spontaneous development of skin cancer was observed during their life span. Application of the two-stage skin carcinogenesis protocol led to a strong increase in skin tumour incidence [28]. However, chronic UV irradiation of K10 HPV38 E6/E7 Tg did not lead to development of any type of skin lesions (Dong et al. unpublished data). The failure of HPV38 E6 and E7 to cooperate with UV irradiation in skin tumour development in this animal model could be explained by the fact that the viral genes were expressed in the suprabasal layers of the epidermis, while in humans beta HPV types infect and initiate the transcription of the early genes in the basal layer. To explore this hypothesis, we developed a novel transgenic mouse model for HPV38 with K14 promoter-driven expression of E6 and E7 in the basal and proliferative rather then the differentiated compartment of skin epidermis. Here, we show that ectopic HPV38 E6 and E7 expression in this location strongly enhances the susceptibility to chemical - and UV-induced carcinogenesis. Most importantly, chronic UV irradiation of K14 HPV38 E6/E7 Tg mice results in the development of actinic keratosis-like lesions and SCC, closely resembling the scenario observed in humans.
Results
Generation and characterization of K14 HPV38 E6/E7-Tg mice
To evaluate the transforming properties of E6 and E7 from HPV38 in the proliferative compartment of skin epidermis, we generated Tg mouse lines expressing the two viral oncogenes under the control of the human keratin 14 promoter that is active in the basal layer of the epidermis [29]. A schematic representation of the transgene construct used is shown in Figure 1A. Transgene-positive offspring were identified by PCR of tail DNA using HPV38 specific primers. Two independent Tg mouse lines (183 and 187) were identified and bred successfully. Viral oncogene expression was determined by RT-qPCR in different epithelia, i.e. ear, dorsal skin, tongue, and esophagus. Line 183 expressed higher HPV38 E6/E7 levels than line 187 in all four examined epithelia (Figure 1B). In each Tg line, HPV38 E6/E7 expression also differed in the four epithelia, being highest in the dorsal skin and the ear, and comparably low in the tongue and esophagus (Figure 1B). As expected, no viral oncogene expression was observed in liver tissue that was included as a negative control (Figure 1B). No HPV38 E6 and E7 expression was detected in the same tissues of the wild-type animals (data not shown).

HPV38 E6 and E7 induce cellular proliferation in the epidermis of Tg mice
Next, we examined whether HPV38 E6/E7 expression induced morphological alterations in the epithelia analyzing HE-stained sections of skin, ear, tongue and esophagus of FBN/V and K14 HPV38 E6/E7-Tg mice. Epidermal hyperplasia in the ear skin was observed in approximately 5% of 6–8 week-old mice from both Tg lines, as representatively shown in Figure 2A. These alterations, although also detected, were much less evident in dorsal skin (Figure 2A). No significant morphological changes were observed in epithelia of the esophagus and tongue of both Tg lines (data not shown).

The morphological alterations observed in ear skin were even more severe in older animals of lines 183 and 187. Approximately 10–15% of 12-month-old mice from both lines presented dysplasia and, hyperkeratosis. A representative section is shown in Figure 2B, where severe dysplastic keratinocytes, hyperkeratosis, endophytic papillomatous epidermis and a pronounced inflammation could be observed.
To determine whether the expression of HPV38 E6 and E7 oncoproteins resulted in a deregulation of cellular proliferation, we next analysed the expression of the proliferation marker Ki-67 by immunohistochemistry. A significant increase of Ki-67 positivity was observed in the ear (line 187 Vs FVB/N p<0,001, line 183 Vs FVB/N p<0.05) and dorsal skin (line 187 Vs FVB/N p<0.001, line 183 Vs FVB/N p<0.01) epidermis of the two Tg mouse lines (Figures 3A and 3B). Although morphological changes were not observed in tongue and esophagus up to the age of 12 months, an increased Ki-67 index was detected in the epithelia of these tissues at similar levels to those observed in the ear and dorsal skin (data not shown). To corroborate these data, we determined the levels of the positive cell cycle regulator cyclin A in protein extracts from dorsal skin of wild-type and Tg mice by immunoblotting. Cyclin A levels were higher in the two Tg mouse lines as compared to the control animals (data not shown).

Together, these data show that ectopic overexpression of HPV38 E6 and E7 oncogenes in the basal layer of the mouse epithelia significantly increased cellular proliferation.
Enhanced formation of SCC in skin of K14 HPV38 E6/E7 transgenic mice upon DMBA/TPA treatment
Previous studies reported an increased incidence of papillomas and SCC in Tg mouse models expressing E6 and E7 from human or animal papillomaviruses [28] [30] [31] when exposed to chemical carcinogens. Therefore, we compared tumour susceptibility of wild-type and K14 HPV38 E6/E7 Tg mice in the multi-stage skin carcinogenesis protocol using DMBA as initiator and TPA as tumour promoter. Wild-type and Tg animals were exposed to a single treatment of DMBA followed by repeated TPA treatments for 20 weeks and subsequent examination for a further five weeks (Figure 4A). Seven weeks after initiation, 100% of Tg animals of both lines had developed tumours, while at the same time wild-type animals showed no skin lesions and became 100% tumour positive three weeks later (Figure 4B). At week 10, the DMBA/TPA-treated back skin of Tg mice was entirely covered by tumours, in contrast to the wild-type animals that developed only a small number of skin lesions (Figure 4C). Due to the high number of skin lesions including large and multiple SCC per mouse, all Tg animals of lines 183 and 187 were sacrificed at week 12 and 14, respectively, while this event was delayed until week 24 in the wild-type group (Figure 4D). The number of tumours per animal was significantly higher in the Tg cohort as compared to the wild-type cohort (Figures 4E). After completion of the experiment at week 24, histological examination of skin lesions from all control and Tg mice was performed. Accordingly, Tg mice of both lines developed SCC more rapidly (Figure 4F) and at higher incidence than the wild-type mice (Figure 4F). Representative sections of the tumours at week 10 from control and Tg animals are shown in Figure 4G. The shown tumour of the control animal was at an early stage and disruption of the basement membrane and extension of tumour islands into the underlying dermis started to be visible (Figure 4G). The representative sections from Tg animals evidenced SCC characterized by tumour cells with prominent intercellular bridges, abundant eosinophilic cytoplasm and a large and vesicular nucleus, plus aberrant accumulations of keratin (keratin pearls) (Figure 4G, line 183), as well as by irregularly elongated rete pegs with atypia, defined as vacuolization and nuclear abnormalities of cells of the stratified squamous epithelium invading the connective tissue (Figure 4G, line 187).

In summary, these results show that ectopic expression of HPV38 E6 and E7 in the proliferative compartment of skin epidermis significantly increases the tumour burden including papillomas and SCCs in a DMBA/TPA multi-step skin carcinogenesis approach.
Reduced UVB-induced cell-arrest and enhanced UVB carcinogenicity in K14 HPV38 E6/E7 Tg mice
UV irradiation is a key risk factor for NMSC in humans. Therefore, we next determined whether K14 HPV38 E6/E7-Tg mice had an enhanced susceptibility to UVB irradiation. Induction of DNA damage by UV irradiation normally leads to the activation of cellular defense mechanisms, mainly mediated by p53 activation that in turn induces cell cycle arrest prior to the S phase or apoptosis. The cell cycle block is primarily mediated by accumulation of the cyclin-dependent kinase inhibitor (CDK) p21WAF1, whose gene is positively regulated by p53. Short-term UVB irradiations of the skin of wild-type mice resulted in an accumulation of p21WAF1 in keratinocytes of the basal layer of the epidermis (Figure 5A). In contrast, this phenomenon was significantly less evident in the skin of Tg animals (Figure 5A, right panel, P<0,001 after the third UVB irradiation). In agreement with the p21WAF1 expression levels, staining of the Ki-67 proliferative marker was stronger in the skin of the Tg mice in comparison to the wild-type animals, even after several doses of UV irradiation (Figure 5B). These data show that HPV38 E6 and E7 have the ability to interfere with the regulation of cellular checkpoints activated by genomic stress, such as UV-induced DNA damage. Thus, it is likely that HPV38 enhances the carcinogenicity of UV irradiation.

To evaluate this hypothesis, long-term UVB experiments, in which FVB/N and Tg mice of lines 183 and 187 were exposed to multiple and increasing doses of UVB, were carried out (Figure 6A). After 20–25 weeks of treatment, the majority of the Tg mice from both lines showed thick, scaly, crusty and reddish patches in dorsal skin exposed to the UVB light, while no lesions were observed in wild-type animals (Figure 6B). Histological analyses revealed that these lesions resemble the precancerous condition of actinic keratosis. The representative sections in Figure 6 C show downward prolongations and slight atypia of the rete pegs without stromal invasion (top panels) and parakeratosis, acanthosis and broadened elongated rete ridges with atypia (bottom panels), which are all features of the SCC precursor, actinic keratosis. In later weeks (25–30), SCC become visible on the UV-irradiated skin of Tg mice from both lines, while still no lesions were detected in the control mice (Figure 6D). Histological analysis of skin sections revealed that more than 80% of the Tg animals from line 183 developed SCC during the 30 weeks of UV irradiation (Figure 6E). SSC were also detected in the dorsal skin of line 187 Tg animals, but after a longer latency period than in line 183 (Figure 6E). The different incidence of SCC in the two Tg lines tightly correlates with the HPV38 E6 and E7 expression levels (Figure 1B). Representative images of SSC lesions observed in K14 HPV38 E6/E7 Tg mice after 30 weeks of treatment are shown in Figures 6F. These lesions show the presence of tumour cells with atypia, horn formation (Figure 6F, top panels) and tumour cell invasion deep into the dermis (Figure 6F, bottom panels). In contrast, histological examination of dorsal skin sections from FVB/N animals at weeks 30 evidenced only irritation and slight atypia in the epidermis (Figure 6G).
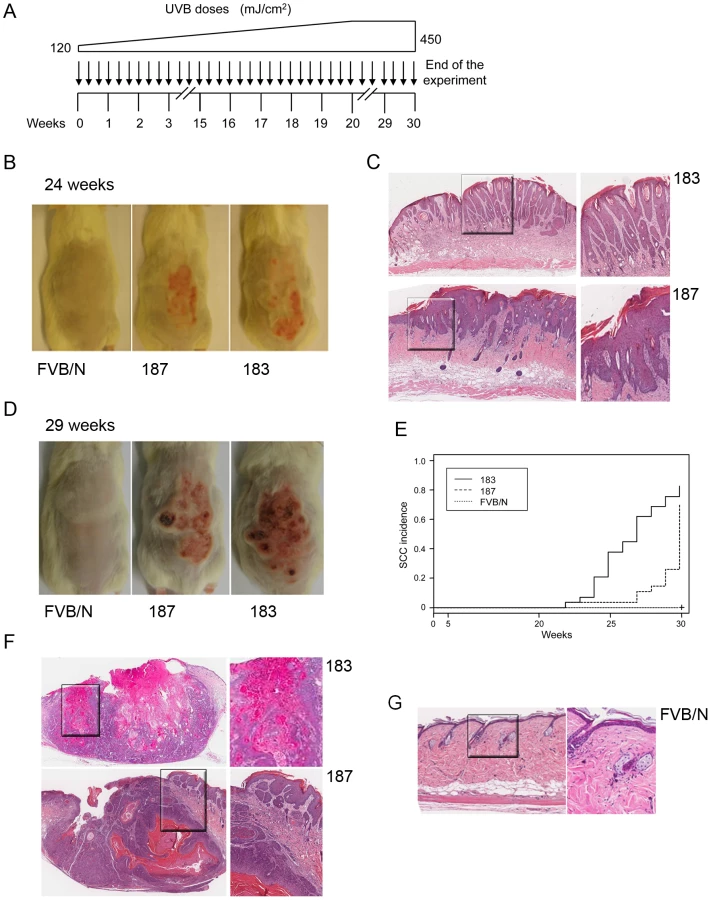
Together, these data show that HPV38 oncoproteins and UVB irradiation cooperate in the development of actinic keratosis and SCC.
Discussion
The role of beta HPV types in NMSC is still not conclusively fully established. Functional studies revealed that beta HPV E6 and E7 proteins, similarly to their homologues from the mucosal HR HPV types, have the ability to deregulate fundamental cellular events, such as cell cycle, apoptosis and senescence [20] [21] [22] [23] [24] [32]. However, despite the functional similarities between E6 and E7 from beta and mucosal HR HPV types, the two subgroups of HPV types may induce cancer development by two distinct mechanisms. It is well demonstrated that the mucosal HR HPV types play a key role in cancer initiation and maintenance. In fact, their genomes and E6 and E7 expression are detected in all cervical cancer cells, and inhibition of the expression of these viral oncogenes in those cells resulted in a rapid induction of apoptosis and/or senescence [33]. In contrast, analyses of skin lesions suggest that beta HPV types may be involved only at early stage of skin carcinogenesis. This hypothesis is mainly based on two findings: (i) higher viral load was found in the SCC-precursor lesion AK than in SCC and (ii) not all cancer cells resulted positive for beta HPV DNA [34]. Taking into consideration also the properties of beta HPV oncoproteins to interfere with the regulation of cell cycle and apoptosis, it is conceivable to hypothesize that beta HPV E6 and E7 enhance the carcinogenicity of sunlight, facilitating the accumulation of DNA damages induced by UV and consequently cancer development. In normal cells, DNA damage induced by UV irradiation activates cellular defense processes leading to p53 activation, which in turn induces cell-cycle arrest or apoptosis to allow repair or elimination of the damaged cells, respectively. In contrast in beta HPV infected cells, E6 and E7 expression circumvent the activation of the cellular defense processes by the UV irradiation, maintaining the cells in a proliferative state and allowing efficient viral DNA replication. As a side-effect, these events favour the accumulation of UV-induced DNA damages and cellular transformation. Due to the irreversible nature of the UV-induced damages, e.g. mutation of tumour suppressor genes, the maintenance of the transformed phenotype may become independent of the viral gene expression.
Our data obtained with K14 HPV38 E6/E7 Tg mice support this model. Indeed, while in normal mice UV irradiation led to accumulation of the cell-cycle inhibitor p21WAF1 and cell cycle arrest, in the Tg animals these UV-induced phenomena were strongly inhibited. In addition, although HPV38 E6 and E7 expression per se did not lead to significant morphological alterations of the epidermis, it strongly facilitated the induction of SCC by chemical carcinogens or chronic UV irradiation. Most importantly, SCC development upon chronic UV exposure of K14 HPV38 E6/E7 Tg mice irradiation was preceded by lesions that closely resemble actinic keratosis, the precursor lesions of SCC also observed in humans.
The different susceptibility of the K10 and K14 HPV38 E6/E7 Tg models to UV-mediated carcinogenesis indicates that the expression of HPV38 E6 and E7 in the basal layer of the epidermis is an essential event for the development of skin cancer induced by chronic UV irradiation. This conclusion is supported by the fact that the natural HPV infection and expression of the viral oncoproteins initiate in the cells of the basal layer. However, it is also possible that the different behavior of the K10 and K14 Tg mice after exposure to chronic UV irradiation is due to the different efficiency of the two keratinocyte promoters, K10 and K14, in expressing the viral genes. Indeed, K10 Tg animals express approximately 3–4 lower levels of HPV38 E6 and E7 than K14 Tg animals (Viariso et al, unpublished data). Thus, additional studies are required to elucidate the different cancer susceptibility of Tg mice expressing the viral oncogenes in the basal or supra-basal layers of the skin. To evaluate the importance of the HPV38 E6 and E7 expression levels in UV-induced carcinogenesis, we are currently generating a novel K14 Tg line that expresses similar levels to the K10 Tg animals.
Previous transgenic models used to study the role of beta HPV in cutaneous cancer express the entire early region (ER) of HPV8 or E6 gene under control of the K14 promoter [25] [26]. The HPV8 ER and HPV8 E6, showed a remarkable similarity in the development of skin lesions. Indeed, a single dose of UV led to a rapid development of papillomas and SCC in both Tg models [26], which also spontaneously developed benign tumours and, in a small percentage, SCC [25] [26]. Although these data support the role of HPV8 in skin carcinogenesis, the HPV8 animal models do not closely correspond to the situation observed in humans, where beta HPV infection is normally asymptomatic and SCC development appears to be strongly associated with chronic UV exposure. The difference observed in K14 HPV8 E6 or HPV8 ER and K14 HPV38 E6/E7 Tg mice may simply reflect the more aggressive properties of the oncoproteins from HPV8 in comparison to HPV38. However, studies in in vitro experimental models do not support this hypothesis. In fact, HPV8 E6 and E7 displayed lower in vitro transforming activities when compared to the oncoproteins from the mucosal HR HPV types [35] [36] [37]. In contrast, HPV38 E6 and E7 were able to immortalize primary human keratinocytes [22] [23], to deregulate p53 functions [24] and up-regulate the expression of the catalytic subunit of the telomerase hTERT [23] [32], all features shared with E6 and E7 from the mucosal HR HPV types. The different phenotype of the K14 HPV8 ER and K14 HPV38 E6/E7 Tg mice could be explained by a different number of integrated copies of HPV DNA and expression levels of the viral oncoprotein in the two Tg models. In addition, regarding the K14 HPV8 ER Tg mice, it is likely that the product of other early genes cooperate with E6 and E7 in promoting cancer development. For instance, HPV8 E2 has been shown to display transforming properties in in vitro and in vivo models [38] [39]. However, as already described above, K14 HPV8 E6 and HPV8 ER Tg mice showed a very similar phenotype, indicating that E2 may play a less important role than E6 in the induction of skin lesions [25] [26]. Thus, it is not yet clear why these two animal models, K14 HPV8 E6/E7 and K14 HPV38 E6/E7, showed different phenotypes.
Independently of these differences, both Tg animal models provided evidence for a cooperation of the beta HPV types and UV irradiation in skin carcinogenesis. Interestingly, the cooperation of infectious agents and environmental factors in carcinogenesis has also been shown in previous studies on bovine papillomavirus type 4 (BPV4). In cows grazed on grass or fed on hay, BPV4 infection leads to development of papillomas of the upper gastrointestinal tract that are spontaneously rejected in a relative short time, e.g. 12 months. In contrast, in cows kept on a diet of bracken fern, BPV4-induced papillomas persist and progress to cancer. This phenomenon is explained by the fact that bracken fern contains several molecules that induce immunosuppression or mutagenesis. These immunosuppressants favour the persistence of the viral infection, while mutagenic substances, e.g. quercetin, promote DNA damage, rendering the infected cell more susceptible to transformation. Thus, studies on beta HPV types and BPV4 underline the importance of environmental factors in virus-mediated carcinogenesis.
Methods
Plasmid construction and generation of Tg mice
The E6 and E7 ORFs of HPV38 were amplified by polymerase chain reaction (PCR) using as template the entire HPV38 genome, and were cloned in a pGEM-3Z vector containing the K14 promoter, a β-globin intron 2, and the K14 polyadenylation sequence (kindly provided by Professor Herbert Pfister, University of Cologne). The complete insert was isolated (see Figure 1A) and microinjected, at a concentration of 3 ng/µl, into the pronuclei of fertilized eggs to generate Tg mice, as described previously [40]. HPV38 E6 and E7 positivity was determined by PCR using specific primers located in the 5′ (5′-ATG GAA CTA CCA AAA CCT CA-3′) and 3′(5′-TTA TCG TCC GCC ATT GCG-3′) regions of the E6 and E7 genes, respectively.
We identified two lines (183 and 187) of HPV38 E6/E7 Tg mice in a FVB/N genetic background. Experiments were performed with K14 HPV38 E6/E7 transgenic lines 183tg/wt, and 187tg/wt and wild-type FVB/N littermates. The animals were kept in the central animal unit of the DKFZ, Heidelberg, Germany, under an artificial day/night rhythm and were fed Kliba 3437 standard food pellets and water ad libitum if not stated otherwise.
Ethics statement
All experiments described in this study were performed in strict accordance to federal law and the standard ethical guidelines (NIH, 1985; European Communities Directives, 1986 86/609/EEC) and approved by local government authorities (Regierungspräsidium Karlsruhe, Germany) under license G162-08. Animal treatments, e.g. UV irradiation, were performed under Sevorane anesthesia, and all efforts were made to minimize suffering.
Total RNA isolation and reverse transcription PCR analyses
Total RNA was isolated from dorsal skin, ear, esophagus, tongue, and liver of 6–8-week-old Tg animals using the Qiagen RNeasy isolation kit (Quiagen, Hilden, Germany). cDNAs were synthesized from 1 µg of total RNA using the M-MLV reverse transcriptase (Invitrogen, Darmstadt, Germany), 18 bp length polydT were used as primers. Quantitative reverse transcription PCRs (RT-qPCRs) were performed in a 25 µl mixture containing 1 µl of 1∶5 diluted cDNA and SYBR-green master mix (SA bioscience, Frederick, Maryland) with specific HPV38 E6 primers (5′-TGC TTA TGC TTC TGC TCA ATA TG-3′ and 5′-GTC TGT TGC TCC ACC TGT TC-3′) or mouse GAPDH primers to amplify a housekeeping gene as internal control (5′ –AAG AAG GTG GTG AAG CAG GCA TC-3′ and 5′-CGA AGG TGG AAG AGT GGG AGT TG-3′), using an Applied Biosystems 7300 machine (Applied Biosystems, Darmstadt, Germany). The fluorescence threshold value was calculated using the SDS analysis software from Applied Biosystems.
Histological and immunohistochemical analysis
Tissue samples from 6–8-week-old mice were fixed in 4% formaldehyde for 24 h at room temperature, and embedded in paraffin. Five µm sections were either stained with hematoxylin/eosin (HE) or used for immunostaining using anti Ki-67 (1∶200) (MM1, Novocastra, Wetzlar, Germany) or p21WAFI antibody (1∶250) (556431, BD Pharmingen, Heidelberg, Germany). Staining was performed using biotin-labeled goat anti-mouse immunoglobulin G and ABC agent from M.O.M kit (Vector Peterborough, UK). The percentages of positive cells were determined by counting 400 hematoxylin-stained cells under 40× magnification in four different fields of the epithelium.
For histological analyses, tissue samples from wild-type and Tg mice were fixed in 4% formaldehyde for 24 h at room temperature, and embedded in paraffin and five µm sections were stained with hematoxylin/eosin (HE).
UVB treatments
UVB irradiation was performed with a Bio-Spectra system (Vilber Lourmat, Marne La Vallee, France) at a wavelength of 312 nm. Each animal was anesthetized with 3% Sevorane (Abbott, Wiesbaden, Germany) in an inhalation anesthetizer (Provet, Lyssach, Switzerland) and placed in a covered compartment with an upper square opening (3×2 cm) at a distance of 40 cm from the UVB lamp. To determine the impact of viral proteins on UV-induced checkpoints, 7-week-old mice where shaved on the dorsal skin, and irradiated up to 5 times in a row, maximum 2 times a day, with UVB at 450 mJ/cm2. Twenty-four hours after the last dose, mice were sacrificed and skin sections stained as described above.
To study UV-induced carcinogenesis, groups of n = 30 7-week-old female FVB/N wild-type or transgenic mice of lines 183tg/wt and 187tg/wt were shaved on the dorsal skin with electric clippers and irradiated 3 times a week for 20 weeks with increasing doses of UVB, starting from 120 mJ/cm2 to a final dose of 450 mJ/cm2, with a constant weekly increase to allow skin thickening. For the following 10 weeks mice were irradiated 3 times a week with 450 mJ/cm2. The tumour incidence (tumour bearers/group) was recorded weekly. Tumours were identified first macroscopically and by histological diagnosis. After thirty weeks, or earlier if the tumour reached the ethically allowed maximal size, the animals were sacrificed and HE-stained sections of dorsal skin served for histological diagnosis.
Initiation-promotion experiments
Five weeks after birth, mice were shifted to Altromin 1324 diet. For epicutaneous applications of initiator, the dorsal skin was shaved with electric clippers 7 days before treatment. Experimental groups of n = 20 (acetone/acetone) or n = 23–27 (7,12-dimethylbenz-anthracene (DMBA)/12-O-tetradecanoyl-phorbol-13-acetate (TPA)) 7-week-old female FVB/N wild-type or transgenic mice of lines 183tg/wt and 187tg/wt were initiated either by a single epicutaneous application of 0,2 ml acetone or 400 nmol DMBA in 0.2 ml acetone. Beginning one week later, the mice were treated each twice weekly with 0.1 ml acetone or with 5 nmol TPA in 0.1 ml acetone for maximally 20 weeks. Papilloma and carcinoma development was monitored up to week 24 without further treatment. Animals were monitored every three days throughout the experiment. The tumour incidence (tumour bearers/survivors in percent) and yield (number of tumours/survivors) were recorded weekly. Tumours were first identified macroscopically and later on by histological diagnosis.
Statistical analysis
Percentages of positive cells in immunostained sections were compared between the different lines with the Student's t-test and statistical analysis were performed with GraphPad Prism (version 4.00, GraphPad Software Inc., La Jolla, CA, USA) Time to first tumor, time to death and time to SCC were displayed with Kaplan-Meier plots or 1 - Kaplan-Meier plots. Animals sacrificed for analysis were considered censored. Time to first tumor, time to death and time to SCC were compared between the different groups with the logrank test. For the CoCa experiment, maximal tumor burden was determined for every animal and groups were compared with the Wilcoxon rank sum test. Statistical analyses were performed with R (version 2.12.0, Copyright (C) 2010 The R Foundation for Statistical Computing) and SAS (Version 9.2, SAS Institute Inc., Cary, NC, USA).
Zdroje
1. PisaniPBrayFParkinDM 2002 Estimates of the world-wide prevalence of cancer for 25 sites in the adult population. Int J Cancer 97 72 81
2. AnanthaswamyHNLoughlinSMCoxPEvansRLUllrichSE 1997 Sunlight and skin cancer: inhibition of p53 mutations in UV-irradiated mouse skin by sunscreens. Nat Med 3 510 514
3. ArmstrongBKKrickerA 2001 The epidemiology of UV induced skin cancer. J Photochem Photobiol B 63 8 18
4. PrestonDSSternRS 1992 Nonmelanoma cancers of the skin. N Engl J Med 327 1649 1662
5. BoyleJMacKieRMBriggsJDJunorBJAitchisonTC 1984 Cancer, warts, and sunshine in renal transplant patients. A case-control study. Lancet 1 702 705
6. KiviatNB 1999 Papillomaviruses in non-melanoma skin cancer: epidemiological aspects. Semin Cancer Biol 9 397 403
7. WalderBKJeremyDCharlesworthJAMacdonaldGJPussellBA 1976 The skin and immunosuppression. Australas J Dermatol 17 94 97
8. WalderBKRobertsonMRJeremyD 1971 Skin cancer and immunosuppression. Lancet 2 1282 1283
9. PfisterH 2003 Chapter 8: human papillomavirus and skin cancer. J Natl Cancer Inst Monogr 31 52 56
10. BouvardVGabetASAccardiRSyllaSBTommasinoM 2006 The cutaneous human papillomavirus types and non-melanoma-skin cancer. Papillomavirus Research: From Natural History to Vaccine and Beyond Caister Academic Press, Norfolk, UK 269 277
11. BerkhoutRJBouwes BavinckJNTer ScheggetJ 2000 Persistence of human papillomavirus DNA in benign and (pre)malignant skin lesions from renal transplant recipients. J Clin Microbiol 38 2087 2096
12. de Jong-TiebenLMBerkhoutRJSmitsHLBouwes BavinckJNVermeerBJ 1995 High frequency of detection of epidermodysplasia verruciformis-associated human papillomavirus DNA in biopsies from malignant and premalignant skin lesions from renal transplant recipients. J Invest Dermatol 105 367 371
13. HarwoodCASurentheranTMcGregorJMSpinkPJLeighIM 2000 Human papillomavirus infection and non-melanoma skin cancer in immunosuppressed and immunocompetent individuals. J Med Virol 61 289 97
14. AnderssonKWaterboerTKirnbauerRSlupetzkyKIftnerT 2008 Seroreactivity to cutaneous human papillomaviruses among patients with nonmelanoma skin cancer or benign skin lesions. Cancer Epidemiol Biomarkers Prev 17 189 195
15. WaterboerTAbeniDSampognaFRotherAMasiniC 2008 Serological association of beta and gamma human papillomaviruses with squamous cell carcinoma of the skin. Br J Dermatol 159 457 459
16. CasabonneDMichaelKMWaterboerTPawlitaMForslundO 2007 A prospective pilot study of antibodies against human papillomaviruses and cutaneous squamous cell carcinoma nested in the Oxford component of the European Prospective Investigation into Cancer and Nutrition. Int J Cancer 121 1862 1868
17. KaragasMRNelsonHHSehrPWaterboerTStukelTA 2006 Human papillomavirus infection and incidence of squamous cell and basal cell carcinomas of the skin. J Natl Cancer Inst 98 389 395
18. KaragasMRWaterboerTLiZNelsonHHMichaelKM 2010 Genus beta human papillomaviruses and incidence of basal cell and squamous cell carcinomas of skin: population based case-control study. BMJ 341 c2986
19. BavinckJNNealeREAbeniDEuvrardSGreenAC 2010 Multicenter Study of the Association between Betapapillomavirus Infection and Cutaneous Squamous Cell Carcinoma. Cancer Res 70 9777 9786
20. JacksonSHarwoodCThomasMBanksLStoreyA 2000 Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev 14 3065 3073
21. UnderbrinkMPHowieHLBedardKMKoopJIGallowayDA 2008 E6 proteins from multiple human betapapillomavirus types degrade Bak and protect keratinocytes from apoptosis after UVB irradiation. J Virol 82 10408 10417
22. CaldeiraSZehbeIAccardiRMalanchiIDongW 2003 The E6 and E7 proteins of cutaneous human papillomavirus type 38 display transforming properties. J Virol 77 2195 2206
23. GabetASAccardiRBellopedeAPoppSBoukampP 2008 Impairment of the telomere/telomerase system and genomic instability are associated with keratinocyte immortalization induced by the skin human papillomavirus type 38. Faseb J 22 622 632
24. AccardiRDongWSmetACuiRHautefeuilleA 2006 Skin human papillomavirus type 38 alters p53 functions by accumulation of deltaNp73. EMBO Rep 7 334 340
25. SchaperIDMarcuzziGPWeissenbornSJKasperHUDriesV 2005 Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res 65 1394 1400
26. MarcuzziGPHufbauerMKasperHUWeissenbornSJSmolaS 2009 Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J Gen Virol 90 2855 2864
27. MichelAKopp-SchneiderAZentgrafHGruberADde VilliersEM 2006 E6/E7 expression of human papillomavirus type 20 (HPV-20) and HPV-27 influences proliferation and differentiation of the skin in UV-irradiated SKH-hr1 transgenic mice. J Virol 80 11153 11164
28. DongWKlozUAccardiRCaldeiraSTongWM 2005 Skin hyperproliferation and susceptibility to chemical carcinogenesis in transgenic mice expressing E6 and E7 of human papillomavirus type 38. J Virol 79 14899 14908
29. FuchsE 1995 Keratins and the skin. Annu Rev Cell Dev Biol 11 123 153
30. HelfrichIChenMSchmidtRFurstenbergerGKopp-SchneiderA 2004 Increased incidence of squamous cell carcinomas in Mastomys natalensis papillomavirus E6 transgenic mice during two-stage skin carcinogenesis. J Virol 78 4797 805
31. SongSLiemAMillerJALambertPF 2000 Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology 267 141 150
32. BedardKMUnderbrinkMPHowieHLGallowayDA 2008 The E6 oncoproteins from human betapapillomaviruses differentially activate telomerase through an E6AP-dependent mechanism and prolong the lifespan of primary keratinocytes. J Virol 82 3894 3902
33. GhittoniRAccardiRHasanUGheitTSyllaB 2010 The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes 40 1 13
34. WeissenbornSJNindlIPurdieKHarwoodCProbyC 2005 Human papillomavirus-DNA loads in actinic keratoses exceed those in non-melanoma skin cancers. J Invest Dermatol 125 93 97
35. IftnerTBierfelderSCsapoZPfisterH 1988 Involvement of human papillomavirus type 8 genes E6 and E7 in transformation and replication. J Virol 62 3655 3661
36. YamashitaTSegawaKFujinagaYNishikawaTFujinagaK 1993 Biological and biochemical activity of E7 genes of the cutaneous human papillomavirus type 5 and 8. Oncogene 8 2433 2441
37. SchmittAHarryJBRappBWettsteinFOIftnerT 1994 Comparison of the properties of the E6 and E7 genes of low - and high-risk cutaneous papillomaviruses reveals strongly transforming and high Rb-binding activity for the E7 protein of the low-risk human papillomavirus type 1. J Virol 68 7051 7059
38. IftnerTFuchsPGPfisterH 1989 Two independently transforming functions of human papillomavirus 8. Curr Top Microbiol Immunol 144 167 173
39. PfefferleRMarcuzziGPAkgulBKasperHUSchulzeF 2008 The human papillomavirus type 8 E2 protein induces skin tumors in transgenic mice. J Invest Dermatol 128 2310 2315
40. AuewarakulPGissmannLCidarreguiA 1994 Targeted expression of the E6 and E7 oncogenes of human papillomavirus type 16 in the epidermis of transgenic mice elicits generalized epidermal hyperplasia involving autocrine factors. Mol Cell Biol 14 8250 8258
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Requires Glycerol for Maximum Fitness During The Tick Phase of the Enzootic Cycle
- Comparative Genomics Yields Insights into Niche Adaptation of Plant Vascular Wilt Pathogens
- The Role of IL-15 Deficiency in the Pathogenesis of Virus-Induced Asthma Exacerbations
- “Persisters”: Survival at the Cellular Level
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy