
A Hereditary Enteropathy Caused by Mutations in the Gene, Encoding a Prostaglandin Transporter
Advanced diagnostic innovations such as capsule endoscopy and balloon endoscopy have provided better understanding of endoscopic findings of small bowel diseases. However, it remains difficult to diagnose small intestinal diseases such as Crohn’s disease, intestinal tuberculosis, and nonsteroidal anti-inflammatory drug-induced enteropathy by the endoscopic findings alone. We previously reported a rare autosomal recessive inherited enteropathy characterized by persistent blood and protein loss from the small intestine. This enteropathy has an intractable clinical course with ineffectiveness of immunosuppressive treatment. In this study, we identified recessive mutations in the SLCO2A1 gene, encoding a prostaglandin transporter, as causative variants of this disorder by exome sequencing of four families, and showed that this disease is distinct from Crohn’s disease. We also showed that the mutations found in the patients caused functional impairment of prostaglandin E2 uptake within cells. The present findings suggest that genetic analysis together with detailed clinical information is invaluable for diagnosis of the disease, and that there may be a concept of enteropathy referred to as “prostaglandin-associated enteropathy”, irrespective of ethnic background.
Published in the journal:
. PLoS Genet 11(11): e32767. doi:10.1371/journal.pgen.1005581
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005581
Summary
Advanced diagnostic innovations such as capsule endoscopy and balloon endoscopy have provided better understanding of endoscopic findings of small bowel diseases. However, it remains difficult to diagnose small intestinal diseases such as Crohn’s disease, intestinal tuberculosis, and nonsteroidal anti-inflammatory drug-induced enteropathy by the endoscopic findings alone. We previously reported a rare autosomal recessive inherited enteropathy characterized by persistent blood and protein loss from the small intestine. This enteropathy has an intractable clinical course with ineffectiveness of immunosuppressive treatment. In this study, we identified recessive mutations in the SLCO2A1 gene, encoding a prostaglandin transporter, as causative variants of this disorder by exome sequencing of four families, and showed that this disease is distinct from Crohn’s disease. We also showed that the mutations found in the patients caused functional impairment of prostaglandin E2 uptake within cells. The present findings suggest that genetic analysis together with detailed clinical information is invaluable for diagnosis of the disease, and that there may be a concept of enteropathy referred to as “prostaglandin-associated enteropathy”, irrespective of ethnic background.
Introduction
The use of capsule endoscopy and balloon endoscopy has provided a better understanding of the features of small bowel ulcers among various gastrointestinal disorders, such as Crohn’s disease (CD), intestinal tuberculosis, and nonsteroidal anti-inflammatory drug (NSAID)–induced enteropathy [1,2]. Previously, we proposed a rare clinicopathologic entity characterized by multiple small intestinal ulcers of nonspecific histology and chronic persistent gastrointestinal bleeding as chronic nonspecific multiple ulcers of the small intestine (CNSU) [3,4]. The macroscopic findings of CNSU are characterized by multiple thin ulcers in a linear or circumferential configuration and concentric stenosis, and apparently mimic those of NSAID-induced enteropathy [4–7]. CNSU predominantly occurs in females and the symptoms, such as general fatigue, edema, and abdominal pain, appear during adolescence. The clinical course of the disease is chronic and intractable with reduced effects of immunosuppressive treatment including prednisolone and azathioprine.
Although CNSU predominantly occurs in females, it also appears to be an autosomal recessive inherited disorder because of frequent parental consanguinity [8]. To identify the causative gene for this disorder, we performed whole-exome sequencing and identified recessive mutations in the SLCO2A1 gene, encoding a prostaglandin transporter, as causative variants. Furthermore, we replicated our findings in other patients with CNSU and established a genetic cause for this inherited disease.
Results
Whole-exome sequencing
We performed whole-exome sequencing in five affected females with CNSU (A-V–2, B-IV–3, C-IV–3, D-II–4, and D-II–5) and one unaffected individual (A-V–3) (Figs 1 and 2). Parental consanguinity was noted in families A, B, and C. The average depth of sequence coverage in the whole-exome sequencing data was 68.9× (S1 Table). We identified a total of 368,403 variants, of which 20,271 were non-synonymous or splice-site mutations.


By filtering the data with dbSNP135, we found 2,406 variants located in 1,578 genes. Based on the parental consanguinity of the patients, we focused on the shared genes with homozygous variants among three affected individuals (A-V–2, B-IV–3, and C-IV–3) and found nine candidate genes, PCSK9, ASPM, DAG1, SLCO2A1, MCPH1, EFEMP2, DDHD1, PKD1L3, and SYNGR1. After consideration of the results for the unaffected individual (A-V–3) and another two sibling patients (D-II–4 and D-II–5), only SLCO2A1 remained as a candidate gene. The three patients with parental consanguinity (A-V–2, B-IV–3, and C-IV–3) had a homozygous splice-site mutation in the SLCO2A1 gene, c.1461+1G>C or c.940+1G>A (Fig 1 and Table 1). The two sibling patients had compound heterozygous mutations, c.664G>A (p.Gly222Arg) and c.1807C>T (p.Arg603X). All four mutations were predicted to be loss-of-function or damaging mutations by SIFT and PolyPhen–2.

Confirmation of the identified mutations
The four identified SLCO2A1 mutations were confirmed to be present in five affected individuals (A-V–2, B-IV–3, C-IV–3, D-II–4, and D-II–5) by Sanger sequencing (Fig 1). Segregation analysis of patient A-V–2 revealed that her unaffected parents, sister, brother, daughter, and son carried the heterozygous c.1461+1G>C mutation (Fig 1A).
To compensate for bias in our analysis, such as the possibility of ethnic-specific variants, we genotyped the four candidate variants in 747 unaffected Japanese subjects from our previous genome-wide association study [9]. All mutations except for the c.940+1G>A mutation were absent in controls (Table 2). The c.940+1G>A mutation was found in the heterozygous state in 3 of 747 controls, showing a similar allele frequency of 0.0022 to the public exome database for the Japanese population (HGVD database).

Sequencing of other CNSU patients
Subsequently, we screened all 14 coding exons and intron-exon boundaries using Sanger sequencing in 12 other CNSU patients, and identified two novel mutations, c.421G>T (p.Glu141X) and c.1372G>T (p.Val458Phe) (Table 1). Eleven of the 12 patients were found to have homozygous (nine patients) or compound heterozygous (two patients) SLCO2A1 mutations that were rare and predicted to be deleterious by SIFT, PolyPhen–2, and PROVEAN (Table 1). The remaining patient (66-year-old female), who did not have an SLCO2A1 mutation, was diagnosed as CNSU because of multiple ulcerations in the duodenum and jejunum. Although anti-tumor necrosis factor-α antibody therapy was ineffective, clinical improvement was achieved by enteral nutrition.
Genetic screening of the SLCO2A1 gene in CD patients
Because CNSU can be misdiagnosed as CD in some cases, we searched for the six identified mutations of SLCO2A1 in CD patients to identify concealed CNSU patients. Among 603 patients previously diagnosed as CD [10], two individuals (patients 17 and 18 in Table 1) were found to carry a pair of compound heterozygous SLCO2A1 mutations, c.940+1G>A/c.547G>A and c.940+1G>A/c.421G>T, respectively. The c.547G>A mutation (p.Gly183Arg), was a novel mutation at a highly conserved site and predicted to be deleterious by in silico analysis. The clinical information for the two individuals was reviewed retrospectively, and the diagnosis of CNSU was confirmed. In total, we identified seven deleterious SLCO2A1 mutations in 18 patients (Table 2).
Clinical features of genetically confirmed CNSU patients
In total, we found 18 patients with CNSU confirmed by genetic analysis (Table 1); 14 of them were female. In all patients, the ulcers occurred in the ileum (Fig 2A–2D). The stomach and duodenum were affected in five (27.8%) and eight (44.4%) patients, respectively.
Because mutations in the SLCO2A1 gene, encoding a prostaglandin transporter, have been reported to be the pathogenic cause of primary hypertrophic osteoarthropathy (PHO; OMIM 614441) [11–13], we investigated whether CNSU patients had clinical manifestations of PHO. Although no patients had any clinical manifestations of PHO requiring treatment, mild digital clubbing or periostosis was present in seven of 18 patients (S2 Table). Moreover, three male patients (patients 12, 16, and 17) fulfilled the major clinical criteria for PHO, having digital clubbing, periostosis, and pachydermia. There were no female patients who fulfilled the major clinical criteria (Fig 2E and 2F).
Among the identified SLCO2A1 mutations, a splice-site mutation of intron 7 (c.940+1G>A) was the most frequent, and nine of the 18 patients were homozygous for this mutation. There were no obvious correlations between the types of mutations and the clinical phenotypes.
Because the SLCO2A1 gene encodes a prostaglandin transporter that mediates the uptake and clearance of prostaglandins, the urinary levels of prostaglandin E metabolite (PGE-M) were measured. The urinary PGE-M levels in CNSU patients were significantly higher than those in unaffected individuals (p = 0.00013; S1 Fig).
mRNA analysis of SLCO2A1
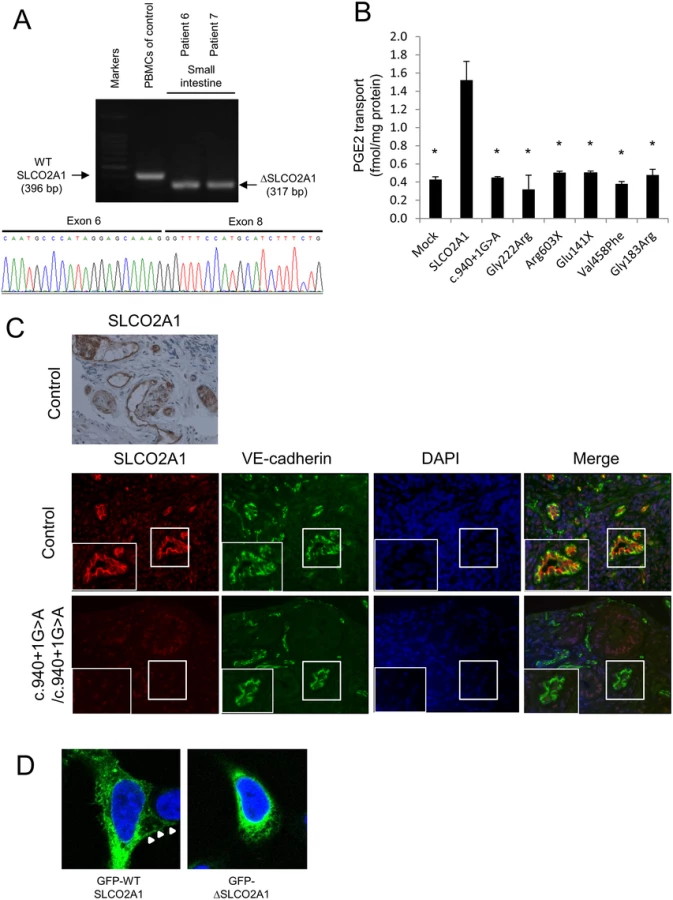
Using RT-PCR, we demonstrated that splicing of the SLCO2A1 mRNA, derived from biopsy specimens of the small intestine, was altered in affected siblings with the homozygous c.940+1G>A mutation (patients 6 and 7) compared with a control individual (Fig 3A). Sequencing of the RT-PCR products revealed deletion of the whole exon 7 of SLCO2A1, leading to a frameshift at amino acid position 288 and introduction of a premature stop codon after six amino acid residues (p.R288Gfs*7). Sequencing of the RT-PCR products of the transcripts in peripheral blood mononuclear cells from patient A-V–2 revealed that the homozygous c.1461+1G>C mutation led to a 23-bp frameshift insertion into intron 10, resulting in a premature stop codon (p.I488Lfs*11) (S2 Fig).
Functional studies of SLCO2A1 protein
For functional analysis of the intact and truncated SLCO2A1 proteins, we investigated the 3H-labeled prostaglandin E2 (PGE2) transport ability in HEK293 cells transfected with intact SLCO2A1 and mutant SLCO2A1 proteins for each identified mutation (c.940+1G>A, p.Gly222Arg, p.Arg603X, p.Glu141X, p.Val458Phe, and p.Gly183Arg). HEK293 cells transfected with intact SLCO2A1 showed the ability for PGE2 transport. In contrast, HEK293 cells transfected with the mutant SLCO2A1 proteins were unable to uptake 3H-labeled PGE (p < 0.0001; Fig 3B). These findings demonstrated that the mutant SLCO2A1 proteins identified in patients lost their functional ability as a PGE transporter.
In control sections of normal small intestinal mucosa, SLCO2A1 was expressed on the cellular membrane of vascular endothelial cells in the small intestine, as evaluated by immunohistochemistry and immunofluorescence staining with a specific anti-SLCO2A1 antibody recognizing the fifth extracellular domain coded by exons 9–11 of the SLCO2A1 gene (Fig 3C). We then analyzed the expression of SLCO2A1 in the small intestine of affected individuals with the homozygous c.940+1G>A mutation (patients 6 and 7) by immunofluorescence staining. However, the immunofluorescence staining did not detect any SLCO2A1 protein in the vascular endothelial cells of the patients (Fig 3C). These results indicated that the entire SLCO2A1 protein was unexpressed in affected individuals with the homozygous c.940+1G>A mutation, consistent with the results of the mRNA transcript sequencing.
To investigate the subcellular localization of SLCO2A1 and the truncated SLCO2A1 protein (ΔSLCO2A1) corresponding to the c.940+1G>A mutation, we constructed expression vectors for GFP-SLCO2A1 and GFP-ΔSLCO2A1 fusion proteins and transfected them into HEK293 cells. GFP-SLCO2A1 was localized on the cellular membrane (Fig 3D, arrows) as well as in the cytoplasm of transfected HEK293 cells, while GFP-ΔSLCO2A1 did not accumulate on the cellular membrane (Fig 3D).
Discussion
In this study, we performed whole-exome sequencing in five Japanese patients with CNSU and one unaffected individual, and identified the SLCO2A1 gene as the candidate for this disorder. We further confirmed that SLCO2A1 gene mutations were involved in the pathogenesis of CNSU by genotyping of control subjects and other CNSU patients. Moreover, a genetic analysis of 603 patients previously diagnosed as CD revealed that two CNSU patients had been included in this disease group. In total, we identified seven different mutations in the SLCO2A1 gene, comprising two splicing-site mutations, two truncating mutations, and three missense mutations, as the causative gene defects for CNSU. Therefore, we propose a more appropriate nomenclature, “chronic enteropathy associated with SLCO2A1 gene” (CEAS), for this disease.
The SLCO2A1 gene encodes a prostaglandin transporter that may be involved in mediating the uptake and clearance of prostaglandins in numerous tissues [14–16]. This gene has already been reported as a causative gene for a subtype of PHO [11]. In fact, three of the seven identified mutations, c.664G>A, c.940+1G>A, and c.1807C>T, have also been reported as causative mutations for PHO [11–13,17,18]. We found that three male patients with CEAS had all of the major clinical features of PHO, such as digital clubbing, periostosis, and pachydermia. Moreover, either digital clubbing or periostosis was present in seven of 18 patients. These findings indicate that CEAS and PHO share a causative gene and that their clinical features are profoundly influenced by other modifiers. Taken together with the facts that that CEAS predominantly occurs in females and PHO predominantly occurs in males [8,17], a sex-influenced gene or hormone may be the main disease modifier. Zhang et al. [17] reported that two female family members of a PHO patient had no clinical features of PHO, despite having a homozygous SLCO2A1 mutation. Moreover, it is interesting to note that these two siblings both had anemia and hypoalbuminemia, suggesting that they had CEAS.
PHO is also known to be caused by mutations of HPGD, encoding 15-hydroxyprostaglandin dehydrogenase (15-PGDH), as well as SLCO2A1 [19]. The transmembrane prostaglandin transporter encoded by the SLCO2A1 gene delivers prostaglandins to cytoplasmic 15-PGDH, resulting in their degradation [14,20]. Because 15-PGDH is the main enzyme for prostaglandin degradation, systemic PGE2 levels are increased in patients with HPGD deficiency. Consistent with the findings in our present investigation, Zhang et al. [11] reported that the urinary levels of PGE2 and PGE-M in SLCO2A1-deficient individuals with PHO are significantly higher than those in controls. In fact, the clinical features of PHO were assumed to be caused by excessive PGE2. Meanwhile, although elevated levels of PGE2 in gastrointestinal tissues are commonly known to protect against mucosal inflammation via the prostaglandin receptor EP3/EP4 [21–23], multiple intestinal ulcers occur in CEAS. This discrepancy and the pathogenesis of intestinal ulcers need to be clarified in future studies.
Although CEAS is presumed to be unaccompanied by immunological inflammation in its pathogenesis, a portion of CEAS patients can be misdiagnosed as CD because of the shared common clinical features, such as multiple small intestinal ulcers, anemia, and hypoalbuminemia. In this study, two of 603 patients previously diagnosed as CD were found to be affected with CEAS by genetic analysis. Because corticosteroid and anti-tumor necrosis factor-α antibody therapies are ineffective for CEAS, recognition and precise diagnosis of CEAS are critical to avoid unnecessary therapies. The findings of our investigation lead us to conclude that genetic analysis in addition to detailed clinical information including digital clubbing, blood tests, and gastrointestinal examinations are invaluable for distinguishing CEAS from CD.
Cases of a similar enteropathy referred to as cryptogenic multifocal ulcerous stenosing enteritis (CMUSE) have been reported in Western populations [24–26]. This enteropathy has been shown to be an autosomal recessive inherited disease caused by mutations in the PLA2G4A gene [27]. CEAS and CMUSE share common clinicopathologic features with respect to age of onset, chronic and recurrent clinical course, and nonspecific stenosing small intestinal ulcers [4,25]. However, the sex predominance, response to steroid therapy, and lesion sites are obviously different between the two conditions. The PLA2G4A gene encodes cytoplasmic phospholipase A2-α (cPLA2α), which catalyzes the release of arachidonic acid from membrane phospholipids. CMUSE patients with compound heterozygous mutations of PLA2G4A have been reported to show globally decreased production of eicosanoids such as PGE2 and thromboxane A2, resulting in multiple ulcers of the small intestine and platelet dysfunction [27,28]. Because impaired prostaglandin use underlies CEAS, CMUSE, and NSAID-induced enteropathy, we propose a new entity of gastrointestinal disorders, namely “prostaglandin-associated enteropathy”.
In conclusion, we have identified loss-of-function mutations in the SLCO2A1 gene as the cause of CEAS. The present findings clearly indicate that CEAS is a genetically distinct entity independent of other gastrointestinal disorders including CD, NSAID-induced enteropathy, and CMUSE. Further studies are needed to elucidate the pathogenesis of CEAS and identify new therapeutic molecular targets for “prostaglandin-associated enteropathy”.
Materials and Methods
Ethics statement
Written informed consent for genetic studies was obtained from each individual. The study was approved by the institutional review board at each collecting site in accordance with the Declaration of Helsinki Principles.
Study participants
We obtained blood samples and family pedigrees from 17 Japanese patients with CNSU and eight unaffected family members in 15 families. The diagnosis of CNSU was based on the published clinical criteria and clinical courses (S3 Table) [8,29]. Genomic DNA samples from 747 participants in our previous genome-wide association study for ulcerative colitis [9] and 603 patients with CD [10,30] were used after excluding subjects who recalled their consent.
Genetic analysis
DNA was extracted from peripheral blood using standard methods. Whole-exome sequencing in five affected individuals (A-V–2, B-IV–3, C-IV–3, D-II–4, and D-II–5) and one unaffected individual (A-V–3) was performed to identify candidate genetic variants (Fig 1). Genomic DNA was enriched using a TruSeq Exome Enrichment Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions, and paired-end sequencing was carried out with an Illumina HiSeq 2000 instrument. Reads were aligned to the human genome reference sequence (hg19 NCBI build 37.1) and decoy sequences using BWA software [31]. Duplicate reads were removed with the Picard program (http://picard.sourceforge.net/). Recalibration and realignment of the data were accomplished with Genome Analysis Toolkit (GATK) [32,33]. Single nucleotide variants and small insertions and deletions (indels) were identified by GATK Unified Genotyper. The effect of each missense mutation was predicted using SIFT (http://sift.jcvi.org/) [34], PolyPhen–2 (http://genetics.bwh.harvard.edu/pph2/) [35], and PROVEAN (http://provean.jcvi.org/) [36].
To compensate for bias in our analysis, such as the possibility of ethnic-specific variants, we genotyped the four candidate variants identified by exome sequencing in 747 unaffected Japanese subjects by Sanger sequencing and restriction fragment length polymorphism analysis (S4 Table). For further validation, Sanger sequencing of all exons of the SLCO2A1 gene in other CNSU patients was performed using standard protocols. Finally, we genotyped the six identified mutation sites in the SLCO2A1 gene in clinically diagnosed CD patients, because CNSU can be misdiagnosed as CD.
Urinary prostaglandin measurement
Urine samples were collected from 15 CNSU patients and 13 unaffected individuals. The PGE-M levels were measured in duplicate using competitive enzyme-linked immunosorbent assays (Cayman Chemical, Ann Arbor, MI, USA).
RNA analysis
We analyzed the exon 7 and exon 10 boundary mutations using RT-PCR to examine the effects of the splice-site mutations on SLCO2A1 transcription. Total RNA was extracted from biopsy specimens of the small intestine and peripheral blood mononuclear cells using a NucleoSpin RNA Kit (Macherey-Nagel, Düren, Germany) or PAXgene Blood RNA Kit (Qiagen, Hilden, Germany). cDNA was synthesized using a PrimeScript First Strand cDNA Synthesis Kit (TaKaRa, Otsu, Japan). The PCR products obtained from the cDNAs were sequenced (S5 Table).
Construction of expression vectors for the SLCO2A1 gene
A full-length cDNA (NM_005630) expression vector and C-terminally GFP-tagged cDNA expression vector were purchased from OriGene Technologies (Rockville, MD, USA). To construct vectors carrying a mutated cDNA, a KOD -Plus - Mutagenesis Kit (Toyobo, Osaka, Japan) was used according to the manufacturer’s instructions. The expression vectors were amplified by inverse PCR with specific primer sets (S6 Table). The PCR products were self-ligated, and transformed into Escherichia coli chemically competent DH5α cells. To correct a frameshift in the downstream of exon 7, a C-terminally GFP-tagged cDNA expression vector with deletion of exon 7 was amplified again.
Tracer PGE2 uptake analysis
On the day before transfection, HEK293 cells were trypsinized, counted, and plated onto 12-well plates at a density of 4×105 cells/well. The cells were transfected by adding a premixed solution containing 0.4 μg of expression vectors and 2 μl of ScreenFectA (Wako, Osaka, Japan). After 24 hours of incubation, the medium was exchanged twice with warmed Waymouth’s medium (Life Technologies, Carlsbad, CA, USA), and the cells were incubated for 30 minutes at 37°C in uptake medium containing [5,6,8,11,12,14,15-3H(N)]-PGE2 (PerkinElmer, Waltham, MA, USA) at 0.6 nM. The cells were washed four times with cold Waymouth’s medium, and lysed with 200 μl of RIPA Buffer (Thermo Fisher Scientific, Hemel Hempstead, UK) containing a protease inhibitor (Roche, Basel, Switzerland). The total protein concentration was quantified using a BCA Protein Assay Kit (Thermo Fisher Scientific). Next, 150 μl of cell lysate was mixed with 5 ml of MicroScint–20 (PerkinElmer), and scintillation counting was performed in a Tri-Carb 3100TR liquid scintillation spectrometer (PerkinElmer).
Immunohistochemistry and immunofluorescence staining
Formalin-fixed paraffin-embedded tissues were sectioned at 3-μm thickness. After antigen unmasking in 10 mM sodium citrate buffer (pH 6) for 15 minutes at 121°C, the sections were blocked with Protein Block Serum-Free (Dako, Glostrup, Denmark) for 30 minutes at room temperature. The sections were then incubated with a diluted anti-SLCO2A1 antibody (HPA013742; Sigma-Aldrich, St. Louis, MO, USA; antigen sequence: PSTSSSIHPQSPACRRDCSCPDSIFHPVCGDNGIEYLSPCHAGCSNINMSSATSKQLIYLNCSCVTGGSASAKTGSCPVPCAH) overnight at 4°C, followed by MAX-PO (MULTI) (Nichirei, Tokyo, Japan) for 30 minutes at room temperature. DAB solution (Nichirei) was applied for color development. After the immunocytochemistry, the sections were counterstained with Mayer’s hematoxylin solution (Nichirei). For immunofluorescence, the sections were incubated with a primary antibody mixture of the anti-SLCO2A1 antibody (HPA013742) and an anti-VE-cadherin antibody (LS-B3780; LifeSpan BioSciences, Seattle, WA, USA) overnight at 4°C, followed by a secondary antibody mixture of Alexa Fluor 568-conjugated goat anti-rabbit IgG (H&L) antibody and Alexa Fluor 488-conjugated goat anti-mouse IgG (H&L) antibody (Life Technologies) for 30 minutes at room temperature. The stained sections were analyzed using an ECLIPSE TE2000-U (Nikon, Tokyo, Japan).
For observation of HEK293 cells expressing GFP-fusion proteins, cells were fixed with 4% paraformaldehyde phosphate buffer solution (Wako) for 20 minutes, and then permeabilized with 0.1% Triton X–100 (Sigma-Aldrich) in D-PBS(-) solution (Wako) for 20 minutes. Nuclei were stained with 16.2 μM Hoechst 33342 (Life Technologies) in D-PBS(-) solution for 5 minutes. GFP and nuclei were visualized using a 40× objective on an LSM710 Laser Scanning Confocal Microscope (Carl Zeiss, Oberkochen, Germany).
Statistical analysis
The chi-square test and Fisher’s exact test, where appropriate, were used to analyze categorical data. Student’s t-test was used to compare quantitative data between two groups. Dunnett’s method was used for multiple comparisons with a control group. The analyses were performed using JMP Pro statistical package 11.2.0 (SAS Institute, Cary, NC, USA). Values of p < 0.05 were regarded as statistically significant.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Iddan G, Meron G, Glukhovsky A, Swain P (2000) Wireless capsule endoscopy. Nature 405 : 417.
2. Yamamoto H, Sekine Y, Sato Y, Higashizawa T, Miyata T, et al. (2001) Total enteroscopy with a nonsurgical steerable double-balloon method. Gastrointest Endosc 53 : 216–220. 11174299
3. Okabe H, Sakimura M (1968) Nonspecific multiple ulcer of the small intestine. Stomach and Intestine 3 : 1539–1549.
4. Matsumoto T, Iida M, Matsui T, Yao T (2007) Chronic nonspecific multiple ulcers of the small intestine: a proposal of the entity from Japanese gastroenterologists to Western enteroscopists. Gastrointest Endosc 66: S99–107. 17709045
5. Bjarnason I, Hayllar J, MacPherson AJ, Russell AS (1993) Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology 104 : 1832–1847. 8500743
6. Matsumoto T, Iida M, Matsui T, Yao T, Watanabe H, et al. (2004) Non-specific multiple ulcers of the small intestine unrelated to non-steroidal anti-inflammatory drugs. J Clin Pathol 57 : 1145–1150. 15509673
7. Matsumoto T, Nakamura S, Esaki M, Yada S, Koga H, et al. (2006) Endoscopic features of chronic nonspecific multiple ulcers of the small intestine: comparison with nonsteroidal anti-inflammatory drug-induced enteropathy. Dig Dis Sci 51 : 1357–1363. 16868823
8. Matsumoto T, Kubokura N, Matsui T, Iida M, Yao T (2011) Chronic nonspecific multiple ulcer of the small intestine segregates in offspring from consanguinity. J Crohns Colitis 5 : 559–565. doi: 10.1016/j.crohns.2011.05.008 22115375
9. Asano K, Matsushita T, Umeno J, Hosono N, Takahashi A, et al. (2009) A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet 41 : 1325–1329. doi: 10.1038/ng.482 19915573
10. Hirano A, Yamazaki K, Umeno J, Ashikawa K, Aoki M, et al. (2013) Association study of 71 European Crohn's disease susceptibility loci in a Japanese population. Inflamm Bowel Dis 19 : 526–533. doi: 10.1097/MIB.0b013e31828075e7 23388546
11. Zhang Z, Xia W, He J, Ke Y, Yue H, et al. (2012) Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am J Hum Genet 90 : 125–132. doi: 10.1016/j.ajhg.2011.11.019 22197487
12. Busch J, Frank V, Bachmann N, Otsuka A, Oji V, et al. (2012) Mutations in the prostaglandin transporter SLCO2A1 cause primary hypertrophic osteoarthropathy with digital clubbing. J Invest Dermatol 132 : 2473–2476. doi: 10.1038/jid.2012.146 22696055
13. Diggle CP, Parry DA, Logan CV, Laissue P, Rivera C, et al. (2012) Prostaglandin transporter mutations cause pachydermoperiostosis with myelofibrosis. Hum Mutat 33 : 1175–1181. doi: 10.1002/humu.22111 22553128
14. Kanai N, Lu R, Satriano JA, Bao Y, Wolkoff AW, et al. (1995) Identification and characterization of a prostaglandin transporter. Science 268 : 866–869. 7754369
15. Lu R, Kanai N, Bao Y, Schuster VL (1996) Cloning, in vitro expression, and tissue distribution of a human prostaglandin transporter cDNA(hPGT). J Clin Invest 98 : 1142–1149. 8787677
16. Schuster VL (1998) Molecular mechanisms of prostaglandin transport. Annual Review of Physiology 60 : 221–242. 9558462
17. Zhang Z, He JW, Fu WZ, Zhang CQ, Zhang ZL (2013) Mutations in the SLCO2A1 gene and primary hypertrophic osteoarthropathy: a clinical and biochemical characterization. J Clin Endocrinol Metab 98: E923–933. doi: 10.1210/jc.2012-3568 23509104
18. Niizeki H, Shiohama A, Sasaki T, Seki A, Kabashima K, et al. (2013) The novel SLCO2A1 heterozygous missense mutation p.E427K and nonsense mutation p.R603* in a female patient with pachydermoperiostosis with an atypical phenotype. Br J Dermatol.
19. Uppal S, Diggle CP, Carr IM, Fishwick CW, Ahmed M, et al. (2008) Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat Genet 40 : 789–793. doi: 10.1038/ng.153 18500342
20. Nomura T, Lu R, Pucci ML, Schuster VL (2004) The two-step model of prostaglandin signal termination: in vitro reconstitution with the prostaglandin transporter and prostaglandin 15 dehydrogenase. Mol Pharmacol 65 : 973–978. 15044627
21. Kunikata T, Tanaka A, Miyazawa T, Kato S, Takeuchi K (2002) 16,16-Dimethyl prostaglandin E2 inhibits indomethacin-induced small intestinal lesions through EP3 and EP4 receptors. Digestive Diseases and Sciences 47 : 894–904. 11991626
22. Rao R, Redha R, Macias-Perez I, Su Y, Hao C, et al. (2007) Prostaglandin E2-EP4 receptor promotes endothelial cell migration via ERK activation and angiogenesis in vivo. Journal of Biological Chemistry 282 : 16959–16968. 17401137
23. Takeuchi K, Kato S, Amagase K (2010) Prostaglandin EP receptors involved in modulating gastrointestinal mucosal integrity. J Pharmacol Sci 114 : 248–261. 21041985
24. Shoesmith JH, Tate GT, Wright CJ (1964) Multiple Strictures of the Jejunum. Gut 5 : 132–135. 14159400
25. Perlemuter G, Chaussade S, Soubrane O, Degoy A, Louvel A, et al. (1996) Multifocal stenosing ulcerations of the small intestine revealing vasculitis associated with C2 deficiency. Gastroenterology 110 : 1628–1632. 8613071
26. Perlemuter G, Guillevin L, Legman P, Weiss L, Couturier D, et al. (2001) Cryptogenetic multifocal ulcerous stenosing enteritis: an atypical type of vasculitis or a disease mimicking vasculitis. Gut 48 : 333–338. 11171822
27. Brooke MA, Longhurst HJ, Plagnol V, Kirkby NS, Mitchell JA, et al. (2014) Cryptogenic multifocal ulcerating stenosing enteritis associated with homozygous deletion mutations in cytosolic phospholipase A2-alpha. Gut 63 : 96–104. doi: 10.1136/gutjnl-2012-303581 23268370
28. Adler DH, Cogan JD, Phillips JA 3rd, Schnetz-Boutaud N, Milne GL, et al. (2008) Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J Clin Invest 118 : 2121–2131. doi: 10.1172/JCI30473 18451993
29. Yao T, Iida M, Matsumoto T (2004) Chronic hemorrhagic ulcers of the small intestine or chronic nonspecific multiple ulcers of the small intestine. In: T Y, M I, editors. Diseases of the small intestine. Tokyo: Igaku-Shoin. pp. 176–186.
30. Yamazaki K, Umeno J, Takahashi A, Hirano A, Johnson TA, et al. (2013) A genome-wide association study identifies 2 susceptibility loci for Crohn's disease in a Japanese population. Gastroenterology 144 : 781–788. doi: 10.1053/j.gastro.2012.12.021 23266558
31. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 : 1754–1760. doi: 10.1093/bioinformatics/btp324 19451168
32. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 : 1297–1303. doi: 10.1101/gr.107524.110 20644199
33. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43 : 491–498. doi: 10.1038/ng.806 21478889
34. Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31 : 3812–3814. 12824425
35. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7 : 248–249. doi: 10.1038/nmeth0410-248 20354512
36. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS One 7: e46688. doi: 10.1371/journal.pone.0046688 23056405
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 11
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development
- Metabolomic Quantitative Trait Loci (mQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis
- Genus-Wide Comparative Genomics of Delineates Its Phylogeny, Physiology, and Niche Adaptation on Human Skin
- Encodes Dual Oxidase, Which Acts with Heme Peroxidase Curly Su to Shape the Adult Wing
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy