
Tissue-Specific Effects of Reduced β-catenin Expression on Mutation-Instigated Tumorigenesis in Mouse Colon and Ovarian Epithelium
Enhanced Wnt signaling contributes to colorectal and other cancers. β-catenin functions in Wnt signaling as a T cell factor (TCF) transcriptional co-activator. Previous studies showed specific β-catenin dosage favors Wnt signaling-dependent tumorigenesis for some tumor types. However, earlier studies emphasized the role of constitutional Ctnnb1 and Apc gene variations, rather than somatic gene targeting, and the work focused on small intestine tumors and no effects on colon tumor phenotypes were described. Furthermore, definitive insights were lacking into how reduced Ctnnb1 gene dosage affected Apc mutation-dependent tumorigenesis. Here, we show somatic inactivation of one Ctnnb1 allele dramatically inhibits mouse colon adenomatous polyposis induced by somatic bi-allelic Apc inactivation. In contrast, Ctnnb1 hemizygous inactivation does not affect mouse ovarian endometrioid adenocarcinoma development arising from Apc - and Pten-inactivation. Ctnnb1 hemizygous gene dose dramatically reduces the active pool of β-catenin, leading to the significant inhibition of β-catenin/TCF-regulated target gene expression, including those encoding key stem cell regulatory and crypt compartmentalization factors in colon epithelium. Tissue-specific differences for expression of selected β-catenin/TCF-regulated genes, such as Myc, may contribute to the context-dependent effects of Ctnnb1 gene dosage in Apc mutation-driven colon and ovarian tumors.
Published in the journal:
. PLoS Genet 11(11): e32767. doi:10.1371/journal.pgen.1005638
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005638
Summary
Enhanced Wnt signaling contributes to colorectal and other cancers. β-catenin functions in Wnt signaling as a T cell factor (TCF) transcriptional co-activator. Previous studies showed specific β-catenin dosage favors Wnt signaling-dependent tumorigenesis for some tumor types. However, earlier studies emphasized the role of constitutional Ctnnb1 and Apc gene variations, rather than somatic gene targeting, and the work focused on small intestine tumors and no effects on colon tumor phenotypes were described. Furthermore, definitive insights were lacking into how reduced Ctnnb1 gene dosage affected Apc mutation-dependent tumorigenesis. Here, we show somatic inactivation of one Ctnnb1 allele dramatically inhibits mouse colon adenomatous polyposis induced by somatic bi-allelic Apc inactivation. In contrast, Ctnnb1 hemizygous inactivation does not affect mouse ovarian endometrioid adenocarcinoma development arising from Apc - and Pten-inactivation. Ctnnb1 hemizygous gene dose dramatically reduces the active pool of β-catenin, leading to the significant inhibition of β-catenin/TCF-regulated target gene expression, including those encoding key stem cell regulatory and crypt compartmentalization factors in colon epithelium. Tissue-specific differences for expression of selected β-catenin/TCF-regulated genes, such as Myc, may contribute to the context-dependent effects of Ctnnb1 gene dosage in Apc mutation-driven colon and ovarian tumors.
Introduction
Colorectal cancers (CRCs) harbor accumulated mutations in tumor suppressor genes and oncogenes along with epigenetic alterations. Many CRCs arise from precursor lesions, such as adenomatous polyps or serrated epithelial lesions with dysplasia. Inactivating mutations in the APC (adenomatous polyposis coli) and TP53 tumor suppressor genes are found in roughly 80% and 60% of CRCs, respectively [1]. Oncogenic mutations activating the functions of the KRAS and PI3KCA (phosphoinositide-3-kinase, catalytic, alpha polypeptide) proteins are found in about 40% and 20% of CRCs, respectively [1]. Constitutional mutations inactivating one APC allele underlie the familial adenomatous polyposis (FAP) syndrome, where affected individuals often develop hundreds to thousands of colon adenomas during their second or third decades of life. The wild type APC allele is somatically inactivated in adenomas arising in those with FAP [1, 2]. Mice carrying certain heterozygous, constitutional mutations inactivating Apc, such as the ApcMin mutation, may develop 50–100 small intestinal tumors and occasional colon tumors by 140 days of age and nearly all of the tumors are adenomas. Similar to the situation in FAP tumors, intestinal tumors in ApcMin mice show somatic inactivation of the wild type Apc allele [3].
The best understood function of the roughly 300 kD APC protein is regulation of the pool of β-catenin protein that functions in the canonical (β-catenin-dependent) Wnt signaling pathway [4–6]. In the absence of an activating Wnt ligand signal, the β-catenin destruction complex—comprised by the APC, AXIN, casein kinase I, and glycogen synthase kinase-3β factors and other proteins—promotes phosphorylation of conserved serine/threonine residues in the β-catenin amino (N)-terminal region. The N-terminally phosphorylated β-catenin can then be β ubiquitinated and degraded by the proteasome. Activating Wnt ligands inhibit degradation of the “free” or Wnt signaling pool of β-catenin via binding at the cell surface to the frizzled and LRP5/6 (low density lipoprotein-related proteins 5 and 6) cognate receptor complex, resulting in inhibition of β-catenin phosphorylation and/or ubiquitination by the destruction complex [4, 6]. In colon adenomas and CRCs where both APC alíeles are defective, destruction of the free pool of β-catenin is impaired and active β-catenin accumulates in the cytoplasm and nucleus, where it can complex with DNA binding proteins of the TCF (T-cell factor family)/Lef (lymphoid enhancer family) family. β-catenin functions as a transcriptional co-activator for TCFs [7]. Normally, β-catenin/TCF transcriptional activation is restricted to the crypt base, especially in the so-called crypt base columnar stem cells characterized by expression of the Wnt-regulated Lgr5 presumptive stem cell marker protein [8]. Constitutive activation of β-catenin/TCF transcription in Wnt pathway-defective adenomas and CRCs may promote a stem or progenitor cell phenotype in epithelial cells independent of cell position in the crypt [9, 10]. Activation of β-catenin/TCF-dependent transcription also alters crypt compartmentalization and coordinated migration of cells, apparently through increased expression of the EphB2 and EphB3 receptors and via inhibition of the expression of their ligands ephrin B1 and B2 [11, 12]. The MYC gene has been highlighted as a potentially key target gene regulated by β-catenin/TCF in CRCs. Genes encoding negative-feedback inhibitor proteins functioning in the Wnt/β-catenin/TCF pathway, such as AXIN2, DKK1, and NKD1, are also activated by β-catenin/TCF (see http://www.stanford.edu/~rnusse/pathways/targets.html for a list of candidates). In APC-mutant neoplastic cells, the ability of these induced regulator proteins to inhibit the Wnt signaling pathway is abrogated because the factors function upstream of or at the level of the APC protein in the pathway [13].
Besides these findings, other evidence indicates that APC inactivation may promote cancer development through β-catenin dysregulation. For instance, while most CRCs harbor APC mutations, a subset of CRCs and other cancers lacking APC mutations have CTNNB1 gene mutations resulting in production of oncogenic β-catenin proteins that are resistant to regulation by the destruction complex and that activate β-catenin/TCF transcription [6, 13]. Also, some prior studies have used genetic approaches to study effects of Ctnnb1 gene dosage on liver, small intestine, and mammary gland tumor phenotypes in mouse models as well as effects of Ctnnb1 hemizygous inactivation state (Ctnnb1+/-) in Apc-mutation induced mouse embryonic development phenotypes [14, 15]. The prior studies indicated the Ctnnb1+/- constitutional state can inhibit intestinal and liver tumorigenesis in mice carrying mutations in the Apc gene (Apc1638N, ApcMin, or Apcfl) [14, 15]. In contrast, mammary gland tumorigenesis was enhanced in Apc1638N Ctnnb1+/- mice, perhaps because Ctnnb1 functions as a tumor suppressor gene in the mammary gland tumors via β-catenin’s role in E-cadherin-dependent tumor suppression [14]. Nonetheless, while the prior studies yielded evidence that β-catenin signaling dosage impacts Apc mutation-induced tumorigenesis in some tissues, the prior work did not assess the role of Ctnnb1 dosage in Apc mutation-induced colon tumorigenesis, the chief site of APC mutation-dependent tumorigenesis in humans. Moreover, the work used mice constitutionally deficient in β-catenin, not just in Apc-mutant epithelial cells, and the findings did not highlight specific factors and mechanisms that might account for effects of Ctnnb1 dosage in Apc mutation-instigated tumorigenesis in different contexts. We report here on studies of the effects of Ctnnb1 gene dosage on β-catenin protein expression and β-catenin/TCF transcription in Apc mutation-induced colon and ovarian mouse tumors and cell culture models. We provide evidence that Apc mutation-induced tumorigenesis in the colon is inhibited by Ctnnb1 hemizygous gene status through marked effects on the free pool of β-catenin in the cytoplasm and nucleus and its ability to activate key β-catenin/TCF-regulated target genes, including those encoding key stem factors, such as Lgr5, and regulators of crypt compartmentalization, such as the EphB2/B3 receptors. We also uncovered a novel feed-forward mechanism where β-catenin protein stabilization and β-catenin/TCF transcription appear critical in regulating Ctnnb1/CTNNB1 transcription in the setting of Apc inactivation in mouse colon and human colon cancer cells. Moreover, we found that differences in the ability to activate Myc expression may underlie colon versus ovary tissue-specific differences in Apc mutation-instigated tumorigenesis in the setting of Ctnnb1 hemizygous gene dosage.
Results
Inactivation of a Ctnnb1 allele extends survival and inhibits adenomatous polyposis and epithelial abnormalities induced by somatic bi-allelic Apc inactivation in mouse colon
We previously described CDX2P-G22Cre transgenic mice, in which human CDX2 regulatory sequences and an out-of-frame Cre transgene allele, carrying a 22-basepair guanine nucleotide repeat tract affecting the Cre open reading frame, manifest mosaic Cre recombinase expression in caudal embryonic tissues and in epithelium of the distal ileum, cecum, colon, and rectum during adult life [16]. We also previously described CDX2P-CreERT2 transgenic mice that express a tamoxifen (TAM)-regulated Cre protein (CreERT2) under control of human CDX2 regulatory sequences, allowing for TAM-inducible targeting of loxP-containing alleles in adult terminal ileum, cecum, colon, and rectal epithelium [17]. Using the CDX2P-G22Cre or CDX2P-CreERT2 transgenic mice, we have described the phenotypic consequences in colon epithelium of somatic, bi-allelic, inactivating mutations in Apc [16, 17]. Consistent with our prior studies, we found CDX2P-G22Cre Apcfl/fl mice lived only for 8–20 days after birth (median survival = 13 d; Fig 1A). After three daily doses of TAM to inactivate both Apc alleles in distal intestinal epithelial tissues, CDX2P-CreERT2 Apcfl/fl adult mice lived on average for 22 days (Fig 1B). In marked contrast, concurrent somatic inactivation of one Ctnnb1 allele along with both Apc alleles, using either the CDX2P-G22Cre or CDX2P-CreERT2 transgene for somatic gene targeting, led to a dramatically increased life span relative to that seen in mice with Apc bi-allelic targeting, with median survival of 168 d of age in CDX2P-G22Cre Apcfl/fl Ctnnb1fl/+ mice and for 134 d after TAM treatment in the CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice (Fig 1A and 1B).

Consistent with our prior reports [16, 17], the proximal colon and cecum of both CDX2P-G22Cre Apcfl/fl mice (when moribund at 8–20 d of age) and CDX2P-CreERT2 Apcfl/fl mice (only 20 days after TAM induction) were dramatically thickened and many polypoid lesions were seen (S1 Fig). Histological analysis of proximal colon epithelial tissues from these mice showed significant hyperplastic and dysplastic (adenomatous) changes along with frequent crypt fission and branching (Fig 1C). The dramatic polyposis in cecum and colon seen following bi-allelic Apc inactivation was significantly inhibited at both early and later time points by concurrent inactivation of one Ctnnb1 allele, with no grossly discernable epithelial phenotype seen in the proximal colon and only two to four polyps in the cecum per mouse as the CDX2P-G22Cre Apcfl/fl Ctnnb1fl/+ and CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice were aged (S1 Fig). The cecal polyps arising in CDX2P-G22Cre Apcfl/fl Ctnnb1fl/+ and CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice may contribute to their premature mortality relative to control mice, as no other grossly detectable intestinal lesions or pathology were noted in the mice. The Cre-mediated somatic inactivation of both Apc alleles and one Ctnnb1 allele in proximal colon epithelium was confirmed by genotyping. The rare cecal adenomas arising in CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice were found to have significant fractions of cells that escaped Cre-mediated Ctnnb1 targeting, even though Cre-mediated somatic inactivation of both Apc alleles occurred to the same extent in the rare adenomas and proximal colon mucosa (S1 Fig). Compared to the situation in CDX2P-CreERT2 Apcfl/fl mice, microscopic examination of proximal colon tissues of CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice revealed modest hyperplastic changes and minimal crypt branching (Fig 1C). Our efforts to inactivate both Ctnnb1 alleles in colon epithelium via either CDX2P-G22Cre - or CDXP-CreERT2-mediated targeting with or without Apc inactivation indicated that colon epithelial cells completely lacking β-catenin expression and function could not be generated. This likely reflects a required role for β-catenin function in colon epithelium, perhaps not limited to Wnt signaling, but also in cadherin-mediated adhesion, centrosome assembly or other functions.
Immunohistological analysis of colon sections from CDX2P-CreERT2 Apcfl/fl mice showed strong cytoplasmic and nuclear β-catenin expression in many epithelial cells, compared to the nearly uniform membrane β-catenin staining in colon epithelium of wild-type mice (Fig 2A). In CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice, we observed infrequent cells with elevated cytoplasmic and/or nuclear β-catenin expression (Fig 2A). Paneth cells, a specialized secretory cell linage that expresses lysozyme and other markers, are found at the crypt base in normal mouse small intestinal epithelium, but are absent in normal mouse colon epithelium. Paneth cells have been proposed to have a key role in generation and/or maintenance of the intestinal crypt stem cell niche [18]. Bi-allelic Apc inactivation has been associated with the generation of many ectopic lysozyme-expressing Paneth-like cells throughout the crypts of small intestine and colon [12, 17, 19, 20]. We confirmed this finding in CDX2P-CreERT2 Apcfl/fl mice (Fig 2A). Whereas no Paneth-like cells were seen in normal mouse colon, modest numbers of lysozyme-expressing cells were seen in the colons of CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice (Fig 2A). We also used a transgenic mouse line carrying a Cre-activated enhanced yellow fluorescence protein (EYFP) reporter gene at the ubiquitously expressed Rosa26 locus to monitor colon epithelial cells and glands where Cre-mediated targeting had occurred. Ectopic lysozyme-expressing cells were found in nearly all of the EYFP-positive crypts in CDX2P-G22Cre Apcfl/fl Ctnnb1fl/+ and CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice (Fig 2B and 2C). The rare occurrence of Paneth-like cells in crypts without EYFP expression likely reflects the possibility that Cre may more efficiently target the loxP sites at the Apc locus than at the Rosa26 locus.

Prior studies from other groups and ours have shown that Apc bi-allelic inactivation increases both cell proliferation and apoptosis in intestine and colon epithelium [12, 17, 19, 21]. Following TAM-induced Apc bi-allelic inactivation in proximal colon epithelium, we confirmed significantly elongated crypts and increased cell proliferation and apoptosis relative to control epithelial tissues (Fig 2D and 2E). In contrast, only modestly increased crypt height, cell proliferation and apoptosis relative to control epithelium were seen in epithelium of CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice following combined Apc and Ctnnb1 gene inactivation (Fig 2D and 2E). Furthermore, although bi-allelic Apc inactivation induced extensive cell proliferation in the upper half of targeted colon crypts, cell proliferation following gene targeting in CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice was largely restricted to the bottom half of each crypt, with only a slight increase in cell proliferation compared to control colon epithelium (Fig 2D and 2E). The cell proliferation and apoptosis results were well correlated with the immunohistochemical studies of β-catenin levels and localization (Fig 2A), suggesting differences in the strength of β-catenin-dependent Wnt signaling in cells with bi-allelic Apc defects underlie the observed effects on colon epithelial morphology, cell fate and differentiation, and cell proliferation and apoptosis.
The orientation of the mitotic spindle axis may impact on cell fate decisions in intestinal epithelium. At cytokinesis, the orientation of the spindle axis in a planar fashion (i.e., parallel to the crypt axis) is thought to generate two daughter cells with equivalent luminal (apical) and basement (extracellular matrix) surfaces. If the spindle axis is not oriented parallel to the crypt axis, cytokinesis generates daughter cells with differences in luminal and basement membrane surfaces and the potential for resultant differences in the fates adopted by the two daughter cells. We previously reported significant increases in the percentage of epithelial cells where the mitotic spindle axis was oriented orthogonal to the planar axis in Apc-mutant mouse colon crypts relative to wild type crypts [17]. Consistent with our prior results, in colon epithelium of CDX2P-CreERT2 Apcfl/fl mice treated with TAM to inactivate both Apc alleles, roughly 50% of the cells in mitosis had mitotic spindle axes ≥30° degrees out of the planar axis, with nearly 20% showing spindle axes between 60° and 90° out of planar alignment. In contrast, in epithelium of CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice and control mice, >75% of mitotic colon epithelial cells had their mitotic spindles aligned within 30° of the planar (crypt) axis (S2 Fig). The findings indicate β-catenin levels have a key role in the altered mitotic spindle axis phenotype of Apc-mutant colon epithelium.
Ctnnb1 inactivation inhibits Apc mutation-induced colon tumorigenesis via maintenance of EphB/ephrinB signaling and restriction of presumptive stem cells to the crypt base
As described above, bi-allelic Apc inactivation acutely induces hyperproliferation and dysplastic alterations in mouse proximal colon epithelium, with the altered epithelium arising in part from expansion of the crypt progenitor compartment at the expense of the differentiated compartment, along with frequent crypt fission/branching [12, 17, 21]. The EphB/ephrinB signaling axis has been implicated in control of intestinal epithelial cell compartmentalization along the crypt axis and in cell migration [11, 22]. The EphB2 and EphB3 receptors are two key effectors of compartmentalization and cell migration in the crypt, and EphB2 and EphB3 are each encoded by a gene activated in intestinal tissues by β-catenin/Tcf transcription. The EphB2/B3 receptor ligands, ephrinB1 and ephrinB2, show highest expression levels in differentiated cells at the crypt surface, and expression of ephrins B1 and B2 is negatively regulated by β-catenin/Tcf activity [11, 23]. Of note, EphB-ephrinB interactions generate repulsive forces that separate and compartmentalize the EphB - and ephrinB-expressing cells to maintain crypt architecture [11, 23]. In normal mouse colon epithelium, the EphB2 and EphB3 receptors were expressed only in progenitor cells at the crypt base (Fig 3A and 3B). We found bi-allelic Apc inactivation in colon epithelium not only increased EphB2 and EphB3 expression, but also perturbed the gradient of EphB2 and B3 receptor expression along the crypt axis, with EphB2/B3 expression seen even at the crypt surface in Apc-mutant crypts (Fig 3A and 3B). In the case of ephrin ligand expression, our studies demonstrated strong expression of ephrinB1 and B2 in normal colon surface epithelial cells and normal colon crypt cells other than the crypt base. The normal pattern of ephrinB1/B2 expression remained largely unaffected in Apc-mutant crypts with one Ctnnb1 allele inactivated (Fig 3C). In contrast, in Apc-mutant crypts where Ctnnb1 dosage was intact, ephrinB1/B2 expression was markedly down-regulated in colon surface epithelial cells and throughout the crypt (Fig 3C). Our findings are consistent with those in a prior study that showed increased expression of EphB2/B3 and loss of ephrinB1/B2 expression in colon adenomas of Apcmin/+ mice [23]. Although expression of EphB2/B3 was moderately elevated in some colon epithelial cells of CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice compared to control mice, elevated EphB2/B3 expression remained restricted to the crypt base region, rather than spreading throughout the crypt as was seen in Apc-mutant crypts with intact Ctnnb1 gene dosage (Fig 3A and 3B). This observation suggests the reduced β-catenin levels in CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice leads to a failure to induce enough β-catenin/TCF-regulated EphB2/B3 expression to overcome the repulsive effects of the retained expression of ephrinB1/B2 ligands in Apc-mutant crypts with reduced Ctnnb1 dosage.

A similar expression pattern to that seen for EphB2 and EphB3 was also found for Sox9, a transcription factor encoded by a β-catenin/Tcf target gene. Sox9 expression is restricted to stem/progenitor cells at the normal colon crypt base (S3 Fig). Sox9 expression was only modestly increased and expanded following gene targeting in crypts of CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice relative to the marked changes in Sox9 levels and the number of Sox9-expressing cells in crypts from CDX2P-CreERT2 Apcfl/fl mice (S3 Fig). In spite of the reduced increase in β-catenin levels in colon crypts of CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice relative to CDX2P-CreERT2 Apcfl/fl mice, the resultant signaling was still sufficient to generate some ectopic Paneth-like cells (Fig 3A and 3B). In addition, the modest increase in β-catenin levels in targeted crypts of CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice was sufficient to induce expression in targeted crypts of a β-galactosidase reporter gene integrated into the β-catenin/TCF-regulated Axin2 locus. However, β-galactosidase expression was reduced in crypts and few if any colon surface epithelial cells expressed β-galactosidase in Apc-mutant epithelium with hemizgyous Ctnnb1 dosage (S3 Fig). In contrast, uniformly strong β-galactosidase expression was seen throughout Apc-mutant colon crypts and surface epithelial cells with intact Ctnnb1 dosage (S3 Fig). Taken together, the findings indicate distinct β-catenin/Tcf target genes in colon epithelium display differing transcriptional responses to β-catenin levels, with Axin2 perhaps representing a target gene capable of being activated by modest to moderate levels of β-catenin in colon epithelium. The Sox9, EphB2, and EphB3 genes appear dependent on higher levels of β-catenin for transcriptional activation in colon epithelium.
To address mechanisms underlying suppression of crypt fission and branching in Apc-deficient colon epithelium when one Ctnnb1 allele was inactivated, we compared expression of presumptive stem cell markers in Apc-deficient colon crypts where both Ctnnb1 alleles were intact or where only one Ctnnb1 allele was active. Consistent with our prior work [17], 20 days after TAM-induced bi-allelic Apc inactivation, we detected strong induction of enhanced green fluorescent protein (EGFP) expressed from the Lgr5 locus (Lgr5-EGFP) (Fig 4A) in Apc-deficient colon epithelium generated by CDX2P-CreERT2 targeting. Lgr5 is a β-catenin/TCF-regulated gene and a marker of presumptive crypt base columnar stem cells in normal colon, and the Lgr5 allele that we used has a EGFP open reading frame integrated in the locus to allow for monitoring of endogenous Lgr5 expression [8]. In Apc-mutant epithelium, we also confirmed strong induction of the Msi1 RNA-binding protein (Fig 4B), another presumptive intestinal stem cell marker [24, 25]. In contrast to a prior study where it was reported that Lgr5-expressing cells were only expanded at the lower part of the crypts in colon epithelium following mutant β-catenin induction [21], we detected EYFP-positive and Msi-positive cells essentially throughout the Apc-mutant dysplastic colon crypts when both Ctnnb1 alleles were active, though expression of EYFP was more prominent near the crypt base region, including in the de novo crypts. While the net number of EYFP - and Msi1-expressing cells per crypt were slightly increased (e.g. from 3–4 to 5–8 Lgr5-positive cells per crypt) in colon epithelium of TAM-treated CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice compared to the control mice, paralleling the subtle increase in crypt fission/budding seen, the expanded population of EYFP-positive cells remained restricted to the crypt base region (Fig 4A), consistent with the EphB2 and EphB3 data described above. EYFP expression patterns in colon similar to those seen in TAM-treated CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice were also obtained when we used TAM-treatment to activate the Lgr5-driven CreERT2 transgene to target Apc and Ctnnb1 alleles and EYFP expression was used to mark Lgr5-expressing cells (Fig 4A).

Consistent with our studies of Lgr5 and Msi expression patterns in colon epithelium, the levels of transcripts encoding presumptive stem cell markers, including Lgr5, CD44, Msi1, and Hopx, were also found to increase dramatically in the colon tissues of CDX2P-CreERT2 Apcfl/fl mice (S4 Fig). The induction of genes encoding stem cell markers and other selective β-catenin/Tcf target genes (such as Axin2, Nkd1, Ccnd1 and Irs1) observed in Apc-deficient colon epithelium was significantly suppressed in colon epithelium from CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice (S4 Fig). Taken together, our data indicate that the robust induction of many β-catenin/Tcf-regulated genes that is seen response to Apc inactivation was variably inhibited in reduced Ctnnb1 gene dosage and β-catenin protein levels in mouse colon epithelium. In the setting of inactivation of one Ctnnb1 allele, the inability of Apc inactivation to substantially activate certain key β-catenin/TCF-regulated genes with functions in colon crypt compartmentalization and cell migration (e.g., EphB2 and EphB3) or stem cell fate (e.g., Lgr5 and Msi) is likely to underlie the dramatic abrogation of adenoma formation in CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice.
Ctnnb1 gene dosage effects on Myc and Ctnnb1 gene expression in Apc-mutant mouse colon tissues and an apparent feed-forward mechanism for β-catenin/TCF in regulating Ctnnb1 transcription
Myc is a well-known β-catenin/TCF-regulated target gene [10, 26], and we found that the strong induction of Myc gene expression in mouse colon epithelium seen following Apc bi-allelic inactivation was abrogated when Apc bi-allelic inactivation occurred concurrently with somatic inactivation of one Ctnnb1 allele (Fig 5A). Another interesting observation was that Ctnnb1 transcripts were increased roughly 2-fold in proximal colon tissues following Apc bi-allelic inactivation in colon epithelium with wild type Ctnnb1 gene dosage, compared to the levels of Ctnnb1 transcript in untargeted colon tissues of Apcfl/fl mice (Fig 5B). Hemizygous Ctnnb1 gene dosage was associated with an inability of Apc bi-allelic inactivation to activate Ctnnb1 transcript levels in proximal colon tissues (Fig 5B). The effects of Apc inactivation and Ctnnb1 gene dosage in mouse colon epithelium on Myc and Ctnnb1 transcript levels did not appear to simply reflect a change in the epithelial cell numbers in Apc-mutant colon epithelium, because transcripts for the epithelial markers epithelial cell adhesion molecule (Epcam) and E-cadherin (Cdh1) were similar in the mouse colon tissues independent of genotype (Fig 5C and 5D). The findings indicate that not only does Apc inactivation lead to increased β-catenin protein levels in murine colon tissues, but Ctnnb1 transcript levels in the colon tissues are also increased by Apc inactivation, consistent with an apparent feed-forward mechanism for up-regulation of Ctnnb1 transcripts following Apc inactivation. Because the Apc mutation-dependent induction of Ctnnb1 transcripts in mouse colon epithelium was not seen in the setting of reduced Ctnnb1 gene dosage, the findings imply that the feed-forward mechanism for Ctnnb1 induction may require sufficient levels of β-catenin and β-catenin/TCF-dependent transcription.

Of interest with regard to a role for β-catenin and β-catenin/TCF transcription in regulating Ctnnb1 transcription is that chromatin immunoprecipitation (ChIP) studies from the ENCODE project indicate that the TCF4 protein, encoded by the TCF7L2 gene, is bound in the proximal promoter and exon 1 region of the CTNNB1 gene in selected cell lines. The mouse Ctnnb1 and human CTNNB1 promoter regions lack known TCF family protein consensus binding elements. Nonetheless, to further explore the role of CTNNB1 transcript and β-catenin levels in regulating CTNNB1 transcription, we generated a reporter gene construct in which a 555 bp fragment of human CTNNB1 upstream and exon 1 sequences (-336 to +219 relative to the transcription start site) were cloned upstream of a firefly luciferase sequence (Fig 6A). This region of the CTNNB1 gene corresponds to the region that the ENCODE ChIP data indicates is occupied by the TCF family member TCF4 in some cell lines. We used two different doxycycline-induced shRNAs against CTNNB1 to study effects of antagonizing CTNNB1 endogenous transcript levels in the DLD1 human colon cancer cell line, which harbor APC defects leading to constitutive activation of β-catenin/TCF signaling (Fig 6B). The doxycycline-mediated shRNA-mediated inhibition of CTNNB1 transcript levels in DLD1 cells also led to marked inhibition of MYC expression (Fig 6B). In addition, we found CTNNB1 reporter luciferase activity was inhibited by about 30–40% by the reduction in CTNNB1 transcripts in the cells, whereas, the prototypical Wnt/β-catenin/TCF reporter gene construct TOPflash was more robustly inhibited by the reduction in CTNNB1 levels (Fig 6C). These studies and data complement the primary mouse colon tissue findings presented above by showing that reduction of CTNNB1 levels in colon cancer cells has a demonstrable effect on CTNNB1 transcriptional activity.
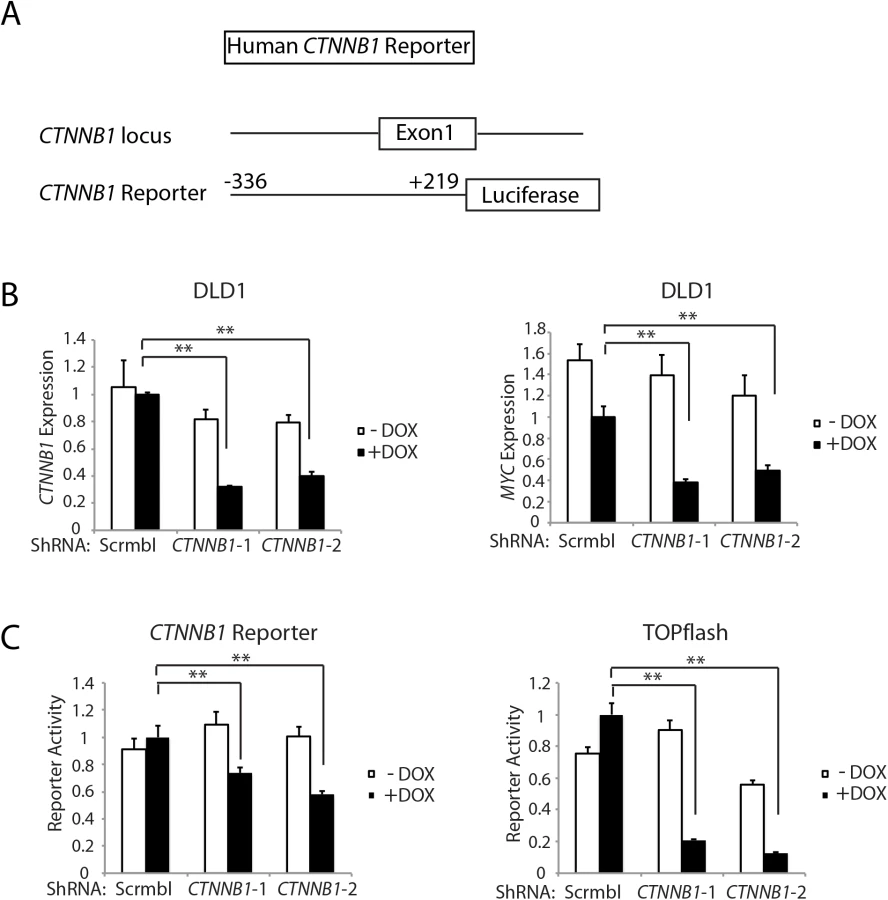
The active pool of β-catenin protein and β-catenin-regulated target gene transcription are highly sensitive to changes in β-catenin transcript levels in colon epithelial cells
Canonical (β-catenin-dependent) Wnt signaling is dependent on increases in the levels and localization of a hypo - or un-phosphorylated pool of β-catenin, which is often termed the “free” or “active” pool of β-catenin in the cytoplasm and nucleus of cells with Wnt pathway activation [27, 28]. Active β-catenin can function as a co-activator for TCF-dependent transcription of endogenous Wnt/β-catenin/TCF target genes [27]. To further address how reduced CTNNB1 gene expression affects β-catenin protein levels and β-catenin/TCF-regulated target gene induction in colon epithelial cells, we used an shRNA approach to antagonize CTNNB1 transcript and β-catenin protein levels in colon cell lines. In the immortalized, non-neoplastic human colon epithelial cell (HCEC) line, through use of a doxycycline (DOX)-regulated shRNA against APC, we reduced endogenous APC gene and protein expression in the cells to less than 10% of control levels (S5 Fig). Following APC shRNA induction, the levels of active β-catenin, as detected with a previously described antibody against the hypo-phosphorylated or active form of β-catenin, were significantly increased, whereas only a minor increase in total β-catenin levels was seen (Fig 7A and S6 Fig). Concurrent DOX-mediated induction of the APC shRNA and either of the two independent CTNNB1 shRNAs, which reduced CTNNB1 transcript levels to about 20–30% of control levels in HCECs, led to dramatic inhibition of the APC inactivation-stimulated effects on active β-catenin protein levels, but only modest to moderate reduction in the levels of total β-catenin protein (Fig 7A and S6 Fig). The marked effects of the APC and CTNNB1 shRNA approaches on the active β-catenin pool, with only more modest to moderate effects on total β-catenin levels in HCEC cells were reproducible (S6 Fig). Following APC shRNA induction by DOX treatment, expression of multiple β-catenin/TCF-regulated target genes, such as AXIN2, BMP4, NKD1 and IRS1, was significantly induced in HCECs (Fig 7B–7E). These APC shRNA-mediated increases in β-catenin/TCF-regulated target gene expression were almost completely abolished by shRNA-mediated inhibition of β-catenin (Fig 7B–7E). We also studied sub-cellular localization of β-catenin in the HCEC cells following APC shRNA induction and combined APC and CTNNB1 shRNA induction by DOX. Consistent with the marked increase in active β-catenin levels following APC shRNA induction, we found β-catenin protein mainly accumulated in the cytosol and nucleus of HCECs (S7 Fig). Concurrent induction of both the APC and CTNNB1 shRNAs in HCECs dramatically reduced the levels of β-catenin protein in the nucleus and cytoplasm (S7 Fig), consistent with the notion that the active, signaling pool of β-catenin in the cytoplasm and nucleus is highly sensitive to changes in Ctnnb1 transcript levels.

The strong inhibitory effect on the active pool of β-catenin compared to that for total β-catenin when CTNNB1 transcript levels were reduced in HCECs was further studied in three human colon cancer cell lines stably transduced with the two DOX-inducible CTNNB1 shRNAs. These included a colon cancer cell line with a gain-of-function mutation in CTNNB1 (HCT116) and two colon cancer cell lines with APC loss-of-function mutations (DLD1 and SW480). At 7 days after DOX-induction of the CTNNB1 shRNAs, in the three colon cancer cell lines, we found moderate (HCT116) to dramatic (DLD1 and SW480) decreases in the active pool of β-catenin protein with only modest changes in total β-catenin protein levels (S8 Fig). Expression of the CTNNB1 shRNAs led to potent inhibition of the expression of Wnt/β-catenin/TCF-regulated target genes in the DLD1 and HCT116 cells, including AXIN2, BMP4, NKD1, LGR5, and CD44 (S9 Fig).
Ctnnb1 hemizygous inactivation does not affect Apc - and Pten-mutation-dependent mouse ovarian endometrioid adenocarcinoma (OEA) development
To assess the role of β-catenin function in another Apc mutation-dependent tumor model, we explored the role of Ctnnb1 gene dosage in a mouse model of ovarian endometrial adenocarcinoma (OEA) arising from bi-allelic inactivation of both the Apc and Pten genes [29]. Prior studies have shown that the Wnt/β-catenin/Tcf signaling pathway is deregulated by mutations in 16%–38% of human OEAs, and PTEN mutations are often seen in the OEAs with Wnt pathway mutations [29–32]. In the mouse OEA model, tumors are initiated by conditional inactivation of the Apc and Pten genes following injection of AdCre into the right ovarian bursa of Apcfl/fl Ptenfl/fl mice [29]. Interestingly, in both Apcfl/fl Ptenfl/fl mice and Apcfl/fl Ptenfl/fl Ctnnb1fl/+ mice, adenocarcinomas morphologically similar to human OEAs formed following AdCre injection, with 100% penetrance and no difference in tumor latency between mice with two wild type Ctnnb1 alleles or one wild type and one floxed Ctnnb1 allele (Table 1). In addition, no significant differences in survival rates, tumor volumes, and rates of liver metastasis were found between AdCre-injected Apcfl/fl Ptenfl/fl mice and Apcfl/fl Ptenfl/fl Ctnnb1fl/+ littermates (Table 1), and OEAs arising in both lines of mice shared similar histological features and immunohistochemical staining patterns for cytokeratin-8 (CK8), E-cadherin and α-inhibin (Fig 8A and Table 1). Efficient Cre-mediated deletion of Ctnnb1 and Apc was confirmed in tumors from these mice, and no OEAs arose in the AdCre-injected right ovaries in Apcfl/fl Ptenfl/fl Ctnnb1fl/fl mice, indicating OEAs could not arise from cells completely lacking β-catenin.


The findings on the lack of a demonstrable effect of Ctnnb1 hemizygous gene dosage in the mouse OEA model contrast with the findings above, where Apc-mutation-dependent polyposis in colon epithelium was dramatically suppressed by Ctnnb1 hemizygous inactivation. Nonetheless, similar to the situation in mouse colon, based on immunohistochemical staining, the presumptive Wnt pathway signaling-competent pool of β-catenin in the nucleus and cytoplasm was significantly reduced in the OEAs in Apcfl/fl Ptenfl/fl Ctnnb1fl/+ mice compared to OEAs in the Apcfl/fl Ptenfl/fl mice (Fig 8B). We also examined the β-catenin/TCF-mediated gene transcription in the OEAs arising in the Apcfl/fl Ptenfl/fl mice and Apcfl/fl Ptenfl/fl Ctnnb1fl/+ mice. Consistent with the β-catenin dosage-dependent effects of Ctnnb1 transcripts seen in mouse Apc-mutant colon tissues described above, Ctnnb1 transcripts were significantly reduced in the OEAs arising in Apcfl/fl Ptenfl/fl Ctnnb1fl/+ mice compared to the OEAs in Apcfl/fl Ptenfl/fl mice (Fig 8C). Interestingly, although the Ctnnb1 hemizygous state in the Apc - and Pten-mutant OEAs markedly suppressed the induction of some β-catenin/TCF-regulated target genes, such as Axin2 and Nkd1 (Fig 8C), hemizygous Ctnnb1 function did not abrogate induction of Myc transcripts in the OEAs (Fig 8C). Therefore, our findings showing that Ctnnb1 hemizgyous state did not prevent development of Apc - and Pten-mutant OEAs even though there was a reduction in β-catenin levels and expression of some β-catenin/TCF-regulated genes suggest that retention of Myc induction in OEAs with hemizygous Ctnnb1 function, but not in Apc-deficient colon epithelium with hemizygous Ctnnb1 function, may be a contributing factor in the observed differences in tumor development in the two tissues. We also studied the consequences of shRNA-mediated inhibition of CTNNB1 on MYC gene expression in the TOV112D human ovarian endometrioid carcinoma cell line that harbors a CTNNB1 oncogenic mutation leading to β-catenin/TCF dysregulation [33]. We found that doxycycline-mediated induction of the two CTNNB1 shRNAs in TOV112D cells reduced CTNNB1 levels to about 50% of baseline (Fig 8D), but no statistically significant effect on MYC transcript levels was seen.
Discussion
Mutations inactivating the APC tumor suppressor gene are believed to be critical initiating lesions in the majority of colon adenomas and carcinomas [6, 34, 35]. APC mutations are likely key contributing factors in the development of some other cancer types, including a subset of human OEAs. The best understood function of the APC protein is to act as a component of a phosphorylation - and ubiquitination-dependent destruction complex that regulates the free or active pool of β-catenin. This pool of β-catenin functions as a regulator of TCF transcription in the Wnt pathway signaling [1]. In the studies described above, we assessed the effects of Ctnnb1 gene dosage on Apc mutation-instigated tumorigenesis in mouse genetically engineered colon and ovarian tumor models and in cultured cells. We found the florid polyposis phenotype resulting from somatic Apc bi-allelic inactivation in mouse colon epithelium is potently inhibited by concurrent somatic inactivation of one Ctnnb1 allele. The few polyps arising in CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice were found to have escaped Ctnnb1 targeting, though Cre-mediated somatic inactivation of both Apc alleles occurred in the lesions, likely reflecting strong positive selection for maintenance of wild type Ctnnb1 gene dosage for Apc-mutant colon adenomas to arise and persist. In contrast to the situation in colon epithelium, in a mouse model of the human OEAs that harbor inactivating mutations in the APC and PTEN genes, we found that, regardless of whether the mice had two wild type Ctnnb1 alleles or one wild type and one targeted Ctnnb1 allele, adenocarcinomas morphologically similar to human OEAs formed with 100% penetrance and no differences in latency, size, morphology, or metastatic potential of the lesions arising from AdCre-mediated targeting of the Apc and Pten genes.
In-depth mutational analyses of the germline and somatic mutations in adenomas arising in patients with FAP led to the proposal there was strong biological selection for a “just-right” level of β-catenin signaling that would be optimal for tumor formation [36]. Some prior studies have used genetic approaches to study experimentally the effects of Ctnnb1 gene dosage on Apc mutation-dependent tumorigenesis in the small intestine, liver, and mammary gland in mouse models [14, 15]. The earlier work indicated that the Ctnnb1+/- constitutional hemizygous state can inhibit intestinal and liver tumorigenesis in mice carrying Apc mutations. In mammary gland tumorigenesis, tumorigenesis was enhanced in Apc1638N Ctnnb1+/- mice relative to Apc1638N Ctnnb1+/+ mice, perhaps because Ctnnb1 functions as a tumor suppressor gene in Apc1638N mammary gland tumors via β-catenin’s role in E-cadherin-dependent tumor suppression [14]. While our findings in a mouse Apc mutation-dependent colon tumorigenesis model are consistent with the prior work on Apc mutation-instigated small intestine and liver tumorigenesis, some significant differences in the studies should be noted. The prior work emphasized models where the mice carried constitutional mutations in one Apc allele and tumors arose following stochastic loss or inactivation of the remaining wild type Apc allele. In addition, mice in the prior small intestine work were constitutionally hemizygous for Ctnnb1. In our Apc mutation-dependent colon tumorigenesis model, both Apc alleles are somatically inactivated in colon epithelium by Cre-mediated targeting, and the Ctnnb1 hemizygous deficiency state was also somatically generated only in the colon epithelial cells by Cre-mediated targeting. In addition, our OEA model work contrasts with the prior published work, as it also relies on somatic targeting of Apc and Ctnnb1. The OEA results also differ from our own colon tumorigenesis results, as the findings indicate Ctnnb1 hemizygous gene dosage had no demonstrable effect on cancer latency, size, or morphology or the metastatic potential of mouse OEAs arising from combined somatic inactivation of Apc and Pten.
Besides highlighting tissue-specific differences for Ctnnb1 gene dosage in Apc mutation-instigated colon and ovarian tumorigenesis, our studies and data have provided several unique and in-depth insights into cell and tissue mechanisms by which Ctnnb1 gene dosage likely contributes to Apc mutation-dependent phenotypes in mouse colon epithelium. Inactivation of one Ctnnb1 allele markedly inhibited the increases in β-catenin cytoplasmic and nuclear levels that result from bi-allelic Apc inactivation in mouse colon epithelium. In turn, there was strikingly attenuated expression of key β-catenin/TCF-regulated target genes, including those encoding the EphB2/B3 receptors, and the stem cell markers Lgr5, Msi1, and Hopx. Of significant interest in terms of a likely key mechanisms through which Ctnnb1 gene dosage inhibits adenoma formation, the inability of the Apc-mutant colon epithelial cells to up-regulate and alter the crypt (high)-surface (low) gradient of EphB2/B3 expression appears to restrict high levels of EphB2/3 expression to the crypt base. Activated β-catenin/TCF transcription has been implicated in repression of ephrinB expression [11, 23]. We found that the robust ephrinB1/B2 expression seen in the upper two-thirds of normal colon crypts as well as in the normal colon surface epithelium was maintained in Apc-mutant crypts when one Ctnnb1 allele was inactivated. In contrast, Apc-mutant crypts with intact Ctnnb1 dosage markedly down-regulated ephrinB1/B2 expression and dramatically upregulated and expanded EphB2/B3 expression throughout the colon crypts. Because the EphB and ephrin molecules mediate critical repulsive interactions in intestinal crypts, the maintenance of the normal inverse EphB/ephrinB gradient from crypt base to cell surface in Apc-mutant crypts where one Ctnnb1 allele is inactive restricts the expansion of the Lgr5-positive crypt stem cell pool and the crypt fission/branching that would result from unrestrained crypt stem cell expansion and altered migration [11, 23]. As a result of the preservation of the inverse gradient of EphB/ephrinB expression in Apc mutant crypts with reduced Ctnnb1 dosage, stem cell expansion and the dysplastic and adenomatous changes induced by Apc inactivation in colon epithelium are potently inhibited, even though Lgr5 and some other stem cell marker genes are modestly increased in expression in the targeted crypts of CDX2P-CreERT2 Apcfl/fl Ctnnb1fl/+ mice relative to crypts in normal mice.
Our findings showing that Ctnnb1 transcripts are up-regulated in Apc-mutant mouse colon epithelium as well as in Apc-mutant mouse OEAs, together with our findings that Ctnnb1 hemizygous gene dosage inhibited Apc mutation-dependent Ctnnb1 transcript induction in colon and ovarian tumor models imply that the Ctnnb1 gene is subject to feed-forward activation by β-catenin levels and β-catenin/TCF-regulated transcription. Of interest with regard to a role for β-catenin and β-catenin/TCF transcription in regulating Ctnnb1 transcription are chromatin immunoprecipitation (ChIP) studies from the ENCODE project reporting that the TCF4 protein, encoded by the TCF7L2 gene, is bound in the promoter region of the CTNNB1 gene in selected cell lines. Based on our mouse colon tissue studies and the ENCODE project findings, we generated a CTNNB1 reporter gene construct containing 555 bp of human CTNNB1 upstream and exon 1 sequences and found that shRNA-mediated inhibition of CTNNB1 endogenous gene expression in the APC-mutant DLD1 human colon cancer cell line led to inhibition of the activity of the CTNNB1 reporter gene. These data demonstrate that CTNNB1 transcript levels affected CTNNB1 transcription in colon cancer cells. The lack of known TCF consensus element binding sites in the mouse Ctnnb1 and human CTNNB1 promoter regions currently limits support for the argument that β-catenin/TCF transcription directly regulates Ctnnb1 transcription, though further studies to address the point will need to be pursued.
In contrast to the near complete abrogation of Myc induction in Apc-mutant colon epithelium with one Ctnnb1 allele, Myc induction was retained in the Apc-mutant mouse OEAs with one functional Ctnnb1 allele. Of note, in prior studies, it has been shown that hemizyous inactivation of Myc dramatically inhibited Apc mutation-induced small intestine tumor phenotypes, but not Apc mutation-induced effects on liver cell proliferation and size [37, 38]. Hence, the findings from our work and these prior studies [37–39] highlight Myc as perhaps one of the key β-catenin/TCF-regulated genes with tissue-specific differences in its regulation by β-catenin/TCF that may account for why Ctnnb1 hemizygous state abrogates Apc mutation-induced effects in some tissues (e.g., small intestine and colon epithelium) but not in other tissues (e.g., liver and ovarian epithelium). The identification of a possible feed-forward mechanism for β-catenin and β-catenin/TCF transcription in regulating Ctnnb1 transcript levels following Apc inactivation are also potentially interesting with regard to Myc, because the ENCODE project work also indicates that the Myc protein is bound in the promoter and intron one regions of the Ctnnb1 gene in selected cell lines. As such, β-catenin/TCF transcription may cooperate in some fashion with Myc, itself encoded by a β-catenin/TCF target gene, in a more complex feed-forward loop to activate Ctnnb1 transcription in certain cell types when Apc is inactivated.
Further studies are needed to better understand the details of the apparent feed-forward mechanisms through which β-catenin and β-catenin/TCF transcription may regulate Ctnnb1/CTNNB1 transcription in the setting of Apc/APC inactivation. Besides Myc, other β-catenin/TCF target genes may also be differentially regulated in a tissue - and context-dependent fashion, perhaps contributing in some fashion to the tissue-specific differences of Ctnnb1 hemizygous gene dosage on Apc mutation-instigated tumorigenesis observed. In addition, the basis for the dramatic changes in the free or active pool of β-catenin protein relative to the more modest effects on total β-catenin protein levels when Ctnnb1/CTNNB1 transcript levels are reduced in colon epithelial cells with Wnt pathway dysregulation remains to be elucidated. Nonetheless, our findings highlight the possibility that novel approaches and/or agents that can reduce CTNNB1 transcript levels and/or the free pool of β-catenin protein might have quite dramatic effects on the development and perhaps persistence of neoplastic cells with Wnt pathway defects.
Materials and Methods
Mice
To target Apc and/or Ctnnb1 alleles in colon tissues, CDX2P-G22Cre transgenic mice [16], or CDX2P-CreERT2 transgenic mice [17], or Lgr5-EGFP-IRES-CreERT2 (B6.129P2-Lgr5tm1(cre/ERT2)Cle/J) transgenic mice [8] (The Jackson Laboratory, Bar Harbor, ME), were first intercrossed with mice homozygous for Apc targeted alleles (Apcfl/fl, 580S) [40] and Ctnnb1-targeted alleles (Ctnnb fl/fl, B6.129-Ctnnb1tm2Kem/KnwJ) [41]. The resulting Cre positive Apcfl/+ Ctnnb1 fl/+ mice were then crossed to Apcfl/fl mice in order to target two alleles of Apc and one allele of Ctnnb1 (Apcfl/fl Ctnnb1 fl/+) or only alleles of Apc (Apcfl/fl), respectively. The Cre positive Apcfl/fl Ctnnb1 fl/+ and Apcfl/fl littermates were compared and the Cre negative littermates served as normal control. The CDX2P-CreERT2 Lgr5-EGFP-IRES-CreERT2 Apcfl/fl compound mice or CDX2P-CreERT2 Lgr5-EGFP-IRES-CreERT2 Apcfl/fl Ctnnb1 fl/+ compound mice were constructed by crossing Lgr5-EGFP-IRES-CreERT2 Apcfl/fl mice to CDX2P-CreERT2 Apcfl/fl mice and CDX2P-CreERT2 Apcfl/fl Ctnnb fl/fl littermates, respectively. To assess Cre-mediated recombination or Wnt signaling in colon epithelium, mice carrying the Gt(ROSA)26Sor tm1(EYFP)Cos/J reporter allele (EYFP) [42] or the B6.129P2-Axin2tm1Wbm/J allele (Axin2-LacZ) [43] (The Jackson Laboratory) were bred into CDX2P-CreERT2 Apcfl/fl mice or CDX2P-CreERT2 Apcfl/fl Ctnnb1 fl/+ mice. To assess the role of β-catenin function in another Apc mutation-dependent mouse tumor model, we used the previously described mouse model of ovarian endometrioid adenocarcinoma (OEA), arising from bi-allelic inactivation of both the Apc and Pten genes (Apcfl/fl Ptenfl/fl) [29]. To introduce the floxed Ctnnb1 allele, Ctnnb fl/fl mice were first crossed to Apcfl/fl Ptenfl/fl mice to generate Apcfl/+ Ptenfl/+ Ctnnb fl/+ mice, and then Apcfl/+ Ptenfl/+ Ctnnb fl/+ mice were bred to Apcfl/fl Ptenfl/fl mice to generate Apcfl/fl Ptenfl/fl, and Apcfl/fl Ptenfl/fl Ctnnb fl/+ mice. All mice were on a mixed C57BL/6 and 129 background, which were backcrossed to C57BL/6 mice for at least 10 generations, except the EYFP reporter mice and the CDX2P-CreERT2 transgenic mice, which were backcrossed for 7 and 3 generations, respectively. All experimental compound mice were on a mixed C57BL/6 and 129 background, and littermates with similar genetic background and different genotypes were used for comparison (see breeding scheme above). Animal husbandry and experimental procedures were carried out under approval from the University Committee on Use and Care of Animals, University of Michigan and according to Michigan state and US federal regulations. All the mice were housed in specific-pathogen free (SPF) conditions. After weaning, rodent 5001 chow and automatically supplied water were provided ad libitum to mice. Animals were euthanized and analyzed at the specified time points, based on particular study design parameters or defined humane treatment and euthanasia guidelines.
Cell lines and RNA interference
Human colonic epithelial cells (HCEC) [44] were kindly provided by Dr. Jerry Shay (UT Southwestern Medical School, Dallas, TX) and routinely grown on media made up with Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Grand Island, NY) and medium 199 (Thermo Scientific HyClone, Waltham, MA) at the ratio of 4 : 1, supplemented with EGF (25 ng/mL) (PeproTech, Inc, Rocky Hill, NJ), insulin (10 μg/mL, Life Technologies), hydrocortisone (1 μg/mL), transferrin (2 μg/mL), sodium selenite (5 nm) (all from Sigma-Aldrich, St Louis, MO), and 2% cosmic calf serum (Thermo Scientific HyClone). Cells were cultured on Primaria dishes (BD Biosciences, San Jose, CA) or chamber slides (Lab-Tek II, Vernon Hills, IL) and grown in 2% oxygen and 7% carbon dioxide. HCEC cells were infected with a TRIPZ inducible lentiviral vector (GE Dhamacon, Lafayette, CO) carrying a shRNA against APC (targeting sequence: 5’-CAAATCATATGGATGATAA-3’) or a non-silencing scramble shRNA (Scrmbl). Cells were selected with 1μg/mL of puromycin (Sigma-Aldrich) for 5 days. The resulting stable cell lines (HCEC/APC shRNA or HCEC/Scrmble) were further transduced with TRIPZ lentiviruses driving expression of two different shRNAs targeting CTNNB1 (CTNNB1-1 and CTNNB1-2; targeting sequence for CTNNB1-1 : 5’-TGGGTGGTATAGAGGCTCT-3’; and targeting sequence for CTNNB1-2 : 5’ - AGCTGATATTGATGGACAG-3’) or a non-silencing scramble shRNA (Scrmbl). Human colon cancer cell lines, HCT116, SW480, and DLD1, and human OEA-derived cell line, TOV-112D, were grown in 5% CO2 with DMEM containing 10% fetal bovine serum and penicillin/streptomycin. HCT116, SW480, DLD1 and TOV-112D cells stably expressing the shRNAs targeting CTNNB1 (CTNNB1-1 and CTNNB1-2) or a non-silencing scramble shRNA (Scrmbl) were made in the same way as HCEC cells. Expression of shRNAs was induced by incubation of cells with doxycycline (DOX; Sigma-Aldrich) at 2 μg/ml or a solvent control for 3 days (for HCEC cells) or 7 days (for HCT116, SW480, and DLD1 cells). The degree of inhibition of the shRNAs on APC transcripts and protein and CTNNB1 transcripts and the respective β-catenin protein was assessed by qRT-PCR and Western blotting assays.
Plasmids
DNA fragment containing human CTNNB1 sequences from −336 to +219 relative to the transcription start site was obtained by PCR amplification of genomic DNA, and was subcloned upstream from the luciferase reporter gene in the pGL3Basic reporter vector (Promega, Madison, WI), using the MluI and XhoI sites. The forward primer for generating the CTNNB1 reporter construct was 5′-ACGCGTGCTGCTCTCCCGGTTCG -3′; the reverse primer for generating the CTNNB1 reporter construct was 5′ - CTCGAGCAGGGGAACAGGCTCCTC-3′.
Tamoxifen (TAM) treatment and AdCre injection
Mice with the CDX2P-CreERT2 transgene or Lgr5-EGFP-IRES-CreERT2 were injected intraperitoneally with TAM (Sigma-Aldrich) dissolved in corn oil (Sigma-Aldrich). For two TAM daily dosing, we used 150mg/kg weight per dose; for three consecutive daily doses, we administered TAM at 100mg/kg weight per dose. Mice were injected with TAM at 2 - to 3-months of age. For OEA induction, 5 x 107 plaque-forming units of replication-incompetent recombinant adenovirus expressing Cre recombinase (AdCre, from the University of Michigan’s Vector Core) were injected into the right ovarian bursal cavities of 6–10 week old female mice as previously described [29].
Immunohistochemistry, immunofluorescence and β-gal analysis
Mouse tissues were prepared for paraffin-embedding or cryosectioning as described previously [16]. For assessment of cell proliferation, mice were pulsed with 5-bromo-2-deoxyuridine (BrdU; Sigma-Aldrich) for 1 hr before euthanasia. Sections of paraffin-embedded human or mouse tissues were subjected to immunohistochemical analysis as previously described [45]. The following primary antibodies were used for immunohistochemical analysis with sections of paraffin-embedded tissues: mouse anti-BrdU (1 : 500; BD Biosciences); rabbit anti-lysozyme (1 : 2000; Dako, Carpinteria, CA); mouse anti-β-catenin (1 : 800; BD Biosciences); rat anti-CK8 (1 : 100, The Developmental Studies Hybridoma Bank, Iowa City, IA); goat anti-E-cadherin (1 : 100, R&D Systems, Minneapolis, MN); mouse anti-α-inhibin (1 : 200, Bio-Rad Laboratories, Inc., Raleigh, NC). For BrdU staining, tissue sections were treated with 2N HCl at 37°C for 30 min after performing antigen retrieval with citrate buffer (pH 6.0, Biogenex, San Ramon, CA). For immunofluorescence using frozen sectioned tissues, mouse colon and intestinal tissues were fixed in 4% paraformaldehyde (PFA) overnight, cryo-protected and frozen in O.C.T. (Fisher HealthCare, Houston, TX 77038). Standard immunofluorescence staining was performed on 6-μm frozen sections with rabbit anti-lysozyme antibody (1 : 1000; Dako). For immunofluorescence using paraffin-embedded tissues, the following primary antibodies were used: rabbit anti-lysozyme (1 : 1000; Dako), rabbit anti-Sox9 (1 : 200; Millipore, Temecula, CA), rat anti-Msi1 (1 : 500; a gift from Dr. Hideyuki Okano [46, 47]), goat anti-EphB2 (1 : 100; R&D Systems), goat anti-EphB3 (1 : 100; R&D Systems), goat anti-ephrinB1 (1 : 200; R&D Systems), goat anti-ephrinB2 (1 : 100; R&D Systems), mouse anti-α-tublin (1 : 1000; Sigma-Aldrich), and rabbit anti-Crb3 (1 : 1000; kindly provided by Dr. Benjamin Margolis at University of Michigan). The secondary antibodies used were Alexa fluor 488-conjugated donkey anti-goat, Alexa fluor 488-conjugated donkey anti-rabbit, Alexa fluor 488-conjugated goat anti-rabbit, Alexa fluor 594-conjugated goat anti-mouse, Alexa fluor 488-conjugated goat anti-mouse, Alexa fluor 594-conjugated goat anti-rabbit, and Alexa fluor 488-conjugated goat anti-rat (Molecular Probes, Life Technologies, Carlsbad, CA), diluted at 1 : 1000. DNA was labeled by Hoechst 33342 (Molecular Probes, Life Technologies) by adding to the washing buffer at 5 μg/ml. β-gal analysis for mouse with Axin2-LacZ reporter was performed as described previously [16].
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) assay
To assess apoptosis, TUNEL assays were undertaken using 4-μm sections of formalin-fixed, paraffin-embedded mouse colon tissues, after the tissue sections were deparaffinized, rehydrated and treated with 20 μg/ml protease K (Roche Applied Sciences, Indianapolis, IN) at 37°C for 15 min. The nicked DNA was labeled by using terminal transferase (TdT) (New England Biolabs, Ipswich, MA) and Biotin-16-UTP (Roche Applied Sciences) according to the manufacturer’s recommendation. The signal was detected by using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) according to the manufacturer’s suggestion.
Mitotic spindle axis assessment
The spindle angles were defined by the orientation of mitotic spindles, based on α-tubulin staining, relative to the most adjacent apical membrane, as indicated by Crb3 staining. The mitotic spindle axis angle relative to the planar axis of the cells (defined by the most adjacent apical membrane) was measured by ImageJ (NIH).
Western blot analysis
Western blot analyses on lysates from HCEC, HCT116, SW480, DLD1 and TOV-112D cells were performed as described [45]. The following antibodies were used: mouse anti-active β-catenin (1 : 2000; Millipore, Temecula, CA), mouse anti-total β-catenin (1 : 10,000; BD Biosciences), rabbit anti-APC (clone C-20, 1 : 1000; Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-APC (clone Ab-5, 1 : 1000; Millipore), and mouse anti-β-actin (1 : 10,000; Sigma). The density of Western blotting bands was quantified using AlphaImager HP system (ProteinSimple, San Jose, CA).
Quantitative reverse transcription (RT)-PCR (qRT-PCR)
cDNA was synthesized using a high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). qRT-PCR was performed with an ABI Prism 7300 Sequence Analyzer using a SYBR green fluorescence protocol (Applied Biosystems). See S1 Table for primer sequences used in qRT-PCR.
Reporter gene assays
DLD1 cells, stably expressing two different doxycycline-inducible shRNAs targeting CTNNB1 (CTNNB1-1 and CTNNB1-2) or a non-silencing scramble shRNA (Scrmbl), were treated for 4 days with DOX at 2 μg/ml or a solvent control. At second day during DOX treatment, cells were plated in 35-mm six-well plates. After 12—24h cells were then transfected with 0.5 μg of CTNNB1 reporter or TOPflash, 1 μg of pCDNA3 (Invitrogen) and 0.05 μg of PRL-CMV Renilla luciferase reporter vector (Promega) using Mirus TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI) according to the manufacturer's protocol. Cells were harvested 45h later and luciferase activities were measured using a Dual-luciferase kit and GloMax-Multi Detection System from Promega.
Statistical analysis
All data for qRT-PCR were evaluated by Student's t test and asterisks denote significance with P < 0.05. Error bars denote standard deviations (S.D.). Kaplan-Meier survival curves were compared by log-rank (Mantel-Cox) test. Chi-Square test was used to determine significance when mitotic spindle angles were compared among mice with different genotypes. P < 0.05 is considered statistically significant.
Ethics statement
Animal husbandry and experimental procedures were carried out under approval from the University Committee on Use and Care of Animals, University of Michigan (PRO00005075) and according to Michigan state and US federal regulations. All the mice were housed in specific-pathogen free (SPF) conditions. After weaning, rodent 5001 chow and automatically supplied water were provided ad libitum to mice. Animals were euthanized and analyzed at the specified time points, based on particular study design parameters or defined humane treatment and euthanasia guidelines.
Supporting Information
Zdroje
1. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6 : 479–507. Epub 2010/11/26. eng. doi: 10.1146/annurev-pathol-011110-130235 21090969
2. Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21(20):2525–38. Epub 2007/10/17. eng. 17938238
3. Moser AR, Shoemaker AR, Connelly CS, Clipson L, Gould KA, Luongo C, et al. Homozygosity for the Min allele of Apc results in disruption of mouse development prior to gastrulation. Developmental dynamics: an official publication of the American Association of Anatomists. 1995;203(4):422–33.
4. Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120(Pt 19):3327–35. Epub 2007/09/21. eng.
5. Brocardo M, Henderson BR. APC shuttling to the membrane, nucleus and beyond. Trends in cell biology. 2008;18(12):587–96. doi: 10.1016/j.tcb.2008.09.002 18848448
6. Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17(1):45–51. Epub 2007/01/09. eng. 17208432
7. Mosimann C, Hausmann G, Basler K. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nature reviews Molecular cell biology. 2009;10(4):276–86. doi: 10.1038/nrm2654 19305417
8. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–7. Epub 2007/10/16. eng. 17934449
9. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205. Epub 2012/06/12. eng. doi: 10.1016/j.cell.2012.05.012 22682243
10. van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111(2):241–50. Epub 2002/11/01. eng. 12408868
11. Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111(2):251–63. Epub 2002/11/01. eng. 12408869
12. Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18(12):1385–90. Epub 2004/06/17. eng. 15198980
13. Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25(57):7531–7. 17143297
14. Bakker ER, Hoekstra E, Franken PF, Helvensteijn W, van Deurzen CH, van Veelen W, et al. beta-Catenin signaling dosage dictates tissue-specific tumor predisposition in Apc-driven cancer. Oncogene. 2013;32(38):4579–85. doi: 10.1038/onc.2012.449 23045279
15. Buchert M, Athineos D, Abud HE, Burke ZD, Faux MC, Samuel MS, et al. Genetic dissection of differential signaling threshold requirements for the Wnt/beta-catenin pathway in vivo. PLoS genetics. 2010;6(1):e1000816. doi: 10.1371/journal.pgen.1000816 20084116
16. Akyol A, Hinoi T, Feng Y, Bommer GT, Glaser TM, Fearon ER. Generating somatic mosaicism with a Cre recombinase-microsatellite sequence transgene. Nat Methods. 2008;5(3):231–3. Epub 2008/02/12. eng. doi: 10.1038/nmeth.1182 18264107
17. Feng Y, Sentani K, Wiese A, Sands E, Green M, Bommer GT, et al. Sox9 induction, ectopic Paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. The American journal of pathology. 2013;183(2):493–503. doi: 10.1016/j.ajpath.2013.04.013 23769888
18. Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–8. Epub 2010/11/30. eng. doi: 10.1038/nature09637 21113151
19. Andreu P, Colnot S, Godard C, Gad S, Chafey P, Niwa-Kawakita M, et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development. 2005;132(6):1443–51. Epub 2005/02/18. eng. 15716339
20. Andreu P, Peignon G, Slomianny C, Taketo MM, Colnot S, Robine S, et al. A genetic study of the role of the Wnt/beta-catenin signalling in Paneth cell differentiation. Dev Biol. 2008;324(2):288–96. Epub 2008/10/25. eng. doi: 10.1016/j.ydbio.2008.09.027 18948094
21. Hirata A, Utikal J, Yamashita S, Aoki H, Watanabe A, Yamamoto T, et al. Dose-dependent roles for canonical Wnt signalling in de novo crypt formation and cell cycle properties of the colonic epithelium. Development. 2013;140(1):66–75. Epub 2012/12/12. eng. doi: 10.1242/dev.084103 23222438
22. Clevers H, Batlle E. EphB/EphrinB receptors and Wnt signaling in colorectal cancer. Cancer research. 2006;66(1):2–5. 16397205
23. Cortina C, Palomo-Ponce S, Iglesias M, Fernandez-Masip JL, Vivancos A, Whissell G, et al. EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nature genetics. 2007;39(11):1376–83. 17906625
24. Nishimura S, Wakabayashi N, Toyoda K, Kashima K, Mitsufuji S. Expression of Musashi-1 in human normal colon crypt cells: a possible stem cell marker of human colon epithelium. Dig Dis Sci. 2003;48(8):1523–9. Epub 2003/08/20. eng. 12924647
25. Potten CS, Booth C, Tudor GL, Booth D, Brady G, Hurley P, et al. Identification of a putative intestinal stem cell and early lineage marker; musashi-1. Differentiation. 2003;71(1):28–41. Epub 2003/02/01. eng. 12558601
26. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–12. 9727977
27. Staal FJ, Noort Mv M, Strous GJ, Clevers HC. Wnt signals are transmitted through N-terminally dephosphorylated beta-catenin. EMBO reports. 2002;3(1):63–8. 11751573
28. van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wnt signaling controls the phosphorylation status of beta-catenin. The Journal of biological chemistry. 2002;277(20):17901–5. 11834740
29. Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer cell. 2007;11(4):321–33. 17418409
30. Moreno-Bueno G, Gamallo C, Perez-Gallego L, de Mora JC, Suarez A, Palacios J. beta-Catenin expression pattern, beta-catenin gene mutations, and microsatellite instability in endometrioid ovarian carcinomas and synchronous endometrial carcinomas. Diagnostic molecular pathology: the American journal of surgical pathology, part B. 2001;10(2):116–22.
31. Sagae S, Kobayashi K, Nishioka Y, Sugimura M, Ishioka S, Nagata M, et al. Mutational analysis of beta-catenin gene in Japanese ovarian carcinomas: frequent mutations in endometrioid carcinomas. Japanese journal of cancer research: Gann. 1999;90(5):510–5. 10391090
32. Wright K, Wilson P, Morland S, Campbell I, Walsh M, Hurst T, et al. beta-catenin mutation and expression analysis in ovarian cancer: exon 3 mutations and nuclear translocation in 16% of endometrioid tumours. International journal of cancer Journal international du cancer. 1999;82(5):625–9. 10417756
33. Wu R, Zhai Y, Fearon ER, Cho KR. Diverse mechanisms of beta-catenin deregulation in ovarian endometrioid adenocarcinomas. Cancer research. 2001;61(22):8247–55. Epub 2001/11/24. eng. 11719457
34. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–67. Epub 1990/06/01. eng. 2188735
35. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–70. Epub 1996/10/18. eng. 8861899
36. Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ, et al. The 'just-right' signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Human molecular genetics. 2002;11(13):1549–60. 12045208
37. Athineos D, Sansom OJ. Myc heterozygosity attenuates the phenotypes of APC deficiency in the small intestine. Oncogene. 2010;29(17):2585–90. doi: 10.1038/onc.2010.5 20140021
38. Reed KR, Athineos D, Meniel VS, Wilkins JA, Ridgway RA, Burke ZD, et al. B-catenin deficiency, but not Myc deletion, suppresses the immediate phenotypes of APC loss in the liver. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(48):18919–23. doi: 10.1073/pnas.0805778105 19033191
39. Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446(7136):676–9. 17377531
40. Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278(5335):120–3. Epub 1997/10/06. eng. 9311916
41. Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128(8):1253–64. Epub 2001/03/23. eng. 11262227
42. Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1 : 4. Epub 2001/04/12. eng. 11299042
43. Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22(4):1184–93. Epub 2002/01/26. eng. 11809809
44. Roig AI, Eskiocak U, Hight SK, Kim SB, Delgado O, Souza RF, et al. Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology. 2010;138(3):1012–21 e1–5. Epub 2009/12/08. eng. doi: 10.1053/j.gastro.2009.11.052 19962984
45. Feng Y, Bommer GT, Zhai Y, Akyol A, Hinoi T, Winer I, et al. Drosophila split ends homologue SHARP functions as a positive regulator of Wnt/beta-catenin/T-cell factor signaling in neoplastic transformation. Cancer research. 2007;67(2):482–91. Epub 2007/01/20. eng. 17234755
46. Kaneko Y, Sakakibara S, Imai T, Suzuki A, Nakamura Y, Sawamoto K, et al. Musashi1: an evolutionally conserved marker for CNS progenitor cells including neural stem cells. Dev Neurosci. 2000;22(1–2):139–53. Epub 2000/02/05. eng. 10657706
47. Sakakibara S, Imai T, Hamaguchi K, Okabe M, Aruga J, Nakajima K, et al. Mouse-Musashi-1, a neural RNA-binding protein highly enriched in the mammalian CNS stem cell. Dev Biol. 1996;176(2):230–42. Epub 1996/06/15. eng. 8660864
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development
- Metabolomic Quantitative Trait Loci (mQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis
- Genus-Wide Comparative Genomics of Delineates Its Phylogeny, Physiology, and Niche Adaptation on Human Skin
- Encodes Dual Oxidase, Which Acts with Heme Peroxidase Curly Su to Shape the Adult Wing
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy