
Dynamic Evolution of Pathogenicity Revealed by Sequencing and Comparative Genomics of 19 Isolates
Closely related pathogens may differ dramatically in host range, but the molecular, genetic, and evolutionary basis for these differences remains unclear. In many Gram - negative bacteria, including the phytopathogen Pseudomonas syringae, type III effectors (TTEs) are essential for pathogenicity, instrumental in structuring host range, and exhibit wide diversity between strains. To capture the dynamic nature of virulence gene repertoires across P. syringae, we screened 11 diverse strains for novel TTE families and coupled this nearly saturating screen with the sequencing and assembly of 14 phylogenetically diverse isolates from a broad collection of diseased host plants. TTE repertoires vary dramatically in size and content across all P. syringae clades; surprisingly few TTEs are conserved and present in all strains. Those that are likely provide basal requirements for pathogenicity. We demonstrate that functional divergence within one conserved locus, hopM1, leads to dramatic differences in pathogenicity, and we demonstrate that phylogenetics-informed mutagenesis can be used to identify functionally critical residues of TTEs. The dynamism of the TTE repertoire is mirrored by diversity in pathways affecting the synthesis of secreted phytotoxins, highlighting the likely role of both types of virulence factors in determination of host range. We used these 14 draft genome sequences, plus five additional genome sequences previously reported, to identify the core genome for P. syringae and we compared this core to that of two closely related non-pathogenic pseudomonad species. These data revealed the recent acquisition of a 1 Mb megaplasmid by a sub-clade of cucumber pathogens. This megaplasmid encodes a type IV secretion system and a diverse set of unknown proteins, which dramatically increases both the genomic content of these strains and the pan-genome of the species.
Published in the journal:
. PLoS Pathog 7(7): e32767. doi:10.1371/journal.ppat.1002132
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002132
Summary
Closely related pathogens may differ dramatically in host range, but the molecular, genetic, and evolutionary basis for these differences remains unclear. In many Gram - negative bacteria, including the phytopathogen Pseudomonas syringae, type III effectors (TTEs) are essential for pathogenicity, instrumental in structuring host range, and exhibit wide diversity between strains. To capture the dynamic nature of virulence gene repertoires across P. syringae, we screened 11 diverse strains for novel TTE families and coupled this nearly saturating screen with the sequencing and assembly of 14 phylogenetically diverse isolates from a broad collection of diseased host plants. TTE repertoires vary dramatically in size and content across all P. syringae clades; surprisingly few TTEs are conserved and present in all strains. Those that are likely provide basal requirements for pathogenicity. We demonstrate that functional divergence within one conserved locus, hopM1, leads to dramatic differences in pathogenicity, and we demonstrate that phylogenetics-informed mutagenesis can be used to identify functionally critical residues of TTEs. The dynamism of the TTE repertoire is mirrored by diversity in pathways affecting the synthesis of secreted phytotoxins, highlighting the likely role of both types of virulence factors in determination of host range. We used these 14 draft genome sequences, plus five additional genome sequences previously reported, to identify the core genome for P. syringae and we compared this core to that of two closely related non-pathogenic pseudomonad species. These data revealed the recent acquisition of a 1 Mb megaplasmid by a sub-clade of cucumber pathogens. This megaplasmid encodes a type IV secretion system and a diverse set of unknown proteins, which dramatically increases both the genomic content of these strains and the pan-genome of the species.
Introduction
Pseudomonas syringae is a Gram-negative bacterial phytopathogen responsible for worldwide disease on many crop species [1]. Despite a collectively broad pathogenic range for the species, individual isolates of P. syringae display pathogenic potential on a limited set of plant species and either elicit immune responses, or simply fail to thrive on alternative species [2]–[4]. In addition to disease outbreaks, strains can be isolated as epiphytes from non-diseased plants as well as from multiple phases of the water cycle [5], [6]. How species with varied lifestyles like P. syringae maintain the genomic flexibility required to survive across this broad range of ecologies is not known. It remains particularly unclear how evolutionary forces shape the pan-genome of this species, especially virulence-related genes.
The host ranges of many P. syringae isolates or pathovars have not been thoroughly characterized. Research has largely focused on identifying the molecular basis of pathogenesis across three divergent strains with finished genome sequences and investigation of virulence mechanisms for a smattering of strains on a limited number of hosts [7]. These studies have shown that a Type III secretion system (TTSS), which acts like a molecular syringe to translocate a suite of type III effector (TTE) proteins into plant cells, is a key virulence determinant [7]–[9]. Once inside the plant cell, TTEs promote pathogenesis by disrupting and suppressing host defense responses at multiple levels [10]–[13]. TTEs can also be recognized by plant disease resistance proteins and recognition of a single effector is sufficient to trigger successful host immune response. However, the virulence functions of many TTEs are redundant, making these phenotypes potentially robust to host-mediated selection against single TTE genes [14]. Thus, host range is structured by the totality of a strain's TTE repertoire.
A high degree of divergence among commonly investigated isolates makes it nearly impossible to pinpoint all the changes that lead to host differentiation or specialization at the present time. As a result, key questions, such as what determines the overall plasticity of host range, remain unanswered. Deep sampling of diverse genomes within a phylogenetic framework can reveal general evolutionary trends indicative of changes in lifestyle and allow for the identification of genetic changes that differentiate between strains that have recently undergone host range shifts [3], [15].
Isolates of P. syringae are subdivided into approximately 50 pathovars based upon host range and comparison with type strains [16]. These are further subdivided into races based upon differential ability among strains within a pathovar to grow and cause disease across host genotypes [17]. Recent multilocus sequence typing (MLST) segregated P. syringae pathovars into at least 5 distinct phylogenetic clades [6], [16], [18], which largely mirror 9 genomospecies based on DNA hybridization [19], [20]. While the selection pressures determining host range may be similar throughout the species, there has simply not been deep enough phenotypic sampling or sequencing of genomes across the species to uncover trends indicating evolutionary differentiation among the clades [4].
Multiple screens, primarily within the three P. syringae strains with completely sequenced ‘gold standard’ genomes [3], [21]–[30], have suggested that the number of TTEs per genome ranges from ∼20 to 33, with the total number of validated TTE protein families ∼50. However, efforts to catalogue the TTE repertoires from various strains often fall short of capturing a complete picture. For instance, false negatives occur from lack of saturation in functional TTE screens or because sequence divergence confounds hybridization based methods. False positives also occur with hybridization methods when the gene sequences are present but contain frame-shifts or disruptions, or are only partial matches to the known TTE genes (i.e. chimeras [31]). Most of these limitations are obviated by whole genome sequences, especially when combined with orthogonal functional methods to validate candidates as TTEs.
The TTSS is not the sole determinant of virulence and host range for P. syringae; coordination of host physiological responses and metabolic pathways is also necessary for pathogen growth within host tissue [32]. Phytotoxins, which can be coordinately regulated with the TTSS, but secreted independently from the TTEs [33], can disrupt host metabolism or act as mimics of plant hormones. Hence, they may replace or complement virulence functions of TTEs [34]. Indeed, manipulation of stomatal function by coronatine, a structural mimic of the plant hormone jasmonic acid, is essential for invasion of A. thaliana leaves by P. syringae pv. tomato (Pto) DC3000. However, coronatine also possesses independent virulence functions during the colonization of roots [35]. Therefore, pathogenesis of P. syringae on any given plant host species, results from both the absence of avirulence factors (an operational definition of TTEs that activate a host immune receptor) and the presence of multiple virulence factors acting coordinately to promote disease and to suppress host immune responses [4], [7], [13].
To date, genomics studies in P. syringae suggest that virulence mechanisms within this species are evolutionarily dynamic and have experienced strong selective pressures [3], [36]. Complete genome sequences exist for three phylogenetically diverse P. syringae isolates representing MLST groups I, II, and III (Pto DC3000, P. syringae pv. syringae B728a (Psy), and P. syringae pv. phaseolicola 1448a (Pph), respectively; [26]–[28]. Recently, additional draft genome sequences were generated by either Roche/454 or Illumina sequencing technologies (for Pto T1, group I; Pta ATCC11528, pathovar tabaci; Psv NCPP3335, pathovar savastanoi; and multiple strains from pathovars aesculi and glycinea, all group III; [30], [37]–[40]), or a hybrid genome assembly pipeline utilizing both Illumina and Roche sequencing technologies (Por 1_6, pathovar oryzae, group IV; [41]). The genomes of these P. syringae strains differ dramatically in gene and plasmid content and in the presence/absence of many virulence-related genes. Given that these strains represent only a fraction of the known diversity within Pseudomonas isolates, much of the phylogenetic, ecological, and host diversity for this plant pathogen remains unexplored.
We provide a phylogenetically comprehensive genomic view of P. syringae with a focus on TTE repertoire evolution. We analyzed data from draft or complete genome sequences of 19 diverse isolates, including 14 new draft genome sequences. We couple these genome sequences with a functional screen to identify new TTE families from diverse strains. The TTE content within these strains, as well as the presence of other known pathogenesis-related genes, is volatile. We show that cost-efficient genome sequencing placed within a phylogenetic context provides a thorough and unique viewpoint into P. syringae evolution and sheds light on previously unrecognized evolutionary patterns and structural diversity for this important plant pathogen.
Results
High Quality Draft Genome Sequences for Phylogenetically Diverse Strains of P. syringae
We employed a hybrid approach [41] utilizing reads from both Illumina and 454 platforms to generate draft genome sequences for 14 phylogenetically diverse strains of P. syringae (Table 1). These draft genomes are each contained on 32 to 222 scaffolds with the N50 value at 81,010 bp (e.g. half of the total sequenced genome, calculated by summing the lengths of all contigs and scaffolds within a given strain, is found in scaffolds 81,010 bp or greater). Although each genome assembly varies slightly, the size distribution of contigs and scaffolds (Figure S1) is equivalent to what we previously described for our hybrid assembly of re-sequenced Pto DC3000 compared to the published sequence of the same strain [26], [41].

We created a Bayesian phylogeny for the sequenced strains using fragments based on the MLST loci used in [16], but extended as far in these gene sequences as was possible to align (Figure 1). We also built maximum likelihood phylogenies by concatenating 324 protein sequences from a subset of proteins present in all strains, after establishing orthology and producing amino acid alignments using a hidden Markov model (Figure S2), as well as individual phylogenies of these 324 protein sequences (Dataset S8). Our phylogenies are largely congruent with prior work, however, we find that the exact placement of Por and Pma and the resolution of the relationships between Pmo, Pta, Pae, and Pla 107 (see Table 1 for strain key) are sensitive to the phylogenetic method used (data not shown). In cases of discrepancies between the tree inferred from MLST sequences, the tree from 324 concatenated sequences, and the individual protein trees, such as the placement of Pae, the second most probable protein tree invariably supported the topology inferred from MLST sequences (Figure S2, Dataset S8).

Defining the P. syringae Core and Pan Genome
The core genome consists of those genes found in all sequenced genomes of a species. Following automated NCBI and manual Phylo-gene-boost annotation (Materials and Methods; Figure S3; Dataset S1, S2, S3, S4) and using a somewhat liberal approach to defining similarity (40% identity over 40% length), we defined a P. syringae core genome of 3,397 genes (Figure 2A). In contrast, the 12,749 genes of the pan genome are found only in subsets of strains (Figure 2B). We extended this analysis to include genomic information from multiple genome sequences of two related pseudomonad lineages: a plant-associated non-pathogenic bacterium (P. fluorescens; 3 genomes; [42], [43]) and a soil bacterium (P. putida; 4 genomes; [44], strains GB-1, F1, W619 unpublished but available at Genbank) (Figure 2C). A core of 2,501 genes was found within all isolates of all three of these pseudomonad lineages (Figure 2C; Figure S7). The 292 core genes shared between P. syringae and P. fluorescens that are not shared with P. putida are candidate plant-association loci and are enriched with genes predicted to be involved in protein localization and transport (Figure S8). There are 514 genes within the P. syringae core genome absent from the three-species core (Figure S9). These include a disproportionate number of metabolic regulators, protein localization and transport genes (Figure S9). The overall percentage of strain-specific genes, 5–10%, is fairly consistent across the P. syringae phylogeny (Figure 2D), the one exception being Pla 107. Roughly one in seven genes within this genome are strain-specific, and most of these are contained on a single megaplasmid (see below).

We analyzed the core and pan genomes for the three major clades of P. syringae (groups I, II, and III according to [16]) (Figure 2D; Dataset S5, S6, S7). Even though each group possesses similar levels of nucleotide sequence divergence, we found that group I strains have ∼500 more genes within their core genome than groups II and III (Figures 2D, S10). Since the number of sequenced isolates is smaller for group I, we performed bootstrapping analysis of the other two clades to show that the inflated core genome of the group I strains is robust to differences in the number of genome sequences sampled (data not shown).
Plasmids or Megaplasmids Are Found in Most Pathovars
Current assembly methods for short read technologies are poor at assembling across repetitive regions. Thus, we investigated the presence of plasmids within these strains using a multi-phase approach based upon the presence of plasmid structural genes within the draft genomes, similarity of loci present within these suspected plasmids to known plasmids from the NCBI database, and an approximation of plasmid coverage using Illumina read depth. We find that 13 out of 15 draft genomes likely contain plasmids (Table S1, Figure S11), highlighting the importance of extra-chromosomal elements in the evolution of P. syringae [45].
The Pla 107 genome assembled into scaffolds representative of a typical chromosome, as well as a ∼1 Mb scaffold with approximately the same GC content and depth of Illumina read coverage as known chromosomal genes (Figure S11) but with little sequence homology to the other P. syringae genomes. We used PCR and Sanger-based sequencing to confirm that this large scaffold was circular (Figure S12A,B). Hence, Pla 107 contains a ∼1 Mb megaplasmid. PCR-based screening shows that this megaplasmid is present within a closely related strain (Pla N7512), but absent or significantly modified in two other closely related strains (Pla YM7902 and Pla YM8003; Figure S12C). Draft genome sequences of closely related outgroups (Pmo, Pta) also lack the megaplasmid. Both strains that contain this extra-chromosomal element grow more slowly in planta and on plates (Figure S12D,E). Since these four Pla strains possess nearly identical sequences at their MLST loci, this megaplasmid is a recent acquisition. Although this extra-chromosomal element encodes an astonishing fraction of hypothetical proteins according to the NCBI annotation (776 of 1080 genes), as well as 35 additional conserved but uncharacterized proteins, it also contains “housekeeping” genes highly similar to those in other Pseudomonas species, a potential type IV secretion system distantly related to the Legionella Dot/Icm system, and 38 additional tRNA loci (bringing the total in this strain from 47 to 85). Although many type IV secretion system related structural genes do appear to be present, tBLASTn searches using sequences of known effector proteins did not produce likely hits [46], [47]. We also searched both the Conserved Domain Database (CDD) and KEGG to identify potential biochemical pathways on the megaplasmid, but found that no complete pathways were present (Fig. S12F). The “housekeeping” genes do not appear to be essential as there are often P. syringae homologues found on the main chromosome. The recent acquisition of this megaplasmid could signal the potential for a dramatic ecological shift across these closely related strains.
Identification of Eight New Type III Effector Families
The genomes from a subsample of the total sequenced isolates (Pgy R4, Pmo, Pla 107, Pac, Ptt, Ppi R6, Pla 106, Pmo, Pan, Psy B728a) were functionally screened for new TTE genes using a previously established method [23] based on the observation that many important virulence genes (and all known TTE) are regulated by the alternative sigma factor HrpL. Two additional strains that were not sequenced (P. syringae pv. atrofaciens DSM50255, and pv. maculicola M4) were also screened, and novel TTEs identified from these strains were included in all similarity searches (Dataset S9). We report the full results from all screened putative TTEs, as well as the type of locus identified in the screen (ORF only, or ORF including the putative hrp-box) in Dataset S9. From this screen, we identified and functionally validated by translocation assays members of eight new TTE families (Figure 3, Table 2). This increased the number of validated TTE families in P. syringae to 58 (not including avrD, defined according to unified nomenclature rules; [48]).


We are confident that there are now few undiscovered TTE families left to be found that are shared by a majority of these strains. First, we maximized phylogenetic diversity and screened four of the five major phylogenetic groups, using divergent strains within each of these groups. Second, our functional screen is close to saturation as measured by the recovery of known hrpL-regulated TTSS loci from the respective genomes at frequencies similar to previously published reports (Table S2; [23]).
We list instances where homologues of previously known TTE families were identified in the screened genomes (Dataset S9). In this file we also list instances where genes were identified as HrpL-regulated within the screen, but which were not translocated according to our tests. Since these genes are confirmed to be HrpL-regulated, and are therefore linked to the major pathogenic regulon in P. syringae, they could contribute to virulence in a translocation-independent way [29].
Type III Effector and Toxin Content Varies Dramatically between Strains and Clades
We characterized the TTE content for each of the sequenced strains by similarity searches to all known P. syringae TTE (Figure 3; left). Our query list was generated by combining all previously identified P. syringae TTE with the eight new TTE families we identified (Materials and Methods; Dataset S9). We also acknowledge the limitation of our study in that TTEs and phytotoxin pathways may have been contained on plasmids or in other regions lost during sub-passaging of these strains. Overall, the total number of potential TTEs (defined as full length ORFs, confirmed for HrpL-induction, and with at least one family member translocated) varies dramatically between strains, from a minimum of 9 (Pja) to a maximum of 39 (Pto DC3000) (Figure 3; right). Furthermore, we find that the group II strains possess lower numbers of known TTEs on average than the other 4 groups. There are a total of five TTE families (hopAA, avrE, hopM, hopI, hopAH) present in some form (as either full length or truncated ORFs or disrupted by IS elements) within each of the sequenced strains. These represent the core TTEs found within all pathogenic P. syringae strains. These TTE are all found in syntenic regions of each genome and three (hopM, avrE and hopAA) are closely linked to TTSS structural genes, as noted [49]. A second class of TTE families (hopX, hopAE, hopAF, hopR, hopAS, hopAB, hopQ, hopD, hopT, hopO, hopW, hopF, hopV, hopAZ, avrPto) are predominantly absent from group II strains. They are located in a wide variety of genomic locations and can be very diverse in sequence, suggestive of extensive horizontal transfer (see below). Sequence differences among members of these families suggest that this class of TTE families may be under different evolutionary pressures relative to the core TTEs (Table S3, Figure S14).
We investigated the genome dynamics of TTE genes within the three most deeply sampled clades in order to characterize the evolution of TTE repertoires (Figure 4). For each strain, the majority of the TTE ORFs are present within other closely related strains from that clade. Moreover, most strains share almost all of their TTEs with at least one additional strain from within the same clade. This result is particularly striking for Pta, Pla 107, Pmp, Pla 106, Ptt, Pja, Cit7, Pac, and Psy B728a, which only have a small percentage of novel TTEs in relation to the rest of their clade (Figure 4, right). In contrast, a handful of isolates (Pgy R4, Pph 1448a, Pae, Ppi R6, Pan, Pto DC3000) have gained numerous TTE that are not present within any other related strains.

TTE truncations and transposon disruptions are common across the P. syringae phylogeny (Figure 4). However, in only two cases, a truncation of hopAA1 and a transposon disruption of hopAG1, are these events shared by a majority of strains within a clade. Similarly, in only three other cases did events occur that were shared between multiple strains. Conversely, of the 46 total TTE gene truncations or transposon disruptions we identified, 41 appear at the tips of our phylogeny. Given the rarity of “older” truncations and disruptions, and given that many of these altered TTEs are found undisturbed in closely related genomes, we believe TTE loss is recent in most cases. This is consistent with ongoing dynamism in host range determination, whether across plant species or within a species, driven by host immune recognition. Additionally, there is a distinctive proliferation of IS element disruptions amongst the group I strains. The other clades appear to display higher rates of disruption by truncation (mostly via frameshift mutations) than IS elements, with the exception of Pla 107 which possesses a relatively high number of TTE with IS element disruptions. This trend potentially reflects differences in the activity levels of clade specific transposases.
Identifying the Most Evolutionarily Dynamic Type III Effector Families
We investigated diversity for 35 TTE families (Table S3) present within a majority of strains (>12) by calculating measurements of pairwise amino acid diversity among all known alleles (Table S3). The most diverse TTE families are hopW, hopZ, avrB, hopAO, hopT, hopAB and hopF (Table S3). The diversity values for hopW, hopT and hopAO are somewhat misleading, however, as these families contain alleles of vastly different lengths. We built phylogenies from protein sequences of the remaining diverse TTEs (hopAB, hopF and avrB) (Figure S14) and note that similar analyses exist for hopZ [36]. The resulting phylogenies differ extensively from the phylogenies inferred from MLST sequences, hence we infer that these widely distributed TTE families are often lost, but can be regained by horizontal transfer of divergent alleles (Figure S14). This could imply that these TTE families play important roles in virulence across a broad range of host species, and are thus often re-incorporated into a strain's TTE suite. But the dynamism of these families also suggests that they may be evolutionarily costly on certain hosts (again likely through host immune recognition) and are therefore lost at higher rates than other TTE families. In support of this model, we note that host disease resistance genes exist that recognize specific members of each of these four TTE families, and that specific members of each of these TTE families can confer virulence on particular hosts [2], [50], [51].
Phylogenetic Distribution of Non-TTSS Secreted Virulence Factors
We investigated the presence of pathways encoding the best understood P. syringae phytotoxins (coronatine, tabtoxin, syringolin, syringopeptin, syringomycin, phaseolotoxin), a gene (avrD) whose enzymatic product, syringolide, can cause a hypersensitive response on specific soybean genotypes [52], and genes involved in production or modification of the plant hormones ethylene and auxin (Figure 3). It should be noted, however, that allelic diversification within these pathways can lead to the production of slightly different toxins [53]. In most cases, pathways coding for toxins found together in Psy B728a (syringomycin, syringopeptin, syringolin) are found in the genomes of group II strains, with the exception of Ppi R6. In only one other case did a strain contain genes known to be involved in the production of multiple toxins (coronatine and tabtoxin in Por). Moreover, although all strains in all groups appear capable of producing the plant hormone auxin, only the group II strains and Pph 1448a lack a gene to modify auxin once it is made (iaaL). avrD is widespread throughout the phylogeny, although the functional significance of different avrD alleles remains unresolved [52]. Only Pgy R4 and Ppi R6 appear to be capable of ethylene production by known pathways. The relative wealth of phytotoxins and the reduced TTE suites of group II strains, compared to the other phylogenetic groups, suggest that the genetic basis of pathogenicity within this clade has diverged from the rest of the P. syringae species.
Associations in the Distribution of Virulence Genes
We analyzed the relationships of virulence gene suites across strains by hierarchical clustering of strains with respect to the distributions of individual virulence genes and pathways (TTE, phytotoxin pathways, plant hormone mimics) (Figure S13). Although we hoped to uncover novel associations between virulence genes, small sample sizes for numerous virulence genes provide little resolution to identify correlations that are independent of phylogeny (i.e. hopN, hopS, hopY) or known proximity on the chromosome (i.e. hopO and hopT). However, clustering of strains by their virulence gene repertories does highlight some interesting trends. Despite phylogenetic assignment to group I, both Pan and Pmp diverge from Pto T1/Pto DC3000/Pla 106 in their virulence gene repertoires. Such patterns reflect the classification of these strains within different genomospecies [20]. Indeed, Pan clusters more closely with group III strains, likely due to the presence of scaffolds and TTE's related to those found on the large virulence plasmid of Pph 1448a, as well as the pathway for the production of phaseolotoxin. This could signal underlying similarity in the virulence strategies of P. syringae pathogens of beans and kiwi. Likewise, two group III strains (Pta and Pla 107) cluster with group II strains based on virulence gene profiles, suggesting that there is a fundamental difference in virulence gene repertoire for these two strains compared to their group III relatives. Lastly, hierarchical clustering clearly demonstrates divergence in virulence genes suites between Ppi R6 and other group II strains.
Identifying Evolutionarily Dynamic TTEs Based on DNA Sequence Diversity within Clades
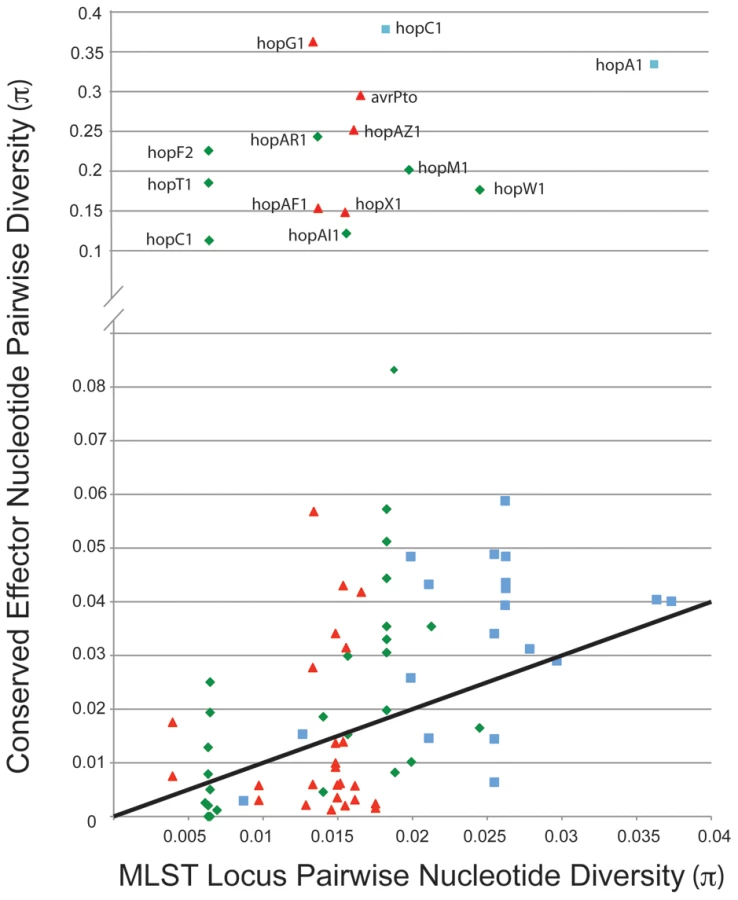
We compared values for π (pairwise nucleotide diversity) for shared TTE subfamilies within the three clades of P. syringae for which there are genome sequences from multiple strains (Figure 5). As a baseline, we compared this value to the π values for the same groups of strains calculated from the concatenated MLST loci used to construct the overall phylogeny in Hwang et al. [16]. This metric requires accurate placement of TTEs into subfamilies ([48]; Materials and Methods). Thus, there is an upper limit to π values for the shared TTEs because extremely divergent alleles will be placed into different subfamilies. A majority of the π values for the TTE subfamilies match, or are slightly higher than, values for the housekeeping genes, consistent with vertical inheritance or low levels of diversifying selection. However, there are numerous instances where diversity within a TTE subfamily far exceeds the diversity values for the housekeeping genes within the same comparison. Future work will show whether horizontal transfer or mutation has enabled functional diversification of these protein families.
The Evolutionary Dynamics of hopM1 within Group I Strains
Our sequence diversity analysis showed that hopM1 has experienced unusual evolutionary dynamics (Figure 5), especially within the group I strains. We aligned the largest contiguous genomic region (bordered by scaffold breaks) including hopM1 for all group I strains that contain a full-length hopM1 allele. We computed π values for the nucleotide sequence of this region for these strains. A small fraction of this genomic region, which includes hopM1 as well as fragments of the TTSS helper protein hrpW and the TTE chaperone shcE, displays inflated π values relative to the bordering regions (Figure 6A). Therefore, the observed inflation of nucleotide diversity for hopM1 (Figure 5) is localized. The phylogenies of both TTSS linked TTEs avrE1 (Figure 6B) and hopAA1 (data not shown) match those created from MLST loci. However, the hopM1 phylogeny shows that a recombination event involving the hopM1 locus splits group I strains into two divergent groups (Pmp/Pan or Pto DC3000/Pla 106/Pto T1) (Figure 6B, right, in green). This result underscores how localized homologous recombination of existing sequences can drive diversification and adaptation of P. syringae TTE repertoires [54].

Allelic Variants of hopM1 are Functionally Diverged
We tested the virulence function of both of the diverged group I hopM1 variants, from Pto DC3000 and from Pmp using a previously published assay [55]. Briefly, a strain carrying a deletion of the Conserved Effector Locus (CEL) that eliminates hopM1 and avrE1 from Pto DC3000 displays attenuated disease symptoms and less growth on Arabidopsis. avrE1 and hopM1 are likely redundant for this virulence function [14], [56], [57]. We found that hopM1Pmp, expressed from a constitutive promoter, did not complement the virulence defect of Pto DC3000 ΔCEL (Figure 6C), even though this effector is translocated (Dataset S9). Therefore, allelic variants of hopM1 display functional divergence for virulence on tomato.
A Natural Allelic Series of AvrPto Orthologs Naively Recapitulates Identification of the GINP Loop as a Region Critical for AvrPto/Pto Avirulence
To show the utility of deep phylogenetic sequencing, we asked if a diverse collection of orthologs could be used to predict functional information about a TTE protein. AvrPto is a widely distributed and well-characterized TTE that interacts with a tomato host cellular target, the protein kinase Pto, in a well-defined manner that results in disease resistance mediated by Pto and the NB-LRR immune receptor Prf [58], [59]. AvrPto also confers added virulence to P. syringae strains that lack it when assayed on pto or prf genotypes of tomato [60]. We assayed 10 AvrPto orthologs (Figure S16A, Figure 7A) for their ability to trigger Pto dependent HR using a standard assay in N. benthamiana [61]. As expected, AvrPto from Pto DC3000 and closely-related orthologs triggered Pto-dependent HR. More distant orthologs did not (Figure 7B). A negative control, AvrPtoDC3000 with a G2A mutation, previously reported to be mislocalized [62], failed to elicit HR in the presence of Pto. Expression of AvrPto orthologs and Pto was verified by Western blotting (Figure S16B).

One allele that was not recognized by Pto, AvrPtoPgyR4, differs in only 2 residues from the recognized allele AvrPtoLac107 (Figure S16A). We used phylogeny-directed mutagenesis to create AvrPtoPgyR4 I85M and AvrPtoPgyR4 S95G, recreating the conserved sequence of the recognized AvrPto orthologs in each case. We tested the ability of these alleles to be recognized by Pto. AvrPtoPgyR4 I85M was not recognized, while AvrPtoPgyR4 S95G triggered Pto-dependent cell death (Figure 7C). In a second assay for HR, we found that, while AvrPtoPgyR4 was inactive, AvrPtoPgyR4 S95G induced ion leakage to levels indistinguishable from AvrPtoPtoDC3000 (Figure 7E). Glycine 95 is common to all the active AvrPto alleles and lies within the GINP loop, a region required for the AvrPto-Pto physical interaction and hence avirulence function [58], [63]–[65]; Figure S16C). Recently, Glycine 95 has been shown to be required for recognition of AvrPto by Pto [66]. Both Serine 94 and Isoleucine 96 are required for avirulence [64]. Isoleucine 96 has been previously shown to tolerate mutation to valine (but not alanine) [64]. Accordingly, while both AvrPtoLac107 and AvrPtoPgyR4 S95G contain valine at position 96, they both trigger avirulence. A more distantly related, non-recognized allele, AvrPtoPmo also contains a non-consensus arginine residue at position 95. Mutation of AvrPtoPmo R95G did not restore recognition (Figure 7D). Thus, other AvrPtoPmo-specific polymorphisms contribute to loss of recognition. The non-recognized ortholog AvrPtoPgyR4 retains its virulence function on tomato leaves that lack Pto function (Figure 7F), suggesting that the two amino acid differences that distinguish it from AvrPto alleles that are recognized are dispensable for virulence. A similar separation of AvrPto avirulence and virulence functions has been previously reported for missense mutations of the canonical AvrPto allele from Pto JL1065 [64]. The virulence effect of AvrPtoPgyR4 was consistently greater than that of AvrPtoPtoC3000 (Figure 7F). This relative difference could be due to either residual avirulence of the Pto DC3000 ortholog dependent on glycine 95, or to uncharacterized residues polymorphic between AvrPtoPgyR4/Lac107 and AvrPtoPtoC3000.
Discussion
Bacterial genomes are dynamic. Large-scale changes occur rapidly and differentiate even closely related isolates within the same species. P. syringae, an important pathogen of many plant species, is a diverse assemblage of strains isolated from different host plants as well as from the environment. To reveal the evolutionary history of pathogenesis within this species, we catalogued the virulence gene repertoires for 19 isolates using genome sequencing coupled with a nearly saturating screen for novel TTE families for a subset of the strains. These phylogenetically diverse genome sequences provide a comprehensive comparative tool to investigate pathogenicity and virulence across plant hosts and a means to gain insight into host range and adaptation of this important phytopathogen.
Genome Structure and Diversity
Although individual isolates of P. syringae contain upwards of 6000 genes, only 3397 are shared amongst all 19 sequenced strains (Figure 2A). While estimates of core genomes typically decrease with further sampling from diverse isolates, we do not expect the P. syringae core to significantly decrease, given the slope of the data in Figure 2 and our sampling of much of the known phylogenetic diversity of pathovars. For comparison, we identified species-specific core genomes, using the same methods, from multiple sequenced genomes of both P. fluorescens (a plant-associated microbe) and P. putida (a soil bacterium) as well as the subsets of genes shared between different combinations of these species (Figures 3C, S7, S8). Both the unique portion of the P. syringae core and the core genes shared between P. syringae and P. fluorescens, are enriched for proteins involved in protein localization and transport (comparing Figures S7, S8, S9), highlighting the potential role for such processes in adaptation to plant hosts. Surprisingly, the core genomes for both P. putida and P. fluorescens are larger than that for P. syringae (Figure 2C), which could reflect differences in evolutionary pressures for the pathogenic strains or the smaller number of sequenced genomes for P. putida and P. fluorescens (the number of core genes could drop substantially with further sampling). P. syringae group I strains share ∼500 more genes than either of the other two well sampled groups (Figure 2D). The majority of these clade-specific genes encode proteins of unknown function (Figure S10).
The 19 P. syringae strains define a larger core genome than do 20 strains of Escherichia coli (3397 vs. 1976) but a substantially smaller pan-genome (12,749 vs. 17,838) [67]. These numbers are surprising given the larger overall genome for pseudomonads in general (average of ∼6000 genes compared to ∼4700 for E. coli). Therefore, even though pseudomonads are ubiquitous across many environments, it is possible that E. coli fills more diverse ecological niches, requiring lower numbers of shared genes but correspondingly higher numbers of unique pathways. However, we have only sequenced isolates from crops and the size of the core genome may be reduced when sampling from more diverse environments.
Plasmids, which contain many pathogenicity genes and have the potential for horizontal transfer across strains, are known determinants of virulence evolution within P. syringae [45]. However, plasmids are often filled with repetitive regions that make assembly from short-read sequencing data difficult. We have attempted to identify plasmid regions using a combination of presence of conserved plasmid genes as well as sequencing coverage levels. However, we ultimately found that it was difficult to truly identify presence of plasmids using assembly information alone. The best strategy for sequencing of these difficult regions may still be to isolate individual plasmids and sequence them separately.
Not surprisingly, 13 of the 15 pathovars show evidence of plasmids that are of the same approximate size and genomic composition as previously identified or sequenced P. syringae plasmids (Table S1). They contain TTE loci and therefore likely contribute to virulence, as noted previously [45]. Moreover, the backbone and many virulence genes found on the large plasmid of Pph 1448a are present within Pgy R4, Pmo, and Pan and this could reflect a larger role for this plasmid as a virulence factor in multiple host species than previously recognized. Indeed, the virulence gene suite of Pan is more similar to these group III strains than other more closely related strains (Figure S13).
We also found a recently acquired 1 Mb megaplasmid in the cucumber isolate Pla 107, and in a closely related strain also isolated from diseased cucumber. This megaplasmid is absent from two other cucumber isolates (Figure S12) and appears to possess the same copy number as the chromosome (Figure S11). This finding was both unexpected and unprecedented, as previously identified megaplasmids are typically conserved within all isolates of a species [68]. Megaplasmids can facilitate dramatic ecological shifts within bacteria [68], but we have not been able to predict phenotypic changes from pathways present on pMPPla107. Additionally, this megaplasmid contains a complete type IV secretion system (TFSS) most closely related to the Dot/ICM system from L. pneumophila. It is unknown whether this TFSS is used strictly for self-transmission and conjugation or if it actively secretes effector proteins. Although we did not find evidence for known type IV effectors on the megaplasmid, presence of this TFSS could represent a completely new contributor to virulence within P. syringae.
Identification and Distribution of Pathogenicity Factors
The primary determinants of virulence in P. syringae are TTEs and phytotoxins. Combining a high-throughput promoter trap screen with draft genome sequences for a subset of strains, we identified eight new TTE families (Table 2, Figure 3). In sum there are now 58 (not including avrD) TTE families across these 19 strains [48]. As we identified only nine new TTE families by screening these phylogenetically diverse strains, we believe that we have nearly saturated the discovery of TTE families found within P. syringae. Furthermore, additional candidate loci identified in our functional screen as HrpL-regulated were not translocated (Dataset S9). Each of these loci with non-translocated proteins possesses a functional hrp-box, linking gene expression to a known pathogenicity regulon, and therefore implicating these genes as virulence factors. Moreover, some of these loci have not been identified in previously sequenced genomes or by previous screens. We are confident that we have an exhaustive list of potential effectors for most of the sequenced strains. However, there is still likely to be substantial undiscovered diversity in the HrpL regulon across P. syringae.
Comparisons of evolutionary rates for TTE families shared throughout the P. syringae phylogeny could reveal specific TTEs important for virulence on a particular host. Yet, the virulence activity of any TTE can drive strong selection against its presence in a pathovar if that activity leads to recognition by a plant immune receptor. To capture this dynamic, we analyzed two classes of TTEs present within the 19 genomes. First, TTE effector families with wide distribution and very little divergence likely perform conserved virulence functions in a range of plants, and may additionally be evolutionarily ‘unrecognized’ across a wide range of plant hosts. Surprisingly, there are only five core TTE genes present in all pathogenic strains (Figure 3), and at least 4 of these have known virulence functions in A. thaliana [14], [55], [69], [70]. By virtue of their positional orthology in each genome, these few TTE potentially provide basic virulence functions.
Second, TTE genes found at different genomic locations in many strains, encoding proteins that are highly divergent (Table S3), could be under intense host selection driving diversification. This may mean that these TTEs have great potential to limit growth or help a pathovar expand across hosts. These widely distributed yet diverse TTE families could represent a class of virulence genes specialized to target rapidly evolving plant genes or pathways. They could, therefore, be most responsible for large-scale differences and limitations in host range. Interestingly, two of these, avrPto and hopAB are known to interact with common, and highly diverged, host PRR kinase domains to suppress host defense [71]–[74]. The rapid evolution within these TTE genes suggests that these TTEs are also widely recognized by the host immune system, leading to rapid loss, replacement, gain and, potentially, diversification. The most divergent TTE families are also experiencing high levels of horizontal gene transfer since their evolutionary histories do not mirror the phylogenies of the respective housekeeping genes (Figure S14).
Broadly, group II strains (including the completely sequenced isolate Psy B728a) contain fewer TTEs on average than the other clades (Figure 3). We hypothesize that strains within group II rely more heavily on non-TTSS based virulence factors for virulence as almost all members of this group share two of three known phytotoxin pathways. Indeed, the one strain from this group with the most TTEs (Ppi R6) is the only strain lacking genes for these pathways from this group. Furthermore, although all strains contain genes for the production of the plant hormone auxin, which can be an important virulence factor, only group II strains and the bean pathogen Pph 1448a lack a gene for auxin modification. Taken together, strains in this clade have apparently shifted their mechanisms of pathogenesis through TTE loss coupled with acquisition of phytotoxins by an ancestor of group II. In support of this hypothesis, we note a recent report where syringolin A modifies stomatal function in a manner that is phenotypically similar to, but mechanistically independent of, coronatine [75].
The smaller TTE repertoire of group II strains is not a sampling artifact. First, the gold standard genome of strain Psy B728a has been searched for the presence of TTEs by both experimental and bioinformatic methods [25]. Only 16 TTEs are found within this strain, still significantly less than most of the strains from the other phylogenetic clades. Secondly, we thoroughly sampled Psy B728a and other strains from this group (Pac, Ppi R6, Ptt, Pat) in our screen for novel TTE (Table S2). Thirdly, strains within group IIC have lost the canonical type III secretion system and most associated TTEs but maintain phytotoxin pathways [18]. It is unlikely that the progenitor of this group lacked the canonical TTSS, because structural genes of the TTSS (data not shown) as well as linked TTEs (avrE1, hopAA1, hopM1) appear to have been vertically inherited within this group, including strain Cit7 which diverges earlier than strains with an atypical TTSS (Figure 6B).
There is no pattern in the distribution of the remaining phytotoxin pathways in these sequenced strains. Excluding the group II clade and Por 1_6, remaining strains do not harbor multiple pathways for the production of known phytotoxins (Figure 3). As previously reported, only Pph 1448a and Pan share the genes involved in phaseolotoxin production. These pathovars also share many TTE loci commonly found in group III (Figure S13). Since Pan appears to have recently acquired these TTE genes (many of which may be present on a plasmid that resembles the large virulence plasmid of Pph 1448a), it is possible that phaseolotoxin and these shared TTE families target complementary host defense functions.
Virulence genes that modify plant hormonal pathways are also evolutionarily dynamic (Figure 3). Both Pgy R4 and Ppi R6 have recently acquired a gene involved in ethylene production. Ethylene production is thought to suppress host responses to biotrophic pathogens [76]. P. syringae strains are biotrophic, at least early in their life cycle on plants. The importance of coronatine, a structural mimic of the plant hormone jasmonic acid, as a virulence factor during Pto DC3000 infection of Arabidopsis has been noted [34]. Our data suggest that coronatine biosynthesis is a recent import into the Pto DC3000 genome and its HrpL regulon. Pto DC3000 mutants deficient in the production of coronatine are impaired during invasion via stomata, but are capable of causing disease if delivered directly into the host tissue [34]. Thus, absence of the coronatine pathway may partially explain why the Pla 106 and the tomato pathogen, Pto T1, are not virulent in Arabidopsis.
Host Range Evolution
Host range is notoriously difficult to define because strains could be pathogenic on unrelated, unknown, and invariably untested, host plant species. Additionally, there may be quantitative differences in pathogen growth on a given host species, even among strains grouped as non-pathogens. Furthermore, basic evolutionary questions such as the plasticity of host range and the number of steps involved in adaptation to a new host remain unanswered [4]. However, as the HrpL regulon is important in structuring host range and promoting virulence [4], [8], [29], understanding the evolution of TTE repertoires can reveal the potential for host range evolution across the P. syringae phylogeny.
Across three well-sampled clades of the phylogeny, the majority of the TTE suites for each strain are shared between at least two other strains within each group (Figure 4). In only a few cases do singleton TTEs make up a significant part of the total TTE repertoire of each strain. Although only a small number of strains have a high proportion of singleton TTEs, singletons may indicate recent shifts in the host ranges of these strains mediated by TTE gain. They are, therefore, important targets of future research into virulence mechanisms. Other isolates form well-defined groups according to conservation of TTEs (Figure 4, S12). These strains potentially utilize similar virulence strategies during infection, which limits the potential for host shifts among their particular host plants. For instance, Pla 106 was isolated from diseased cucumbers but shares much of its TTE suite with two tomato pathogens. Thus, host shifts might be more likely between tomato and cucumber because these strains carry similar suites of TTEs. Furthermore, as noted above, host range shifts or pathogenicity across bean and kiwi plants may be correlated due to the recent acquisition by Pan of a plasmid likely containing many virulence genes found in Pph 1448a as well as the pathway for phaseolotoxin production.
In striking contrast to the conservation patterns of functional TTE is the diversity of TTE inactivation due to truncations and transposon insertions (Figure 4). Because any given TTE can trigger a specific immune response during infection, inactivation of TTEs (i.e. removing the trigger) may play an important role in broadening and maintaining host range [25], [77]. Inactivated and truncated TTEs are more frequently found at the tips of the phylogeny than at internal nodes. Two non-mutually exclusive possibilities can explain this trend; the majority of TTE disruptions occurred recently or inactivated TTE are rapidly purged from the genome [78]. If the rate of TTE truncation and pseudogenization is truly higher at the tips of the phylogeny, then the lack of recognized TTE may be more important for recent changes in host range than the presence of functional TTE.
Recent studies using Xanthomonas suggested that isolates convergently evolved to infect the same hosts have acquired very similar sets of TTE [79]. To test this idea in P. syringae, we compared the TTE repertoires of Pla 106 and Pla 107, two distantly related strains from designated as the same pathovar (Figure S15). Five shared TTE families are common to these two strains that could act as general cucumber virulence factors. Three of these appear to be fairly recent acquisitions within both of these strains, in that they are only present within a limited number of other strains within the clade (hopE1, hopA1, hopBD1). Furthermore, hopAG1 is a TTE that has been convergently lost in each of these strains, suggesting that HopAG1 is recognized by cucumber. We tested for recognition of the Psy B728a hopAG1 allele in Pla 107 during growth in planta, but found no effect (Figure S15). Although generalizations of the role of hopAG1 in limiting host range on cucumber should include tests of multiple alleles on multiple cultivars of cucumber, these results suggest that hopAG1 does not play a broad role in limiting host range for pv. lachrymans and such gene loss may just be coincident pseudogenization of unnecessary proteins.
Type III Effector Function and Evolution
Host range could also be modified by diversification of shared TTE [36], [80]. Our draft genome data enable the identification of evolutionary signatures of diversification across shared alleles. Although pairwise diversity is slightly higher than the housekeeping loci for most TTE subfamilies, a handful of shared alleles display elevated levels of divergence, suggesting dramatic changes in specificity or function of TTE families.
In the hopM1 subfamily, allelic diversification may contribute to host range. This is consistent with the positional conservation of hopM1 across strains, its linkage to the TTSS-encoding pathogenicity island, and its defined virulence function in Pto DC3000. A fragment of HopM1 interacts with the A. thaliana protein AtMIN7, an ARF-GEF protein likely to be involved in vesicle transport and potentially in secretion of anti-microbial products [55]. As shown in Figure 6, it is striking that this TTE has undergone a clean gene conversion event while divergent alleles of other shared TTE are horizontally transferred to different places within the genome. While the hopM1 allele from Pto DC3000 complements virulence deficiencies of the Pto DC3000 ΔCEL mutant, the divergent hopM1 allele from Pmp does not (Figure 6C). Therefore, sequence divergence of hopM1 within the group I strains leads to functional divergence during Arabidopsis infection. These diverse alleles could target different host proteins, have host-dependent specificities of interaction, for example with diverged AtMIN7 orthologs, or have functionally co-evolved with other virulence-related genes in these strains. Interestingly, avrE1 from the Pmp/Pan clade appears to be vertically inherited from the ancestor of the group I strains suggesting that it would complement the Pto DC3000 ΔCEL virulence deficiencies (Figure 6B). In this case, the functions of the Pmp alleles of hopM1 and avrE1 are not likely to be redundant, in contrast to the finding that either allele from Pto DC3000 can complement Pto DC3000 ΔCEL [14], [55], [56]. Given the high levels of divergence across the hopM1 sub-family, it is difficult to pinpoint causal amino acid changes for the virulence defects. As this example illustrates, evolutionary divergence among shared TTE could structure changes in host range and pathogenicity. Our unbiased measurements of diversity are a first step towards identifying TTE families with interesting evolutionary signatures.
Our deep phylogenetic sequencing generated a large collection of orthologs. These orthologs are a natural allelic series. As a test case, we used AvrPto to see if its natural diversity could uncover important functional information. AvrPto has been extensively studied, and its physical interaction with Pto has been characterized by both mutagenesis and co-crystallization [58], [60], [64]. We found that the orthologs of AvrPto most closely related to AvrPtoPtoDC3000 were able to trigger a Pto-dependent HR. The most informative orthologs were AvrPtoPla107 and AvrPtoPgyR4. AvrPtoPla107 triggered a Pto-dependent HR, while AvrPtoPgyR4 did not. Both of these alleles are divergent from the Pto DC3000, Psy B728a and Pla 106 group, but relative to each other they are only polymorphic at 2 residues (positions 85 and 95); these are conserved in all AvrPto proteins that cause Pto-dependent HR. Both residues lie in or near the previously characterized AvrPto-Pto interaction surface. By individually mutating these two residues back to the consensus residue, we demonstrated that G95 is required for recognition, while M85 is not. G95 is in the GINP loop critical for AvrPto-Pto interaction. Thus, isolation of a natural allelic series allowed us to locate the binding surface on a TTE required for recognition by a host protein. This approach is generalizable to uncharacterized TTEs, given the identification of assayable host response.
The hypothesis that the G95 residue of an ancestor of the Pgy R4 ortholog has evolved to avoid recognition by a Pto/Prf-like system is consistent with the AvrPto phylogeny. Our virulence assay on tomato indicates that, consistent with previous studies, AvrPto is capable of mutation away from avirulence, while retaining virulence. The Pgy R4 ortholog is a striking evolutionary confirmation of the generation of avirulence-compromised, but virulence-competent mutants of AvrPto [64]. These data suggest that, at least in the case of AvrPto/Pto, P. syringae may be capable of quickly evolving at the level of a single amino acid to evade host R-gene recognition.
Conclusions
P. syringae, a phytopathogen responsible for crop loss throughout the world, has evolved to live in a diversity of environments and infect a broad range of host plants. Although the evolutionary basis of host range determination is unknown for many pathogen systems, TTE and phytotoxins are thought to be the primary contributors within P. syringae. Here we uncover the evolutionary conservation of these virulence determinants across diverse strains of P. syringae using cost-efficient genome sequencing coupled with screening a subset of these strains for the presence of novel TTE proteins. We only found a small core of five TTEs, one of which is often disrupted, that were conserved across strains. We show that although TTE and phytotoxin repertoires change rapidly, overarching trends emerge for both TTE and phytotoxin content – such as the trade off between complexity of TTE content and the presence of phytotoxin pathways. These evolutionary trends are only apparent in the context of broad phylogenetic sampling of genome sequences. Furthermore, complete genome sequencing of diverse strains enables more refined investigation of shared virulence genes and also provides a framework to inform and identify novel structure-function relationships.
Materials and Methods
Strains
We chose strains for sequencing to maximize the sampling of genetic diversity and the variety of hosts of isolation across the phylogeny of P. syringae (Strain names and pathovar designations can be found in Table 1). It should be noted that Pma was originally misidentified as pathovar maculicola and actually should be classified as P. cannabina pv. alisalensis, but we maintain Pma nomenclature for consistency with previous reports [81]. It should also be noted that Pae is also known as strain NCPPB3681 [40]. Most surveyed strains have been categorized [3], [16]. Our characterizations of host range throughout the paper are largely inferred from pathovar designation and previously published results, and we did not extensively characterize host range for any strain. We minimized the amount of laboratory passage before sequencing (although we are unsure of exactly how long each strain has been passaged under laboratory conditions since isolation, and some of the sequenced strains are rifampicin resistant derivatives). Genomic DNA for construction of all sequencing libraries was from a single colony of each strain that was picked and grown overnight in 250 mL of King's B media shaking at 28°C. Genomic DNA was isolated using a CTAB genomic protocol.
Sequencing, Assembly, and Annotation
Genome sequencing for all strains consisted of a minimum of 1 lane of unpaired 36 bp Illumina reads, in addition to ¼ plate of 454 Flex reads. For six of the strains (Pgy R4, Pma, Pja, Pan, Ppi R6, Pla 106) this ¼ plate of 454 was supplemented with an additional ¼ plate of 454 paired ends from a separate library. Paired ends for the remaining strains (Pac, Pae, Ptt, Pmp, Pta, Cit7, Pla 107, Pmo) were created using the Roche 454 Long Paired end protocol, and were thus part of the initial ¼ plate of 454. For Pla 107, additional Illumina runs (1 lane of each at 48 bp, 56 bp, 72 bp) were used to fill in scaffolds. Genome sequences of Pta and Pma were supplemented with genomic data from the same strains publically available from Genbank (Pta: NZ_ACHU00000000, Pma: NC_005922; NC_005921; NC_005920; NC_005919; NC_t005918) [30], [82]. A draft genome was also recently published for Pae [40]. All analyses for this strain were performed using only our own assembly, but the final assembly of this strain in GenBank includes genomic sequence from the other published sequence of the same isolate (NZ_ACXS00000000). A draft genome sequence was also recently published [38] for Pgy R4 (AEGH00000000), too late to be included in our assembly or analysis. Publically available genomic sequences for all assemblies can be considered at least high quality drafts [83].
The genomes of each strain were assembled using a modified version of the pipeline described in Reinhardt et al. 2009 [41]. All genomes were subject to all steps of the pipeline downstream of, and including, Newbler assembly. In some cases (Pgy R4, Ppi R6, Pja, Cit7, Ptt) VCAKE was used to build initial contigs from Illumina reads, while in other cases (Pma, Pan, Pla 106, Pla 107, Pmo, Pmp, Pta, Pae, Pac) EDENA was used to build these initial contigs [84]. We found that there was no large-scale difference in size or quality of contigs built with VCAKE or EDENA. Annotation was carried out by submitting all contigs and scaffolds for each of the draft genomes to the NCBI PGAAP pipeline (http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html). NCBI accession numbers for each genome sequence are in Table 1 and protein sequences for all called ORFs as well as those fixed by Phylo-gene-boost consolidation (see below) are listed in Dataset S1, S2, S3, S4. Mauve alignments of draft genomes to complete genomes within each MLST group are presented for group I (Figure S6), group II (Figure S5), and group III (Figure S4).
Phylogenetics
Fragments of nucleotide sequences for 7 genes previously used for MLST analysis (gyrB, gapA, fruK, pgi, rpoD, acnB, gltA) were extracted from each draft genome as well as from the three completely sequenced P. syringae genomes (Pto DC3000, Pph 1448a, Psy B728a) and from P. fluorescens Pf-0 as an outgroup [26]–[28], [42]. Sequences were individually aligned using default parameters in ClustalX, trimmed, and concatenated. The program Mr. Bayes (http://mrbayes.csit.fsu.edu/) was used to create a Bayesian phylogeny for these sequences while parsimony-based and maximum likelihood phylogenies were created using programs within the Phylip package (http://evolution.genetics.washington.edu/phylip.html). For both parsimony and maximum likelihood, a consensus tree was created from 100 independent phylogenies. For the maximum likelihood trees, nucleotide frequencies and model were chosen using the program jModeltest (http://darwin.uvigo.es/software/modeltest.html). Similar methods were used to derive phylogenies for the avrE, hopM, avrPto, avrB, hopF, hopH, and hopAB families except that phylogenetic analyses were performed on protein sequences. Phylogenies built using all three methods for the MLST genes were generally congruent.
We choose to resolve problematic nodes by building amino acid sequence based trees using a database of 324 orthologous genes shared by all strains. We required reciprocal best hits (RBH), with 80% amino acid identity for greater than 80% of the length, an e-value <1×10−200 and no evidence of being on a plasmid. For each apparent ortholog, we used ProbCons, a probabilistic consistency algorithm that combines sum-of-pairs scoring and HMM-derived posterior probabilities, to produced a consensus alignment for all alleles of a gene [85]. For each gene we performed a model test to determine the best amino acid substitution model. We then concatenated all aligned sequences and, using the majority best-fit model from the individual loci, constructed a tree using RAXML (Figure S2). RAXML is a maximum likelihood-based tool for large phylogenetic trees, and was optimized for running on our computers [86], [87]. We also produced individual trees for each locus using a similar strategy, except that gene specific substitution models were used. We visually inspected and categorized a random selection of 15% of the trees to ensure consistence and to investigate discrepancies at several nodes. We observed that in cases where the tree inferred from MLST sequences differed from the protein consensus tree, the trees inferred from MLST sequences were the second common topology among trees for that gene. Individual trees are contained in Dataset S8.
Plasmid Identification
Each draft genome sequence was surveyed with BLASTn (evalue = 1e−5) for the presence of structural genes associated with previously identified P. syringae plasmids (Table S1). Whole scaffolds/contigs containing a BLAST hit identifying that fragment as coming from a plasmid were compared to the NCBI database to identify other plasmid distinctive elements within that scaffold. Depth of coverage of Illumina reads for the entire potential plasmid scaffold/contig was compared to coverage for known housekeeping genes within each genome. Those with higher (>2×) coverage were considered ‘plasmid-like’. In strain Pla 107, two large (∼1 Mb total) fragments of sequence did not assemble with other contigs. Further manual assembly showed that these two genomic regions formed one large contig, and primer sets were designed to confirm manual assembly. Three primer pairs were also designed to PCR out from the ends of this potential megaplasmid, followed by Sanger based sequencing of the PCR fragments, in order to demonstrate circularization. Furthermore, six primer sets were designed to investigate the presence of this megaplasmid within closely related P. syringae strains.
We used the Conserved Domain Database and KEGG [88], [89] to evaluate the potential functions and metabolic pathways associated with genes harbored on the plasmids (Figure S12).
Defining the Core and Pan Genome
We defined the core genome for all isolates using an iterative BLASTx (querying with the Pto DC3000 proteome, E = 10−6, 40% homology, 40% length hit) approach. Starting with ORFs from Pto DC3000 [26], we performed iterative tBLASTn versus all nucleotides of other assembled draft genomes sequentially. The other two ‘gold standard’ genomes of Psy B728a and Pph 1448a were used for the first and second iterations. During each iteration, the ‘core genome’ set – cumulatively derived to that point – was compared to an additional draft genome. In the end, ORFs from Pto DC3000 found within all genomes were retained as the core genome set. In addition, we performed all possible pair-wise tBLASTns, within each clade and among all clades, to determine genes unique to each isolate, clade, and sub-clade. The same procedure was performed with the genomes four P. putida, and three P. fluorescens isolates [42]–[44] to generate a ‘genus level’ core. Gene lists for the core, using the Pto DC3000 annotations, is found in Dataset S5, S6, S7.
To determine the pan-genome (that is, the set of genes found in at least one strain) for all isolates, we used the same iterative BLAST strategy. Only contiguous, full length ORFs (as annotated by NCBI) were used. Pseudogenes and incomplete annotations were ignored. Starting with the P. syringae core genome, we performed BLASTp analysis versus Pph 1448a, to determine which of this strain's genes were not present in the Pseudomonas core. These genes, combined with the core genome, are our initial pairwise “pan genome”. Subsequently this pan genome was BLASTed against all other isolates in the phylogenetic order presented in Figure 1. During each iteration, isolate genes missing (BLASTp non-hits) from the current version of the pan-genome were identified. These genes were then added to the pan-genome before the next isolate was considered. As before, we then identified clade and taxon specific “pan” genomes (pathovar unique genes are in Dataset S5, S6, S7).
Phylogenetic Gene Gain
Starting from the most closely related isolate pair in each clade, we stepped through the phylogeny for every member of each clade, identifying the number of genes gained upon branch bifurcation, using a strategy similar to that used for the core genome above. We started by comparing genes shared by the pair of most closely related isolates (e. g. Ptt and Pja, see Figure 1) to those contained within the next closest relative (e.g. Ppi R6). Resulting BLASTp hits (that is, the genes shared between Ppi R6 and the Ptt-Pja pair) were removed, leaving the genes exclusively shared by the Ptt-Pja pair. We subsequently stepped deeper into the clade phylogeny, taking the Ppi R6, Pja and Ptt shared genes and comparing them to genes shared by Psy-Pac pair, thus identifying genes exclusive to the Psy-Pac pair and the Ppi R6-Pja-Ptt triplet respectively (Figure 2D).
Phylo-Gene-Boost ORF Consolidation Strategy
To repair mis-annotated genes produced by the NCBI ORF annotation pipeline, we subjected all sequenced genomes to the ‘Phylo-gene-boost’ algorithm, which is similar to the “gene-boost” strategy [90]. The key difference between these two approaches is the use of phylogenetic information to inform orthology and identify suitable sequence comparisons. The NCBI annotation pipeline produces a list of continuous ORFs, along with fractured ORFs and hypothetical protein sequences. The fractured ORFs may in fact identify protein fragments from the same Cluster of Orthologous Groups (COGs, which indicate phylogenetic relatedness among the ORFs). We exploited this possibility by comparing (BLASTp e = 10−6, b = 1) fractured ORFs from each of the sequenced genomes to the previously assembled NCBI P. syringae reference genome appropriate for each clade. Resulting hits sharing a COG ID with one or more of the other hits were grouped (COG clusters). Subsequently, we aligned (ClustalW) these hits against the reference gene they matched. Locations of COG cluster ORFs with respect to the reference gene were characterized as (1) overlapping each other and the reference, (2) overlapping the reference but not each other, (3) two sequences overlapping the reference but not each other, with a linker sequence overlapping both. Group 1 sequences were concatenated, however these are potentially problematic as they may be duplication events that result in assembly errors. Group 2 sequences were concatenated as long as the unmatched region was not larger than either of the sequences. Group 3 sequences were annotated as a single ORF, retaining as much similarity to the reference protein as possible. The annotation for each isolate was updated with newly defined continuous ORFs. In some cases up to 30% of the fractured ORFs were successfully joined and annotated.
Identification of Novel TTE Families
A subset of the sequenced strains were screened for the presence of novel TTEs using the protocol described in Chang et al [23]. Briefly, genomic libraries were created from these strains by cloning fragments into a vector upstream of promoterless GFP. These libraries were screened for the presence of active hrp-boxes by selecting for GFP expression using FACS sorting under conditions where the hrpL sigma factor was expressed. Potential TTE and hrp-boxes were identified after sequencing the clones. Additionally, strains pv. maculicola M4 (Pma M4, which is very closely related to Pma) and pv. atrofaciens DSM50255 (Pat) were screened by this method and their novel TTE sequences included in all similarity searches. TTE chimeras were also independently identified from all of the draft genomes through similarity (tBLASTn with no e-value cutoff over a significant portion of the effector) with known TTEs families or experimentally confirmed TTEs from the screen. Full length potential TTEs were cloned from at least one genome into plasmid pJC532, including sequences likely to contain the type III hrp-box upstream regulatory element. If hrp boxes could not be identified or there was a scaffold break upstream of the ORF, only the ORF sequence of the putative effector was cloned into plasmid pBAV178 where expression of an ‘Δ79AvrRpt2 fusion protein could be driven off a constitutive promoter [91]. Representatives from each putative TTE family were tested for their ability to translocate the active C-terminal fragment of AvrRpt2 to cause a hypersensitive response (HR) in Arabidopsis accession Col-0 [24]. All HR tests were performed on ∼5 week old plants and included a positive control of a known TTE cloned into either pJC532 or pBAV178 [23], [91]. The results of all tested loci are found in Dataset S9.
Type III Effector and Phytotoxin Content
Draft genome sequences for each strain were searched by tBLASTn (at first with an with e-value cutoff of 10−5, but later with no cutoff) for the presence of known TTEs. This list was constructed by combining protein sequences for known P. syringae TTEs (http://pseudomonas-syringae.org/) with our list of novel TTEs identified from a subset of these genomes (see previous section). The potential TTE sequence was then pulled out of the draft genome sequence to the next possible stop codon and, if there was no identifiable start codon based on similarity to known TTEs, up to the earliest possible start codon after an upstream stop codon. The position of the start codon was further refined by relationship to an identifiable hrp-box or by comparison to other known sequences. When possible, if there was a frameshift or early stop codon that led to early protein termination (such that the locus was split into two halves that were each orthologous to a given TTE) or if there was a scaffold break disrupting a potential TTE, genomic sequences were bridged or verified by PCR-based sequencing. In some cases, the presence of TTEs could not be verified because PCR-based sequencing failed on 3 separate attempts, or because only a partial sequence of the TTE was present on a contig with no ability to bridge a gap. In some cases, there were loci in the genomes that matched by BLAST, but were significantly diverged from previously identified TTEs or were novel chimeras. In these cases, at least one subfamily member from these TTE families were cloned and tested for translocation (Dataset S9). Sequences identified as HrpL-regulated by screen or as potential TTE by similiarity, but which were not tested for translocation, are also listed in Dataset S9.
Draft genome sequences were also searched for pathways involved in construction of six well known phytotoxins associated with P. syringae (coronatine, phaseolotoxin, tabtoxin, syringomycin, syringopeptin, syringolin) as well as genes for ethylene production (efe), auxin production and modification (iaaM, iaaH, and iaaL) and an enzyme whose activity leads to secretion of an HR inducing factor, syringolide (avrD). Protein sequences for loci involved in toxin metabolic pathways in various strains were obtained from NCBI and used as a tBLASTn query on each draft genome sequence. A strain was considered to possess a toxin or gene if a majority of the protein sequences for each pathway had significant BLAST hits (<1e−5) with an average similarity of 80% or greater. If some, but not all, of the pathway for a particular toxin was present, the strain was considered to potentially possess the toxin.
Analyzing the Distributions of Type III Effectors and Toxins
Within the three main clades of P. syringae with five or more genome sequences sampled, full length and partial TTE for each strain were assigned to bins according to prevalence within the clade. TTEs were defined as shared between strains as long as other strains within the same clade possessed the same subfamily of TTE either in the functional form or as a pseudogene/truncation. TTEs from the same subfamily, but of clearly divergent alleles within a phylogenetic group, were not classified as shared. Duplicated TTEs with no orthologous duplication in other strains were not classified as shared. For truncated or transposon disrupted TTEs, the phylogenetic node where such a change took place was identified by parsimony. If two strains within the same phylogenetic clade shared a truncated or pseudogenized TTE, positional (synteny) and sequence information was used to determine the independence of these events.
Virulence gene suites were hierarchically clustered using average linkage in Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm). A value of 1 was given for each potentially full-length TTE and complete phytotoxin pathways per strain. If a TTE gene or toxin biosynthetic pathway were potentially present, but unconfirmed, a value of 0.5 was given. A value of −1 was given for truncated/disrupted TTE as well as absence of a TTE. For truncated or transposon disrupted TTEs, the phylogenetic node where such a change took place was identified by parsimony. If two strains within the same phylogenetic clade shared a truncated or pseudogenized TTE, positional (synteny) and sequence information was used to determine the independence of these events.
Pairwise Diversity Calculations for Effector Families
To calculate pairwise amino acid diversity for TTE families found within a majority of strains, TTE families were aligned using ClustalX with the default parameters. Pairwise diversity was calculated using a custom Perl script and was performed by both including deletions/insertions as divergent sites or excluding them completely. If a TTE subfamily was found to be present within multiple strains within a clade of P. syringae, nucleotide sequences for each member of the TTE subfamily within the clade were aligned using Revtrans 1.4 [92] with Dialign 2 alignment. The same custom Perl script was then used to calculate pairwise nucleotide diversity (π). Sites that contained insertions or deletions were not scored for these nucleotide pairwise diversity calculations. As a control, π was also calculated for the concatenated MLST gene fragments (gyrB, gapA, fruK, pgi, rpoD, acnB, gltA) for the same group of strains as the TTE of interest. For further investigation of hopM1 within the group I clade, we aligned as much contiguous nucleotide sequence as possible from within five group I strains using ClustalX using default parameters without including scaffold breaks. The pairwise diversity was calculated for each site using the same custom script as above, and was averaged over 25 nucleotide windows to provide the values for Figure 6A.
Testing the Virulence Function of hopM1Pmp
hopM1 and its chaperone, shcM were cloned from both Pto DC3000 and Pmp into a broad host range plasmid (pBV226) for constitutive expression in P. syringae. Plasmids were mated into a strain [56] in which the CEL (Conserved Efector Locus, including hopM1) was deleted. Deletion of the CEL produces a noticeable decrease in growth on Arabidopsis [55] and loss of disease symptoms of Pto DC3000 during growth on tomato cv. Moneymaker [56]. In Arabidopsis, we tested the ability of hopM1 from Pmp to complement the virulence defect by dip inoculating of 5×106 bacterial cells per mL in 10 mM MgCl2 into 2 week old plants. Lids were kept off of the plants for all 3 days of the experiment. Dip infection assays were performed three independent times, with 6 replicates for each genotype. Statistical analysis (ANOVA and Tukey's HSD, p≤0.05) of growth data was performed using JMP7 (SAS). The cloned fragment of Pmp consisting of hopM1, shcM, and the upstream hrp box was confirmed for HopM1 translocation using the Δ79avrRpt2 translocation assay [23].
Agrobacterium-Mediated Transient Expression Assays in Nicotiana Benthamiana
The tomato (Solanum pimpinellifolium) Pto gene and avrPto family members were cloned into pDONR207 as ORF clones without a stop codon. avrPto ORF clones were recombined into pGWB14 (35S∶GW∶HA) using LR clonase (Invitrogen). Similarly, Pto was recombined into pGWB17 (35S∶GW∶MYC). The final destination clones were moved into C58C1 (gift of Brad Day) by triparental mating. For transient expression in N. benthamiana, overnight cultures of Agrobacterium strains carrying avrPto, Pto or the p19 viral suppressor (to enhance transient expression; [93]) were all washed in induction media (10 mM MgCl2, 10 mM MES pH 5.6, 150 uM acetosyringone) for 1 hour, diluted to OD 0.8 and mixed 1∶1∶1 and syringe inoculated. Four days later the inoculated regions were assessed for HR by visual symptoms of chlorosis/necrosis. Inoculations were performed at least three times. Expression of AvrPto and Pto was verified by Western blotting. For ion leakage assays, four 7 mm leaf disks from transient assays were taken and rinsed in distilled water for 30 min and then transferred to tubes containing 6 mL distilled water. Ion leakage was quantified over time using an Orion conductivity meter.
P. Syringae Based Virulence Assays
Virulence assays using this strain on tomato were performed as described [3]. avrPto ORF clones were expressed in P. syringae Pto T1 (gift of John Rathjen) from pDLtrpGW. pDLtrpGW is a modified version of pBBR1-MCS [94] that expresses ORF clones under the constitutive trp promoter with a C-terminal HA tag (gifts of Derek Lundberg). Tomato plants (76R prf3, gift of Greg Martin) were vacuum infiltrated with OD = 0.0002 bacteria in 10 mM MgCl2 and 0.02% Silwet L-77. Four days after inoculation leaf discs were cored (8 replicates, each 4 cores), ground in 10 mM MgCl2, serially diluted and plated on KB/Rif. Statistical analysis (ANOVA and Tukey's HSD, p≤0.05) of growth data was performed using JMP7 (SAS). This experiment was performed 3 times with similar results. Newly described avrPto orthologs were all translocated when tested from pBAV178 (data not shown) [91].
Accession Numbers
All genome accession numbers are listed in Table 1, at bottom. Protein accession numbers for new TTE families are listed in Table 2.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FatmiMBCollmerASante IacobellisN 2008 Pseudomonas syringae Pathovars and Related Pathogens - Identification, Epidemiology, and Genomics. Springer 433
2. LinN-CMartinGB 2005 An avrPto/avrPtoB mutant of Pseudomonas syringae pv. tomato DC3000 does not elicit Pto-mediated resistance and is less virulent on tomato. MPMI 18 43 51
3. SarkarSFGordonJSMartinGBGuttmanDS 2006 Comparative genomics of host-specific virulence in Pseudomonas syringae. Genetics 174 1041 1056
4. LindebergMCunnacSCollmerA 2009 The evolution of Pseudomonas syringae host specificity and type III effector repertoires. Mol Plant Pathol 10 767 775
5. MohrTJLiuHYanSMorrisCECastilloJA 2008 Naturally occurring nonpathogenic isolates of the plant pathogen Pseudomonas syringae lack a type III secretion system and effector gene orthologues. J Bacteriol 190 2858 2870
6. MorrisCESandsDCVannesteJLMontarryJOakleyB 2010 Inferring the Evolutionary History of the Plant Pathogen Pseudomonas syringae from Its Biogeography in Headwaters of Rivers in North America, Europe, and New Zealand. mBio 1 e00107-00110 e00107-00120
7. CunnacSLindebergMCollmerA 2009 Pseudomonas syringae type III secretion system effectors: repertoires in search of functions. Curr Opin Microbiol 12 53 60
8. CollmerABadelJLCharkowskiAODengWLFoutsDE 2000 Pseudomonas syringae Hrp type III secretion system and effector proteins. Proc Natl Acad Sci U S A 97 8770 8777
9. MansfieldJW 2009 From bacterial avirulence genes to effector functions via the hrp delivery system: an overview of 25 years of progress in our understanding of plant innate immunity. Mol Plant Pathol 10 721 734
10. MudgettMB 2005 New insights to the function of phytopathogenic bacterial type III effectors in plants. Annu Rev Plant Biol 56 509 531
11. GrantSRFisherEJChangJHMoleBMDanglJL 2006 Subterfuge and manipulation: type III effector proteins of phytopathogenic bacteria. Annu Rev Microbiol 60 425 449
12. JonesJDGDanglJL 2006 The plant immune system. Nature 444 323 329
13. ZhouJChaiJ 2008 Plant pathogenic bacterial type III effectors subdue host responses. Curr Opin Microbiol 11 179 185
14. KvitkoBHParkDHVelásquezACWeiC-FRussellAB 2009 Deletions in the Repertoire of Pseudomonas syringae pv. tomato DC3000 Type III Secretion Effector Genes Reveal Functional Overlap among Effectors. PLoS Pathog 5 e1000388
15. FerrantePClarkeCRCavanaughKAMichelmoreRWBuonaurioR 2009 Contributions of the effector gene hopQ1-1 to differences in host range between Pseudomonas syringae pv. phaseolicola and P. syringae pv. tabaci. Mol Plant Pathol 10 837 842
16. HwangMSHMorganRLSarkarSFWangPWGuttmanDS 2005 Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae. Appl Environ Microbiol 71 5182 5191
17. TaylorJTeversonDAllenDPastor-CorralesM 1996 Identification and origin of races of Pseudomonas syringae pv. phaseolicola from Africa and other bean growing areas. Plant Pathol 45 469
18. ClarkeCRCaiRStudholmeDJGuttmanDSVinatzerBA 2010 Pseudomonas syringae strains naturally lacking the classical P. syringae hrp/hrc Locus are common leaf colonizers equipped with an atypical type III secretion system. Mol Plant Microbe Interact 23 198 210
19. BullCTClarkeCRCaiRVinatzerBJardiniTM 2011 Multilocus Sequence Typing of Pseudomonas syringae sensu lato confirms previously described genomospecies and permits rapid identification of P. syringae pv. coriandricola and P. syringae pv. apii causing bacterial leaf spot on parsley. Phytopathology In press
20. GardanLShafikHBelouinSBrochRGrimontF 1999 DNA relatedness among the pathovars of Pseudomonas syringae and description of Pseudomonas tremae sp. nov. and Pseudomonas cannabina sp. nov. (ex Sutic and Dowson 1959). Int J Syst Bacteriol 49 Pt 2 469 478
21. BochJJoardarVGaoLRobertsonTLLimM 2002 Identification of Pseudomonas syringae pv. tomato genes induced during infection of Arabidopsis thaliana. Mol Microbiol 44 73 88
22. Zwiesler-VollickJPlovanich-JonesAENomuraKBandyopadhyaySJoardarV 2002 Identification of novel hrp-regulated genes through functional genomic analysis of the Pseudomonas syringae pv. tomato DC3000 genome. Mol Microbiol 45 1207 1218
23. ChangJHUrbachJMLawTFArnoldLWHuA 2005 A high-throughput, near-saturating screen for type III effector genes from Pseudomonas syringae. Proc Natl Acad Sci U S A 102 2549 2554
24. GuttmanDSVinatzerBASarkarSFRanallMVKettlerG 2002 A functional screen for the type III (Hrp) secretome of the plant pathogen Pseudomonas syringae. Science 295 1722 1726
25. VinatzerBATeitzelGMLeeM-WJelenskaJHottonS 2006 The type III effector repertoire of Pseudomonas syringae pv. syringae B728a and its role in survival and disease on host and non-host plants. Mol Microbiol 62 26 44
26. BuellCRJoardarVLindebergMSelengutJPaulsenIT 2003 The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc Natl Acad Sci U S A 100 10181 10186
27. FeilHFeilWSChainPLarimerFDiBartoloG 2005 Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc Natl Acad Sci U S A 102 11064 11069
28. JoardarVLindebergMJacksonRWSelengutJDodsonR 2005 Whole-genome sequence analysis of Pseudomonas syringae pv. phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition. J Bacteriol 187 6488 6498
29. FoutsDEAbramovitchRBAlfanoJRBaldoAMBuellCR 2002 Genomewide identification of Pseudomonas syringae pv. tomato DC3000 promoters controlled by the HrpL alternative sigma factor. Proc Natl Acad Sci U S A 99 2275 2280
30. StudholmeDJIbanezSGMacLeanDDanglJLChangJH 2009 A draft genome sequence and functional screen reveals the repertoire of type III secreted proteins of Pseudomonas syringae pathovar tabaci 11528. BMC Genomics 10 395
31. StavrinidesJMaWGuttmanDS 2006 Terminal reassortment drives the quantum evolution of type III effectors in bacterial pathogens. PLoS Pathog 2 e104
32. LindebergMMyersCRCollmerASchneiderDJ 2008 Roadmap to new virulence determinants in Pseudomonas syringae: insights from comparative genomics and genome organization. Mol Plant Microbe Interact 21 685 700
33. BenderCLAlarcón-ChaidezFGrossDC 1999 Pseudomonas syringae phytotoxins: mode of action, regulation, and biosynthesis by peptide and polyketide synthetases. Microbiol Mol Biol Rev 63 266 292
34. MelottoMUnderwoodWKoczanJNomuraKHeSY 2006 Plant stomata function in innate immunity against bacterial invasion. Cell 126 969 980
35. MilletYADannaCHClayNKSongnuanWSimonMD 2010 Innate immune responses activated in Arabidopsis roots by microbe-associated molecular patterns. Plant Cell 22 973 990
36. MaWDongFFTStavrinidesJGuttmanDS 2006 Type III effector diversification via both pathoadaptation and horizontal transfer in response to a coevolutionary arms race. PLoS Genet 2 e209
37. AlmeidaNFYanSLindebergMStudholmeDJSchneiderDJ 2009 A draft genome sequence of Pseudomonas syringae pv. tomato T1 reveals a type III effector repertoire significantly divergent from that of Pseudomonas syringae pv. tomato DC3000. Mol Plant Microbe Interact 22 52 62
38. QiMWangDBradleyCAZhaoY 2011 Genome Sequence Analyses of Pseudomonas savastanoi pv. glycinea and Subtractive Hybridization-Based Comparative Genomics with Nine Pseudomonads. PLoS ONE 6 e16451
39. Rodríguez-PalenzuelaPMatasIMMurilloJLópez-SolanillaEBardajiL 2010 Annotation and overview of the Pseudomonas savastanoi pv. savastanoi NCPPB 3335 draft genome reveals the virulence gene complement of a tumour-inducing pathogen of woody hosts. Environ Microbiol 12 1604 1620
40. GreenSStudholmeDJLaueBEDoratiFLovellH 2010 Comparative genome analysis provides insights into the evolution and adaptation of Pseudomonas syringae pv. aesculi on Aesculus hippocastanum. PLoS ONE 5 e10224
41. ReinhardtJABaltrusDANishimuraMTJeckWRJonesCD 2009 De novo assembly using low-coverage short read sequence data from the rice pathogen Pseudomonas syringae pv. oryzae. Genome Res 19 294 305
42. SilbyMCerdeño-TárragaAVernikosGGiddensSJacksonR 2009 Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol 10 R51
43. PaulsenITPressCMRavelJKobayashiDYMyersGSA 2005 Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol 23 873 878
44. NelsonKEWeinelCPaulsenITDodsonRJHilbertH 2002 Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ Microbiol 4 799 808
45. VivianAMurilloJJacksonRW 2001 The roles of plasmids in phytopathogenic bacteria: mobile arsenals? Microbiology 147 763 780
46. BackertSMeyerTF 2006 Type IV secretion systems and their effectors in bacterial pathogenesis. Curr Opin Microbiol 9 207 217
47. HubberARoyCR 2010 Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol 26 261 283
48. LindebergMStavrinidesJChangJHAlfanoJRCollmerA 2005 Proposed guidelines for a unified nomenclature and phylogenetic analysis of type III Hop effector proteins in the plant pathogen Pseudomonas syringae. Mol Plant Microbe Interact 18 275 282
49. AlfanoJRCharkowskiAODengWLBadelJLPetnicki-OcwiejaT 2000 The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proc Natl Acad Sci U S A 97 4856 4861
50. TsiamisGMansfieldJWHockenhullRJacksonRWSesmaA 2000 Cultivar-specific avirulence and virulence functions assigned to avrPphF in Pseudomonas syringae pv. phaseolicola, the cause of bean halo-blight disease. EMBO J 19 3204 3214
51. OngLEInnesRW 2006 AvrB mutants lose both virulence and avirulence activities on soybean and Arabidopsis. Mol Microbiol 60 951 962
52. KeithLWBoydCKeenNTPartridgeJE 1997 Comparison of avrD alleles from Pseudomonas syringae pv. glycinea. Mol Plant Microbe Interact 10 416 422
53. SorensenKNKimKHTakemotoJY 1998 PCR Detection of Cyclic Lipodepsinonapeptide-Producing Pseudomonas syringae pv. syringae and Similarity of Strains. Appl Environ Microbiol 64 226 230
54. YanSLiuHMohrTJJenretteJChiodiniR 2008 Role of recombination in the evolution of the model plant pathogen Pseudomonas syringae pv. tomato DC3000, a very atypical tomato strain. Appl Environ Microbiol 74 3171 3181
55. NomuraKDebroySLeeYHPumplinNJonesJ 2006 A bacterial virulence protein suppresses host innate immunity to cause plant disease. Science 313 220 223
56. BadelJNomuraKBandyopadhyaySShimizuRCollmerA 2003 Pseudomonas syringae pv. tomato DC 3000 HopPtoM(CEL ORF 3) is important for lesion formation but not growth in tomato and is secreted and translocated by the Hrp type III secretion system in a chaperone-dependent manner. Mol Microbiol 49 1239 1251
57. DebroySThilmonyRKwackY-BNomuraKHeSY 2004 A family of conserved bacterial effectors inhibits salicylic acid-mediated basal immunity and promotes disease necrosis in plants. Proc Natl Acad Sci U S A 101 9927 9932
58. XingWZouYLiuQLiuJLuoX 2007 The structural basis for activation of plant immunity by bacterial effector protein AvrPto. Nature 449 243 247
59. ZipfelCRathjenJP 2008 Plant immunity: AvrPto targets the frontline. Curr Biol 18 R218 220
60. ChangJHRathjenJPBernalAJStaskawiczBJMichelmoreRW 2000 avrPto enhances growth and necrosis caused by Pseudomonas syringae pv. tomato in tomato lines lacking either Pto or Prf. Mol Plant Microbe Interact 13 568 571
61. RathjenJPChangJHStaskawiczBJMichelmoreRW 1999 Constitutively active Pto induces a Prf-dependent hypersensitive response in the absence of avrPto. EMBO J 18 3232 3240
62. de VriesJSAndriotisVMEWuAJRathjenJP 2006 Tomato Pto encodes a functional N-myristoylation motif that is required for signal transduction in Nicotiana benthamiana. Plant J 45 31 45
63. ChangJHTobiasCMStaskawiczBMichelmoreRW 2001 Functional studies of the bacterial avirulence protein AvrPto by mutational analysis. Mol Plant Microbe Interact 14 451 459
64. ShanLHePZhouJTangX 2000 A cluster of mutations disrupt the avirulence but not the virulence functions of AvrPto. Mol Plant Microbe Interact 13 592 598
65. WulfJPascuzziPEFahmyAMartinGBNicholsonLK 2004 The Solution Structure of Type III Effector Protein AvrPto Reveals Conformational and Dynamic Features Important for Plant Pathogenesis. Structure 12 1257 1268
66. KunkeawSTanSCoakerG 2010 Molecular and evolutionary analyses of Pseudomonas syringae pv. tomato race 1. Mol Plant Microbe Interact 23 415 424
67. TouchonMHoedeCTenaillonOBarbeVBaeriswylS 2009 Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5 e1000344
68. HarrisonPWLowerRPJKimNKDYoungJPW 2010 Introducing the bacterial ‘chromid’: not a chromosome, not a plasmid. Trends Microbiol 18 141 148
69. MunkvoldKRRussellABKvitkoBHCollmerA 2009 Pseudomonas syringae pv. tomato DC3000 type III effector HopAA1-1 functions redundantly with chlorosis-promoting factor PSPTO4723 to produce bacterial speck lesions in host tomato. Mol Plant Microbe Interact 22 1341 1355
70. JelenskaJYaoNVinatzerBAWrightCMBrodskyJL 2007 A J domain virulence effector of Pseudomonas syringae remodels host chloroplasts and suppresses defenses. Curr Biol 17 499 508
71. XiangTZongNZouYWuYZhangJ 2008 Pseudomonas syringae effector AvrPto blocks innate immunity by targeting receptor kinases. Curr Biol 18 74 80
72. ShanLHePLiJHeeseAPeckSC 2008 Bacterial effectors target the common signaling partner BAK1 to disrupt multiple MAMP receptor-signaling complexes and impede plant immunity. Cell Host Microbe 4 17 27
73. NtoukakisVMucynTSGimenez-IbanezSChapmanHCGutierrezJR 2009 Host inhibition of a bacterial virulence effector triggers immunity to infection. Science 324 784 787
74. BollerT 2008 Stabbing in the BAK–an original target for avirulence genes of plant pathogens. Cell Host Microbe 4 5 7
75. SchellenbergBRamelCDudlerR 2010 Pseudomonas syringae virulence factor syringolin A counteracts stomatal immunity by proteasome inhibition. Mol Plant Microbe Interact 23 1287 1293
76. WeingartHUllrichHGeiderKVölkschB 2001 The Role of Ethylene Production in Virulence of Pseudomonas syringae pvs. glycinea and phaseolicola. Phytopathology 91 511 518
77. LinN-CMartinGB 2007 Pto - and Prf-mediated recognition of AvrPto and AvrPtoB restricts the ability of diverse Pseudomonas syringae pathovars to infect tomato. Mol Plant Microbe Interact 20 806 815
78. KuoCHOchmanH 2010 The extinction dynamics of bacterial pseudogenes. PLoS Genet 6 e1001050
79. HajriABrinCHunaultGLardeuxFLemaireC 2009 A “repertoire for repertoire” hypothesis: repertoires of type three effectors are candidate determinants of host specificity in Xanthomonas. PLoS ONE 4 e6632
80. StevensCBennettMAAthanassopoulosETsiamisGTaylorJD 1998 Sequence variations in alleles of the avirulence gene avrPphE.R2 from Pseudomonas syringae pv. phaseolicola lead to loss of recognition of the AvrPphE protein within bean cells and a gain in cultivar-specific virulence. Mol Microbiol 29 165 177
81. BullCTManceauCLydonJKongHVinatzerBA 2010 Pseudomonas cannabina pv. cannabina pv. nov., and Pseudomonas cannabina pv. alisalensis (Cintas Koike and Bull, 2000) comb. nov., are members of the emended species Pseudomonas cannabina (ex Sutic & Dowson 1959) Gardan, Shafik, Belouin, Brosch, Grimont & Grimont 1999. Syst Appl Microbiol 33 105 115
82. StavrinidesJGuttmanDS 2004 Nucleotide sequence and evolution of the five-plasmid complement of the phytopathogen Pseudomonas syringae pv. maculicola ES4326. J Bacteriol 186 5101 5115
83. ChainPSGGrafhamDVFultonRSFitzgeraldMGHostetlerJ 2009 Genomics. Genome project standards in a new era of sequencing. Science 326 236 237
84. HernandezDFrançoisPFarinelliLOsteråsMSchrenzelJ 2008 De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res 18 802 809
85. DoCB 2005 ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res 15 330 340
86. StamatakisA 2004 RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21 456 463
87. StamatakisA 2006 RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22 2688 2690
88. KanehisaMGotoS 2000 KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28 27 30
89. Marchler-BauerA 2003 CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res 31 383 387
90. SalzbergSLSommerDDPuiuDLeeVT 2008 Gene-boosted assembly of a novel bacterial genome from very short reads. PLoS Comput Biol 4 e1000186
91. VinatzerBAJelenskaJGreenbergJT 2005 Bioinformatics correctly identifies many type III secretion substrates in the plant pathogen Pseudomonas syringae and the biocontrol isolate P. fluorescens SBW25. Mol Plant Microbe Interact 18 877 888
92. WernerssonRPedersenAG 2003 RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res 31 3537 3539
93. VoinnetORivasSMestrePBaulcombeD 2003 An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. Plant J 33 949 956
94. KovachMEPhillipsRWElzerPHRoopRM2ndPetersonKM 1994 pBBR1MCS: a broad-host-range cloning vector. Biotechniques 16 800 802
95. LindowSE 1985 Ecology of Pseudomonas syringae relevant to the field use of Ice - deletion mutants constructed in vitro for plant frost control. HalvorsonHOPramerDRogulM Engineered organisms in the environment: scientific issues 23 35 Washington, D.C.
96. ZumaqueroAMachoAPRufiánJSBeuzónCR 2010 Analysis of the role of the type III effector inventory of Pseudomonas syringae pv. phaseolicola 1448a in the interaction with the plant. J Bacteriol 192 4474 4488
97. HuangDWShermanBTLempickiRA 2009 Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 44 57
98. DelanoWL 2008 The PyMOL Molecular Graphics System DeLano Scientific LLC, Palo Alto, CA USA
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Requires Glycerol for Maximum Fitness During The Tick Phase of the Enzootic Cycle
- Comparative Genomics Yields Insights into Niche Adaptation of Plant Vascular Wilt Pathogens
- The Role of IL-15 Deficiency in the Pathogenesis of Virus-Induced Asthma Exacerbations
- “Persisters”: Survival at the Cellular Level
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy