
Genetic Interactions Implicating Postreplicative Repair in Okazaki Fragment Processing
Genome duplication via the process of DNA replication is a prerequisite for cell division and underlies the propagation of all living organisms. This fundamentally important mechanism has been highly conserved throughout eukaryotic evolution, allowing us to use the relatively simple and genetically tractable Saccharomyces cerevisiae as a model to better understand DNA replication in human cells. Furthermore, there is strong evidence to suggest that defects in DNA replication are prominent contributors to mutation and genome instability, a hallmark of cancer. Not surprisingly, evolution has selected for mechanisms to mitigate the effects of defective replication and avoid the most harmful outcomes. Postreplicative repair (PRR) pathways are two such mechanisms with well described functions in promoting the completion of replication under adverse conditions. In this study, we utilized a non-biased genome wide genetic screen to systematically identify conditions under which PRR is required. Our findings indicate that in addition to previously described roles in rescuing DNA synthesis defects, PRR is also required in response to aberrant DNA processing. Specifically, we report a requirement for PRR in cells lacking RAD27, the yeast homolog of the tumor suppressor FEN1. These findings expand the known functions of PRR and reveal their importance in promoting the viability of cells lacking a known tumor suppressor.
Published in the journal:
. PLoS Genet 11(11): e32767. doi:10.1371/journal.pgen.1005659
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005659
Summary
Genome duplication via the process of DNA replication is a prerequisite for cell division and underlies the propagation of all living organisms. This fundamentally important mechanism has been highly conserved throughout eukaryotic evolution, allowing us to use the relatively simple and genetically tractable Saccharomyces cerevisiae as a model to better understand DNA replication in human cells. Furthermore, there is strong evidence to suggest that defects in DNA replication are prominent contributors to mutation and genome instability, a hallmark of cancer. Not surprisingly, evolution has selected for mechanisms to mitigate the effects of defective replication and avoid the most harmful outcomes. Postreplicative repair (PRR) pathways are two such mechanisms with well described functions in promoting the completion of replication under adverse conditions. In this study, we utilized a non-biased genome wide genetic screen to systematically identify conditions under which PRR is required. Our findings indicate that in addition to previously described roles in rescuing DNA synthesis defects, PRR is also required in response to aberrant DNA processing. Specifically, we report a requirement for PRR in cells lacking RAD27, the yeast homolog of the tumor suppressor FEN1. These findings expand the known functions of PRR and reveal their importance in promoting the viability of cells lacking a known tumor suppressor.
Introduction
The accurate copying of a cellular genome and subsequent transmission of genetic material to two daughter cells occurs on a microscopic scale, but is nonetheless a prodigious task. Considering the difficulty of accomplishing this fundamental process for living cells, it is hardly surprising that evolution has selected for a complex and multi-layered system of checkpoints and redundancies that promote its completion under sub-optimal conditions [1,2]. Many of these processes are regulated by post-translational modification of proteins that act as molecular switches to regulate downstream responses.
The replication clamp, proliferating cell nuclear antigen (PCNA), is one such target for a variety of post-translational modifications that trigger an array of downstream effects. Known modifications include the covalent attachment of ubiquitin and the small ubiquitin-like modifier (SUMO) to specific lysine (K) residues [3]. SUMO modification of chromatin-bound PCNA, or sumoylation, occurs during an unperturbed S phase at K164 and–to a lesser extent–at K127 [3]. Sumoylation acts primarily to recruit the helicase/anti-recombinase suppressor of rad six 2 (Srs2), which prevents illegitimate recombination at replication forks [4–6].
Ubiquitination of PCNA occurs predominately at K164, however, alternative attachment sites have been mapped in yeast and human cells [7–11]. In contrast to sumoylation, ubiquitination is induced by replication stress [3]. PCNA-K164 ubiquitination was initially identified as a response to template strand lesions, which stall the highly selective processive polymerases (Pol-) δ and Pol-ε [3]. Polymerase stalling leads to the accumulation of single-stranded (ss) DNA, which quickly becomes coated with replication protein A (RPA) [12]. This allows for the recruitment of the E2-E3 ubiquitin ligase complex radiation sensitive-6 and -18 (Rad6-Rad18) to mono-ubiquitinate PCNA-K164 [13]. Mono-ubiquitin can be subsequently extended to K63-linked poly-ubiquitin chains by methyl methanesulfonate sensitive 2-ubiquitin conjugating 13-radiation sensitive 5 (Mms2-Ubc13-Rad5) [3]. The length of the ubiquitin chain plays a crucial role in determining which of two PRR pathways is activated. Mono-ubiquitin facilitates an error-prone pathway for lesion bypass dependent on translesion polymerase activity [3,14–16]. Recent data indicates that replication past lesions by the mutagenic translesion polymerase Pol-ζ may continue for up to 1 kilobase beyond the lesion [17]. Alternatively, poly-ubiquitin chains enable a template switching pathway in which the nascent DNA of the sister chromatid acts as a template, allowing for lesion bypass and filling of ssDNA gaps [18]. This process is considered to be “error-free”, because it does not rely on the intrinsically mutagenic translesion polymerases of the error-prone pathway [19]. The precise mechanism of this branch is not yet well understood.
In addition to the originally described function of PRR in DNA damage tolerance and lesion bypass, recent work has suggested that mutants with impaired replisome function also activate these pathways for replication of undamaged template strands [20–23]. We have previously demonstrated that PRR promotes the viability of mcm10 mutants in the absence of DNA damage [23]. To systematically explore the role of PCNA-K164 in response to intrinsic cellular dysfunction, we performed SGA analysis of a PCNA-K164 to arginine (PCNA-K164R) mutant. Interestingly, we found that the genetic interaction profile of the PCNA-K164R mutant closely resembled that of many alleles of lagging strand replication factors, including those involved in Okazaki fragment processing. This observation was particularly intriguing, as PRR has not been implicated in tolerating Okazaki fragment processing defects. As a result, we further investigated the activity of PCNA-K164-dependent pathways in mutants disrupting normal lagging strand replication. Specifically, we focused on the role of PCNA-K164 in cells deficient for the flap endonuclease radiation sensitive 27 (Rad27) or enhanced level of genomic instability 1 (Elg1), a homolog of replication factor C (RFC) subunit Rfc1 [24].
Rad27 processes 5’ flaps generated during lagging strand replication when DNA synthesis by Pol-δ collides with the 5’ end of the RNA-DNA primer of the previous Okazaki fragment, displacing it into a small <10-nucleotide (nt) flap [25,26]. If the 5’ flap escapes processing by Rad27 and grows long enough to bind replication protein A (RPA), it is then cleaved by Dna2 [26–28]. RPA binding of the flap serves both to inhibit Rad27, and to recruit Dna2 [27]. Dna2 cleaves the long ~30-nt flap to a short flap (5–10 nt), which can then be further processed by Dna2 or Rad27 into a ligatable nick [25,27–29]. Although processing of long flaps must be relatively efficient under normal conditions, rad27Δ mutants exhibit a temperature dependent slow-growth phenotype [30]. This is best explained by an increased rate in DNA replication and concomitant increase in the formation of long flaps [30,31]. At the restrictive temperature of 37°C, rad27Δ mutants are unable to meet flap processing demands, resulting in lethality, whereas at the semi-permissive temperature of 35°C growth is merely impaired [30,32]. In the absence of complete flap removal–even at lower temperatures–Pol δ-exonuclease (exo) activity can resect the nascent 3’ end allowing the small 5’ flap to re-anneal and form a ligatable nick [26,33]. After ligation of the nick by DNA ligase I (Cdc9), PCNA is unloaded from chromatin by the Elg1:Rfc2-5 complex [34–36]. In the present study, we report that PCNA is ubiquitinated in rad27Δ and elg1Δ mutants. Whereas ablation of PRR is inconsequential in the elg1Δ strain, both translesion synthesis (TLS) and template switching promote rad27Δ viability, possibly by enabling alternative flap processing. Furthermore, the long RPA-coated flaps generated in the absence of Rad27 play an active role in promoting the ubiquitination of PCNA at K164 and initiating PRR.
Results
PCNA-K164R mutants resemble lagging strand replication mutants
To examine the global role of PCNA-K164 in the absence of exogenous DNA replication stressors, we performed SGA analysis of two independently isolated PCNA-K164R mutant clones (identified as PCNA-K164R clone 1 and PCNA-K164R clone 2, respectively) against a library of temperature-sensitive (TS) alleles and a full genome (FG) array as previously described (S1–S4 Tables) [37,38]. Parallel analyses were performed against the TS array using a decreased abundance by mRNA perturbation (DAmP) allele of PCNA or a wild type (WT) allele as the query strain [39,40]. Since ubiquitination or sumoylation of K164 facilitates only a subset of PCNA functions, we anticipated that interactions identified in the PCNA-K164R SGA screens should represent a small part of those identified in the PCNA-DAmP analysis. Indeed the vast majority of hits identified in the K164R mutant screen with the TS array were also identified with the PCNA-DAmP allele (for PCNA-K164R clone 1 : 18/26 hits overlapped with PCNA-DAmP with p-value < 10−11, and for PCNA-K164R clone 2 : 11/14 hits overlapped with p-value < 10−8 as determined by Fisher’s test) (S1 and S2 Tables and Fig 1A). Furthermore, the negative genetic interactions were largely consistent with previous reports, including a requirement for PRR when thiol-specific antioxidant 1 (TSA1) is deficient [41]. Mutants of TSA1 have reduced ability to neutralize reactive oxygen species, leading to increased DNA damage and synthetic sickness with PRR mutation [41]. We also observed a modest requirement for homologous recombination (HR) components in K164R mutants (Fig 1A) [42]. This gave us confidence that genes identified in the PCNA-K164R screens represented bona fide genetic interactions.

The most informative results were revealed when examining the similarity of the PCNA-K164R SGA profiles with the interaction signatures of other mutants. Strong Pearson correlation coefficient (PCC) values between PCNA-K164R clones and rad5Δ and rad6Δ mutants were consistent with the known functions of K164 in facilitating PRR (Fig 1B and Table 1). Strikingly, PCNA-K164R also exhibited strong correlation with many mutant alleles of genes involved in lagging strand replication, suggesting that those mutants have replication defects similar to those in the PCNA-K164R mutants (Fig 1B and Table 1). To validate this observation in an unbiased manner, we performed a Gene Ontology (GO) enrichment analysis (http://www.ebi.ac.uk/QuickGO/). Alleles with SGA profiles similar to that of PCNA-K164R against the FG array (PCC > 0.15) were significantly associated with leading and lagging strand replication GO terms (Fig 1C). Interestingly, the list of enriched GO terms included “Okazaki fragment processing”, which has not been associated with PCNA-K164-dependent pathways (Fig 1C). GO enrichment of SGA profiles similar to PCNA-K164R against the TS array (PCC > 0.2) also showed nearly 25-fold enrichment with the “Okazaki fragment processing” term (S6 Table). Because this initial analysis relied on existing GO annotations, we manually compiled an informed list of genes associated with leading and lagging strand replication for further analysis (S7 Table). We found that PCNA-DAmP and PCNA-K164R profiles against the TS array were highly similar (PCC > 0.2) to profiles of genes implicated in leading and lagging strand replication (Fig 2A–2D). We confirmed these results when we compared the profile of a second PCNA-K164R query strain (Table 1 and S1 Fig). The PCNA-WT profile did not show any significant similarities (Table 1 and S1 Fig).


Altogether, these findings suggested that PCNA-K164 may have an active role in lagging strand replication, even in the absence of exogenous DNA damage. Strong correlations with PCNA-K164R included pol1 mutants, which we previously described to activate PRR pathways, and pol3 mutants (deficient in Pol-δ) which have also been described to elicit PCNA ubiquitination and TLS (Table 1) [20,23]. Additional strong correlations were observed for rfc5, pol31, rad27, and elg1 mutants (Fig 1B and Table 1). These genes have been implicated in different steps of Okazaki fragment synthesis and processing, suggesting that PCNA-K164 is required at multiple junctions when lagging strand replication is impaired. This was surprising, as K164 ubiquitination of PCNA is dependent on the formation of ssDNA and is typically associated with DNA synthesis defects only [13]. The source of ssDNA–particularly in Okazaki fragment processing mutants, such as rad27Δ –was thus not immediately obvious. Nonetheless, these results led us to hypothesize that PCNA-K164-dependent pathways may be required to tolerate defects in lagging strand maturation. Because the function of K164 in PRR is dependent on its modification by ubiquitin, we hypothesized that lagging strand defects would result in chronic PCNA ubiquitination and activation of PRR pathways. To experimentally address this question, we assayed PCNA ubiquitination in rad27Δ and elg1Δ mutants, both of which had interaction profiles that correlated strongly with the PCNA-K164R alleles (Fig 1B and Table 1).
rad27Δ and elg1Δ mutants constitutively ubiquitinate PCNA at K164
rad27Δ is a temperature-sensitive allele, and for all subsequent experiments we shifted these mutants to 37°C for 3 h prior to analysis. To determine whether PCNA is ubiquitinated at K164 in rad27Δ and elg1Δ mutants, we purified histidine tagged PCNA (His6-PCNA) on Ni-NTA agarose and analyzed the eluates with PCNA-, ubiquitin - and SUMO-specific antibodies by western blot (Fig 3A and 3B). PCNA was indeed ubiquitinated in both mutants and ubiquitin attachment was completely ablated when PCNA carried a K164R substitution (Fig 3A and 3B), indicating that alternative attachment sites were not targeted. PCNA-K164R mutants also exhibited loss of K164-dependent sumoylation, consistent with previous reports [3,44]. Interestingly, when we introduced the PCNA-K164R mutation in elg1Δ cells, we reproducibly observed a minor PCNA-SUMO species of a slightly lower molecular weight than K164-SUMO (marked by an asterisk), consistent with an earlier study that revealed an alternative sumoylation site (Fig 3B) [44]. As shown previously, K127-SUMO migrated markedly slower than K164-SUMO. Moreover, levels of K127-SUMO were elevated in PCNA-K164R mutants [3]. Both rad27Δ and elg1Δ exhibited increased PCNA sumoylation at K127 and K164 compared to wild type (Fig 3A and 3B).
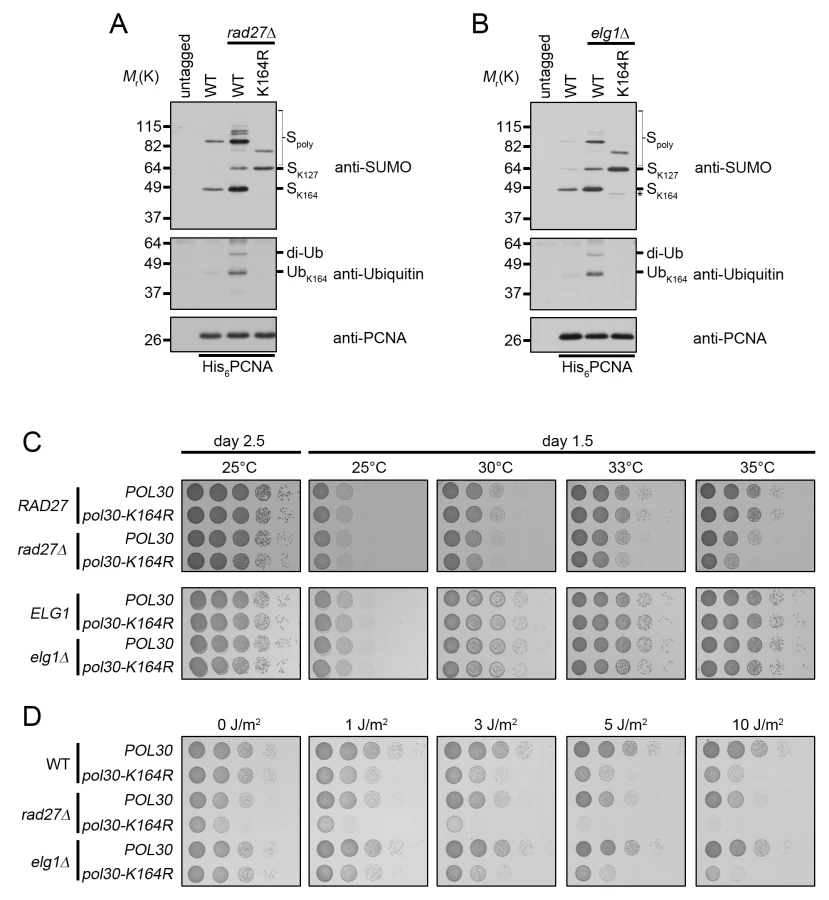
Next we asked whether PCNA-K164 modifications were important for viability of these two strains. Spotting analysis revealed that rad27Δ mutants had a significant reduction in growth at the semi-permissive temperature of 35°C when combined with the PCNA-K164R (pol30-K164R) mutation, whereas elg1Δ cells exhibited no such sensitivity at any temperature tested (Fig 3C), nor when they were exposed to UV light (Fig 3D). In contrast, rad27Δ pol30-K164R double mutants were acutely sensitive even to low doses of UV, showing a severe reduction in growth after exposure to 1J/m2 (Fig 3D). Together, these results suggested that K164-dependent pathways are important for the growth of rad27Δ, but not elg1Δ cells.
TLS and template switching play redundant roles in rad27Δ mutants
Because ubiquitination of PCNA at K164 is necessary for both TLS and template switching, we sought to determine which of these pathways are active in rad27Δ cells. Spotting analysis revealed that pol30-K164R and rad18Δ mutations each significantly reduced the viability of rad27Δ cells at 35°C (Fig 4A). The rad27Δ rad18Δ double mutant reproducibly appeared to have a slightly more severe growth defect than the rad27Δ pol30-K164R strain (Fig 4A). We attribute this finding to the known fact that PCNA-K164 sumoylation suppresses HR, and therefore substitution of K164 upregulates HR [4,5]. To address whether sumoylation of PCNA-K164 affected the viability of rad27Δ mutants, we deleted SIZ1, which encodes the SUMO E3 ligase that catalyzes this reaction. Consistent with published reports, rad27Δ siz1Δ double mutants did not exhibit any increased temperature sensitivity [42,45]. These results strongly suggest that the PCNA-K164 dependent phenotype is solely due to the loss of ubiquitination.

To estimate the relative contribution of TLS and template switching to rad27Δ viability, we generated rad27Δ strains with rev3Δ or rad5Δ mutations rendering them deficient in TLS and template switching, respectively. In addition, we analyzed a rad27Δ rev3Δ rad5Δ triple mutant, defective in both branches. We found that rad27Δ rev3Δ double mutants did not display any significant growth defects, whereas the rad27Δ rad5Δ cells exhibited a mild but noticeable growth delay, suggesting that the template switching pathway is the more prominent of the two (Fig 4B). However, loss of both pathways in the rad27Δ rev3Δ rad5Δ triple mutant resulted in a synergistic effect, resembling that of the rad27Δ rad18Δ double mutant (Fig 4A and 4B). These results argue that the two branches of PRR are both active in rad27Δ cells and have partially redundant roles in promoting viability.
The finding that REV3 affected the survival of rad27Δ mutants in the absence of RAD5 encouraged us to further examine the activity of the TLS branch of PRR. To accomplish this, we took advantage of the fact that TLS employs intrinsically mutagenic polymerases, which have a higher rate of nucleotide misincorporation combined with a lack of proofreading activity [46]. We predicted that TLS induced mutations would be dependent on K164. Mutation of K164 to arginine disables DNA synthesis by pol-ζ and its binding partner Rev1, which are responsible for the vast majority of TLS induced mutations [47]. Consistent with previous reports, fluctuation analysis revealed that rad27Δ mutants have a drastically increased rate of mutation (Fig 4C) [31,48]. Addition of the pol30-K164R allele led to a significant decrease in the mutation rate that accounted for 20–25% of total alterations, confirming that TLS was active in these cells (Fig 4C). Because the pol30-K164R mutation also removes the suppressive effect of PCNA-SUMO on HR, an increase in gross chromosomal rearrangements may mask the decrease in mutation rate due to the loss of TLS. Therefore, the K164-dependent reduction is likely an underestimation of the contribution by TLS [4–6]. In agreement with this notion, deletion of the pol-ζ catalytic subunit REV3 results in a more severe reduction in the mutation rate than the pol30-K164R mutation (Fig 4C). Nonetheless, our observations are consistent with a recent report that had estimated point mutations in rad27Δ mutants to account for ~40% of all genomic aberrations [48]. Our results support the idea that the majority of these single nucleotide variations are a result of translesion polymerase activity.
PCNA-K164 suppresses rad27Δ replication defects
To further explore how PCNA-K164 aids in cell survival, we analyzed activation of the Rad53 kinase, a downstream target of the mitotic entry checkpoint kinase 1 (Mec1), the homolog of human ATR (ataxia telangiectasia mutated - and Rad3-related) [49]. rad27Δ pol30-K164R double mutants showed increased phosphorylation of Rad53 relative to rad27Δ cells after they were shifted to the restrictive temperature of 37°C for 3 and 4 h. This was indicative of enhanced replication stress (Fig 5A). Consistently, the double mutants also displayed a robust late S/G2 phase arrest at those time points (Fig 5B). Since rad27Δ cells passed more proficiently through mitosis (indicated by the higher G1 peak marked with a red arrow at 3 and 4 h in Fig 5B), we concluded that PRR pathways likely facilitated the completion of S phase and ultimately allowed for entry into mitosis. Therefore, without PRR cells have a reduced ability to tolerate Rad27 deficiency. Altogether, our findings support a role for PRR pathways in suppressing replication defects when Rad27 is absent. In contrast, elg1Δ pol30-K164R double mutants did not display any Rad53 activation or observable alterations in cell cycle distribution relative to elg1Δ (S2 Fig).

Overexpression of EXO1 eliminated PCNA ubiquitination in rad27Δ
Previous work demonstrated that ubiquitination of PCNA at K164 by Rad6-Rad18 requires persistent regions of RPA-coated ssDNA [13]. These normally accumulate if the replicative polymerases are impeded [12]. However, in the context of Rad27 deficiency, the source of ssDNA was not readily apparent. Earlier studies established that in the absence of Rad27, Okazaki fragment flaps are processed through a “long flap” pathway by the combined activities of Dna2 and Pol3-exo [25,26,33,50,51]. In this pathway short flaps become longer through enhanced strand displacement until they are sufficiently large to be bound by RPA [52,53]. Binding by RPA serves to recruit Dna2 and stimulate its nuclease activity, reducing the flap to approximately 5 nt [27,28]. Although Dna2 has been shown to be competent to subsequently cleave the remaining short flap in vitro [28], Pol3-exo activity is clearly essential in rad27Δ mutants at all temperatures [33]. Pol3-exo is thought to resect the 3’ end of the preceding Okazaki fragment, allowing the remaining 5’ flap to re-anneal and form a ligatable nick [26,33]. It is conceivable that both Dna2 and Pol3-exo contribute to the resolution of short flaps in rad27Δ cells. The binding of RPA to long flap intermediates prior to processing by Dna2 led us to consider that long ssDNA flaps themselves could provide the stimulus for PCNA ubiquitination. We inferred that the close proximity of these RPA-bound structures to PCNA should allow for recruitment of the Rad6-Rad18 complex and subsequent PCNA ubiquitination. To test this hypothesis, we sought to modulate flap processing by overexpression of DNA2 [29]. Because the current model for Dna2 processing of 5’ flaps proposes that RPA binding occurs prior to cleavage, we expected that recruitment of Rad6 and Rad18 may not be significantly reduced upon DNA2 overexpression (Fig 6A) [27,54], unless it would directly compete with the E2-E3 complex. Notably, overexpression of DNA2 did not reduce PCNA ubiquitination in rad27Δ (Fig 6B). We also considered the possibility that Pol3-exo activity during long flap processing could generate ssDNA regions sufficiently large to bind RPA (S3A Fig) However, overexpression of an exonuclease-dead allele of POL3 (pol3-01) failed to reduce PCNA ubiquitination in rad27Δ (S3B Fig). Consistent with previous reports, pol3-01 expression was lethal in combination with rad27Δ (S3C Fig) [55].

We next sought to modulate flap processing in a manner that reduced the formation of long RPA-bound flaps. Multiple studies have demonstrated that overexpression of exonuclease 1 (EXO1) rescues the DNA damage sensitivity of rad27Δ mutants [56–59]. Exo1 and Rad27 are both Rad2 family nucleases and crystal structures of their human homologs, FEN1 and EXO1, reveal highly conserved mechanisms of substrate binding and cleavage [60–62]. Thus, we hypothesized that Exo1, like Rad27, may cleave flaps before RPA can bind to them. If this were true, Exo1 overexpression should reduce PCNA ubiquitination in rad27Δ cells (Fig 6C). Indeed, overexpression of EXO1 from a galactose inducible promoter eliminated PCNA ubiquitination in rad27Δ mutants (Fig 6D). Furthermore, this phenotype was dependent on the catalytic activity of EXO1, as overexpression of a nuclease-dead exo1-D173A allele had no impact on PCNA ubiquitination (Fig 6D) [59,63]. Furthermore, EXO1 overexpression did not rescue the temperature sensitivity of rad27Δ (S4 Fig). In summary, our results suggest that the majority of PCNA ubiquitination in rad27Δ is dependent on RPA-coated ssDNA intermediates, which recruit the Rad6-Rad18 complex and are degraded by Exo1.
PCNA ubiquitination caused by ssDNA gaps is not impacted by EXO1 overexpression
To examine whether the effect of EXO1 overexpression on PCNA ubiquitination in rad27Δ mutants could be due to indirect suppression of ssDNA gap formation, we exposed EXO1 overexpressing cells to 50 J/m2 of UV light, which has been proven to cause ssDNA gaps (Fig 7A) [12]. Overexpression of EXO1 had no impact on the level of PCNA ubiquitination under these conditions (Fig 7B). Moreover, we did not observe any differences in ubiquitination when cells were treated with 100 J/m2 of UV light, arguing that Exo1 did not act directly or indirectly to eliminate ssDNA regions (Fig 7B). In support of this conclusion, overexpression of EXO1 had no impact on PCNA ubiquitination in cells harboring the temperature sensitive pol1-1 allele (Fig 7C and 7D). This allele is thought to generate ssDNA regions as a result of reduced efficiency in the priming of Okazaki fragments (Fig 7C) [64,65], and causes ubiquitination of PCNA at K164 at the non-permissive temperature of 35°C [23]. Taken together, these findings indicate that Exo1 does not suppress the formation of ssDNA gaps.

Discussion
The power of SGA analysis to identify networks of genetic interactors has greatly increased our knowledge of cellular pathway control and carries the considerable advantage of being an unbiased systems-level approach to complex biological questions [67]. However, such screens are useful not only for identifying direct genetic interactors, but also in revealing functional relationships between genes through comparison of their SGA signatures [37,38,68]. Using this type of comparative analysis we identified a pattern of correlation between the profiles of PCNA-K164R mutants and the profiles of several lagging strand replication and Okazaki fragment processing mutants, including rad27Δ (Fig 1B and Table 1). The high degree of similarity between the rad27Δ and PCNA-K164R profiles suggested to us that PCNA-K164-mediated pathways may counteract replication defects in rad27Δ. Proof of PCNA ubiquitination in rad27Δ cells and synthetic sickness in rad27Δ pol30-K164R and rad27Δ rad18Δ, but not rad27Δ siz1Δ double mutants further substantiated this notion and led us to focus on the role of K164 ubiquitination-dependent pathways in the absence of flap endonuclease (Figs 3A and 4A).
Long ssDNA flaps are a platform for PRR activation
The primary replication defect in rad27Δ cells is caused by impaired processing of 5’ flaps generated during Okazaki fragment processing [31]. At elevated temperatures, DNA replication proceeds more rapidly, likely leading to an increase in the formation of long flap intermediates, which must bind RPA before they can be efficiently processed [27]. We speculated that these long RPA-coated flaps may serve as a platform to promote PCNA ubiquitination. To examine this hypothesis, we modulated flap length by overexpression of EXO1, a close relative of RAD27 with a highly conserved mechanism of substrate binding and cleavage [60,61]. A number of prior studies have demonstrated that overexpression of EXO1 suppresses the intrinsic mutagenicity of the rad27Δ allele [56,57]. In particular, EXO1 overexpression suppresses the duplication of short direct repeats that have been hypothesized to result from longer flap structures generated in rad27Δ, which is consistent with Exo1 activity preventing long RPA-coated flap formation [56,57]. Thus, our finding that catalytically active Exo1 counteracts PCNA ubiquitination in rad27Δ, but has no effect on ubiquitination in pol1-1 cells or after UV treatment, argues that the DNA structures mediating ubiquitination in flap endonuclease deficient cells are different from ssDNA gaps (Figs 6 and 7).
It has been speculated that long unprocessed flaps could participate in the replication stress response [10,52,69]. Campbell and colleagues found that constitutive Mec1 activation is responsible for dna2Δ lethality [69]. They hypothesized that Mec1 activation originated from long RPA-coated flaps that accumulate in these mutants [69]. Interestingly, EXO1 overexpression partially rescues the temperature sensitivity of a viable dna2-1 mutant, consistent with the notion that Exo1 acts upstream of long flap formation [70].
Another well-documented hallmark of Okazaki fragment processing mutants is profound instability of trinucleotide repeat (TNR) regions [71,72]. This raises the question as to whether a requirement for PCNA-K164 in rad27Δ mutants is specific to the replication of TNR regions. A previous study had linked error-free PRR to the suppression of TNR expansion in rad27Δ cells [73]. However, error-prone TLS did not appear to regulate TNR expansion at all [73,74]. Our finding that both TLS and error-free PRR are active in rad27Δ cells therefore leads us to conclude that the role of PCNA-K164 in these mutants is not restricted to genomic regions that encompass TNRs (Fig 4B).
PCNA ubiquitination is a sensor of Okazaki fragment processing defects
Historically, PCNA ubiquitination and PRR were considered rescue pathways for template strand lesions that impair polymerase progression and require TLS or template switching for bypass [3]. Later work from the Shcherbakova group demonstrated that intrinsic replisome deficiencies in hypomorphic alleles of the replicative polymerases POL2 and POL3 also lead to PCNA ubiquitination and activation of TLS on undamaged DNA templates [20]. This important finding described ubiquitination of PCNA and activation of PRR in the absence of replication stressors that damage DNA. Nevertheless, the essential effect of DNA damaging agents and hypomorphic polymerases on replication is a disruption of DNA synthesis. Both therefore intuitively lead to ssDNA gaps, triggering PCNA ubiquitination and subsequent gap-filling by PRR.
In contrast, our study describes PCNA ubiquitination under conditions in which DNA synthesis is not impaired. Rad27 catalyzes Okazaki fragment flap cleavage, which does not occur until after Okazaki fragment synthesis, yet we see a requirement for PRR to support the viability of Rad27 deficient cells. This distinction suggests that PCNA-K164 is active in mediating DNA processing defects that are unlinked to problems in DNA synthesis. It is tempting to speculate that PRR pathways are promoting processing of Okazaki fragments by Dna2 or potentially an alternative mechanism. Error-free template switching could allow for synthesis past multiple Okazaki fragments using the sister chromatid as a template and reducing the overall requirement for flap endonuclease. A role for PRR in promoting flap processing and thereby reducing the half-life of long flaps is consistent with our observation of elevated Rad53 phosphorylation in rad27Δ pol30-K164R double mutants (Fig 5A). The mechanism by which PRR promotes flap processing or bypass is currently unclear.
PRR does not sustain survival of elg1Δ mutants
Similar to RAD27, ELG1 has been described as an important protector of genome stability [24,75–78]. Recent reports have identified Elg1 as a crucial component of an alternative RFC complex that unloads PCNA from double-stranded DNA [34,35]. Our SGA screen revealed that the genetic interaction profile of elg1Δ correlates strongly with the PCNA-K164R and PCNA-DAmP profiles, leading us to investigate ubiquitination of PCNA at K164 in this mutant (Figs 1B and 3B and Table 1). Unlike rad27Δ, elg1Δ cells did not exhibit synthetic sickness with the K164R mutation and displayed no acute requirement for PRR pathways to tolerate intrinsic replication stress or mild UV treatment (Fig 3C and 3D). We speculate that replication defects in elg1Δ are mimicking those present in PCNA-DAmP mutants in that both are limiting the amount of free PCNA in the nucleus that is available to load onto chromatin and engage in replication. In the case of the PCNA-DAmP allele this is simply the result of lower steady-state levels of PCNA protein, whereas in elg1Δ, PCNA is likely sequestered on DNA due to diminished unloading [34,35,79].
In summary, our results suggest that during the processing of Okazaki fragments via the long flap pathway, the flap itself is likely not an inert DNA processing intermediate, but may play an active role in signaling replication stress through PCNA. It is conceivable that under normal conditions a division of labor between long and short flap processing is required for efficient Okazaki fragment maturation [27,80–82]. In rad27Δ cells, the balance is pushed severely to the long flap pathway, leading to the accumulation of RPA bound ssDNA structures that can be eliminated by Exo1. This becomes particularly problematic at elevated temperatures when the kinetics of DNA replication are increased and flaps are produced at a higher rate. The mechanism by which PRR is suppressing replication stress under these conditions is not clear at this time, but we speculate that it is helping to circumvent flap processing.
These findings are salient in light of the relationship between the regulation of flap processing and cancer. Homozygous deletion of the RAD27 homolog FEN1 in mice is lethal, but heterozygous deletion in combination with mutation of the adenomatous polyposis coli (Apc) gene results in increased numbers of adenocarcinomas, enhanced tumor progression and decreased survival [83]. Mutations which reduce FEN1 activity have also been demonstrated to vastly increase cancer incidence in mouse models [84]. This study provides molecular evidence for the pathways contributing to mutagenesis when flap endonuclease function is compromised and gives insight into how cells sustain viability under these conditions.
Materials and Methods
Strains and plasmids
All yeast strains used in this study are isogenic derivatives of wild type E133 cells, which were derived from CG379 [85], with the exception of pol1-1 strains which are derived from SSL204 [23]. Strains with gene deletions were generated by PCR mediated gene disruption [86]. All clones were verified by PCR and sequencing of the modified locus. Strains carrying pol30-K164R mutations were generated by PCR mediated gene disruption as follows: continuous PCR fragments consisting of PCNA, its endogenous promoter and a LEU2 marker were amplified from pCH1654 (a gift from L Prakash, UTMB) and integrated at the endogenous PCNA locus. Integration and clonal homogeneity were verified by PCR and sequencing. All strains generated in this study are listed in S8 Table.
His6-tagged PCNA strains were constructed using Yip128-P30-POL30wt (gift from HD Ulrich, IMB Mainz). Plasmid variants with lysine mutations were constructed using the QuikChange Lightning (Agilent Technologies) site-directed mutagenesis kit. Briefly, the plasmid was linearized at an AflII restriction site in the LEU2 coding sequence and transformed. Clones were screened by PCNA western blot to ensure that His6-PCNA expression was equivalent to endogenous (untagged) expression levels. The endogenous copy of PCNA was then knocked out via PCR mediated gene disruption.
In experiments using galactose inducible gene expression, liquid cultures were grown to OD600 = 0.600 at 25°C in raffinose containing medium. Galactose was then added to a final concentration of 2% and the cultures were shifted to 37°C for 3 h before collecting. Overexpression of POL3 and pol3-01 was induced by adding galactose to cells carrying the plasmids pBL336 and pBL336-01, respectively (gifts from D. Gordenin, NIEHS, originally constructed in P.M.J. Burgers laboratory, Washington University in St. Louis) [26]. Expression of DNA2 was induced with galactose in cells carrying pgal-DNA2 (a gift from R. Wellinger, Université de Sherbrooke, Québec) [87]. EXO1 overexpression was induced by adding galactose to cells carrying pcDNA50.1, a derivative of pRS316 that was referred to as gal-EXO1 (a gift from K. Lewis, Texas State University) [58]. The exo1-D173A variant was generated using the QuikChange Lightning (Agilent Technologies) site-directed mutagenesis kit.
UV treatment (254nm) of liquid cultures was applied using a UV crosslinker (CL-1000, UVP). Cultures were transferred to a sterile tray and treated in the crosslinker before being returned to flasks and cultured an additional 40 min before harvesting.
Synthetic Genetic Array (SGA) screen
A genome-wide screen for genetic interactions with four POL30 alleles as query strains (PCNA-DAmP, PCNA-WT, PCNA-K164R Cl.1, and PCNA-K164R Cl.2) was conducted at 30°C as previously described [38]. Because the screens are performed at 30°C they did uncover a synthetic interaction between PCNA-K164R and rad27Δ. Briefly, the query strains, marked with a nourseothricin (NatMX4) resistance cassette and harboring the SGA haploid specific markers and reporter [38], were mated to an array of 786 temperature-sensitive and 175 viable deletion mutants (TS array: manuscript in preparation) (S1 and S2 Tables). Additionally, PCNA-K164R Cl.1 and Cl.2 were mated to an array of 3827 viable deletion mutants (FG array). Nourseothricin - and geneticin-resistant heterozygous diploid mutants were selected and sporulated with MATa pol30 double mutants as described [38]. Results of this screen are also included in the supplementary material (S3 and S4 Tables). Different PCC cutoffs were applied to the FG and TS array data (0.15 and 0.2, respectively) in order to enrich for the top 2% of all profile correlations.
His6-PCNA purification
Cultures were grown to OD600 = 0.600 at 25°C and then shifted to 37°C for 3 h before harvesting (with the exception of elg1Δ strains which remained at 25°C for 3 h after reaching OD600 = 0.600). Cell pellets were frozen at -80°C. Briefly, cells were lysed under denaturing conditions and protein extracts were prepared as previously described [88]. Extracts were incubated rotating overnight at room temperature with Ni-NTA conjugated agarose (Qiagen) to bind His6PCNA. After incubation, His6PCNA-bound beads were washed with buffers of decreasing pH to increase stringency with successive washes. His6-PCNA was eluted from the beads with an EDTA-containing buffer and eluates were fractionated by SDS-PAGE. Purified PCNA and modified forms were then analyzed by western blot using specific antibodies against PCNA, ubiquitin, and SUMO.
Protein extraction and western blotting
Whole cell protein extraction was accomplished by TCA precipitation as previously described and fractionated by SDS-PAGE [89]. Western blots were probed using the following antibodies; anti-PCNA at 1 : 4000 dilution (S871, a gift from B. Stillman, CSHL), anti-SUMO at 1 : 3000 dilution (A gift from X. Zhao, MSKCC), anti-ubiquitin at 1 : 1000 dilution(P4D1, Covance), anti-Rad53 at 1 : 1000 dilution (A gift from JFX Diffley, LRI, UK), anti-phospho-S129 H2A at 1 : 1000 dilution (ab15083, Abcam), and anti-tubulin at 1 : 5000 dilution (MMS-407R, Covance).
Mutation rate analysis
Mutation rates were estimated by measuring the forward rate of mutations at the CAN1 locus that confer resistance to canavanine [90]. Determinations were made from the median of at least 14 independent cultures for each strain. Cultures were inoculated from single colonies in 5 ml of YPD medium and grown to saturation for 5 days at 30°C. Cells were then washed and diluted to appropriate concentrations before plating on medium lacking arginine and containing canavanine (60 mg/L). Dilutions were also plated on non-selective YPD to obtain a viable cell count. Mutation rates were calculated using Drake’s formula as previously described [91,92]. Significance was determined by the Mann Whitney U test as previously described [43].
Cell viability analysis
Relative cell viability was measured using an assay referred to as the “spotting assay”. In this assay, 10 ml cultures were grown to saturation for 4 days at 25°C. Cells were then harvested, washed with water, quantified, and diluted to equal volumes containing 2x107 cells. 10-fold serial dilutions were made from these 2x107 cells in a 96-well plate and then “spotted” on rich medium using a multi-pronged spotting manifold. Replicates were generated for incubation at various temperatures. UV treatment (254nm) was applied where indicated after plating using a UV crosslinker (CL-1000, UVP). Plates were imaged after 1.5 days of growth except where indicated.
Cell cycle analysis
Cell cycle progression was measured by flow cytometry as previously described [8]. Briefly, 1 ml of liquid culture was pelleted and fixed in ice-cold 70% ethanol overnight. DNA was stained with Sytox Green (Invitrogen) and cells were analyzed using a BD Accuri C6 flow cytometer (BD Biosciences). Peaks were quantified using the quantification feature of the BD Accuri C6 software.
Supporting Information
Zdroje
1. Myung K, Chen C, Kolodner RD (2001) Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411 : 1073–1076. 11429610
2. Cremona CA, Sarangi P, Yang Y, Hang LE, Rahman S, et al. (2012) Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the Mec1 checkpoint. Molecular Cell 45 : 422–432. doi: 10.1016/j.molcel.2011.11.028 22285753
3. Hoege C, Pfander B, Moldovan G-L, Pyrowolakis G, Jentsch S (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419 : 135–141. 12226657
4. Pfander B, Moldovan G-L, Sacher M, Hoege C, Jentsch S (2005) SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436 : 428–433. 15931174
5. Papouli E, Chen S, Davies AA, Huttner D, Krejci L, et al. (2005) Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Molecular Cell 19 : 123–133. 15989970
6. Haracska L, Torres-Ramos CA, Johnson RE, Prakash S, Prakash L (2004) Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Molecular and Cellular Biology 24 : 4267–4274. 15121847
7. Xu G, Paige JS, Jaffrey SR (2010) Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nature Biotechnology 28 : 868–873. doi: 10.1038/nbt.1654 20639865
8. Das-Bradoo S, Nguyen HD, Wood JL, Ricke RM, Haworth JC, et al. (2010) Defects in DNA ligase I trigger PCNA ubiquitylation at Lys 107. Nature Cell Biology 12 : 74–79. doi: 10.1038/ncb2007 20010813
9. Povlsen LK, Beli P, Wagner SA, Poulsen SL, Sylvestersen KB, et al. (2012) Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nature Cell Biology 14 : 1089–1098. doi: 10.1038/ncb2579 23000965
10. Nguyen HD, Becker J, Thu YM, Costanzo M, Koch EN, et al. (2013) Unligated Okazaki fragments induce PCNA ubiquitination and a requirement for Rad59-dependent replication fork progression. PloS One 8: e66379. 23824283
11. Elia AE, Boardman AP, Wang DC, Huttlin EL, Everley RA, et al. (2015) Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Molecular Cell 59 : 867–881. doi: 10.1016/j.molcel.2015.05.006 26051181
12. Lopes M, Foiani M, Sogo JM (2006) Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Molecular Cell 21 : 15–27. 16387650
13. Davies AA, Huttner D, Daigaku Y, Chen S, Ulrich HD (2008) Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Molecular Cell 29 : 625–636. doi: 10.1016/j.molcel.2007.12.016 18342608
14. Stelter P, Ulrich HD (2003) Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425 : 188–191. 12968183
15. Garg P, Burgers PM (2005) Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases η and Rev1. Proceedings of the National Academy of Sciences of the United States of America 102 : 18361–18366. 16344468
16. Freudenthal BD, Gakhar L, Ramaswamy S, Washington MT (2010) Structure of monoubiquitinated PCNA and implications for translesion synthesis and DNA polymerase exchange. Nature Structural & Molecular Biology 17 : 479–484.
17. Kochenova OV, Daee DL, Mertz TM, Shcherbakova PV, Jinks-Robertson S (2015) DNA Polymerase ζ-dependent lesion bypass in Saccharomyces cerevisiae is accompanied by error-prone copying of long stretches of adjacent DNA. PLoS Genetics 11: e1005110 doi: 10.1371/journal.pgen.1005110 25826305
18. Branzei D (2011) Ubiquitin family modifications and template switching. FEBS Letters 585 : 2810–2817. doi: 10.1016/j.febslet.2011.04.053 21539841
19. Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D (2010) Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genetics 6: e1001205. doi: 10.1371/journal.pgen.1001205 21085632
20. Northam MR, Garg P, Baitin DM, Burgers PM, Shcherbakova PV (2006) A novel function of DNA polymerase ζ regulated by PCNA. The EMBO Journal 25 : 4316–4325. 16957771
21. Northam MR, Robinson HA, Kochenova OV, Shcherbakova PV (2010) Participation of DNA polymerase ζ in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics 184 : 27–42. doi: 10.1534/genetics.109.107482 19841096
22. Karras GI, Jentsch S (2010) The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141 : 255–267. doi: 10.1016/j.cell.2010.02.028 20403322
23. Becker JR, Nguyen HD, Wang X, Bielinsky A - K (2014) Mcm10 deficiency causes defective-replisome-induced mutagenesis and a dependency on error-free postreplicative repair. Cell Cycle 13 : 1737–1748. doi: 10.4161/cc.28652 24674891
24. Bellaoui M, Chang M, Ou J, Xu H, Boone C, et al. (2003) Elg1 forms an alternative RFC complex important for DNA replication and genome integrity. The EMBO Journal 22 : 4304–4313. 12912927
25. Ayyagari R, Gomes XV, Gordenin DA, Burgers PM (2003) Okazaki fragment maturation in yeast I. Distribution of functions between FEN1 and DNA2. Journal of Biological Chemistry 278 : 1618–1625. 12424238
26. Jin YH, Ayyagari R, Resnick MA, Gordenin DA, Burgers PM (2003) Okazaki Fragment Maturation in Yeast II. Cooperation between the polymerase and 3'-5'-exonuclease activities of Pol δ in the creation of a ligatable nick. Journal of Biological Chemistry 278 : 1626–1633. 12424237
27. Bae S-H, Bae K-H, Kim J-A, Seo Y-S (2001) RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature 412 : 456–461. 11473323
28. Levikova M, Cejka P (2015) The Saccharomyces cerevisiae Dna2 can function as a sole nuclease in the processing of Okazaki fragments in DNA replication. Nucleic Acids Research: gkv710.
29. Bae S-H, Seo Y-S (2000) Characterization of the enzymatic properties of the yeast DNA2 helicase/endonuclease suggests a new model for Okazaki fragment processing. Journal of Biological Chemistry 275 : 38022–38031. 10984490
30. Reagan MS, Pittenger C, Siede W, Friedberg EC (1995) Characterization of a mutant strain of Saccharomyces cerevisiae with a deletion of the RAD27 gene, a structural homolog of the RAD2 nucleotide excision repair gene. Journal of Bacteriology 177 : 364–371. 7814325
31. Tishkoff DX, Filosi N, Gaida GM, Kolodner RD (1997) A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell 88 : 253–263. 9008166
32. Symington LS (1998) Homologous recombination is required for the viability of rad27 mutants. Nucleic Acids Research 26 : 5589–5595. 9837987
33. Jin YH, Obert R, Burgers PM, Kunkel TA, Resnick MA, et al. (2001) The 3′→ 5′ exonuclease of DNA polymerase δ can substitute for the 5′ flap endonuclease Rad27/Fen1 in processing Okazaki fragments and preventing genome instability. Proceedings of the National Academy of Sciences 98 : 5122–5127.
34. Parnas O, Zipin‐Roitman A, Pfander B, Liefshitz B, Mazor Y, et al. (2010) Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. The EMBO Journal 29 : 2611–2622. doi: 10.1038/emboj.2010.128 20571511
35. Kubota T, Nishimura K, Kanemaki MT, Donaldson AD (2013) The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Molecular Cell 50 : 273–280. doi: 10.1016/j.molcel.2013.02.012 23499004
36. Kubota T, Katou Y, Nakato R, Shirahige K, Donaldson AD (2015) Replication-coupled PCNA unloading by the Elg1 complex occurs genome-wide and requires Okazaki fragment ligation. Cell Reports 12 : 774–787. doi: 10.1016/j.celrep.2015.06.066 26212319
37. Li Z, Vizeacoumar FJ, Bahr S, Li J, Warringer J, et al. (2011) Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nature Biotechnology 29 : 361–367. doi: 10.1038/nbt.1832 21441928
38. Baryshnikova A, Costanzo M, Dixon S, Vizeacoumar FJ, Myers CL, et al. (2010) Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae and Schizosaccharomyces pombe. Methods in Enzymology 470 : 145–179. doi: 10.1016/S0076-6879(10)70007-0 20946810
39. Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, et al. (2005) Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123 : 507–519. 16269340
40. Beltrao P, Cagney G, Krogan NJ (2010) Quantitative genetic interactions reveal biological modularity. Cell 141 : 739–745. doi: 10.1016/j.cell.2010.05.019 20510918
41. Huang M-E, Kolodner RD (2005) A biological network in Saccharomyces cerevisiae prevents the deleterious effects of endogenous oxidative DNA damage. Molecular Cell 17 : 709–720. 15749020
42. Motegi A, Kuntz K, Majeed A, Smith S, Myung K (2006) Regulation of gross chromosomal rearrangements by ubiquitin and SUMO ligases in Saccharomyces cerevisiae. Molecular and Cellular Biology 26 : 1424–1433. 16449653
43. Mann HB, Whitney DR (1947) On a test of whether one of two random variables is stochastically larger than the other. The Annals of Mathematical Statistics: 50–60.
44. Windecker H, Ulrich HD (2008) Architecture and assembly of poly-SUMO chains on PCNA in Saccharomyces cerevisiae. Journal of Molecular Biology 376 : 221–231. 18155241
45. Chen XL, Silver HR, Xiong L, Belichenko I, Adegite C, et al. (2007) Topoisomerase I-dependent viability loss in Saccharomyces cerevisiae mutants defective in both SUMO conjugation and DNA repair. Genetics 177 : 17–30. 17603101
46. Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, et al. (2009) Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiology and Molecular Biology Reviews 73 : 134–154. doi: 10.1128/MMBR.00034-08 19258535
47. Nelson JR, Lawrence CW, Hinkle DC (1996) Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science 272 : 1646–1649. 8658138
48. Serero A, Jubin C, Loeillet S, Legoix-Né P, Nicolas AG (2014) Mutational landscape of yeast mutator strains. Proceedings of the National Academy of Sciences 111 : 1897–1902.
49. Osborn AJ, Elledge SJ (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes & Development 17 : 1755–1767.
50. Burgers PM (2009) Polymerase dynamics at the eukaryotic DNA replication fork. Journal of Biological Chemistry 284 : 4041–4045. doi: 10.1074/jbc.R800062200 18835809
51. Garg P, Stith CM, Sabouri N, Johansson E, Burgers PM (2004) Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes & Development 18 : 2764–2773.
52. Budd ME, Reis CC, Smith S, Myung K, Campbell JL (2006) Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase δ. Molecular and Cellular Biology 26 : 2490–2500. 16537895
53. Rossi ML, Pike JE, Wang W, Burgers PM, Campbell JL, et al. (2008) Pif1 helicase directs eukaryotic Okazaki fragments toward the two-nuclease cleavage pathway for primer removal. Journal of Biological Chemistry 283 : 27483–27493. doi: 10.1074/jbc.M804550200 18689797
54. Stewart JA, Miller AS, Campbell JL, Bambara RA (2008) Dynamic removal of replication protein A by Dna2 facilitates primer cleavage during Okazaki fragment processing in Saccharomyces cerevisiae. Journal of Biological Chemistry 283 : 31356–31365. doi: 10.1074/jbc.M805965200 18799459
55. Gary R, Park MS, Nolan JP, Cornelius HL, Kozyreva OG, et al. (1999) A novel role in DNA metabolism for the binding of Fen1/Rad27 to PCNA and implications for genetic risk. Molecular and Cellular Biology 19 : 5373–5382. 10409728
56. Tishkoff DX, Boerger AL, Bertrand P, Filosi N, Gaida GM, et al. (1997) Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proceedings of the National Academy of Sciences 94 : 7487–7492.
57. Sun X, Thrower D, Qiu J, Wu P, Zheng L, et al. (2003) Complementary functions of the Saccharomyces cerevisiae Rad2 family nucleases in Okazaki fragment maturation, mutation avoidance, and chromosome stability. DNA Repair 2 : 925–940. 12893088
58. Lewis LK, Karthikeyan G, Westmoreland JW, Resnick MA (2002) Differential suppression of DNA repair deficiencies of yeast rad50, mre11 and xrs2 mutants by EXO1 and TLC1 (the RNA component of telomerase). Genetics 160 : 49–62. 11805044
59. Moreau S, Morgan EA, Symington LS (2001) Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism. Genetics 159 : 1423–1433. 11779786
60. Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, et al. (2011) Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell 145 : 198–211. doi: 10.1016/j.cell.2011.03.004 21496641
61. Orans J, McSweeney EA, Iyer RR, Hast MA, Hellinga HW, et al. (2011) Structures of human exonuclease 1 DNA complexes suggest a unified mechanism for nuclease family. Cell 145 : 212–223. doi: 10.1016/j.cell.2011.03.005 21496642
62. Miętus M, Nowak E, Jaciuk M, Kustosz P, Studnicka J, et al. (2014) Crystal structure of the catalytic core of Rad2: insights into the mechanism of substrate binding. Nucleic Acids Research 42 : 10762–10775. doi: 10.1093/nar/gku729 25120270
63. Tran PT, Erdeniz N, Dudley S, Liskay RM (2002) Characterization of nuclease-dependent functions of Exo1p in Saccharomyces cerevisiae. DNA Repair 1 : 895–912. 12531018
64. Gutiérrez PJ, Wang TS-F (2003) Genomic instability induced by mutations in Saccharomyces cerevisiae POL1. Genetics 165 : 65–81. 14504218
65. Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D (2015) Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 complex. Molecular Cell 57 : 812–823. doi: 10.1016/j.molcel.2014.12.038 25661486
66. Suzuki M, Niimi A, Limsirichaikul S, Tomida S, Huang QM, et al. (2009) PCNA mono-ubiquitination and activation of translesion DNA polymerases by DNA polymerase α. Journal of Biochemistry 146 : 13–21. doi: 10.1093/jb/mvp043 19279190
67. Tong AHY, Evangelista M, Parsons AB, Xu H, Bader GD, et al. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294 : 2364–2368. 11743205
68. Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, et al. (2008) Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 322 : 597–602. doi: 10.1126/science.1162790 18948542
69. Budd ME, Antoshechkin IA, Reis C, Wold BJ, Campbell JL (2011) Inviability of a DNA2 deletion mutant is due to the DNA damage checkpoint. Cell Cycle 10 : 1690–1698. 21508669
70. Budd ME, Tong AHY, Polaczek P, Peng X, Boone C, et al. (2005) A network of multi-tasking proteins at the DNA replication fork preserves genome stability. PLoS Genetics 1: e61. 16327883
71. Freudenreich CH, Kantrow SM, Zakian VA (1998) Expansion and length-dependent fragility of CTG repeats in yeast. Science 279 : 853–856. 9452383
72. Callahan JL, Andrews KJ, Zakian VA, Freudenreich CH (2003) Mutations in yeast replication proteins that increase CAG/CTG expansions also increase repeat fragility. Molecular and Cellular Biology 23 : 7849–7860. 14560028
73. Daee DL, Mertz T, Lahue RS (2007) Postreplication repair inhibits CAG· CTG repeat expansions in Saccharomyces cerevisiae. Molecular and Cellular Biology 27 : 102–110. 17060452
74. Collins NS, Bhattacharyya S, Lahue RS (2007) Rev1 enhances CAG· CTG repeat stability in Saccharomyces cerevisiae. DNA Repair 6 : 38–44. 16979389
75. Ben-Aroya S, Koren A, Liefshitz B, Steinlauf R, Kupiec M (2003) ELG1, a yeast gene required for genome stability, forms a complex related to replication factor C. Proceedings of the National Academy of Sciences 100 : 9906–9911.
76. Smith S, Hwang J-Y, Banerjee S, Majeed A, Gupta A, et al. (2004) Mutator genes for suppression of gross chromosomal rearrangements identified by a genome-wide screening in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America 101 : 9039–9044. 15184655
77. Kanellis P, Agyei R, Durocher D (2003) Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Current Biology 13 : 1583–1595. 13678589
78. Davidson MB, Katou Y, Keszthelyi A, Sing TL, Xia T, et al. (2012) Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. The EMBO Journal 31 : 895–907. doi: 10.1038/emboj.2011.485 22234187
79. Yu C, Gan H, Han J, Zhou Z-X, Jia S, et al. (2014) Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Molecular Cell 56 : 551–563. doi: 10.1016/j.molcel.2014.09.017 25449133
80. Balakrishnan L, Stewart J, Polaczek P, Campbell JL, Bambara RA (2010) Acetylation of Dna2 endonuclease/helicase and flap endonuclease 1 by p300 promotes DNA stability by creating long flap intermediates. Journal of Biological Chemistry 285 : 4398–4404. doi: 10.1074/jbc.M109.086397 20019387
81. Kao H-I, Veeraraghavan J, Polaczek P, Campbell JL, Bambara RA (2004) On the roles of Saccharomyces cerevisiae Dna2p and Flap endonuclease 1 in Okazaki fragment processing. Journal of Biological Chemistry 279 : 15014–15024. 14747468
82. Rossi ML, Bambara RA (2006) Reconstituted Okazaki fragment processing indicates two pathways of primer removal. Journal of Biological Chemistry 281 : 26051–26061. 16837458
83. Kucherlapati M, Yang K, Kuraguchi M, Zhao J, Lia M, et al. (2002) Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proceedings of the National Academy of Sciences 99 : 9924–9929.
84. Zheng L, Dai H, Zhou M, Li M, Singh P, et al. (2007) Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nature Medicine 13 : 812–819. 17589521
85. Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA (1997) Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Molecular and Cellular Biology 17 : 2859–2865. 9111358
86. Lorenz MC, Muir RS, Lim E, McElver J, Weber SC, et al. (1995) Gene disruption with PCR products in Saccharomyces cerevisiae. Gene 158 : 113–117. 7789793
87. Parenteau J, Wellinger RJ (1999) Accumulation of single-stranded DNA and destabilization of telomeric repeats in yeast mutant strains carrying a deletion of RAD27. Molecular and Cellular Biology 19 : 4143–4152. 10330154
88. Ulrich HD (2009) SUMO protocols: Springer.
89. Ricke RM, Bielinsky A-K (2006) A conserved Hsp10-like domain in Mcm10 is required to stabilize the catalytic subunit of DNA polymerase-α in budding yeast. Journal of Biological Chemistry 281 : 18414–18425. 16675460
90. Whelan WL, Gocke E, Manney TR (1979) The CAN1 locus of Saccharomyces cerevisiae: fine-structure analysis and forward mutation rates. Genetics 91 : 35–51. 372045
91. Drake JW (1991) A constant rate of spontaneous mutation in DNA-based microbes. Proceedings of the National Academy of Sciences 88 : 7160–7164.
92. Foster PL (2006) Methods for determining spontaneous mutation rates. Methods in Enzymology 409 : 195–213. 16793403
93. Ricke RM, Bielinsky A-K (2004) Mcm10 regulates the stability and chromatin association of DNA polymerase-α. Molecular Cell 16 : 173–185. 15494305
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development
- Metabolomic Quantitative Trait Loci (mQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis
- Genus-Wide Comparative Genomics of Delineates Its Phylogeny, Physiology, and Niche Adaptation on Human Skin
- Encodes Dual Oxidase, Which Acts with Heme Peroxidase Curly Su to Shape the Adult Wing
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy