
QTL Mapping of Sex Determination Loci Supports an Ancient Pathway in Ants and Honey Bees
Whether a developing embryo becomes male or female has significant downstream consequences. Depending on the species, sex can be determined by a wide variety of mechanisms. Sex determination systems can evolve rapidly, but how this occurs, and even how widespread the same mechanism is within a given taxonomic group, remains largely unknown. By experimentally mapping the sex determination architecture in the ant, Vollenhovia emeryi, we found that the well-characterized honey bee sex determination locus originated more than 100 million years ago. However, we also found an additional locus that has no homology to the first. Currently uncharacterized, this locus suggests that different species may use a variety of complementary sex determination mechanisms. Yet, core elements of the complementary sex determination machinery appear to be ancient.
Published in the journal:
. PLoS Genet 11(11): e32767. doi:10.1371/journal.pgen.1005656
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005656
Summary
Whether a developing embryo becomes male or female has significant downstream consequences. Depending on the species, sex can be determined by a wide variety of mechanisms. Sex determination systems can evolve rapidly, but how this occurs, and even how widespread the same mechanism is within a given taxonomic group, remains largely unknown. By experimentally mapping the sex determination architecture in the ant, Vollenhovia emeryi, we found that the well-characterized honey bee sex determination locus originated more than 100 million years ago. However, we also found an additional locus that has no homology to the first. Currently uncharacterized, this locus suggests that different species may use a variety of complementary sex determination mechanisms. Yet, core elements of the complementary sex determination machinery appear to be ancient.
Introduction
One of the major developmental events faced by most organisms is whether they become male or female [1,2]. Mechanisms underlying this choice have major implications for a variety of life history traits, and even the risk of extinction. For example, chromosomal sex determination systems, such as XY or ZW, dictate 50 : 50 sex ratios in the offspring. Other systems, such as haplodiploidy or environmental sex determination, can in principle allow females complete control of the sex ratio. Some sex determination mechanisms even impose ecological costs under certain circumstances. For instance, under complementary sex determination (CSD), individuals heterozygous at one or more CSD loci become females, while those homo - or hemizygous become males [3] (e.g., aculeate Hymenoptera, such as ants, bees, and social wasps). Single-locus CSD systems carry a high penalty for inbreeding (up to 50%), since diploid males resulting from homozygosity at these loci are typically sterile [3], and under CSD, small populations can fall into an extinction vortex [4]. Sex determination systems can evolve rapidly in response to biotic and abiotic conditions that alter sex ratios, such as changes in temperature for environmental sex determiners [5], or in response to bacterial feminizing factors [6]. Consequently, one expects a well-modulated interplay between molecular mechanisms of sex determination, and the ecology of a particular species.
While molecular pathways underlying sex determination are diverse, they nonetheless contain highly conserved elements, but their evolutionary dynamics remain poorly understood. In particular, few examples of turnovers in sex determination mechanisms have been rigorously studied [7]. More specifically in insects, although a wide variety of sex determination mechanisms exist, many of them revolve around a core conserved pathway [1,8]. In the fruit fly (Drosophila melanogaster), where sex determination is best understood, this pathway involves the transformer (tra) gene, which regulates doublesex (dsx), a gene that initiates the female developmental pathway [9]. Interestingly, there is evidence that tra plays a role in sex determination in other species as well [1]. For example, in the jewel wasp, Nasonia vitripennis, sex determination relies on genomic imprinting, with tra mRNA being provided maternally [10,11]. By contrast, in honey bees (Apis mellifera), a homolog of tra called feminizer (fem) underwent duplication to give rise to the complementary sex determiner (csd) gene [12]. The two genes are located adjacent to each other, and csd acts upstream of fem, which in turn acts on dsx. However, with the exception of these two species, little is known about the mechanisms of sex determination and their evolution in other hymenopteran insects.
Although tra and dsx are widely distributed phylogenetically among insects [1], most of the evidence implying their importance has been indirect. Recent comparative genomics studies have noted that many hymenopteran species have tandem copies of tra (as does the honey bee) that show signatures of balancing selection and synteny conservation [13]. Because the tandem tra homologs are widespread, csd/fem was even proposed as the ancestral hymenopteran sex determination system [14]. Although the idea that there exists an ancient CSD mechanism is intriguing, thus far no functional data support this hypothesis. Rather, preliminary data from the fire ant (Solenopsis invicta) suggest that tra/fem do not play a role in CSD in this species [15]. Furthermore, even more recent comparative genomics work has disputed the hypothesis that tra/fem are generally involved in aculate hymenopteran CSD, suggesting separate origins of CSD pathways, and pointing out that in the absence of functional studies, comparative data have limitations [16,17]. Here we test the whether tra/fem are involved in sex determination in hymenopteran insects other than the honey bee by mapping CSD loci in the ant Vollenhovia emeryi.
One reason that functional evidence for the role of tra/fem has been hard to obtain is that many ants, bees, and social wasps have complex life cycles. As a result, it is impossible to rear multiple generations or to cross them experimentally for mapping studies. Species that can be crossed are often adapted to routine inbreeding, and do not produce diploid males [18,19], thus lacking phenotypic diversity necessary for mapping studies. V. emeryi is particularly suited to linkage mapping analysis because of its unusual reproductive system (Fig 1), which involves parthenogenetic reproduction by queens, and androgenetic reproduction by males, the latter arising from eggs that lack the queen’s genome [20,21]. In addition, workers and some queens are produced sexually [22]. Sexually produced queens can be crossed with the paternal clone in the genetic equivalent of a classic backcross, an experimental design we used to investigate the genetics of diploid male production in this species (Fig 1).
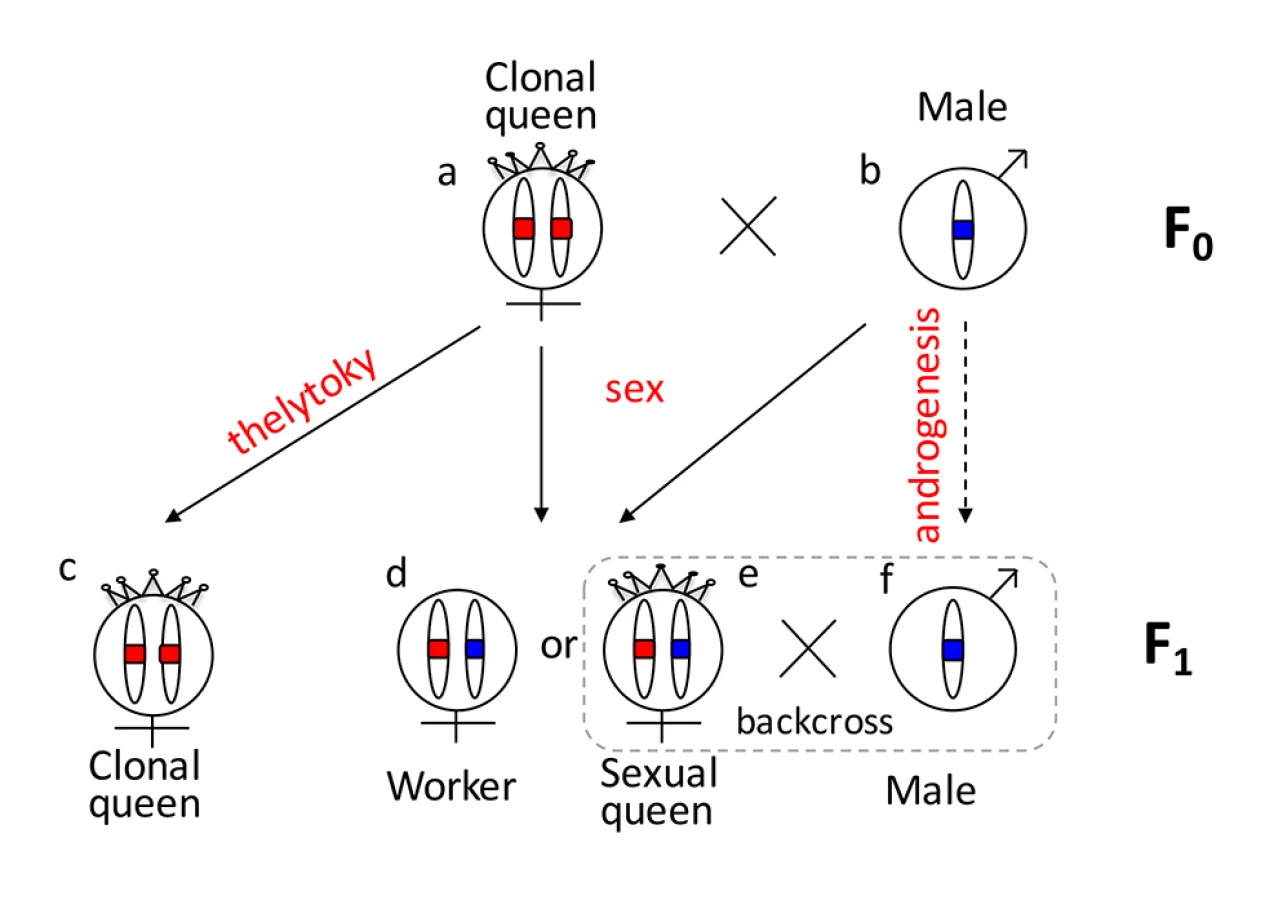
V. emeryi is one of the few ants that can be experimentally crossed in the lab and reared across several generations. It provides an excellent model system to address a range of biological questions, including mechanisms of sex determination, genetic caste determination, and social parasitism [21,23,24]. Here we present a wide range of genomic resources for this species, including a genome, transcriptome, linkage map, and population genomic data, and use them to investigate the mechanism of sex determination. Using QTL mapping we show that worker sex determination in V. emeryi relies on multi-locus CSD (ml-CSD) with two loci, one of them at the same position as duplicated tra homologs, supporting the ancient role of this sex determination mechanism. However, we also uncovered another locus, which is unrelated to the first, and which illustrates the diversity of hymenopteran CSD systems.
Results
Genome assembly and annotation
The genome assembly totaled 287,900,827 bp, including 19,131,583 gaps (47.0X coverage). It had 13,258 scaffolds (N50 1,346,088) and 23,916 contigs (N50 32,417). As evaluated with BUSCO, the genome assembly was largely complete, with only 16 out of 2,675 (0.59%) universal, single-copy orthologs missing, and 64 (2.4%) partial genes. The genome annotation contained 14,870 coding genes, of which 9,239 had a known protein with an alignment covering 50% or more of the query, and 2,671 had an alignment covering 95%. The complete annotation report is available from NCBI (V. emeryi Annotation Release 100). This genome was a major improvement over the previous genome version, which was based only on shotgun Roche 454 data [24], and could be used for linkage mapping.
Diploid male production from inbred and outbred crosses
A quarter (27.1 ± 8.91% SD) of all offspring in inbred crosses were diploid males, while the rest were workers, and a single queen (Table 1). By contrast, of the 1,742 offspring produced by 17 queens mated to males from other populations (outbred crosses) there were no diploid males after ten months of laboratory rearing (S1 Table). The ratio of diploid males produced in inbred crosses was not significantly different from 25%, as predicted for a model with two independent sex determination loci.

Sterility of diploid males
While seminal vesicles of androgenetic haploid males contained sperm, those of diploid males did not, even five months after eclosion, indicating that diploid males were sterile (S1 Fig), and suggesting that inbred queens must invest a quarter of their resources producing reproductively useless males. Correspondingly, inbred queens also produced about 25% fewer workers. Despite the fact that diploid males were immediately removed from the colonies during the experiment, the number of emerged workers per month produced by inbred queens was significantly lower than for outbred queens (8.3 ± 3.06 vs. 13.9 ± 4.80, P < 0.05, F1,21 = 6.81, one-way ANOVA). This suggests that colonies do not compensate for production of diploid males in other ways, such as increasing worker production.
Linkage map and QTL analysis
The cross contained 239 individuals genotyped at 3,541 markers with 1.9% missing data, and a genotype error rate of 0.26%, inferred from quality scores (S1 Data). These markers clustered into 18 linkage groups, consistent with previous karyotype estimates [25]. A genome scan identified two QTLs with large phenotypic effect (CsdQTL1 and CsdQTL2), located on linkage groups 13 and 14, which jointly accounted for almost all of the phenotypic variance in diploid male production (98.0%) (Fig 2). The two regions can be explored interactively using the NCBI genome browser (CsdQTL1: http://goo.gl/oNp5Jq, CsdQTL2: http://goo.gl/8x3MLe). Located on LG14, CsdQTL1 includes three tightly linked [0 centimorgans (cM) apart] markers spanning 38kb and located 20kb from two tandem tra homologs. This arrangement is typical of other ants [13,14]. CsdQTL2 spans 3.2 cM and covers a range of at least 236kb on scaffold NW_011967112.1, which is spanned by eight markers, co-located at two positions on the linkage map. It also includes one marker from scaffold NW_011967235.1, which probably resulted from a scaffolding error, since the rest of that scaffold is on LG2. Although QTL analysis does not explicitly consider the effect of heterozygosity on a phenotype, diploid male phenotypes were associated with homozygosity at these loci (S1 Data), consistent with the proposed mechanism of sex determination. There was no homology between CsdQTL1 and CsdQTL2, suggesting that these loci did not arise by duplication.

Note: The automated NCBI annotation fused the two tra homologs at CsdQTL1. Unfortunately, this error will remain in the genome viewer for the foreseeable future (http://goo.gl/oNp5Jq). However, the NCBI RefSeq database has been updated with the manually corrected nucleotide sequences (traA: NM_001310035.1 and traB: NM_001310036.1).
Population genomic investigation of CSD QTLs
CsdQTL1 contains two tra homologs (traA and traB, NCBI NM_001310035.1 and NM_001310036.1, Fig 3A). Re-sequencing of this QTL region in 10 males and 11 queens showed that the sexual clones differ in protein sequence at both traA and traB (Fig 3B). Divergence of these genes preceded the separation of male and female clones (Fig 3, S2 Fig). In general, the two tra homologs are more similar to each other within a species, relative to other species, making it difficult to determine putative csd and fem genes [13,14], a pattern that also holds true in V. emeryi (S2 Fig). Interestingly traA has much higher diversity than does traB. In honey bees, fem and csd experience stabilizing and diversifying selection, respectively, and copies of csd are more numerous [12,26]. Although this effect is much less pronounced in V. emeryi, MEME analysis found two sites under episodic diversifying selection in traA, but found no such site in traB (S3 Fig). By analogy with honey bees, it seems that given its higher peptidyl variability, and some evidence of diversifying selection, traA may be the homolog of csd. However, queens have no non-synonymous heterozygosity in either traA or traB, and therefore heterozygosity at this locus is not required for female determination in queens. Unlike CsdQTL1, we found no obvious candidate CSD genes for CsdQTL2, which spans 15 annotated genes (Table 2). None of the genes found in this region are homologous to CsdQTL1, indicating that ml-CSD in V. emeryi did not evolve by duplication. A NCBI BLASTN search using default parameters found that the entire scaffold containing CsdQTL2 was highly conserved (79% sequence identity and perfect synteny) in the genome of the little fire ant (Wasmannia auropunctata) (NCBI accession NW_012026774). There were also hits to fire ant scaffolds, but they are not contiguous enough to draw definite conclusions about structural conservation.


Population genetic analysis using microsatellite markers
Sequences of microsatellite markers developed from RAD-tag data, population-level summary statistics, and raw data can be found in S2 Data and S1–S4 Tables. V. emeryi in its native range (in Japan) exists in isolated populations with preponderantly one male and one queen clone in each one (S4 Fig, S4 Table). Even though isolated patches where these ants are found were sometimes separated by only a few meters, each one had a different pair of clones, indicating minimal gene flow between them (S4 Fig, S3 Table). By contrast, male and female clones were the same in experimental invasive US populations, separated by about 10 km, suggesting regionally low genetic diversity (S4 Fig, S3 Table).
Discussion
Sex ratio data and QTL mapping analysis both show that V. emeryi has two unlinked sex determination loci (CsdQTL1 and CsdQTL2) (Figs 3 and 4, Table 1). The CsdQTL1 locus has duplicated tra homologs, as is typical of ants [13] (S2 Fig). This configuration resembles the sex determination locus of honey bees, where csd is located adjacent to fem, with both genes acting in the sex determination pathway [12,27]. As ants and bees diverged more than 100 million years ago [28], sex determination in honey bees and V. emeryi is probably homologous and has been conserved for at least this long. These data are consistent with a hypothesis based on comparative genomic data, which proposed that csd/fem form the core of an ancient pathway in Aculeata, or possibly even a more ancient hymenopteran [14], though investigations of other hymenopteran lineages will be necessary to confirm this hypothesis. It is possible that csd/fem evolves rapidly in species-specific ways as a result of frequent gene conversion [29]. Alternatively, CSD could have evolved separately in both lineages by convergent co-option and duplication of fem for CSD [16]. The latter scenario seems less likely, given the frequent co-occurrence of syntenic tra homologs across a range of hymenoptera [13,14] (S2 Fig), and would require a remarkable convergence in the evolutionary patterns and function of CSD loci in aculeate Hymenoptera.

Population genomic analysis of sex determination loci
Population genomic analysis revealed that V. emeryi males and queens have different protein sequences at CsdQTL1, which ensures that workers are always heterozygous at this locus (Figs 3 and 4). One of the genes, traA, had much higher protein diversity and weak evidence of diversifying selection, suggesting that it may be the csd homolog. Still, levels of allelic diversity in traA and selection were much lower than in the honey bee. This may be the result of V. emeryi’s unusual clonal reproductive system, where males and queens form separate lineages, and clonality fixes diversity at CsdQTL1 between them (Figs 1 and 4). Since every mating is thus guaranteed to produce a heterozygous combination of CSD loci in the F1 offspring, we would not expect diversifying selection to act upon them strongly. Interestingly, the F0 queens did not show protein-level differences at either traA or traB. This suggests either a different sex determination mechanism in the clonal queens vs. their sexually produced daughters, or that the clonal queens are fixed for heterozygosity at some locus linked to CsdQTL2 (Fig 4).
How does the second locus work?
When Crozier proposed the existence of ml-CSD [30], he envisioned that the different loci evolve by duplication and act together as a heteropolymers, similarly to hemoglobin, which consists of paralogous α and β subunits. This does not appear to be the case, as the QTL loci share no homology and appear to act independently (Table 1, Fig 2). However, the actual genomic locus where CsdQTL2 is located appears quite old, being shared with W. auropunctata, representing more than 75 million years of evolutionary divergence [31]. It is presently unclear whether W. auropunctata or other ants use this locus for sex determination, but the fact that it is conserved offers an opportunity for future comparative analysis. QTL data alone cannot provide further functional insight into the actual molecular mechanisms encoded by CsdQTL2. Future work focusing on sex-specific splicing of genes at this locus in F2 workers vs. diploid males during development may shed additional light on the mechanism.
Clonality: A strategy to overcome the limitations of CSD?
Microsatellite analysis shows that in the native range, patches of V. emeryi are highly genetically differentiated (on the scale of meters), with each patch being dominated by different pairs of male and female clones (S4 Fig, S3 Table, S4 Table). Though this should create high potential for inbreeding, a decade of genetic studies has not detected diploid males [21,22,32,33]. The absence of diploid males is not a result of their effective elimination by the colonies, since they are readily produced in our experimental crosses, suggesting that clonality in V. emeryi, and perhaps in other ants with the same reproductive strategy may effectively prevent inbreeding [34]. Completely clonal ants, which often have low rates of recombination, may also retain heterozygosity at sex determination loci [35,36]. Thus, clonality may represent an evolutionary strategy to circumvent limitations posed by ancestral CSD. Many clonal ants are also invasive or live in human-associated habitats [37], suggesting that clonality may also facilitate anthropogenic spread. For instance, most invasions by the highly invasive little fire ant (W. auropunctata) are initiated by single queen clone introductions [38], which may also be the case for the introduction of V. emeryi into the United States. Finally, CSD may explain why queens in species such as V. emeryi and W. auropunctata mate to clonal male lineages rather than producing sons. Separate male and queen clones maintain fixed heterozygosity at CSD loci in workers, allowing them to be produced sexually but without the possibility of inbreeding (although see [39] for alternative view).
Implications for the evolution of CSD loci
More recent work has challenged the original suggestion [14] that csd and fem form the core of an ancient hymenopteran CSD system [16,17]. Critics principally argue that fem and csd homologs do not experience consistent selective pressures across hymenopteran lineages; thus they are not likely part of a conserved CSD pathway. However, multi-locus CSD systems may reconcile the ancient function of csd/fem in CSD with lineage-specific differences. Since only one locus is necessary for sex determination, a ml-CSD system can collapse into a single-locus system if allelic diversity at the other loci is lost [40]. Thus, lineages can potentially converge on different single-locus CSD systems. This may have happened in the fire ant, which does not appear to use tra/fem, at least in the invasive range where it was studied [15], and where there was ample opportunity for allelic loss. Since ml-CSD is known from other Hymenoptera [40], and the CsdQTL2 region is conserved, it is even possible that the mechanism encoded by CsdQTL2 is part of ancestral CSD that was lost in the honey bee. Detailed mapping studies in other Hymenoptera including basal taxa will be needed to fully resolve the evolution of csd/fem and other CSD loci.
Materials and Methods
Samples used in the study
V. emeryi nests in slightly wet, fallen branches in secondary forests. Field surveys confirmed that all sites were composed of colonies containing clonal, short-winged queens [23]. Because queens do not have functional wings, colonies are typically propagated by budding after intra-colony mating, and occur in dense, but isolated patches, which we call ‘sites’ in the present study [41]. We collected live V. emeryi colonies from the native range at one site in Tokyo and four sites in Ishikawa Prefecture in Japan, from May to September 2013. We also genotyped invasive populations from three sites in near Rockville, Maryland, USA, collected in 2012. Experimental colonies were provided dry crickets, sugar water, and distilled water every other day. These colonies were kept in artificial plaster nests at 25 C, 50–60% humidity and a 12-hour light/dark cycle.
Genome and transcriptome sequencing, assembly, and annotation
The reference genome was shotgun sequenced from two sets of PCR-free libraries (GS FLX+ library preparation kit for Roche, and TruSeq DNA PCR-free sample preparation kit for Illumina), and several mate-pair libraries (GS FLX paired-end kit (Roche)). One set of libraries, made from a single male clone, was sequenced on the Roche 454 FLX+ platform, yielding 6,216,952 reads containing 3.6 gigabases of sequence. Another set of libraries was prepared from another male using an Illumina TruSeq kit with 6 amplification cycles, and sequenced using an Illumina MiSeq in paired-end 2x300 mode. The two paired ends were subsequently merged using PEAR (parameters:—min-overlap 10 -n 200 -m 600 -p 0.0001) [42] to produce 23,078,792 (10.7 gigabases) high-quality super-reads (440 ± 80 bp), which are similar to 454 reads in length distribution. Both libraries were assembled using Newbler (v. 2.6), which was developed specifically for the 454’s medium-length reads (parameters: -large -m -cpu 10 -mi 95 -siom 390 -l 1000 -a 500 -urt -novs -a 1000) [43]. We also sequenced mate-paired libraries for scaffolding, which were prepared using Illumina kits (two 3.5 kb libraries, two 5.5 kb libraries, two 8.5 kb libraries and one 14 kb library). We sequenced approximately 600,000 read pairs in every library. The Newbler assembly was scaffolded using SSPACE (3.0) (parameters: -z 1000 -p 1 -x 1 -v 1) [44].
For purposes of annotation, we also sequenced RNA-seq libraries prepared from males, queens, and workers (5 replicates each) collected at sites A-D (S4 Fig). The libraries were prepared as in Aird et al. [45] and were sequenced on an Illumina HiSeq 2000 in paired-end 100 cycle mode. RNA-seq libraries yielded an average 7,086 ± 1,167 mb of data. Scaffolds were annotated using NCBI’s automated genome annotation pipeline, which takes advantage of species-specific RNA-seq data, as well as extensive protein homology data stored in GenBank [46].
Experimental crosses
Colonies collected at site A (S4 Fig) were kept in the laboratory until new reproductives emerged. Wings of new reproductives were genotyped to classify them into two groups: short-winged queens produced through parthenogenesis, and long-winged queens produced sexually [22]. We crossed sexually produced queens, which had both parental genomes, with their brothers, which had only the paternal genetic contribution. This cross was equivalent to a classic backcross between inbred lines, and produced inbred F2 offspring (Fig 4).
For crosses, one to five queens were placed in small artificial nests with one male, and with a few workers for three days under constant light. After that, queens were isolated and kept in new artificial nests with 20 to 30 workers. Six of the twenty experimental queens mated with males and started to lay eggs. Eggs and larvae produced by sib-mated queens were transferred to other nursing colonies containing only workers, until offspring emerged. To compare rates of diploid male production for inbred and outbred colonies, we also set up 21 crosses between sexually produced queens from site A and males from three other sites (S4 Fig, Site B, C & D). Nineteen of the twenty-one experimental queens mated with males and started to lay eggs. Because two of the 19 mated queens died before laying eggs, 17 queens were used for experimental outbred crosses.
Observation of testicular development and sperm production
To determine fertility of diploid males and haploid males, we monitored gonadal and accessory gland development from one to three months after eclosion, paying particular attention to sperm production. Six more diploid males were dissected five months after eclosion. In total, 32 lab-reared diploid and 16 haploid (lab-reared and field collected) males were randomly chosen and dissected.
Dissected testes and accessory glands were treated with 4% PFA for 30 min. After fixation, tissues were washed with 0.1% PBT (PBS and 0.1% Triton X-100) 5 times and mounted on VECTASHIELD Mounting Medium with DAPI (Vector Laboratories, Inc). Tissues under the cover glass were gently compressed during mounting and observed with a Zeiss Axio Scope LED at 400× magnification.
Linkage and QTL mapping
RAD-tag libraries were prepared using the methodology in Tin et al. [47], including removal of duplicate reads. RAD-tag data were sequenced for 288 individuals on an Illumina HiSeq 2500 in single-end 50-bp mode. Raw data were sorted by barcode, and aligned to the V. emeryi genome assembly using bowtie2 [48]. Following alignment, duplicate reads were removed as in Tin et al. [47]. Genotype calls were made with FreeBayes using default parameters [49]. Raw genotypes were then filtered to include only bi-allelic sites with high quality (Q>40) using VCFTools [50]. For every locus we then sequentially dropped individuals with the most missing data, aiming to obtain the largest possible data set subject to the following constraints: (1) all genotype calls should have quality scores of at least 13 (95% accuracy), (2) no more than 5% missing data per locus, (3) minor allele frequency greater than 0.20. This data set was further filtered in R/QTL [51] to eliminate sites with segregation distortion (p<0.01) and four extremely homozygous individuals with (>20% homozygosity). Because our map was made from four families, we also checked to see whether there were families with non-segregating sites, which could skew allele frequencies; there were no such loci. The final data set contained 68 diploid males and 171 workers genotyped at 3,541 loci. The linkage map was computed using MSTMap (parameters: cut_off_p_value 0.00001, no_map_dist 15.0, no_map_size 2, missing_threshold .1, estimation_before_clustering yes, detect_bad_data yes, objective_function COUNT) [52]. QTL analysis was then carried out in R/QTL using a genome scan with a single QTL model and a binary response variable (see S1 Data and S1 Script).
Population genomic analysis of CsdQTL1
In order to investigate species-level variation at this locus, we sequenced 10 male and 11 queen genomes from samples collected in Japan (eight sites including site A and C in S4 Fig), Korea (one site), and the United States (one site in S4 Fig). Libraries were prepared using Nextera XT kits, and sequenced on an Illumina HiSeq 2000 sequencer at an average mapped coverage of 25.5 ± S.D. 4.2. Variants were called using FreeBayes [49], and subjected to several levels of quality filtering. First we used VCFTools [50] to remove sites with quality scores less than 40, and with ≥30% missing data. We then filtered out sites containing indels and sites heterozygous in more than one male genome. We the used the FastaAlternateReferenceMaker module from GATK (v3.3) [53] to convert variant call files to sequence files for subsequent analysis. We then manually annotated traA and traB gene models in Geneious (v. 8.1.3) [54] using its MAFFT (v. 7.017) [55] plugin to create translation alignments of the genes. We then computed the best multiple likelihood protein tree with 100 bootstrap replicates using RAXML (v. 8.1.18) [56] (model PROTCATJTTF). Finally, we conducted MEME [57] analysis for episodic diversifying selection on traA and traB using the DataMonkey server, retaining default settings for the analysis [58].
Development and amplification of microsatellite markers for population genetics
We screened for candidate microsatellite marker loci among perfect repeats showing polymorphism in our backcross RAD-tag data, producing 17 candidate loci. Native and invasive populations were genotyped using these loci, and those from other studies [59]. Because of variable polymorphism, not all loci were useful in all populations, but at least 11 microsatellite markers were used per population (S2 Table).
DNA extractions were performed using 5% Chelex solution, from bodies of queens without their gasters, from whole bodies of males, and from spermathecal contents of mated queens. Samples were incubated in 100 μL Chelex solution for 20 min at 95°C, and stored at 4°C. Instead of using sequence-specific fluorescent primers for each locus, we used universal M13 tails. PCR was performed in 10.05 μL volumes containing 0.2 μL of primer mix (2 μM forward primer with and M13(-21) tail at the 5´-end and 8 μM reverse primer), 1.2 μL universal fluorescent (FAM, HEX, or TAM) labeled M13 primer, 1 μL of diluted DNA, 1 μL of ExTaq 10x buffer, 1 μL of 2.5 mM dNTP, 0.05 μL of ExTaq enzyme, and 5.2 μL of RNAse-free water. PCR amplification conditions are as follows: 94°C (5 min), then 30 cycles at 94°C (30 s) / 58°C (45 s) / 72°C (45 s), followed by 8 cycles 94°C (30 s) / 53°C (45 s) / 72°C (45 s), and a final extension at 72°C for 10 min. Fluorescent PCR fragments were visualized by capillary electrophoresis on an ABI 3100xl Genetic Analyzer (Applied Biosystems). Genotypes were scored manually using GeneMarker [60].
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Verhulst EC, van de Zande L, Beukeboom LW. Insect sex determination: it all evolves around transformer. Curr Opin Genet Dev. 2010;20 : 376–383. doi: 10.1016/j.gde.2010.05.001 20570131
2. Beukeboom LW, Perrin N. The evolution of sex determination. Oxford University Press; 2014.
3. Crozier RH. Heterozygosity and sex determination in haplo-diploidy. Am Nat. 1971;105 : 399–412. doi: 10.2307/2459509
4. Zayed A, Packer L, Michener CD. Complementary sex determination substantially increases extinction proneness of haplodiploid populations. Proc Natl Acad Sci USA. 2005;102 : 10742–10746. doi: 10.2307/3376153 16020532
5. Conover DO, Van Voorhees DA, Ehtisham A. Sex ratio selection and the evolution of environmental sex determination in laboratory populations of Menidia menidia. Evolution. 1992;46 : 1722–1730. doi: 10.2307/2410026
6. Rigaud T, Juchault P, Mocquard JP. The evolution of sex determination in isopod crustaceans. Bioessays. 1997.
7. Bachtrog D, Mank JE, Peichel CL, Kirkpatrick M, Otto SP, Ashman T-L, et al. Sex determination: why so many ways of doing it? PLoS Biol. 2014;12: e1001899. doi: 10.1371/journal.pbio.1001899 24983465
8. Heimpel GE, de Boer JG. Sex determination in the hymenoptera. Annu Rev Entomol. 2008;53 : 209–230. doi: 10.1146/annurev.ento.53.103106.093441 17803453
9. Cline TW, Meyer BJ. Vive la difference: males vs females in flies vs worms. Annu Rev Genet. 1996;30 : 637–702. doi: 10.1146/annurev.genet.30.1.637 8982468
10. Molbo D, Parker ED. Mating structure and sex ratio variation in a natural population of Nasonia vitripennis. Proc R Soc Lond B Biol. 1996;263 : 1703–1709. doi: 10.1098/rspb.1996.0249
11. Beukeboom LW, van de Zande L. Genetics of sex determination in the haplodiploid wasp Nasonia vitripennis (Hymenoptera: Chalcidoidea). J Genet. 2010;89 : 333–339. 20877000
12. Hasselmann M, Gempe T, Schiøtt M, Nunes-Silva CG, Otte M, Beye M. Evidence for the evolutionary nascence of a novel sex determination pathway in honeybees. Nature. 2008;454 : 519–522. doi: 10.1038/nature07052 18594516
13. Privman E, Wurm Y, Keller L. Duplication and concerted evolution in a master sex determiner under balancing selection. Proc R Soc Lond B Biol. 2013;280 : 20122968. doi: 10.1098/rspb.2012.2968
14. Schmieder S, Colinet D, Poirié M. Tracing back the nascence of a new sex-determination pathway to the ancestor of bees and ants. Nat Comm. 2012;3 : 895. doi: 10.1038/ncomms1898
15. Huang Y-C, Nipitwattanaphon M, Lee C-C, Wang J. Mapping a novel sex determination gene in ants. IUSSI Congress, Cairns; 2014. p. 197.
16. Koch V, Nissen I, Schmitt BD, Beye M. Independent evolutionary origin of fem paralogous genes and complementary sex determination in hymenopteran insects. PLoS ONE. 2014;9: e91883. doi: 10.1371/journal.pone.0091883 24743790
17. Biewer M, Schlesinger F, Hasselmann M. The evolutionary dynamics of major regulators for sexual development among Hymenoptera species. Front Genet; 2015;6 : 124. doi: 10.3389/fgene.2015.00124 25914717
18. Schmidt AM, Linksvayer TA, Boomsma JJ, Pedersen JS. No benefit in diversity? The effect of genetic variation on survival and disease resistance in a polygynous social insect. Ecol Entomol. 2011;36 : 751–759. doi: 10.1111/j.1365-2311.2011.01325.x
19. Schrempf A, Aron S, Heinze J. Sex determination and inbreeding depression in an ant with regular sib-mating. Heredity. 2006;97 : 75–80. doi: 10.1038/sj.hdy.6800846 16705320
20. Kobayashi K, Hasegawa E, Ohkawara K. Clonal reproduction by males of the ant Vollenhovia emeryi (Wheeler). Entomol Sci. 2008. doi: 10.1111/j.1479-8298.2008.00272.x/pdf
21. Ohkawara K, Nakayama M, Satoh A, Trindl A, Heinze J. Clonal reproduction and genetic caste differences in a queen-polymorphic ant, Vollenhovia emeryi. Biol Let. 2006;2 : 359–363. doi: 10.1098/rsbl.2006.0491
22. Okamoto M, Kobayashi K, Hasegawa E, Ohkawara K. Sexual and asexual reproduction of queens in a myrmicine ant, Vollenhovia emeryi (Hymenoptera: Formicidae). Myrmecol News. 2015;21 : 13–17.
23. Kinomura K, Yamauchi K. Frequent occurrence of gynandromorphs in the natural population of the ant Vollenhovia emeryi (Hymenoptera: Formicidae). Insect Soc. 1994;41 : 273–278.
24. Smith CR, Helms Cahan S, Kemena C, Brady SG, Yang W, Bornberg-Bauer E, et al. How do genomes create novel phenotypes? Insights from the loss of the worker caste in ant social parasites. Mol Biol Evol. 2015. in press.
25. Imai HT. The chromosome observation techniques of ants and the chromosomes of Formicinae and Myrmicinae. Act Hymenop. 1966; 2 : 119–131
26. Hasselmann M, Lechner S, Schulte C, Beye M. Origin of a function by tandem gene duplication limits the evolutionary capability of its sister copy. Proc Natl Acad Sci USA. 2010;107 : 13378–13383. doi: 10.1073/pnas.1005617107 20624976
27. Beye M, Hasselmann M, Fondrk MK, Page RE Jr, Omholt SW. The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell. 2003;114 : 419–429. 12941271
28. Misof B, Liu S, Meusemann K, Peters RS, Donath A, Mayer C, et al. Phylogenomics resolves the timing and pattern of insect evolution. Science. 2014;346 : 763–767. doi: 10.1126/science.1257570 25378627
29. Privman E, Wurm Y, Keller L. Duplication and concerted evolution in a master sex determiner under balancing selection. Proc R Soc Lond B Biol. 2013;280 : 20122968–20122968. doi: 10.1098/rspb.2012.2968
30. Crozier RH. Heterozygosity and sex determination in haplo-diploidy. Am Nat. 1971;105 : 399–412.
31. Ward PS, Brady SG, Fisher BL, Schultz TR. The evolution of myrmicine ants: phylogeny and biogeography of a hyperdiverse ant clade (Hymenoptera: Formicidae). System Entom. 2015;40 : 61–81. doi: 10.1111/syen.12090
32. Okamoto M, Ohkawara K. Egg production and caste allocation in the clonally reproductive ant Vollenhovia emeryi. Behav Ecol. 2010;21 : 1005–1010. doi: 10.1093/beheco/arq093
33. Kobayashi K, Hasegawa E, Ohkawara K. Clonal reproduction by males of the ant Vollenhovia emeryi (Wheeler). Entomol Sci. 2008;11 : 167–172. doi: 10.1111/j.1479-8298.2008.00272.x
34. Pearcy M, Goodisman MAD, Keller L. Sib mating without inbreeding in the longhorn crazy ant. Proc R Soc Lond B Biol. 2011;278 : 2677–2681. doi: 10.1038/hdy.2009.169
35. Rey O, Loiseau A, Facon B, Foucaud J, Orivel J, Cornuet J-M, et al. Meiotic recombination dramatically decreased in thelytokous queens of the little fire ant and their sexually produced workers. Mol Biol Evol. 2011;28 : 2591–2601. doi: 10.1093/molbev/msr082 21459760
36. Oxley PR, Ji L, Fetter-Pruneda I, McKenzie SK, Li C, Hu H, et al. The genome of the clonal raider ant Cerapachys biroi. Curr Biol. 2014;24 : 451–458. doi: 10.1016/j.cub.2014.01.018 24508170
37. Rabeling C, Kronauer DJC. Thelytokous parthenogenesis in eusocial Hymenoptera. Annu Rev Entomol. 2013;58 : 273–292. doi: 10.1146/annurev-ento-120811-153710 23072461
38. Mikheyev A, Bresson S, Conant P. Single-queen introductions characterize regional and local invasions by the facultatively clonal little fire ant Wasmannia auropunctata. Mol Ecol. 2009;18 : 2937–2944. doi: 10.1111/j.1365-294X.2009.04213.x 19538342
39. Miyakawa MO, Mikheyev AS. Males are here to stay: fertilization enhances viable egg production by clonal queens of the little fire ant (Wasmannia auropunctata). Sci Nat. 2015;102 : 1–7. doi: 10.1007/s00114-015-1265-8
40. de Boer JG, Ode PJ, Rendahl AK, Vet LEM, Whitfield JB, Heimpel GE. Experimental support for multiple-locus complementary sex determination in the parasitoid Cotesia vestalis. Genetics. 2008;180 : 1525–1535. doi: 10.1534/genetics.107.083907 18791258
41. Ohkawara K, Ishii H, Fukushima Y, Yamauchi K, Heinze J. Queen polymorphism and reproductive behaviour in myrmicine ant. Proc XIV Int Congr IUSSI. 2002; p. 206.
42. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30 : 614–620. doi: 10.1093/bioinformatics/btt593 24142950
43. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437 : 376–380. doi: 10.1038/nature03959 16056220
44. Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27 : 578–579. doi: 10.1093/bioinformatics/btq683 21149342
45. Aird SD, Watanabe Y, Villar-Briones A, Roy MC, Terada K, Mikheyev AS. Quantitative high-throughput profiling of snake venom gland transcriptomes and proteomes (Ovophis okinavensis and Protobothrops flavoviridis). BMC Genomics. 2013;14 : 790. doi: 10.1186/1471-2164-14-790 24224955
46. Thibaud-Nissen F, Souvorov A, Murphy T, DiCuccio M, Kitts P. Eukaryotic Genome Annotation Pipeline. The NCBI Handbook. 2nd ed. Bethesda, MD; 2013.
47. Tin MMY, Rheindt FE, Cros E, Mikheyev AS. Degenerate adaptor sequences for detecting PCR duplicates in reduced representation sequencing data improve genotype calling accuracy. Mol Ecol Res. 2014. doi: 10.1111/1755-0998.12314
48. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9 : 357–359. doi: 10.1038/nmeth.1923 22388286
49. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv preprint. 2010.
50. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27 : 2987–2993. doi: 10.1093/bioinformatics/btr509 21903627
51. Broman KW, Wu H, Sen Ś, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19 : 889–890. doi: 10.1093/bioinformatics/btg112 12724300
52. Wu Y, Bhat PR, Close TJ, Lonardi S. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. Kruglyak L, editor. Plos Genet. Public Library of Science; 2008;4: e1000212. doi: 10.1371/journal.pgen.1000212 18846212
53. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2013;11 : 11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
54. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics; 2012;28 : 1647–1649. doi: 10.1093/bioinformatics/bts199 22543367
55. Katoh K, Misawa K, Kuma KI, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30 : 3059–3066. doi: 10.1093/nar/gkf436 12136088
56. Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22 : 2688–2690. doi: 10.1093/bioinformatics/btl446 16928733
57. Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. Detecting individual sites subject to episodic diversifying selection. Plos Genet. 2012;8: e1002764. doi: 10.1371/journal.pgen.1002764 22807683
58. Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26 : 2455–2457. doi: 10.1093/bioinformatics/btq429 20671151
59. Foitzik S, Haberl M, Gadau J, Heinze J. Mating frequency of Leptothorax nylanderi ant queens determined by microsatellite analysis. Insect Soc. 1997;44 : 219–227.
60. Hulce D, Li X, Snyder-Leiby T. GeneMarker genotyping software: tools to increase the statistical power of DNA fragment analysis. J Biomol Tech. 2011;22: S35.
61. Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics. 2012;28 : 2537–2539. doi: 10.1093/bioinformatics/bts460 22820204
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development
- Metabolomic Quantitative Trait Loci (mQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis
- Genus-Wide Comparative Genomics of Delineates Its Phylogeny, Physiology, and Niche Adaptation on Human Skin
- Encodes Dual Oxidase, Which Acts with Heme Peroxidase Curly Su to Shape the Adult Wing
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy