
Sae2 Function at DNA Double-Strand Breaks Is Bypassed by Dampening Tel1 or Rad53 Activity
Genome instability is one of the most pervasive characteristics of cancer cells and can be due to DNA repair defects and failure to arrest the cell cycle. Among the many types of DNA damage, the DNA double strand break (DSB) is one of the most severe, because it can cause mutations and chromosomal rearrangements. Generation of DSBs triggers a highly conserved mechanism, known as DNA damage checkpoint, which arrests the cell cycle until DSBs are repaired. DSBs can be repaired by homologous recombination, which requires the DSB ends to be nucleolytically processed (resected) to generate single-stranded DNA. In Saccharomyces cerevisiae, DSB resection is initiated by the MRX complex together with Sae2, whereas more extensive resection is catalyzed by both Exo1 and Dna2-Sgs1. The absence of Sae2 not only impairs DSB resection, but also leads to the hyperactivation of the checkpoint proteins Tel1/ATM and Rad53, leading to persistent cell cycle arrest. In this manuscript we show that persistent Tel1 and Rad53 signaling activities in sae2Δ cells cause DNA damage hypersensitivity and defective DSB resection by increasing the amount of Rad9 bound at the DSBs, which in turn inhibits the Sgs1-Dna2 resection machinery. As ATM inhibition has been proposed as a strategy for cancer treatment, the finding that defective Tel1 signaling activity restores DNA damage resistance in sae2Δ cells might have implications in cancer therapies that use ATM inhibitors for synthetic lethal approaches that are devised to kill tumor cells with defective DSB repair.
Published in the journal:
. PLoS Genet 11(11): e32767. doi:10.1371/journal.pgen.1005685
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005685
Summary
Genome instability is one of the most pervasive characteristics of cancer cells and can be due to DNA repair defects and failure to arrest the cell cycle. Among the many types of DNA damage, the DNA double strand break (DSB) is one of the most severe, because it can cause mutations and chromosomal rearrangements. Generation of DSBs triggers a highly conserved mechanism, known as DNA damage checkpoint, which arrests the cell cycle until DSBs are repaired. DSBs can be repaired by homologous recombination, which requires the DSB ends to be nucleolytically processed (resected) to generate single-stranded DNA. In Saccharomyces cerevisiae, DSB resection is initiated by the MRX complex together with Sae2, whereas more extensive resection is catalyzed by both Exo1 and Dna2-Sgs1. The absence of Sae2 not only impairs DSB resection, but also leads to the hyperactivation of the checkpoint proteins Tel1/ATM and Rad53, leading to persistent cell cycle arrest. In this manuscript we show that persistent Tel1 and Rad53 signaling activities in sae2Δ cells cause DNA damage hypersensitivity and defective DSB resection by increasing the amount of Rad9 bound at the DSBs, which in turn inhibits the Sgs1-Dna2 resection machinery. As ATM inhibition has been proposed as a strategy for cancer treatment, the finding that defective Tel1 signaling activity restores DNA damage resistance in sae2Δ cells might have implications in cancer therapies that use ATM inhibitors for synthetic lethal approaches that are devised to kill tumor cells with defective DSB repair.
Introduction
Programmed DNA double-strand breaks (DSBs) are formed during meiotic recombination and rearrangement of the immunoglobulin genes in lymphocytes. Furthermore, potentially harmful DSBs can arise by exposure to environmental factors, such as ionizing radiations and radiomimetic chemicals, or by failures in DNA replication. DSB generation elicits a checkpoint response that depends on the mammalian protein kinases ATM and ATR, whose functional orthologs in Saccharomyces cerevisiae are Tel1 and Mec1, respectively [1]. Tel1/ATM is recruited to DSBs by the MRX (Mre11-Rad50-Xrs2)/MRN (Mre11-Rad50-Nbs1) complex, whereas Mec1/ATR recognizes single-stranded DNA (ssDNA) covered by Replication Protein A (RPA) [2]. Once activated, Tel1/ATM and Mec1/ATR propagate their checkpoint signals by phosphorylating the downstream checkpoint kinases Rad53 (Chk2 in mammals) and Chk1, to couple cell cycle progression with DNA repair [2].
Repair of DSBs can occur by either non-homologous end joining (NHEJ) or homologous recombination (HR). Whereas NHEJ directly joins the DNA ends, HR uses the sister chromatid or the homologous chromosome to repair DSBs. HR requires that the 5’ ends of a DSB are nucleolytically processed (resected) to generate 3’-ended ssDNA that can invade an undamaged homologous DNA template [3,4]. In Saccharomyces cerevisiae, recent characterization of core resection proteins has revealed that DSB resection is initiated by the MRX complex, which catalyzes an endonucleolytic cleavage near a DSB [4], with the Sae2 protein (CtIP in mammals) promoting MRX endonucleolytic activity [5]. This MRX-Sae2-mediated DNA clipping generates 5’ DNA ends that are optimal substrates for the nucleases Exo1 and Dna2, the latter working in concert with the helicase Sgs1 [6–9]. In addition, the MRX complex recruits Exo1, Sgs1 and Dna2 to DSBs independently of the Mre11 nuclease activity [10]. DSB resection is also negatively regulated by Ku and Rad9, which inhibit the access to DSBs of Exo1 and Sgs1-Dna2, respectively [11–14].
The MRX-Sae2-mediated endonucleolytic cleavage is particularly important to initiate resection at DNA ends that are not easily accessible to Exo1 and Dna2-Sgs1. For instance, both sae2Δ and mre11 nuclease defective mutants are completely unable to resect meiotic DSBs, where the Spo11 topoisomerase-like protein remains covalently attached to the 5’-terminated strands [15,16]. Furthermore, the same mutants exhibit a marked sensitivity to camptothecin (CPT), which extends the half-life of DNA-topoisomerase I cleavable complexes [17,18], and to methyl methanesulfonate (MMS), which can generate chemically complex DNA termini. The lack of Rad9 or Ku suppresses both the hypersensitivity to DSB-inducing agents and the resection defect of sae2Δ cells [10–14]. These suppression events require Dna2-Sgs1 and Exo1, respectively, indicating that Rad9 increases the requirement for MRX-Sae2 activity in DSB resection by inhibiting Sgs1-Dna2 [13,14], while Ku mainly limits the action of Exo1 [10–12]. By contrast, elimination of either Rad9 or Ku does not bypass Sae2/MRX function in resecting meiotic DSBs [11,13], likely because Sgs1-Dna2 and Exo1 cannot substitute for the Sae2/MRX-mediated endonucleolytic cleavage when this event is absolutely required to generate accessible 5’-terminated DNA strands.
Sae2 plays an important role also in modulating the checkpoint response. Checkpoint activation in response to DSBs depends primarily on Mec1, with Tel1 playing a minor role [19]. On the other hand, impaired Mre11 endonuclease activity caused by the lack of Sae2 leads to increased MRX persistence at the DSB ends. The enhanced MRX signaling in turn causes unscheduled Tel1-dependent checkpoint activation that is associated to prolonged Rad53 phosphorylation [20–22]. Mutant mre11 alleles that reduce MRX binding to DSBs restore DNA damage resistance in sae2Δ cells and reduce their persistent checkpoint activation without restoring efficient DSB resection [23,24], suggesting that enhanced MRX association to DSBs contributes to the DNA damage hypersensitivity caused by the lack of Sae2. Persistently bound MRX might increase the sensitivity to DNA damaging agents of sae2Δ cells by hyperactivating the DNA damage checkpoint. If this were the case, then the DNA damage hypersensitivity of sae2Δ cells should be restored by the lack of Tel1 or of its downstream effector Rad53, as they are responsible for the sae2Δ enhanced checkpoint signaling [20,22]. However, while Rad53 inactivation has never been tested, TEL1 deletion not only fails to restore DNA damage resistance in sae2Δ cells, but it exacerbates their sensitivity to DNA damaging agents [23,24]. Therefore, other studies are required to understand whether the Tel1 - and Rad53-mediated checkpoint signaling has any role in determining the DNA damage sensitivity of sae2Δ cells.
By performing a genetic screen, we identified rad53 and tel1 mutant alleles that suppress both the hypersensitivity to DNA damaging agents and the resection defect of sae2Δ cells by reducing the amount of Rad9 at DSBs. Decreased Rad9 binding at DNA ends bypasses Sae2 function in DNA damage resistance and resection by relieving the inhibition of the Sgs1-Dna2 resection machinery. Altogether our data suggest that the primary cause of the resection defect of sae2Δ cells is Rad9 association to DSBs, which is promoted by persistent Tel1 and Rad53 signaling activities in these cells.
Results
The Rad53-H88Y and Tel1-N2021D variants suppress the DNA damage hypersensitivity of sae2Δ cells
We have previously described our search for extragenic mutations that suppress the CPT hypersensitivity of sae2Δ cells [13]. This genetic screen identified 15 single-gene suppressor mutants belonging to 11 distinct allelism groups. Analysis of genomic DNA by next-generation Illumina sequencing of 5 non allelic suppressor mutants revealed that the DNA damage resistance was due to single base pair substitutions in the genes encoding Sgs1, Top1, or the multi-drug resistance proteins Pdr3, Pdr10 and Sap185 [13]. Subsequent genome sequencing and genetic analysis of 2 more non allelic suppressor mutants allowed to link suppression to either the rad53-H88Y mutant allele, causing the replacement of Rad53 amino acid residue His88 by Tyr, or the tel1-N2021D allele, resulting in the replacement of Tel1 amino acid residue Asn2021 by Asp. Both rad53-H88Y and tel1-N2021D alleles restored resistance of sae2Δ cells not only to CPT, but also to phleomycin (phleo) and MMS (Fig 1A). While both rad53-H88Y and tel1-N2021D fully rescued the hypersensitivity of sae2Δ cells to phleomycin and MMS, the CPT hypersensitivity of sae2Δ cells was only partially suppressed by the same alleles (Fig 1A), suggesting that they did not bypass all Sae2 functions.

Both rad53-H88Y and tel1-N2021D suppressor alleles were recessive, as the sensitivity to genotoxic agents of sae2Δ/sae2Δ RAD53/rad53-H88Y and sae2Δ/sae2Δ TEL1/tel1-N2021D diploid cells was similar to that of sae2Δ/sae2Δ RAD53/RAD53 TEL1/TEL1 diploid cells (S1 Fig), suggesting that rad53-H88Y and tel1-N2021D alleles encode hypomorphic variants. Furthermore, both variants suppressed the hypersensitivity to DNA damaging agents of sae2Δ cells by altering the same mechanism, as sae2Δ rad53-H88Y tel1-N2021D triple mutant cells survived in the presence of DNA damaging agents to the same extent as sae2Δ rad53-H88Y and sae2Δ tel1-N2021D double mutant cells (Fig 1B).
The MRX complex not only provides the nuclease activity for initiation of DSB resection, but also it promotes the binding of Exo1, Sgs1 and Dna2 at the DSB ends [10]. These MRX multiple roles explain the severe DNA damage hypersensitivity and resection defect of cells lacking any of the MRX subunits compared to cells lacking either Sae2 or the Mre11 nuclease activity. As Sae2 has been proposed to activate Mre11 nuclease activity [5], we asked whether the suppression of sae2Δ DNA damage hypersensitivity by Rad53-H88Y and Tel1-N2021D requires Mre11 nuclease activity. Both rad53-H88Y and tel1-N2021D alleles suppressed the hypersensitivity to DNA damaging agents of sae2Δ cells carrying the nuclease defective mre11-H125N allele (Fig 1C). By contrast, sae2Δ mre11Δ rad53-H88Y and sae2Δ mre11Δ tel1-N2021D triple mutant cells were as sensitive to genotoxic agents as sae2Δ mre11Δ double mutant cells (Fig 1D), indicating that neither the rad53-H88Y nor the tel1-N2021D allele can suppress the hypersensitivity to DNA damaging agents of sae2Δ mre11Δ cells. Altogether, these findings indicate that both Rad53-H88Y and Tel1-N2021D require the physical presence of the MRX complex, but not its nuclease activity, to bypass Sae2 function in cell survival to genotoxic agents.
The Rad53-H88Y variant is defective in the interaction with Rad9 and bypasses the adaptation defect of sae2Δ cells by impairing checkpoint activation
A single unrepairable DSB induces a DNA damage checkpoint that depends primarily on Mec1, with Tel1 playing a minor role [19]. This checkpoint response can be eventually turned off, allowing cells to resume cell cycle progression through a process that is called adaptation [25–27]. In the absence of Sae2, cells display heightened checkpoint activation that prevents cells from adapting to an unrepaired DSB [20,22]. This persistent checkpoint activation is due to increased MRX amount/persistence at the DSB that in turn causes enhanced and prolonged Tel1 activation that is associated with persistent Rad53 phosphorylation [20–22,28].
If the rad53-H88Y mutation impaired Rad53 activity, then it is expected to suppress the adaptation defect of sae2Δ cells by lowering checkpoint activation. We addressed this point by using JKM139 derivative strains, where a single DSB at the MAT locus can be generated by expression of the HO endonuclease gene under the control of a galactose-dependent promoter. This DSB cannot be repaired by HR because of the deletion of the homologous donor loci HML and HMR [27]. We measured checkpoint activation by monitoring the ability of cells to arrest the cell cycle and to phosphorylate Rad53 after HO induction. Both rad53-H88Y and sae2Δ rad53-H88Y cells formed microcolonies of more than 2 cells with higher efficiency than either wild type or sae2Δ cells (Fig 2A). Furthermore, the Rad53-H88Y variant was poorly phosphorylated after HO induction both in the presence and in the absence of Sae2 (Fig 2B). Thus, the rad53-H88Y mutation suppresses the adaptation defect of sae2Δ cells by impairing Rad53 activation.
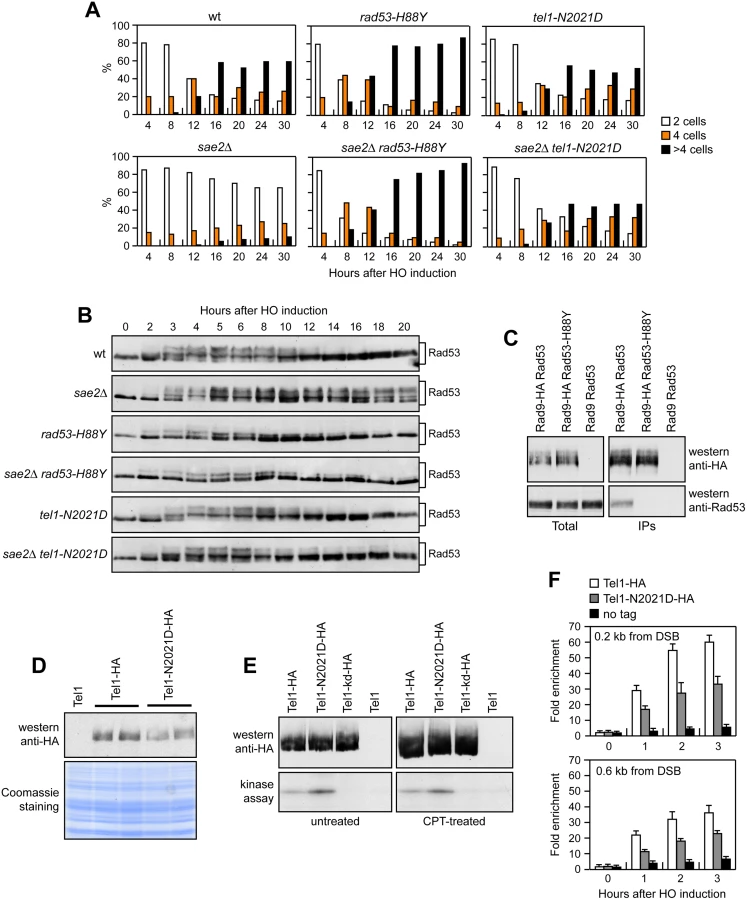
DNA damage-dependent activation of Rad53 requires its phospho-dependent interaction with Rad9, which acts as a scaffold to allow Rad53 intermolecular authophosphorylation and activation [29–31]. Interestingly, the His88 residue, which is replaced by Tyr in the Rad53-H88Y variant, is localized in the forkhead-associated domain 1 of the protein and has been implicated in mediating Rad9-Rad53 interaction [32]. Thus, we asked whether the Rad53-H88Y variant was defective in the interaction with Rad9. When HA-tagged Rad9 was immunoprecipitated with anti-HA antibodies from wild type and rad53-H88Y cells grown for 4 hours in the presence of galactose to induce HO, wild type Rad53 could be detected in Rad9-HA immunoprecipitates, whereas Rad53-H88Y did not (Fig 2C). This defective interaction of Rad53-H88Y with Rad9 could explain the impaired checkpoint activation in sae2Δ rad53-H88Y double mutant cells.
The Tel1-N2021D variant binds poorly to DSBs and bypasses the adaptation defect of sae2Δ cells by reducing persistent Rad53 activation
Tel1 signaling activity is responsible for the prolonged Rad53 activation that prevents sae2Δ cells to adapt to the checkpoint triggered by an unrepairable DSB [20,22]. Although telomere length in tel1-N2021D mutant cells was unaffected both in the presence and in the absence of Sae2 (S2 Fig), the recessivity of tel1-N2021D suppressor effect on sae2Δ DNA damage hypersensitivity suggests that the N2021D substitution impairs Tel1 function. If this were the case, Tel1-N2021D might suppress the adaptation defect of sae2Δ cells by reducing the DSB-induced persistent Rad53 phosphorylation. When G1-arrested cell cultures were spotted on galactose-containing plates to induce HO, wild type, sae2Δ, tel1-N2021D and sae2Δ tel1-N2021D cells accumulated large budded cells within 4 hours (Fig 2A). This cell cycle arrest is due to checkpoint activation. In fact, when the same cells exponentially growing in raffinose were transferred to galactose, Rad53 phosphorylation was detectable about 2–3 hours after galactose addition (Fig 2B). However, while sae2Δ cells remained arrested as large budded cells for at least 30 hours (Fig 2A) and showed persistent Rad53 phosphorylation (Fig 2B), wild type, tel1-N2021D and sae2Δ tel1-N2021D cells formed microcolonies with more than 2 cells (Fig 2A) and decreased the amounts of phosphorylated Rad53 (Fig 2B) with similar kinetics 10–12 hours after HO induction. Therefore, the Tel1-N2021D variant impairs Tel1 signaling activity, as it rescues the sae2Δ adaptation defect by reducing the persistent Rad53 phosphorylation.
The N2021D substitution resides in the Tel1 FAT domain, a helical solenoid that encircles the kinase domain of all the phosphoinositide 3-kinase (PI3K)-related kinases (PIKKs) [33,34], suggesting that this amino acid change might reduce Tel1 kinase activity. Western blot analysis revealed that the amount of Tel1-N2021D was slightly lower than that of wild type Tel1 (Fig 2D). We then immunoprecipitated equivalent amounts of Tel1-HA and Tel1-N2021D-HA variants from both untreated and CPT-treated cells (Fig 2E, top), and we measured their kinase activity in vitro using the known artificial substrate of the PIKKs family PHAS-I (Phosphorylated Heat and Acid Stable protein) [35]. Both Tel1-HA and Tel1-N2021D-HA were capable to phosphorylate PHAS-I, with the amount of phosphorylated substrate being slighly higher in Tel1-N2021D-HA than in Tel1-HA immunoprecipitates (Fig 2E, bottom). This PHAS-I phosphorylation was dependent on Tel1 kinase activity, as it was not detectable when the immunoprecipitates were prepared from strains expressing either kinase dead Tel1-kd-HA or untagged Tel1 (Fig 2E, bottom). Thus, the tel1-N2021D mutation does not affect Tel1 kinase activity.
Interestingly, the FAT domain is in close proximity to the FATC domain, which was shown to be important for Tel1 recruitment to DNA ends [36], suggesting that the Tel1-N2021D variant might be defective in recruitment/association to DSBs. Strikingly, when we analyzed Tel1 and Tel1-N2021D binding at the HO-induced DSB by chromatin immunoprecipitation (ChIP) and quantitative real time PCR (qPCR), the amount of Tel1-N2021D bound at the DSB turned out to be lower than that of wild type Tel1 (Fig 2F). This decreased Tel1-N2021D association was not due to lower Tel1-N2021D levels, as the ChIP signals were normalized for each time point to the amount of immunoprecipitated protein. Thus, the inability of sae2Δ tel1-N2021D cells to sustain persistent Rad53 phosphorylation after DSB generation can be explained by a decreased association of Tel1-N2021D to DSBs.
Checkpoint-mediated cell cycle arrest is not responsible for the DNA damage hypersensitivity of sae2Δ cells
As both Rad53-H88Y and Tel1-N2021D reduce checkpoint signaling in sae2Δ cells, we asked whether the increased DNA damage resistance of sae2Δ rad53-H88Y and sae2Δ tel1-N2021D cells was due to the elimination of the checkpoint-mediated cell cycle arrest. This hypothesis could not be tested by deleting the MEC1, DDC1, RAD24, MEC3 or RAD9 checkpoint genes, because they also regulate DSB resection [37–39]. On the other hand, an HO-induced DSB activates also the Chk1 checkpoint kinase [40], which contributes to arrest the cell cycle in response to DSBs by controlling a pathway that is independent of Rad53 [41]. Importantly, chk1Δ cells do not display DNA damage hypersensitivity and are not defective in resection of uncapped telomeres [38,41]. We therefore asked whether CHK1 deletion restores DNA damage resistance in sae2Δ cells. Consistent with the finding that Chk1 contributes to arrest the cell cycle after DNA damage independently of Rad53 [41], Rad53 was phosphorylated with wild type kinetics after HO induction in both chk1Δ and sae2Δ chk1Δ cells (Fig 3A). Furthermore, CHK1 deletion suppresses the adaptation defect of sae2Δ cells. In fact, both chk1Δ and sae2Δ chk1Δ cells spotted on galactose-containing plates formed microcolonies of more than 2 cells with higher efficiency than wild type and sae2Δ cells (Fig 3B), although they did it less efficiently than mec1Δ cells, where both Rad53 and Chk1 signaling were abrogated [41]. Strikingly, the lack of Chk1 did not suppress the hypersensitivity to DNA damaging agents of sae2Δ cells (Fig 3C), although it overrides the checkpoint-mediated cell cycle arrest.

To rule out the possibility that CHK1 deletion failed to restore DNA damage resistance in sae2Δ cells because it impairs DSB resection, we used JKM139 derivative strains to monitor directly generation of ssDNA at the DSB ends in the absence of Chk1. As ssDNA is resistant to cleavage by restriction enzymes, we followed loss of SspI restriction sites as a measure of resection by Southern blot analysis under alkaline conditions, using a single-stranded probe that anneals to the 3’ end at one side of the break. Consistent with previous indications that Chk1 is not involved in DNA-end resection [38], chk1Δ single mutant cells resected the DSB with wild type kinetics (Fig 3D). Furthermore, CHK1 deletion did not exacerbate the resection defect of sae2Δ cells (Fig 3E). Altogether, these data indicate that the prolonged checkpoint-mediated cell cycle arrest of sae2Δ cells is not responsible for their hypersensitivity to DNA damaging agents.
The Rad53-H88Y and Tel1-N2021D variants restore resection and SSA in sae2Δ cells
As the checkpoint-mediated cell cycle arrest was not responsible for the DNA damage hypersensitivity of sae2Δ cells, we asked whether Rad53-H88Y and/or Tel1-N2021D suppressed the sae2Δ resection defect. We first measured the efficiency of single-strand annealing (SSA), a mechanism that repairs a DSB flanked by direct DNA repeats when sufficient resection exposes the complementary DNA sequences, which can then anneal to each other [3]. The rad53-H88Y and tel1-N2021D alleles were introduced in the YMV45 strain, which carries two tandem leu2 gene repeats located 4.6 kb apart on chromosome III, with a HO recognition site adjacent to one of the repeats [42]. This strain also harbors a GAL-HO construct for galactose-inducible HO expression. Both Rad53-H88Y and Tel1-N2021D bypass Sae2 function in SSA-mediated DSB repair. In fact, accumulation of the SSA repair product after HO induction occurred more efficiently in both sae2Δ rad53-H88Y (Fig 4A and 4B) and sae2Δ tel1-N2021D (Fig 4C and 4D) than in sae2Δ cells, where it was delayed compared to wild type.

To confirm that Rad53-H88Y and Tel1-N2021D suppress the SSA defect of sae2Δ cells by restoring DSB resection, we used JKM139 derivative strains to monitor directly generation of ssDNA at the DSB ends. Indeed, sae2Δ rad53-H88Y (Fig 5A) and sae2Δ tel1-N2021D (Fig 5B) cells resected the HO-induced DSB more efficiently than sae2Δ cells, indicating that both Rad53-H88Y and Tel1-N2021D suppress the resection defect of sae2Δ cells.

The DSB resection defect of sae2Δ cells is thought to be responsible for the increased persistence of MRX at the DSB [43]. Because Rad53-H88Y and Tel1-N2021D restore DSB resection in sae2Δ cells, we expected that the same variants also reduce the amount of MRX bound at the DSB. The amount of Mre11 bound at the HO-induced DSB end turned out to be lower in both sae2Δ rad53-H88Y and sae2Δ tel1-N2021D than in sae2Δ cells (Fig 5C). Therefore, the Rad53-H88Y and Tel1-N2021D variants restore DSB resection in sae2Δ cells and reduce MRX association/persistence at the DSB.
Consistent with the finding that Rad53-H88Y and Tel1-N2021D do not fully restore CPT resistance in sae2Δ cells (Fig 1A), and therefore do not bypass completely all Sae2 functions, the rad53-H88Y and tel1-N2021D mutations were unable to suppress the sporulation defects of sae2Δ/sae2Δ diploid cells (Fig 5D), suggesting that they cannot bypass the requirement for Sae2/MRX endonucleolytic cleavage to remove Spo11 from meiotic DSBs.
Suppression of the DNA damage hypersensitivity of sae2Δ cells by Rad53-H88Y and Tel1-N2021D variants requires Sgs1-Dna2
The MRX complex not only provides the nuclease activity for initiation of DSB resection, but also allows extensive resection by promoting the binding at the DSB ends of the resection proteins Exo1 and Sgs1-Dna2 [6,7,10]. Suppression of the DNA damage hypersensitivity of sae2Δ cells by Rad53-H88Y and Tel1-N2021D requires the physical presence of the MRX complex but not its nuclease activity (Fig 1C and 1D). As the loading of Exo1, Sgs1-Dna2 at DSBs depends on the MRX complex independently of its nuclease activity [10], we asked whether the investigated suppression events require Exo1, Sgs1 and/or Dna2. This question was particularly interesting, as Rad53 was shown to inhibit resection at uncapped telomeres through phosphorylation and inhibition of Exo1 [38,44]. As shown in Fig 6A, sae2Δ suppression by Rad53-H88Y and Tel1-N2021D was Exo1-independent. In fact, although the lack of Exo1 exacerbated the sensitivity to DNA damaging agents of sae2Δ cells, both sae2Δ exo1Δ rad53-H88Y and sae2Δ exo1Δ tel1-N2021D triple mutants were more resistant to genotoxic agents than sae2Δ exo1Δ double mutant cells (Fig 6A).

By contrast, neither Rad53-H88Y nor Tel1-N2021D were able to suppress the sensitivity to DNA damaging agents of sae2Δ cells carrying the temperature sensitive dna2-1 allele (Fig 6B), suggesting that Dna2 activity is required for their suppressor effect. Dna2, in concert with the helicase Sgs1, functions as a nuclease in DSB resection [7]. The dna2-E675A allele abolishes Dna2 nuclease activity, which is essential for cell viability and whose requirement is bypassed by the pif1-M2 mutation that impairs the nuclear activity of the Pif1 helicase [45]. The lack of Sgs1 or expression of the Dna2-E675A variant in the presence of the pif1-M2 allele impaired viability of sae2Δ cells even in the absence of genotoxic agents. The synthetic lethality of sae2Δ sgs1Δ cells, and possibly of sae2Δ dna2-E675A pif1-M2, is likely due to defects in DSB resection, as it is known to be suppressed by either EXO1 overexpression or KU deletion [11]. Thus, we asked whether Rad53-H88Y and/or Tel1-N2021D could restore viability of sae2Δ sgs1Δ and/or sae2Δ dna2-E675A pif1-M2 cells. Tetrad dissection of diploid cells did not allow to find viable spores with the sae2Δ dna2-E675A pif1-M2 rad53-H88Y (Fig 6C) or sae2Δ dna2-E675A pif1-M2 tel1-N2021D genotypes (Fig 6D), indicating that neither Rad53-H88Y nor Tel1-N2021D can restore the viability of sae2Δ dna2-E675A pif1-M2 cells. Similarly, no viable sae2Δ sgs1Δ spores could be recovered, while sae2Δ sgs1Δ rad53-H88Y and sae2Δ sgs1Δ tel1-N2021D triple mutant spores formed very small colonies that could not be further propagated (Fig 6E and 6F). Finally, neither Rad53-H88Y nor Tel1-N2021D, which allowed DNA damage resistance in sae2Δ exo1Δ cells (Fig 6A), were able to suppress the growth defect of sgs1Δ exo1Δ double mutant cells even in the absence of genotoxic agents (Fig 6G). Altogether, these findings indicate that suppression by Rad53-H88Y and Tel1-N2021D of the DNA damage hypersensitivity caused by the absence of Sae2 is dependent on Sgs1-Dna2.
The lack of Rad53 kinase activity suppresses the DNA damage hypersensitivity and the resection defect of sae2Δ cells
The Rad53-H88Y protein is defective in interaction with Rad9 (Fig 2C) and therefore fails to undergo autophosphorylation and activation, prompting us to test whether other mutations affecting Rad53 activity can bypass Sae2 functions. To this end, we could not use rad53Δ cells because they show growth defects even when the lethal effect of RAD53 deletion is suppressed by the lack of Sml1 [46]. We then substituted the chromosomal wild type RAD53 allele with the kinase-defective rad53-K227A allele (rad53-kd), which does not impair cell viability in the absence of genotoxic agents but affects checkpoint activation [47]. The rad53-kd allele rescued the sensitivity of sae2Δ cells to CPT and MMS to an extent similar to Rad53-H88Y (Fig 7A). Furthermore, accumulation of the SSA repair products occurred more efficiently in sae2Δ rad53-kd cells than in sae2Δ (Fig 7B and 7C), indicating that the lack of Rad53 kinase activity bypasses Sae2 function in SSA-mediated DSB repair.

The lack of Tel1 kinase activity bypasses Sae2 function at DSBs, whereas Tel1 hyperactivation increases Sae2 requirement
Suppression of sae2Δ may be peculiar to Tel1-N2021D, which is poorly recruited to DSBs (Fig 2F), or it might be performed also by TEL1 deletion (tel1Δ) or by expression of a Tel1 kinase defective variant (Tel1-kd). Indeed, the Tel1-kd variant, carrying the G2611D, D2612A, N2616K, and D2631E amino acid substitutions that abolish Tel1 kinase activity in vitro (Fig 2E) [35], rescued the hypersensitivity of sae2Δ cells to genotoxic agents to an extent similar to Tel1-N2021D (Fig 8A). The lack of Tel1 kinase activity bypassed also Sae2 function in DSB resection, because sae2Δ tel1-kd cells repaired a DSB by SSA more efficiently than sae2Δ cells (Fig 8B and 8C). By contrast, and consistent with previous studies [23,24], TEL1 deletion was not capable to suppress the hypersensitivity to DNA damaging agents of sae2Δ cells (Fig 8A). Rather, tel1Δ sae2Δ double mutant cells displayed higher sensitivity to CPT than sae2Δ cells (Fig 8A). Altogether, these data indicate that the lack of Tel1 kinase activity can bypass Sae2 function both in DNA damage resistance and DSB resection, but these suppression events require the physical presence of the Tel1 protein.

As impairment of Tel1 function rescued the sae2Δ defects, we asked whether Tel1 hyperactivation exacerbates the DNA damage hypersensitivity of sae2Δ cells. We previously isolated the TEL1-hy909 allele, which encodes a Tel1 mutant variant with enhanced kinase activity that causes an impressive telomere overelongation [48]. As shown in Fig 8D, sae2Δ TEL1-hy909 double mutant cells were more sensitive to DNA damaging agents than sae2Δ single mutant cells. This enhanced DNA damage sensitivity was likely due to Tel1 kinase activity, as sae2Δ cells expressing a kinase defective Tel1-hy909-kd variant were as sensitive to DNA damaging agents as sae2Δ cells (Fig 8D). Thus, impairment of Tel1 activity bypasses Sae2 function at DSBs, whereas Tel1 hyperactivation increases the requirement for Sae2 in survival to genotoxic stress.
The absence of Tel1 failed not only to restore DNA damage resistance in sae2Δ cells (Fig 8A), but also to suppress their SSA defect (Fig 9A and 9B). The difference in the effects of tel1Δ and tel1-kd was not due to checkpoint signaling, as Rad53 phosphorylation decreased with similar kinetics in both sae2Δ tel1-kd and sae2Δ tel1Δ double mutant cells 10–12 hours after HO induction (Fig 9C). Interestingly, SSA-mediated DSB repair occurred with wild type kinetics in tel1-kd mutant cells (Fig 8B and 8C), while tel1Δ cells repaired a DSB by SSA less efficiently than wild type cells (Fig 9A and 9B), suggesting that Tel1 might have a function at DSBs that does not require its kinase activity. Indeed, TEL1 deletion was shown to slight impair DSB resection [19]. Furthermore, it did not exacerbate the resection defect [19] and the hypersensitivity to DNA damaging agents of mre11Δ cells (Fig 9D), suggesting that the absence of Tel1 can impair MRX function. Tel1 was also shown to promote MRX association at DNA ends flanked by telomeric DNA repeats independently of its kinase activity [49], and we are showing that suppression of sae2Δ by Tel1-N2021D requires the physical presence of the MRX complex (Fig 1D). Thus, it is possible that the lack of Tel1 fails to bypass Sae2 function at DSBs because it reduces MRX association at DSBs to a level that is not sufficient to restore DNA damage resistance and DSB resection in sae2Δ cells. Indeed, the amount of Mre11 bound at the HO-induced DSB was decreased in tel1Δ, but not in tel1-kd cells, compared to wild type (Fig 9E). In agreement with a partial loss of Tel1 function, the Tel1-N2021D variant, whose association to DSBs is diminished compared to wild type Tel1 but not abolished (Fig 2F), only slightly decreased Mre11 association to the DSB (Fig 9E). As the rescue of sae2Δ by Tel1-N2021D requires the physical presence of the MRX complex, this Tel1 function in promoting MRX association to DSBs can explain the inability of tel1Δ to bypass Sae2 function in DNA damage resistance and resection.

Tel1 and Rad53 kinase activities promote Rad9 binding to the DSB ends
The suppression of the DNA damage hypersensitivity of sae2Δ cells by Rad53-H88Y and Tel1-N2021D requires Dna2-Sgs1 (Fig 6B–6G). Because Sgs1-Dna2 activity is counteracted by Rad9, whose lack restores DSB resection in sae2Δ cells [13,14], we asked whether suppression of the DSB resection defect of sae2Δ cells by Rad53 or Tel1 dysfunction might be due to decreased Rad9 association to the DSB ends. We have previously shown that wild type and sae2Δ cells have similar amounts of Rad9 bound at 1.8 kb from the DSB (Fig 10A) [43]. However, a robust increase in the amount of Rad9 bound at 0.2 kb and 0.6 kb from the DSB was detected in sae2Δ cells compared to wild type (Fig 10A) [14]. Strikingly, this enhanced Rad9 accumulation in sae2Δ cells was reduced in the presence of the Rad53-kd or Tel1-kd variant, which both decreased the amount of Rad9 bound at the DSB also in otherwise wild type cells (Fig 10A). Thus, Rad9 association close to the DSB depends on Rad53 and Tel1 kinase activity.

Rad9 inhibits DSB resection by counteracting Sgs1 recruitment to DSBs [13] and, as expected, Sgs1 binding to DSBs was lower in sae2Δ cells than in wild type (Fig 10B). By contrast, the presence of Rad53-kd or Tel1-kd variants increased the amount of Sgs1 at the DSB in both wild type and sae2Δ cells (Fig 10B). Together with the observation that the suppression of sae2Δ hypersensitivity to genotoxic agents by Rad53 and Tel1 dysfunctions requires Sgs1-Dna2, these findings indicate that the lack of Rad53 or Tel1 kinase activity restores DSB resection in sae2Δ cells by decreasing Rad9 association close to the DSB and therefore by relieving Sgs1-Dna2 inhibition. Although both rad53-kd and tel1-kd cells showed some lowering of Rad9 binding at DSBs compared to wild type cells (Fig 10A), they did not appear to accelerate SSA, suggesting that this extent of Rad9 binding is anyhow sufficient to limit resection in a wild type context.
Rad9 is known to be enriched at the sites of damage by interaction with histone H2A that has been phosphorylated on serine 129 (γH2A) by Mec1 and Tel1 [50–53]. As the lack of γH2A suppresses the SSA defect of sae2Δ cells [14], Tel1 activity might increase the amount of Rad9 bound at the DSB in sae2Δ cells by promoting generation of γH2A. Indeed, the hta1-S129A allele, which encodes a H2A variant where Ser129 is replaced by a non-phosphorylatable alanine residue, thus causing the lack of γH2A, suppressed the resection defect of sae2Δ cells (S3 Fig). Furthermore, γH2A formation turned out to be responsible for the enhanced Rad9 binding close to the break site, as sae2Δ hta1-S129A cells showed wild type levels of Rad9 bound at the DSB (Fig 10C). Finally, γH2A formation close to the DSB depends on Tel1 kinase activity, as γH2A at the DSB was not detectable in sae2Δ tel1-kd cells (Fig 10D). Altogether, these data indicate that Tel1 promotes Rad9 association to DSB in sae2Δ cells through γH2A generation.
Discussion
Cells lacking Sae2 not only are defective in DSB resection, but also show persistent DSB-induced checkpoint activation that causes a prolonged cell cycle arrest. This enhanced checkpoint signaling is due to persistent MRX binding at the DSBs, which activates a Tel1-dependent checkpoint that is accompanied by Rad53 phosphorylation [20,22]. While failure to remove MRX from the DSBs has been shown to sensitize sae2Δ cells to genotoxic agents [23,24], the possible contribution of the DNA damage checkpoint in determining the DNA damage hypersensitivity and the resection defect of sae2Δ cells has never been studied in detail.
We show that impairment of Rad53 activity either by affecting its interaction with Rad9 (Rad53-H88Y) or by abolishing its kinase activity (Rad53-kd) suppresses the sensitivity to DNA damaging agents of sae2Δ cells. A similar effect can be detected also when Tel1 function is compromised either by reducing its recruitment to DSBs (Tel1-N2021D) or by abrogating its kinase activity (Tel1-kd). These suppression effects are not due to the escape of the checkpoint-mediated cell cycle arrest, as CHK1 deletion, which overrides the persistent cell cycle arrest of sae2Δ cells, does not suppress the hypersensitivity of the same cells to DNA damaging agents. Rather, we found that impairment of Rad53 or Tel1 signaling suppresses the resection defect of sae2Δ by decreasing the amount of Rad9 bound very close to the break site. As it is known that Rad9 inhibits Sgs1-Dna2 [13,14], this reduced Rad9 association at DSBs relieves inhibition of Sgs1-Dna2 activity that can then compensate for the lack of Sae2 function in DSB resection. In this view, active Rad53 and Tel1 increase the requirement for Sae2 in DSB resection by promoting Rad9 binding to DSBs and therefore by inhibiting Sgs1-Dna2. Consistent with a role of Sgs1 in removing MRX from the DSBs [54], the relieve of Sgs1-Dna2 inhibition by Rad53 or Tel1 dysfunction leads to a reduction of MRX association to DSBs in sae2Δ cells.
Our finding that Tel1 or Rad53 inactivation can restore both DNA damage resistance and DSB resection in sae2Δ cells is apparently at odds with previous findings that attenuation of the Rad53-dependent checkpoint signaling by decreasing MRX association to DSBs suppresses the DNA damage hypersensitivity of sae2Δ cells but not their resection defect [23,24]. Noteworthy, the bypass of Sae2 function by Rad53 or Tel1 dysfunction requires the physical presence of MRX bound at DSBs, which is known to promote stable association of Exo1, Sgs1 and Dna2 to DSBs [10]. Thus, we speculate that a reduced MRX association at DSBs allows sae2Δ cells to initiate DSB resection by relieving Rad9-mediated inhibition of Sgs1-Dna2 activity. As DSB repair by HR has been shown to require limited amount of ssDNA at DSB ends [55,56], the ssDNA generated by this initial DSB processing might be sufficient to restore DNA damage resistance in sae2Δ cells even when wild type levels of resection are not restored because DSB-bound MRX is not enough to ensure stable Sgs1 and Dna2 association.
Surprisingly, TEL1 deletion, which relieves the persistent Tel1-dependent checkpoint activation caused by the lack of Sae2, did not restore DNA damage resistance and DSB resection in sae2Δ cells. We found that the lack of Tel1 protein affects the association of MRX to the DSB ends independently of its kinase activity. As the rescue of sae2Δ by Tel1-N2021D requires the physical presence of the MRX complex, this reduced MRX-DNA association can explain the inability of TEL1 deletion to restore DNA damage resistance and resection in sae2Δ cells. Therefore, while an enhanced Tel1 signaling activity in the absence of Sae2 leads to DNA damage hypersensitivity and resection defects, a sufficient amount of Tel1 needs to be present at DSBs to support MRX function at DSBs.
How do Rad53 and Tel1 control Rad9 association to DSB? Rad53-mediated phosphorylation of Rad9 does not appear to promote Rad9 binding to the DSB [57,58]. Because Rad53 and RPA compete for binding to Sgs1 [59], it is tempting to propose that impaired Rad53 signaling activity might shift Sgs1 binding preference from Rad53 to RPA, leading to increased Sgs1 association to RPA-coated DNA that can counteract Rad9 binding and inhibition of resection. In turn, Tel1 and Mec1 can phosphorylate Rad9 [60,61], and abrogation of these phosphorylation events rescues the sensitivity to DNA damaging agents of sae2Δ cells [14], suggesting that Tel1 might control Rad9 association to DSBs directly through phosphorylation. On the other hand, Tel1 promotes generation of γH2A [50–53], which counteracts DSB resection by favoring Rad9 association at the DSB [43]. We show that expression of a non-phosphorylatable H2A variant in sae2Δ cells suppresses their resection defect and prevents the accumulation of Rad9 at the DSB. Furthermore, γH2A generation close to the break site depends on Tel1 kinase activity. Thus, although we cannot exclude a direct control of Tel1 on Rad9 association to DNA ends, our findings indicate that Tel1 acts in this process mostly through γH2A generation.
Altogether, our results support a model whereby Tel1 and Rad53, once activated, limit DSB resection by promoting Rad9 binding to DSBs and therefore by inhibiting Sgs1-Dna2. Sae2 activates Mre11 endonucleolytic activity that clips the 5’-terminated DNA strand, thus generating 5’ and 3’ tailed substrates that can be processed by Exo1/Sgs1-Dna2 and Mre11 activity, respectively (Fig 10E, left). When Sae2 function fails, defective Mre11 nuclease activity causes increased MRX persistence at the DSB that leads to enhanced and prolonged Tel1-dependent Rad53 activation. As a consequence, Tel1 - and Rad53-mediated phosphorylation events increase the amount of Rad9 bound at the DSB, which inhibits DSB resection by counteracting Sgs1-Dna2 activity (Fig 10E, middle). Dysfunction of Rad53 or Tel1 reduces Rad9 recruitment at the DSB ends and therefore relieves inhibition of Sgs1-Dna2, which can compensate for the lack of Sae2 in DNA damage resistance and resection (Fig 10E, right). Altogether, these findings indicate that the primary cause of the resection defect of sae2Δ cells is an enhanced Rad9 binding to DSBs that is promoted by the persistent MRX-dependent Tel1 and Rad53 signaling activities.
ATM inhibition has been proposed as a strategy for cancer treatment [62]. Therefore, the observation that dampening Tel1/ATM signaling activity restores DNA damage resistance in sae2Δ cells might have implications in cancer therapies that use ATM inhibitors for synthetic lethal approaches to threat tumors with deficiencies in the DNA damage response.
Materials and Methods
Yeast strains
The yeast strains used in this study are derivatives of W303, JKM139 and YMV45 strains and are listed in S1 Table. Cells were grown in YEP medium (1% yeast extract, 2% peptone) supplemented with 2% glucose (YEPD), 2% raffinose (YEPR) or 2% raffinose and 3% galactose (YEPRG).
Search for suppressors of sae2Δ sensitivity to CPT
To search for suppressor mutations of the CPT-sensitivity of sae2Δ mutant, 5x106 sae2Δ cells were plated on YEPD in the presence of 30μM CPT. Survivors were crossed to wild type cells to identify by tetrad analysis the suppression events that were due to single-gene mutations. Genomic DNA from two single-gene suppressors was analyzed by next-generation Illumina sequencing (IGA technology services) to identify mutations altering open reading frames within the reference S. cerevisiae genome. To confirm that rad53-H88Y and tel1-N2021D mutations were responsible for the suppression, either URA3 or HIS3 gene was integrated downstream of the rad53-H88Y and tel1-N2021D stop codon, respectively, and the resulting strain was crossed to wild type cells to verify by tetrad dissection that the suppression of the sae2Δ CPT sensitivity co-segregated with the URA3 or HIS3 allele.
DSB resection and repair by SSA
DSB end resection at the MAT locus in JKM139 derivative strains was analyzed on alkaline agarose gels as previously described [63]. DSB formation and repair in YMV45 strain were detected by Southern blot analysis using an Asp718-SalI fragment containing part of the LEU2 gene as a probe as previously described [63]. Quantitative analysis of the repair product was performed by calculating the ratio of band intensities for SSA product with respect to a loading control.
Other techniques
Protein extracts for western blot analysis were prepared by TCA precipitation. ChIP assays were performed as previously described [64]. Data are expressed as fold enrichment at the HO-induced DSB over that at the non-cleaved ARO1 locus, after normalization of each ChIP signals to the corresponding amount of immunoprecipitated protein and input for each time point. Fold enrichment was then normalized to the efficiency of DSB induction. The kinase assay and coimmunoprecipitation were performed as previously described [48]. Rad53 was detected by using anti-Rad53 polyclonal antibodies (ab104232) from Abcam. γH2A was immunoprecipitated by using anti-γH2A antibodies (ab15083) from Abcam.
Supporting Information
Zdroje
1. Gobbini E, Cesena D, Galbiati A, Lockhart A, Longhese MP (2013) Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair 12 : 791–799. doi: 10.1016/j.dnarep.2013.07.009 23953933
2. Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40 : 179–204. doi: 10.1016/j.molcel.2010.09.019 20965415
3. Mehta A, Haber JE (2014) Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 6: a016428. doi: 10.1101/cshperspect.a016428 25104768
4. Symington LS, Gautier J (2011) Double-strand break end resection and repair pathway choice. Annu Rev Genet 45 : 247–271. doi: 10.1146/annurev-genet-110410-132435 21910633
5. Cannavo E, Cejka P (2014) Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514 : 122–125. doi: 10.1038/nature13771 25231868
6. Mimitou EP, Symington LS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455 : 770–774. doi: 10.1038/nature07312 18806779
7. Zhu Z, Chung WH, Shim EY, Lee SE, Ira G (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134 : 981–994. doi: 10.1016/j.cell.2008.08.037 18805091
8. Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, et al. (2010) DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467 : 112–116. doi: 10.1038/nature09355 20811461
9. Niu H, Chung WH, Zhu Z, Kwon Y, Zhao W, Chi P, et al. (2010) Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 467 : 108–111. doi: 10.1038/nature09318 20811460
10. Shim EY, Chung WH, Nicolette ML, Zhang Y, Davis M, Zhu Z, et al. (2010) Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J 29 : 3370–3380. doi: 10.1038/emboj.2010.219 20834227
11. Mimitou EP, Symington LS (2010) Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 29 : 3358–3369. doi: 10.1038/emboj.2010.193 20729809
12. Foster SS, Balestrini A, Petrini JH (2011) Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol Cell Biol 31 : 4379–4389. doi: 10.1128/MCB.05854-11 21876003
13. Bonetti D, Villa M, Gobbini E, Cassani C, Tedeschi G, Longhese MP (2015) Escape of Sgs1 from Rad9 inhibition reduces the requirement for Sae2 and functional MRX in DNA end resection. EMBO Rep 16 : 351–361. doi: 10.15252/embr.201439764 25637499
14. Ferrari M, Dibitetto D, De Gregorio G, Eapen VV, Rawal CC, Lazzaro F, et al. (2015) Functional interplay between the 53BP1-ortholog Rad9 and the Mre11 complex regulates resection, end-tethering and repair of a double-strand break. PLoS Genet 11: e1004928. doi: 10.1371/journal.pgen.1004928 25569305
15. Keeney S, Kleckner N (1995) Covalent protein-DNA complexes at the 5' strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci USA 92 : 11274–11278. 7479978
16. Usui T, Ohta T, Oshiumi H, Tomizawa J, Ogawa H, Ogawa T (1998) Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell 95 : 705–716. 9845372
17. Liu C, Pouliot JJ, Nash HA (2002) Repair of topoisomerase I covalent complexes in the absence of the tyrosyl-DNA phosphodiesterase Tdp1. Proc Natl Acad Sci USA 99 : 14970–14975. 12397185
18. Deng C, Brown JA, You D, Brown JM (2005) Multiple endonucleases function to repair covalent topoisomerase I complexes in Saccharomyces cerevisiae. Genetics 170 : 591–600. 15834151
19. Mantiero D, Clerici M, Lucchini G, Longhese MP (2007) Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep 8 : 380–387. 17347674
20. Usui T, Ogawa H, Petrini JH (2001) A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 7 : 1255–1266. 11430828
21. Lisby M, Barlow JH, Burgess RC, Rothstein R (2004) Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118 : 699–713. 15369670
22. Clerici M, Mantiero D, Lucchini G, Longhese MP (2006) The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep 7 : 212–218. 16374511
23. Chen H, Donnianni RA, Handa N, Deng SK, Oh J, Timashev LA, et al. (2015) Sae2 promotes DNA damage resistance by removing the Mre11-Rad50-Xrs2 complex from DNA and attenuating Rad53 signaling. Proc Natl Acad Sci USA 112 : 1880–1887.
24. Puddu F, Oelschlaegel T, Guerini I, Geisler NJ, Niu H, Herzog M, et al. (2015) Synthetic viability genomic screening defines Sae2 function in DNA repair. EMBO J 34 : 1509–1522. doi: 10.15252/embj.201590973 25899817
25. Sandell LL, Zakian VA (1993) Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell 75 : 729–739. 8242745
26. Toczyski DP, Galgoczy DJ, Hartwell LH (1997) CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90 : 1097–1106. 9323137
27. Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE (1998) Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94 : 399–409. 9708741
28. Fukunaga K, Kwon Y, Sung P, Sugimoto K (2011) Activation of protein kinase Tel1 through recognition of protein-bound DNA ends. Mol Cell Biol 31 : 1959–1971. doi: 10.1128/MCB.05157-11 21402778
29. Sun Z, Hsiao J, Fay DS, Stern DF (1998) Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 281 : 272–274. 9657725
30. Gilbert CS, Green CM, Lowndes NF (2001) Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol Cell 8 : 129–136. 11511366
31. Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D (2005) Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol 15 : 1364–1375. 16085488
32. Durocher D, Henckel J, Fersht AR, Jackson SP (1999) The FHA domain is a modular phosphopeptide recognition motif. Mol Cell 4 : 387–394. 10518219
33. Bosotti R, Isacchi A, Sonnhammer EL (2000) FAT: a novel domain in PIK-related kinases. Trends Biochem Sci 25 : 225–227. 10782091
34. Baretić D, Williams RL (2014) PIKKs—the solenoid nest where partners and kinases meet. Curr Opin Struct Biol 29 : 134–142. doi: 10.1016/j.sbi.2014.11.003 25460276
35. Mallory JC, Petes TD (2000) Protein kinase activity of Tel1p and Mec1p, two Saccharomyces cerevisiae proteins related to the human ATM protein kinase. Proc Natl Acad Sci USA 97 : 13749–13754. 11095737
36. Ogi H, Goto GH, Ghosh A, Zencir S, Henry E, Sugimoto K (2015) Requirement of the FATC domain of protein kinase Tel1 for localization to DNA ends and target protein recognition. Mol Biol Cell 26 : 3480–3488. doi: 10.1091/mbc.E15-05-0259 26246601
37. Lydall D, Weinert T (1995) Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science 270 : 1488–1491. 7491494
38. Jia X, Weinert T, Lydall D (2004) Mec1 and Rad53 inhibit formation of single-stranded DNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics 166 : 753–764. 15020465
39. Ngo GH, Lydall D (2015) The 9-1-1 checkpoint clamp coordinates resection at DNA double strand breaks. Nucleic Acids Res 43 : 5017–5032. doi: 10.1093/nar/gkv409 25925573
40. Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE (2001) Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell 7 : 293–300. 11239458
41. Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, et al. (1999) Control of the DNA damage checkpoint by Chk1 and Rad53 protein kinases through distinct mechanisms. Science 286 : 1166–1171. 10550056
42. Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, et al. (2002) Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10 : 373–385. 12191482
43. Clerici M, Trovesi C, Galbiati A, Lucchini G, Longhese MP (2014) Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J 33 : 198–216. doi: 10.1002/embj.201386041 24357557
44. Morin I, Ngo HP, Greenall A, Zubko MK, Morrice N, Lydall D (2008) Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J 27 : 2400–2410. doi: 10.1038/emboj.2008.171 18756267
45. Budd ME, Choe Wc, Campbell JL (2000) The nuclease activity of the yeast DNA2 protein, which is related to the RecB-like nucleases, is essential in vivo. J Biol Chem 275 : 16518–16529. 10748138
46. Zhao X, Muller EG, Rothstein R (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2 : 329–340. 9774971
47. Fay DS, Sun Z, Stern DF (1997) Mutations in SPK1/RAD53 that specifically abolish checkpoint but not growth-related functions. Curr Genet 31 : 97–105. 9021124
48. Baldo V, Testoni V, Lucchini G, Longhese MP (2008) Dominant TEL1-hy mutations compensate for Mec1 lack of functions in the DNA damage response. Mol Cell Biol 28 : 358–375. 17954565
49. Hirano Y, Fukunaga K, Sugimoto K (2009) Rif1 and Rif2 inhibit localization of Tel1 to DNA ends. Mol Cell 33 : 312–322. doi: 10.1016/j.molcel.2008.12.027 19217405
50. Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, et al. (2004) Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol 14 : 1703–1711. 15458641
51. Javaheri A, Wysocki R, Jobin-Robitaille O, Altaf M, Côté J, Kron SJ (2006) Yeast G1 DNA damage checkpoint regulation by H2A phosphorylation is independent of chromatin remodeling. Proc Natl Acad Sci USA 103 : 13771–13776. 16940359
52. Toh GW, O'Shaughnessy AM, Jimeno S, Dobbie IM, Grenon M, Maffini S, et al. (2006) Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair 5 : 693–703. 16650810
53. Hammet A, Magill C, Heierhorst J, Jackson SP (2007) Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep 8 : 851–857. 17721446
54. Bernstein KA, Mimitou EP, Mihalevic MJ, Chen H, Sunjaveric I, Symington LS et al. (2013) Resection activity of the Sgs1 helicase alters the affinity of DNA ends for homologous recombination proteins in Saccharomyces cerevisiae. Genetics 195 : 1241–1251. doi: 10.1534/genetics.113.157370 24097410
55. Ira G, Haber JE (2002) Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol 22 : 6384–6392. 12192038
56. Jinks-Robertson S, Michelitch M, Ramcharan S (1993) Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol Cell Biol 13 : 3937–3950. 8321201
57. Naiki T, Wakayama T, Nakada D, Matsumoto K, Sugimoto K (2004) Association of Rad9 with double-strand breaks through a Mec1-dependent mechanism. Mol Cell Biol 24 : 3277–3285. 15060150
58. Usui T, Foster SS, Petrini JH (2009) Maintenance of the DNA-damage checkpoint requires DNA-damage-induced mediator protein oligomerization. Mol Cell 33 : 147–159. doi: 10.1016/j.molcel.2008.12.022 19187758
59. Hegnauer AM, Hustedt N, Shimada K, Pike BL, Vogel M, Amsler P, et al. (2012) An N-terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. EMBO J 31 : 3768–3783. doi: 10.1038/emboj.2012.195 22820947
60. Emili A (1998) MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol Cell 2 : 183–189. 9734355
61. Vialard JE, Gilbert CS, Green CM, Lowndes NF (1998) The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J 17 : 5679–5688. 9755168
62. Cremona CA, Behrens A (2014) ATM signalling and cancer. Oncogene 33 : 3351–3360. doi: 10.1038/onc.2013.275 23851492
63. Trovesi C, Falcettoni M, Lucchini G, Clerici M, Longhese MP (2011) Distinct Cdk1 requirements during single-strand annealing, noncrossover, and crossover recombination. PLoS Genet 7: e1002263. doi: 10.1371/journal.pgen.1002263 21901114
64. Viscardi V, Bonetti D, Cartagena-Lirola H, Lucchini G, Longhese MP (2007) MRX-dependent DNA damage response to short telomeres. Mol Biol Cell 18 : 3047–3058. 17538011
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development
- Metabolomic Quantitative Trait Loci (mQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis
- Genus-Wide Comparative Genomics of Delineates Its Phylogeny, Physiology, and Niche Adaptation on Human Skin
- Encodes Dual Oxidase, Which Acts with Heme Peroxidase Curly Su to Shape the Adult Wing
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy