
Translation Enhancing ACA Motifs and Their Silencing by a Bacterial Small Regulatory RNA
GcvB is an archetypal multi-target small RNA regulator of genes involved in amino acid uptake or metabolism in enteric bacteria. Included in the GcvB regulon is the yifK locus, encoding a conserved putative amino acid transporter. GcvB inhibits yifK mRNA translation by pairing with a sequence immediately upstream from the Shine-Dalgarno motif. Surprisingly, we found that some target sequence mutations that disrupt pairing, and thus were expected to relieve repression, actually lower yifK expression and cause it not to respond to GcvB variants carrying the corresponding compensatory changes. Work prompted by these observations revealed that the GcvB target sequence in yifK mRNA includes elements that stimulate translation initiation. Replacing each base of an ACA trinucleotide near the center of the target sequence, by any other base, caused yifK expression to decrease. Effects were additive, with some triple replacements causing up to a 90% reduction. The enhancer activity did not require the ACA motif to be strictly positioned relative to the Shine-Dalgarno sequence, nor did it depend on a particular spacing between the latter and the initiating AUG. The dppA mRNA, another GcvB target, contains four ACA motifs at the target site. Quite strikingly, replacement of all four ACAs by random trinucleotide sequences yielded variants showing over 100-fold reduction in expression, virtually inactivating the gene. Altogether, these data identify the ACA motif as a translation-enhancing module and show that GcvB's ability to antagonize the enhancer function in target mRNAs is quintessential to the regulatory effectiveness of this sRNA.
Published in the journal:
. PLoS Genet 10(1): e32767. doi:10.1371/journal.pgen.1004026
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004026
Summary
GcvB is an archetypal multi-target small RNA regulator of genes involved in amino acid uptake or metabolism in enteric bacteria. Included in the GcvB regulon is the yifK locus, encoding a conserved putative amino acid transporter. GcvB inhibits yifK mRNA translation by pairing with a sequence immediately upstream from the Shine-Dalgarno motif. Surprisingly, we found that some target sequence mutations that disrupt pairing, and thus were expected to relieve repression, actually lower yifK expression and cause it not to respond to GcvB variants carrying the corresponding compensatory changes. Work prompted by these observations revealed that the GcvB target sequence in yifK mRNA includes elements that stimulate translation initiation. Replacing each base of an ACA trinucleotide near the center of the target sequence, by any other base, caused yifK expression to decrease. Effects were additive, with some triple replacements causing up to a 90% reduction. The enhancer activity did not require the ACA motif to be strictly positioned relative to the Shine-Dalgarno sequence, nor did it depend on a particular spacing between the latter and the initiating AUG. The dppA mRNA, another GcvB target, contains four ACA motifs at the target site. Quite strikingly, replacement of all four ACAs by random trinucleotide sequences yielded variants showing over 100-fold reduction in expression, virtually inactivating the gene. Altogether, these data identify the ACA motif as a translation-enhancing module and show that GcvB's ability to antagonize the enhancer function in target mRNAs is quintessential to the regulatory effectiveness of this sRNA.
Introduction
A relevant chapter in the expanding field of RNA-mediated gene regulation is devoted to the activities of multi-target trans-encoded small RNAs in bacteria. Acting in concert with chaperon protein Hfq, these RNA regulators function by base-pairing with short, often imperfectly complementary sequences in the 5′ untranslated regions (UTR) of target messenger RNAs. They can affect translation and turnover of several mRNAs simultaneously thus reprogramming gene expression of whole gene families in a coordinate manner in response to environmental cues (reviewed in [1]–[3]). Archetypal examples of this class of regulators are the RyhB small RNA (sRNA) which represses expression of mRNA for dispensable iron-sequestering proteins when iron is limiting [4]–[8]; RybB, which downregulates several outer membrane protein mRNAs under envelope stress conditions [9]–[13], Spot 42, which amplifies the regulatory range of catabolite repression by targeting several mRNAs involved in sugar uptake and consumption [14] and GcvB, which downregulates dozens of different mRNAs involved in amino acid uptake or metabolism in E. coli and Salmonella [15]–[18].
GcvB, a 200 nucleotide-long sRNA, was identified serendipitously during a study of gcvA, the gene for the main transcriptional regulator of the glycine cleavage operon gcvTHP [18]. The latter encodes the enzymes of the glycine cleavage system, the pathway generating one-carbon units from the oxidative cleavage of glycine [19]. The gcvB gene is located immediately adjacent to gcvA in the opposite orientation with its promoter partially overlapping the gcvA promoter. In the presence of excess glycine, the GcvA protein activates transcription of the gcvTHP operon as well as of gcvB [18]. Initial characterization of GcvB showed this sRNA to downregulate the synthesis of DppA and OppA proteins, main components of dipeptide - and oligopeptide-transport systems, respectively [16], [18]. Since then, the number of genes found to be regulated by GcvB has increased exponentially. A recent transcriptomic study in Salmonella enterica set this number to more than 40, making the GcvB regulon the largest of its kind [17]. The vast majority of these loci are linked directly or indirectly to amino acid metabolism and are negatively controlled by GcvB. Typically, regulation is exerted during exponential growth in nutrient rich environments and possibly aimed at coordinating the expression of interconnected metabolic pathways [16], [17]; however, its precise role remains incompletely understood.
GcvB uses a specific sequence region to pair with most, although not all [20] of its mRNA targets. This pairing domain – named the R1 region [16] – is characterized by its high degree of sequence conservation, the lack of secondary structure and a typical GU-rich sequence bias. Hence, most sequences targeted by GcvB include CA-rich repeats. They are typically found inside, or immediately adjacent to, the ribosome binding sites (RBS) of target mRNAs. In one of these targets - the gltI mRNA for a glutamate-aspartate transport protein – the CA-rich element is located 45 nucleotides (nt) upstream from the translation initiation codon. Removal of this sequence (as part of a 27 nt deletion), besides causing the loss GcvB regulation, affected gltI translation, suggesting that the CA-rich element acts as a translational enhancer. Consistent with this interpretation, crafting the 27 nt segment at the corresponding position of an unrelated mRNA conferred simultaneously GcvB control and increased translational efficiency [16].
Some years ago, our laboratory performed a lac-based genetic screen aimed at identifying genes controlled by trans-encoded small RNAs in Salmonella. A random library of lacZ fusions to chromosomal genes was generated using a phage Mu-derived transposon (MudK) and screened for isolates whose LacZ levels changed (either increased or decreased) upon inactivating Hfq [21]. Among the candidates that were found, two independent isolates upregulated in the hfq mutant background, carried the lacZ insert translationally fused to the yifK gene [21]. Presumptive identification of this gene as an amino acid transporter suggested that yifK might be a GcvB target. We thus proceeded to test this hypothesis and characterize yifK regulation. While this work was underway, Sharma and coworkers identified yifK mRNA as a member of the gcvB regulon by microarray analysis; however, these authors could not confirm direct regulation by GcvB due to low reporter fluorescence of the yifK-gfp fusion used in the study [17]. Since this study also identified global regulator Lrp as a GcvB target [17], the possibility remained that the GcvB effects on yifK expression might be indirect.
Here we present in vivo and in vitro evidence that GcvB downregulates yifK directly by pairing with a sequence immediately preceding the Shine-Dalgarno (SD) motif in yifK mRNA. A surprising observation in the course of this study was that some target sequence mutations that disrupted pairing did not cause yifK expression to increase – as expected for the relief of GcvB repression – but had the opposite effect. The drop in expression was not suppressed by deleting gcvB nor was it accentuated in a GcvB mutant carrying the appropriate compensatory changes. Closer analysis revealed that the GcvB target sequence includes elements that stimulate yifK mRNA translation. In the absence of such elements, the role of GcvB pairing in regulation becomes marginal.
Results
Genetic identification of a GcvB-regulated locus
Our original screen for Hfq-regulated genes yielded two isolates carrying the MudK (lac) transposon in the yifK gene; one predicted to produce a LacZ protein fusion to the 48th amino acid (aa) of the 461 aa YifK (yifK87::MudK); the other with LacZ inserted after the 95th aa of YifK (yifK88::MudK) [21]. Preliminary tests showed both fusions to be regulated in a closely similar manner; however, yifK87::MudK produced significantly higher ß-galactosidase activity and was chosen for the present study. A survey of protein sequence databases showed YifK to be a highly conserved protein with the characteristic signature of amino acid transporters. The known role of GcvB in the regulation of some members of this family made this small RNA the likeliest candidate to control yifK expression. This was confirmed by deleting the gcvB gene and testing the effects of the deletion on the expression of the yifK87::MudK fusion (hereafter referred to as yifK-lacZY). As shown in Figure 1, the gcvB deletion causes a nearly 5-fold increase of ß-galactosidase activity in exponentially growing cells, while effects decline in stationary phase. Somewhat surprisingly, LacZ levels in the gcvB-deleted strain are not as high as the levels measured in a strain deleted for hfq (Figure 1). This might reflect the existence of one or more additional sRNA(s) participating in yifK repression. Alternatively, Hfq could repress yifK directly [22]. The data in Figure 1 show that loss of Hfq is epistatic to the gcvB deletion.

Mutations affecting yifK expression
Primer extension experiments mapped the 5′ end of yifK mRNA to 64 nucleotides upstream from the initiating AUG (Figure 2). This 5′ untranslated region (UTR) includes a 14-nt stretch complementary to the 3′ half of GcvB's R1 region. As an initial step to characterize GcvB involvement in yifK regulation, we tested whether point mutations in the gcvB gene or in the promoter-proximal portion of yifK relieved GcvB-mediated repression. For this, DNA fragments spanning either of these two regions were randomly mutagenized by the polymerase chain reaction (PCR) under error-prone conditions and introduced into the chromosome of a strain harboring the yifK-lacZY reporter fusion via lambda red recombination. Most of the isolates originating from the gcvB mutagenesis carried changes in the gcvB promoter or in the promoter of the adjacent gcvA gene (Figure S1). Thus, these mutations appeared to lower the levels rather than the activity of GcvB and were not further considered.

Mutagenesis of yifK promoter-proximal segment yielded three mutants with elevated yifK-lacZY expression. One isolate carried a C:G to A:T change 33 base-pairs upstream from the 5′ end of yifK mRNA. The position and the nature of the change (producing a -35 promoter consensus match, TTGACA, Figure 2A), strongly suggest that the mutation increases the strength of the yifK promoter. The mutation leads to a sharp rise in the intensity of the primer extension product (lane “-33A” in Figure 2B) and a more than 10-fold increase in ß-galactosidase activity (data not shown). These findings confirmed that the 5′ end identified by primer extension corresponds to yifK transcription initiation site. The remaining two mutations affect residues within the 5′ UTR (Figure 2A). One allele, resulting in a U to C change at position +21, falls within a AU-rich segment (AUAACAAUAA) that might constitute a site for Hfq binding [3], [23]. Consistent with this interpretation, the mutation has no effect in Δhfq background (Figure 2C). Finally the third allele (G to A at +27) affects the CG-rich stem of a presumptive secondary structure immediately adjacent to the AU-rich segment. The change causes a generalized increase of yifK-lacZY expression by an unidentified mechanism.
GcvB activity stimulates RNase E-dependent yifK mRNA decay
Northern blot analysis was used to assess the effects of GcvB regulation on yifK mRNA levels. This study critically benefited from the availability of the -33 promoter mutant (see above), yifK mRNA being otherwise undetectable when expressed from the wild-type promoter (data not shown). The analysis identified two yifK mRNA species, a 1.4 kilobase (Kb) mRNA covering just the yifK coding portion and a longer, 2.0 Kb RNA extending into the adjacent argX-hisR-leuT-proM tRNA operon. As shown in Figure 3A, both RNAs accumulate upon RNase E inactivation, whereas only the shorter species accumulates in cells lacking GcvB or Hfq. This suggested that derepression of yifK translation in ΔgcvB or Δhfq cells protects the 1.4 Kb RNA against RNase E cleavage. To confirm this interpretation, the analysis was repeated with strains that, besides the promoter “up” mutation, carried a mutation in the Shine-Dalgarno sequence (G to Cat position +59; described in more detail below). As shown in Figure 3C, the SD mutation causes the intensity of 1.4 Kb band to sharply decrease in the ΔgcvB or Δhfq strains but not in the rne ts mutant, consistent with the idea that reduced translation renders yifK mRNA susceptible to RNAse E degradation.
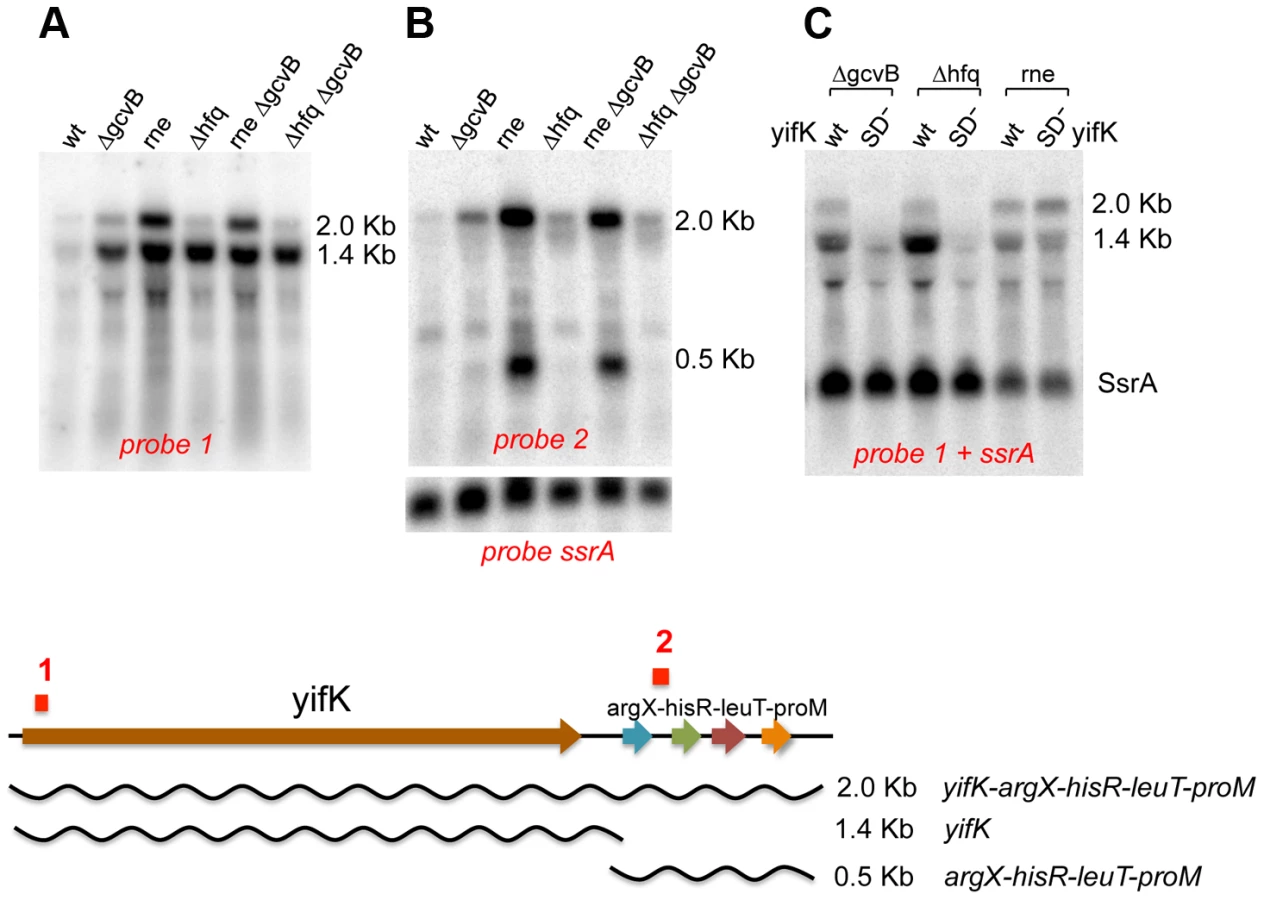
Absence of any obvious transcription termination signals in the intercistronic region between yifK and the tRNA operon suggests that the 1.4 Kb RNA originates from processing of the longer form. Likely, under normal conditions (i.e., wt yifK promoter) yifK transcription contributes only to a small fraction of the four tRNAs, as most the tRNA operon transcription results from a strong promoter located immediately upstream from the argX gene [24]. The approximately 500 nt RNA accumulates in the RNase E mutant (Figure 3B). Previous work in E.coli, showed that this tRNA precursor is processed by the concerted actions of RNase E and RNase P in a pathway that, intriguingly, also sees the participation of Hfq [25].
yifK is repressed by Lrp
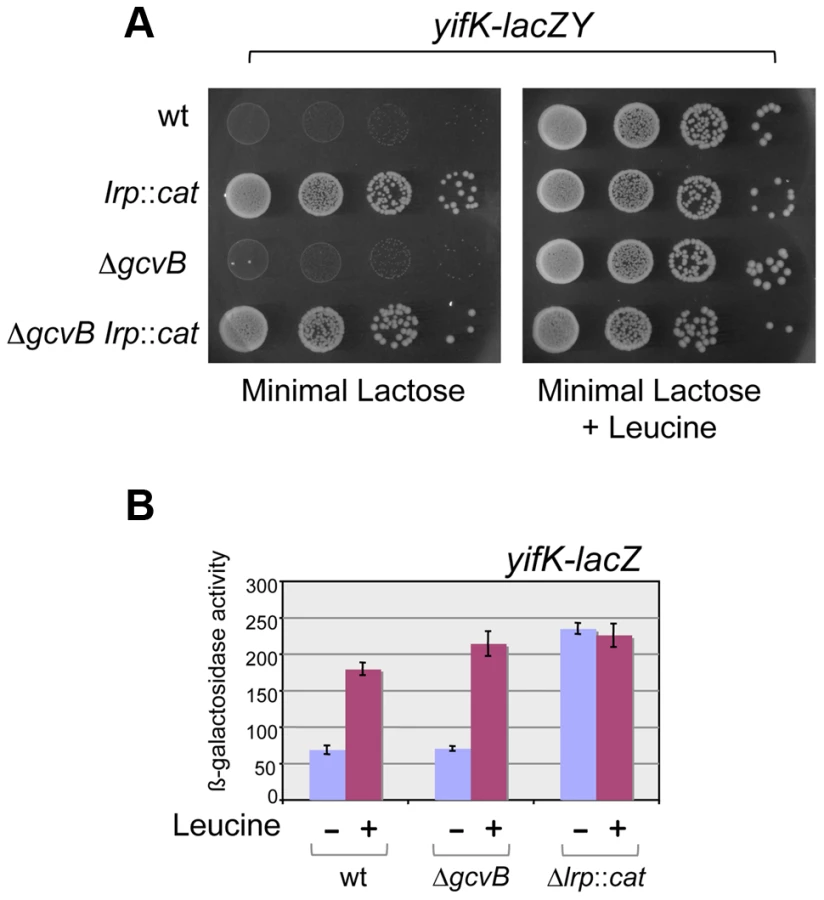
Early on in this study, it became apparent that yifK expression was exquisitely sensitive to the growth medium and virtually silenced in minimal medium. As a result, a strain with the yifK-lacZY fusion is phenotypically Lac− when plated in minimal medium. We exploited this phenotype to positively select for spontaneous Lac+ mutants. The selection yielded two classes of mutations, one genetically linked to the yifK-lacZY locus, the second mapping elsewhere. All of the linked mutants that were analyzed were found to harbor the -33 C:G to A:T promoter change obtained previously (see above). The unlinked mutations mapped in a chromosomal interval encompassing the gene for leucine response regulator, Lrp. Prompted by this observation, we introduced an lrp insertion mutation into the yifK-lacZY-containing strain. The resulting strain acquired a Lac+ phenotype (Figure 4A), indicating that yifK silencing in minimal medium results from Lrp repression. Addition of leucine efficiently relieves repression (Figure 4). The data in Figure 4 also show that GcvB does not contribute to yifK repression to any significant extent in minimal medium. This is not surprising as GcvB is transcribed at very low level under these conditions [16] and inactivating Lrp does not reverse this pattern (Figure S2). The data in Figure S2 differ from those of Modi et al [26] who reported an approximate 30-fold increase in GcvB levels in an lrp deletion mutant of in E.coli. This discrepancy might reflect differences in the organisms used or in media composition.
GcvB inhibits yifK translation by targeting an enhancer element
The above approach yielded no mutations affecting the presumptive pairing sequences of GcvB or yifK. Reasoning that single base changes might not disrupt regulation enough to be revealed by the MacConkey plate screen, we resorted to introducing multiple changes by site-directed mutagenesis. An initial test involved changing a UGUG quadruplet in the GcvB segment thought to pair with yifK mRNA. The alteration caused expression of the yifK-lacZY fusion to increase approximately threefold, thus corroborating the postulated role of this sequence in yifK repression. Unexpectedly, however, when the ACAC sequence at the corresponding position in yifK mRNA was changed, yifK-lacZ expression did not increase but actually declined (data not shown). Trying to clarify this observation, portions of the region of interest were mutagenized separately. As shown in Figure 5A, converting the AAA sequence in the middle of the target sequence to UGU, or making the opposite change (UGU to AAA) in GcvB, similarly relieves yifK-lacZY repression. Repression is restored upon combining the compensatory alleles. Thus, this portion of the target sequence behaves as expected, and the behavior of the compensatory mutant strongly suggests that GcvB represses yifK through a base-pair interaction. In vitro toeprint experiments, showing that GcvB inhibits the binding of ribosomal 30S subunit to yifK translation initiation site, specifically and in a dose-dependent manner (Figure 6), provided independent support to this conclusion.


Again, however, changing the CA doublet on the 3′ side of the AAA sequence to UC produced an unusual pattern: like in the quadruplet mutant above, yifK-lacZY expression decreased rather than increase, becoming insensitive to a GcvB variant carrying the compensatory change (Figure 5B). To verify that the compensatory change did not hamper GcvB's function in an unpredictable way, we took advantage of the fact that the replaced nucleotides do not participate in the pairing with dppA [16] and tested the mutant's ability to repress a dppA-lacZ translational fusion. This analysis showed both GcvB variants to be as efficient as wild-type in repressing dppA, indicating that both alleles remain fully functional (Figure S3).
Besides being insensitive to the compensatory GcvB allele, the yifK CA49,50 to UC49,50 mutant fails to respond to gcvB or hfq deletions (Figure 7A). We interpreted these findings to suggest that the CA to UC conversion lowers translation efficiency and under such conditions, GcvB action is no longer rate-limiting for yifK expression. The effects of the mutation on yifK translation were examined in vitro using a reconstituted system. Results in Figure 7B showed an epitope-tagged Cat protein to accumulate at significantly greater levels when made from a gene fusion to the wt yifK 5′ UTR than from an equivalent construct carrying the CA to UC change. These data confirmed that the CA49,50 doublet stimulates translation and suggested that GcvB effectiveness in regulation reflects the targeting of an activating element.

Anatomy of a translational enhancer
To characterize the enhancer element, an 8-nt segment preceding the SD sequence was modified by systematically changing individual residues to each of the three alternative bases. Effects on expression of the yifK-lacZ fusion were measured in a strain deleted for the gcvB gene. As shown in Figure 8, any change in the ACA sequence at positions +48 to +50 lowers yifK-lacZY expression. Variations range between 23% and 62%, with G residues exerting the most adverse effects at any position. Significantly, the effects appear to be additive since a separate experiment in which all three bases in the ACA sequence were randomized yielded alleles undergoing as much as 92% reduction in yifK-lacZY expression (bottom portion of Figure 8).

Alteration of a second ACA motif between +53 and +55 produced a somewhat different pattern. Changes in the central C were either neutral or stimulatory; in contrast, having a C at the first position was highly deleterious resulting in nearly 95% reduction of ß-galactosidase activity (Figure 8). Altogether, these data suggested that ACA motifs enhance the efficiency of yifK translation. Conservation of these motifs in distant members of the Enterobacteriaceae family (Figure S4) is consistent with their functional importance.
A peculiarity of the yifK translation signal is the unusually short distance (four nucleotides) between the most conserved base of the SD motif [27] and the initiating AUG. We thus envisaged that the role of the enhancer could be to somehow compensate for such suboptimal arrangement. To test this possibility, we generated a 7-nt tandem direct duplication of the SD region and then inactivated either copy of the SD by changing the GAGGA motif to GACGA (Figure 9). Thus, the resulting constructs have the functional SD sequence positioned either 4 or 11 nt from the AUG. As shown in Figure 9, these two variants (n. 4 and n. 5) express ß-galactosidase levels that are similar to each other and to the strain in which both SD are functional (n. 1). However, when the upstream ACA motif is replaced by GGG, yifK-lacZ expression drops sharply in both constructs (compare n. 4 ton. 6, and n. 5 ton. 7). Similar effects were observed in a separate construct where the segment between SD sequence and the initiating AUG was replaced by the sequence found at the corresponding position in the chiP gene [28] where the spacing (9-nt) is optimal (n. 8 and n. 9). While construct n. 8 is tightly repressed by GcvB, its variant lacking the upstream ACA (n. 9) shows a weak response to this sRNA (Figure S5). In conclusion, these results indicate that the enhancer activity does not require strict positioning of ACA relative to the initiation site, nor it depends on the spacing between the SD and the initiation codon. Loss of the enhancer function causes yifK expression to be less sensitive to repression by GcvB.

Essential role of ACA motifs in dppA expression
To assess the generality of the ACA effects, we turned to the dppA gene, a major GcvB target [16], [18]. The target sequence of GcvB in dppA mRNA includes four ACA motifs clustered within a 15 nt segment near the SD sequence (Figure 10). As an initial test, this 15-nt sequence was deleted in a strain carrying a dppA-lacZY translational fusion. The resulting mutant showed 96% lower of ß-galactosidase activity than the parental strain (data not shown). In the next experiment, we randomly mutagenized all four ACA repeats (in the same lacZ fusion background) and screened the mutants on MacConkey lactose indicator plates. Out of 43 mutants analyzed, 18 formed white colonies and had ß-galactosidase activities ranging between 0.5 and 5% of the wild-type levels. Four representative isolates from this group are shown in Figure 10. They are essentially Lac− mutants. 4 of the initial 43 mutants formed red colonies and expressed significant levels of ß-galactosidase (three shown in Figure 10). Interestingly, in two of these strains, the mutagenic process regenerated an ACA sequence. The remaining 21 isolates had an intermediate phenotype (pink colonies) and were not analyzed. Examination of the mutant sequences by the Mfold algorithm [29] showed a complete lack of correlation between presence/absence of secondary structures (or free energy values) and lacZ expression. Hence the most likely conclusion from this analysis is that the variations in lacZ expression levels are solely dictated by primary sequence determinants. Although the possibility that decreased expression in some of the mutants could be due to reduced mRNA stability, independent of ribosome binding, cannot be formally ruled out, it seems most likely that the observed differences reflect variations in translation initiation rates. Thus, on one hand, the data in Figure 10 further corroborate the positive role of the ACA motif in translation initiation; on the other hand, they reiterate the notion that an SD motif and a properly spaced AUG are not sufficient to promote initiation if placed in unfavorable sequence contexts [30].

Discussion
In the present work, we have characterized the regulation of Salmonella's yifK locus encoding a putative amino acid transporter highly conserved in Enterobacteriaceae. Our analysis showed yifK to be negatively controlled at the transcriptional level by the leucine response regulator Lrp, and at the post-transcriptional level by GcvB sRNA. These findings place yifK at the intersection of two global regulatory networks devoted to amino acid management [17], [31]. The relative impacts of two systems on yifK expression vary as a function of growth conditions, with the Lrp control predominating in leucine-deprived poor media and the GcvB control operating when amino acids are plentiful, possibly in excess. The sole condition where yifK appears to escape negative control is leucine-supplemented minimal medium, where Lrp repression is relieved. This response closely parallels that of the oligopeptide permease operon, oppABCDF [31] whose transcript is also a target of GcvB repression [16], [18]. Likely, the overlap of Lrp and GcvB networks reflects the link between amino acid metabolism and one-carbon units production; however, the precise physiological role and the implications of the above responses remain incompletely understood.
Genetic analysis of GcvB:yifK mRNA interactions revealed that the GcvB target sequence in yifK mRNA contains an enhancer element. Intriguingly, mutations that disrupt the enhancer - and lower yifK expression as a result - render yifK expression totally insensitive to GcvB repression. This suggests that the effectiveness of GcvB regulation is dependent on the enhancer function and that when this component is removed, GcvB-mediated repression no longer constitutes a rate-limiting step in yifK expression. Sharma and coworkers (2007) previously showed that GcvB's target sequence in the gltI gene of Salmonella acts as transferable translation enhancer (see Introduction). Unlike in our study, the effects of GcvB as a translational repressor were much greater than the effects of removing the enhancer, leading the authors to conclude that GcvB did not simply block the enhancer effect [16]. It seems possible that the plasmid-borne nature of the gcvB gene in the study by Sharma and coworkers made the GcvB repression tighter than when the sRNA is expressed from the chromosome. Alternatively, the contribution of the enhancer to gltI expression might be less important than in yifK expression. The gltI enhancer, located 45 nt upstream from the initiation codon, was characterized as part of a 27 nt segment and not analyzed in any further detail [16]. Here we found that nucleotide replacement in either of two ACA triplets within GcvB target site in yifK can result in more than 90% reduction in yifK expression. Although our data do not allow defining the contours of the enhancer element, they unequivocally identify the ACA motif as a determinant of its activity. We also found that the enhancer activity is maintained following a 7 nt shift in the position of the initiation site, suggesting the absence of strict spatial requirements for the functioning of the element. This is consistent with the data from the gltI system and with a report showing CA repeats to stimulate translation even when placed downstream from the start codon [32].
Translation initiation efficiencies have been known to vary greatly as a function of the sequence context of the initiation region [30], [33]. Computational analysis of sequences surrounding translation initiation sites of E.coli genes showed that the spacing between the SD and the initiation codon affects SD sequence conservation and its pattern. This study did not reveal significant biases outside these main elements [27]; however, conserved patterns occurring at variable positions might have been difficult to identify by the statistical analysis. Indeed, separates lines of evidence point to the role of the ACA motif in translation initiation. The motif is found in other translation enhancer sequences [34], [35] and, as an ACAA repeat, was shown to promote translation initiation in the absence of a SD sequence [36]. ACA is also found in the loops of pseudoknots formed by RNA ligands to ribosomal protein S1, obtained through Systematic Evolution of Ligands by Exponential Enrichment (SELEX) [37] and is part of the SELEX-determined consensus sequence for binding of protein CsrA, a translational regulator [38]. Finally, ACA is the recognition sequence of the MazF endonuclease that inactivates E.coli mRNAs by preferentially cleaving near the translation initiation codon [39].
The lack of position requirements for the functioning of the enhancer suggests that its role is to provide an anchor point for the 30 S ribosomal subunit so as to facilitate subsequent recognition of the SD sequence. Some of the evidence reviewed above tentatively identifies protein S1 as the possible candidate for the interaction. In vitro S1-binding studies with some of the mutants constructed in the course of this work should allow testing of this idea. Combined with the mutational analysis of other GcvB-regulated mRNAs, this approach might provide further insight into how the ACA motif participates in the translation initiation step.
Materials and Methods
Bacterial strains and culture conditions
Strains used in this study were derivatives of Salmonella enterica serovar Typhimurium strain LT2 [40]. Strain SV4280 was a gift of J. Casadesús. Except for the latter strain and for strain MA7224, all other strains were derived from MA3409, an LT2 derivative cured for the Gifsy-1 prophage [41]. The genotypes of the relevant strains used are listed in Table S1. Bacteria were cultured at 37°C in liquid media or in media solidified by the addition of 1.5% Difco agar. LB broth [42] was used as complex medium. Carbon-free medium (NCE) [43], supplemented with 0.2% glycerol or 0.2% lactose was used as minimal medium. Antibiotics (Sigma-Aldrich) were included at the following final concentrations: chloramphenicol, 10 µg ml−1; kanamycin monosulphate, 50 µg ml−1; sodium ampicillin 100 µg ml−1; spectinomycin dihydrochloride, 80 µg ml−1; tetracycline hydrochloride, 25 µg ml−1. MacConkey agar plates containing 1% lactose [44] were used to monitor lacZ expression in bacterial colonies. Liquid cultures were grown in New Brunswick gyratory shakers and growth was monitored by measuring the optical density at 600 nm with a Shimazu UV-mini 1240 spectrophotometer.
Relevant enzymes and chemicals
T4 polynucleotide kinase and Taq DNA polymerase were from New England Biolabs, Pfu-Turbo DNA polymerase was from Stratagene, T4 DNA ligase was from New England Biolabs. DNA oligonucleotides were custom synthesized by Sigma Aldrich or Eurofins MWG/Operon. The complete list of DNA oligonucleotides used in this study is shown in Table S2. DNA sequencing was performed by GATC biotech. Acrylamide-bisacrylamide and other electrophoresis reagents were from BioRad. Agarose was from Invitrogen. Hybond-N+ membranes and hybridization buffer used for Northern blot analysis were from GE Healthcare and from Applied Biosystems-Ambion, respectively. The rNTPs were from Promega and the 32P-NTPs were from PerkinElmer or Hartmann Analytic. 32P-labeled nucleic acids were detected by phosphorimaging using ImageQuant software.
Genetic techniques
Generalized transduction was performed using the high-frequency transducing mutant of phage P22, HT 105/1 int-201 [45] as described [46]. Chromosomal engineering (recombineering) was carried out by the λ red recombination method [47]–[49] implemented as in [47]. Donor DNA fragments were generated by PCR using plasmid DNA or chromosomal DNA or DNA oligonucleotides as templates. Amplified fragments were electroporated into appropriate strains harboring the conditionally replicating plasmid pKD46, which carries the λ red operon under the control of the PBAD promoter [47]. Bacteria carrying pKD46 were grown at 30°C in the presence of ampicillin and exposed to arabinose (10 mM) for 3 hours prior to preparation of electrocompetent cells. Electroporation was carried out using a Bio-Rad MicroPulser under the conditions specified by the manufacturer. Recombinant colonies were selected on LB plates containing the appropriate antibiotic. Constructs were verified by PCR and DNA sequence analysis (performed by GATC company).
Random PCR mutagenesis
PCR amplification of DNA fragments under error-prone conditions was carried out as previously described [50].
“Scarless” DNA recombineering
Scarless modification of chromosomal DNA sequences at the single base-pair level was achieved with a two-step recombineering procedure as previously described [51]. Briefly, this involved: 1) inserting a tetAR module (produced by PCR) at the chromosomal site to be modified and: 2) replacing the tetAR module by a DNA fragment carrying the desired changed through positive selection tetracycline-sensitive recombinants [52]. Typically, the DNA fragment in the second step was also obtained by PCR using oligonucleotides with complementary sequences at their 3′ ends priming DNA synthesis on each other (“reciprocal priming”). In site-directed mutagenesis experiments, one of the two primers contained the desired nucleotide changes or randomized sequence stretches. All constructs were verified by DNA sequencing. Table S3 shows the list of alleles made by standard or scarless recombineering.
RNA extraction and analysis by primer extension and Northern blotting
RNA was prepared by the acid-hot-phenol method from exponentially growing cells (OD600 of 0.35) as previously described [50]. Reverse transcriptase reactions (enzyme Superscript II from Invitrogen) were carried out using 5 µg of bulk RNA and 32P-labeled primer ppF49. The same DNA primer was used for the sequencing reactions. Reactions were performed with the fmol DNA Cycle Sequencing System from Promega, according to the manufacturer's protocol. Reaction products were fractionated on a 10% polyacrylamide-8 M urea gel. For Northern blot analysis, RNA was fractionated on a 1% agarose-formaldehyde gel, blotted onto a nylon membrane, and hybridized to the appropriate 32P-labeled DNA oligonucleotide probes.
In vitro translation
In vitro coupled transcription/translation was performed using New England Biolabs' PURExpress In vitro Protein Synthesis kit (NEB #E6800) according to the manufacturer instructions. Genes to be analyzed were cloned under T7 promoter control in the DFRH plasmid provided with the kit. The hybrid genes carried yifK wt or mutant 5′ UTR sequences fused to the cat-3×FLAG coding sequence (chloramphenicol acetyl transferase in-frame fusion to the 3×FLAG epitope). Final volume of the transcription/translation reaction was 25 µl in all cases. In addition to kit solutions A and B, reaction mix contained, 10 U of RNase inhibitor SUPERase (Ambion) and template plasmid DNA added to either 0.5 or 5 pM final concentration. Incubation times at 37°C varied from 15 to 90 min. Reactions were stopped by addition of equal volume of 2× Laemmli buffer and immediate freezing. Aliquots were loaded on 12.5% Acrylamide gels and Western analysis performed as previously described [53].
Toeprinting assay
Toeprinting reactions were carried out as described by Darfeuille et al [54] with minor modifications. RNA fragments spanning positions +1 to +135 of yifK mRNA were synthesized in vitro from T7 DNA templates generated by PCR amplification of chromosomal DNA (from strains MA8020 or MA11793) with primers ppI22 and ppI23. 2 pmol of RNA were annealed with 5′end-labeled primer ppI23 (1 pmol) in 10 mM Tris-acetate [pH 7.6], 0.1 M potassium acetate, and 1 mM DTT for 1 min at 90°C and chilled in ice for 5 min. Then, all dNTPs (final concentration 1 mM), Mg Acetate (10 mM final) were added; this was followed by preincubation with 2 pmol of 30S ribosomal subunit (a gift of Dominique Fourmy and Satoko Yoshizawa) at 37°C for 5 min. In experiments involving GcvB, 5, 1 or 0.5 pmol of sRNA were added prior to both, addition of the 30S ribosomal subunit and the preincubation step. After the 5-min period, 2 pmol of tRNAfMet were added and preincubation at 37°C continued for 15 additional min. Finally, Reverse Transcriptase (Superscript II, Invitrogen, 200U) was added and samples incubated for 15 min at 37°C. Following phenol chloroform extraction and ethanol precipitation, resuspended samples were loaded onto a 10% polyacrylamide-8 M urea gel along with the sequencing reaction samples generated with the same primer.
Measurement of β-galactosidase activity
β-galactosidase activity was assayed in toluene-permeabilized cells as described in [55] and is expressed in Miller units throughout this work. Typically, measurements were performed on duplicate or triplicate cultures grown in late exponential phase (OD600≈0.7). All experiments included parental or reference strains as normalization controls. Standard deviations were generally less than 5% of the mean.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GottesmanS, StorzG (2011) Bacterial Small RNA Regulators: Versatile Roles and Rapidly Evolving Variations. Cold Spring Harb Perspect Biol doi:10.1101/cshperspect.a003798
2. StorzG, VogelJ, WassarmanKM (2011) Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43 : 880–891.
3. VogelJ, LuisiBF (2011) Hfq and its constellation of RNA. Nat Rev Microbiol 9 : 578–589.
4. JacquesJF, JangS, PrevostK, DesnoyersG, DesmaraisM, et al. (2006) RyhB small RNA modulates the free intracellular iron pool and is essential for normal growth during iron limitation in Escherichia coli. Mol Microbiol 62 : 1181–1190.
5. MasséE, EscorciaFE, GottesmanS (2003) Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev 17 : 2374–2383.
6. MasséE, GottesmanS (2002) A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc Natl Acad Sci U S A 99 : 4620–4625.
7. MasséE, VanderpoolCK, GottesmanS (2005) Effect of RyhB small RNA on global iron use in Escherichia coli. J Bacteriol 187 : 6962–6971.
8. PrevostK, DesnoyersG, JacquesJF, LavoieF, MasséE (2011) Small RNA-induced mRNA degradation achieved through both translation block and activated cleavage. Genes Dev 25 : 385–396.
9. BalbontínR, FioriniF, Figueroa-BossiN, CasadesúsJ, BossiL (2010) Recognition of heptameric seed sequence underlies multi-target regulation by RybB small RNA in Salmonella enterica. Mol Microbiol 78 : 380–394.
10. GogolEB, RhodiusVA, PapenfortK, VogelJ, GrossCA (2011) Small RNAs endow a transcriptional activator with essential repressor functions for single-tier control of a global stress regulon. Proc Natl Acad Sci U S A 108 : 12875–12880.
11. JohansenJ, RasmussenAA, OvergaardM, Valentin-HansenP (2006) Conserved small non-coding RNAs that belong to the σE regulon: role in down-regulation of outer membrane proteins. J Mol Biol 364 : 1–8.
12. PapenfortK, PfeifferV, MikaF, LucchiniS, HintonJC, et al. (2006) σE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol Microbiol 62 : 1674–1688.
13. ThompsonKM, RhodiusVA, GottesmanS (2007) σE regulates and is regulated by a small RNA in Escherichia coli. J Bacteriol 189 : 4243–4256.
14. BeiselCL, StorzG (2011) The base-pairing RNA spot 42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli. Mol Cell 41 : 286–297.
15. PulvermacherSC, StaufferLT, StaufferGV (2009) Role of the sRNA GcvB in regulation of cycA in Escherichia coli. Microbiology 155 : 106–114.
16. SharmaCM, DarfeuilleF, PlantingaTH, VogelJ (2007) A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev 21 : 2804–2817.
17. SharmaCM, PapenfortK, PernitzschSR, MollenkopfHJ, HintonJC, et al. (2011) Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol Microbiol 81 : 1144–1165.
18. UrbanowskiML, StaufferLT, StaufferGV (2000) The gcvB gene encodes a small untranslated RNA involved in expression of the dipeptide and oligopeptide transport systems in Escherichia coli. Mol Microbiol 37 : 856–868.
19. StaufferLT, StaufferGV (2005) GcvA interacts with both the alpha and sigma subunits of RNA polymerase to activate the Escherichia coli gcvB gene and the gcvTHP operon. FEMS Microbiol Lett 242 : 333–338.
20. CoornaertA, ChiaruttiniC, SpringerM, GuillierM (2013) Post-Transcriptional Control of the Escherichia coli PhoQ-PhoP Two-Component System by Multiple sRNAs Involves a Novel Pairing Region of GcvB. PLoS Genet 9: e1003156.
21. Figueroa-BossiN, LemireS, MaloriolD, BalbontínR, CasadesúsJ, et al. (2006) Loss of Hfq activates the σE-dependent envelope stress response in Salmonella enterica. Mol Microbiol 62 : 838–852.
22. DesnoyersG, MasséE (2012) Noncanonical repression of translation initiation through small RNA recruitment of the RNA chaperone Hfq. Genes Dev 26 : 726–739.
23. BrennanRG, LinkTM (2007) Hfq structure, function and ligand binding. Curr Opin Microbiol 10 : 125–133.
24. BossiL, SmithDM (1984) Conformational change in the DNA associated with an unusual promoter mutation in a tRNA operon of Salmonella. Cell 39 : 643–652.
25. ZhangA, WassarmanKM, RosenowC, TjadenBC, StorzG, et al. (2003) Global analysis of small RNA and mRNA targets of Hfq. Mol Microbiol 50 : 1111–1124.
26. ModiSR, CamachoDM, KohanskiMA, WalkerGC, CollinsJJ Functional characterization of bacterial sRNAs using a network biology approach. Proc Natl Acad Sci U S A 108 : 15522–15527.
27. ShultzabergerRK, BucheimerRE, RuddKE, SchneiderTD (2001) Anatomy of Escherichia coli ribosome binding sites. J Mol Biol 313 : 215–228.
28. Figueroa-BossiN, ValentiniM, MalleretL, FioriniF, BossiL (2009) Caught at its own game: regulatory small RNA inactivated by an inducible transcript mimicking its target. Genes Dev 23 : 2004–2015.
29. ZukerM (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31 : 3406–3415.
30. DreyfusM (1988) What constitutes the signal for the initiation of protein synthesis on Escherichia coli mRNAs? J Mol Biol 204 : 79–94.
31. CalvoJM, MatthewsRG (1994) The leucine-responsive regulatory protein, a global regulator of metabolism in Escherichia coli. Microbiol Rev 58 : 466–490.
32. Martin-FarmerJ, JanssenGR (1999) A downstream CA repeat sequence increases translation from leadered and unleadered mRNA in Escherichia coli. Mol Microbiol 31 : 1025–1038.
33. YarchukO, JacquesN, GuillerezJ, DreyfusM (1992) Interdependence of translation, transcription and mRNA degradation in the lacZ gene. J Mol Biol 226 : 581–596.
34. KomarovaAV, TchufistovaLS, SupinaEV, BoniIV (2002) Protein S1 counteracts the inhibitory effect of the extended Shine-Dalgarno sequence on translation. RNA 8 : 1137–1147.
35. McCarthyJE, SchairerHU, SebaldW (1985) Translational initiation frequency of atp genes from Escherichia coli: identification of an intercistronic sequence that enhances translation. EMBO J 4 : 519–526.
36. TzarevaNV, MakhnoVI, BoniIV (1994) Ribosome-messenger recognition in the absence of the Shine-Dalgarno interactions. FEBS Lett 337 : 189–194.
37. RingquistS, JonesT, SnyderEE, GibsonT, BoniI, et al. (1995) High-affinity RNA ligands to Escherichia coli ribosomes and ribosomal protein S1: comparison of natural and unnatural binding sites. Biochemistry 34 : 3640–3648.
38. DubeyAK, BakerCS, RomeoT, BabitzkeP (2005) RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction. RNA 11 : 1579–1587.
39. VesperO, AmitaiS, BelitskyM, ByrgazovK, KaberdinaAC, et al. (2011) Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell 147 : 147–157.
40. LilleengenK (1948) Typing of Salmonella typhimurium by means of bacteriophage. Acta Pathol Microbiol Scand 77 : 2–125.
41. Figueroa-BossiN, CoissacE, NetterP, BossiL (1997) Unsuspected prophage-like elements in Salmonella typhimurium. Mol Microbiol 25 : 161–173.
42. BertaniG (2004) Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J Bacteriol 186 : 595–600.
43. MaloySR, RothJR (1983) Regulation of proline utilization in Salmonella typhimurium: characterization of put::Mu d(Ap, lac) operon fusions. J Bacteriol 154 : 561–568.
44. MacconkeyA (1905) Lactose-Fermenting Bacteria in Faeces. J Hyg (Lond) 5 : 333–379.
45. SchmiegerH (1972) Phage P22-mutants with increased or decreased transduction abilities. Mol Gen Genet 119 : 75–88.
46. LemireS, Figueroa-BossiN, BossiL (2011) Bacteriophage crosstalk: coordination of prophage induction by trans-acting antirepressors. PLoS Genet 7: e1002149.
47. DatsenkoKA, WannerBL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 : 6640–6645.
48. MurphyKC, CampelloneKG, PoteeteAR (2000) PCR-mediated gene replacement in Escherichia coli. Gene 246 : 321–330.
49. YuD, EllisHM, LeeEC, JenkinsNA, CopelandNG, et al. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97 : 5978–5983.
50. BossiL, Figueroa-BossiN (2007) A small RNA downregulates LamB maltoporin in Salmonella. Mol Microbiol 65 : 799–810.
51. BossiL, SchwartzA, GuillemardetB, BoudvillainM, Figueroa-BossiN (2012) A role for Rho-dependent polarity in gene regulation by a noncoding small RNA. Genes Dev 26 : 1864–1873.
52. BochnerBR, HuangHC, SchievenGL, AmesBN (1980) Positive selection for loss of tetracycline resistance. J Bacteriol 143 : 926–933.
53. UzzauS, Figueroa-BossiN, RubinoS, BossiL (2001) Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A 98 : 15264–15269.
54. DarfeuilleF, UnosonC, VogelJ, WagnerEG (2007) An antisense RNA inhibits translation by competing with standby ribosomes. Mol Cell 26 : 381–392.
55. Miller JH (1992) A Short Course in Bacterial Genetics. A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press.
56. CamachoEM, CasadesúsJ (2002) Conjugal transfer of the virulence plasmid of Salmonella enterica is regulated by the leucine-responsive regulatory protein and DNA adenine methylation. Mol Microbiol 44 : 1589–1598.
57. EllermeierCD, JanakiramanA, SlauchJM (2002) Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290 : 153–161.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- GATA6 Is a Crucial Regulator of Shh in the Limb Bud
- Large Inverted Duplications in the Human Genome Form via a Fold-Back Mechanism
- Differential Effects of Collagen Prolyl 3-Hydroxylation on Skeletal Tissues
- Affects Plant Architecture by Regulating Local Auxin Biosynthesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy