
Down-Regulation of eIF4GII by miR-520c-3p Represses Diffuse Large B Cell Lymphoma Development
Deregulation of the translational machinery is emerging as a critical contributor to cancer development. The contribution of microRNAs in translational gene control has been established however; the role of microRNAs in disrupting the cap-dependent translation regulation complex has not been previously described. Here, we established that elevated miR-520c-3p represses global translation, cell proliferation and initiates premature senescence in HeLa and DLBCL cells. Moreover, we demonstrate that miR-520c-3p directly targets translation initiation factor, eIF4GII mRNA and negatively regulates eIF4GII protein synthesis. miR-520c-3p overexpression diminishes cells colony formation and reduces tumor growth in a human xenograft mouse model. Consequently, downregulation of eIF4GII by siRNA decreases translation, cell proliferation and ability to form colonies, as well as induces cellular senescence. In vitro and in vivo findings were further validated in patient samples; DLBCL primary cells demonstrated low miR-520c-3p levels with reciprocally up-regulated eIF4GII protein expression. Our results provide evidence that the tumor suppressor effect of miR-520c-3p is mediated through repression of translation while inducing senescence and that eIF4GII is a key effector of this anti-tumor activity.
Published in the journal:
. PLoS Genet 10(1): e32767. doi:10.1371/journal.pgen.1004105
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004105
Summary
Deregulation of the translational machinery is emerging as a critical contributor to cancer development. The contribution of microRNAs in translational gene control has been established however; the role of microRNAs in disrupting the cap-dependent translation regulation complex has not been previously described. Here, we established that elevated miR-520c-3p represses global translation, cell proliferation and initiates premature senescence in HeLa and DLBCL cells. Moreover, we demonstrate that miR-520c-3p directly targets translation initiation factor, eIF4GII mRNA and negatively regulates eIF4GII protein synthesis. miR-520c-3p overexpression diminishes cells colony formation and reduces tumor growth in a human xenograft mouse model. Consequently, downregulation of eIF4GII by siRNA decreases translation, cell proliferation and ability to form colonies, as well as induces cellular senescence. In vitro and in vivo findings were further validated in patient samples; DLBCL primary cells demonstrated low miR-520c-3p levels with reciprocally up-regulated eIF4GII protein expression. Our results provide evidence that the tumor suppressor effect of miR-520c-3p is mediated through repression of translation while inducing senescence and that eIF4GII is a key effector of this anti-tumor activity.
Introduction
Control of gene expression at the level of mRNA translation is a crucial step that regulates proper function of major cellular processes such as cell proliferation, growth, differentiation, apoptosis, stress response, and tumorigenesis. A large body of recent research indicates that the deregulation of mRNA translation can promote cellular transformation and a malignant phenotype [1]–[4]. Cellular senescence, resulting in permanent arrest of cell growth is emerging as an intrinsic tumor suppressive mechanism [5]. While it has been established that aging and senescence is associated with lower rates of mRNA translation the linkage between translation deregulation and senescence in malignant cells is poorly described. Therefore, understanding the translational regulation in the framework of senescence program in tumor cells may provide an important approach in cancer therapy.
Translation of most mRNAs is primarily regulated at the level of initiation, a process that requires the protein complex known as eukaryotic initiation factor 4F (eIF4F), consisting of three proteins: cap-binding protein eIF4E, scaffolding protein eIF4G, and ATP-dependent RNA helicase eIF4A [6], [7]. The aberrant expression of eIF4F components has been shown to be involved in many cancers, particularly in B-cell lymphoma [8]–[10]. Thus, targeting of the translation initiation complex is emerging as a potential cancer therapy [11]–[13]. The eukaryotic translation initiation factor 4, gamma (eIF4G) is expressed in mammalian cells in the form of the two homologs, eIF4GI and eIF4GII [14], [15]. Recent studies have found that eIF4GI and eIF4GII despite the biochemical and functional similarities can fulfill different roles in mammalian cells [7]. Elevated eIF4GI levels have been shown to correlate with a malignant cell transformation [16]–[19]. However, little is currently known about eIF4GII expression and its function in translation initiation.
MicroRNAs (miRNAs) are endogenous regulatory RNA molecules that modulate protein expression based on sequence complementation with their target messenger RNAs (mRNAs) [20]–[24]. Given that these small regulatory molecules are frequently deregulated in various human cancers, they have become potential candidates as biomarkers and therapeutic intervention. Gene expression profiling studies and bioinformatics analysis have discovered numerous miRNAs differentially expressed in several types of human cancers, including B-cell malignancies [25], [26]. Among the miRNAs expressed by hematopoietic cells, several were implicated in different types of human B-cell lymphomas [27], [28] and suggest a promising option for lymphoma/leukemia therapy [29]–[31]. However, the mechanisms by which these small regulatory RNAs deregulation contribute to lymphomagenesis have not been well established. Therefore, the identification of the genes and pathways directly targeted by microRNAs remains to be fully elucidated.
Herein, we discovered that elevated expression of miR-520c-3p results in decreased global translation and cell proliferation, and induced premature senescence in HeLa and diffuse large B-cell lymphoma (DLBCL). Furthermore, miR-520c-3p negatively regulates the protein synthesis of eIF4G3/eIF4GII (known from here out as eIF4GII) through the direct targeting of its mRNA. Overexpression of miR-520c-3p resulted in diminished clonogenic capacity of DLBCL cells and reduced tumor growth in a human lymphoma xenograft mouse model. Importantly, we found that DLBCL cell lines and patient samples expressed reduced levels of miR-520c-3p and elevated levels of eIF4GII as compared to normal donor B-cell lymphocytes. Our findings presented here provide a previously unknown tumor suppressor function for miR-520c-3p elicited by decreasing eIF4GII expression, resulting in repressed global protein synthesis and induction of premature cellular senescence.
Results
Overexpression of miR-520c-3p Decreased Global Translation and Proliferation and Induced Senescence in HeLa and DLBCL Cells
The contribution of microRNAs in control of post-transcriptional gene regulatory events in human cancers is becoming increasingly evident. In a recent survey identifying the microRNAs associated with elevated levels of the oncoprotein MCT-1 (data not shown), we found that overexpression of miR-520c-3p greatly repressed global protein synthesis, suggesting that miR-520c-3p could contribute broadly to regulating protein abundance. Given that miR-520c was found to have decreased expression in the series of DLBCL cases used for miRNA profiling [32] and deregulation of protein synthesis has been recognized as a critical part of cancer development [33]–[35] we focused on miR-520c-3p function in human cancers with a particular interest in DLBCL. The human cervical carcinoma HeLa cell line was used for the mechanistic studies because these cells can be easily transfected at a very high efficiency, an important requirement for such experiments.
First, we explored the abundance of miR-520c-3p in several primary normal B-cells, primary DLBCL cells, cultured DLBCL and Burkitt Lymphoma (BL) cell lines by RT-qPCR analysis. As shown in Figure 1A, miR-520c-3p levels were consistently lower in both primary DLBCL samples and DLBCL cell lines as compared to normal B-cells. Forty-eight and seventy-two hours after transfection to elevate the levels of miR-520c-3p, significant reduction in the global protein synthesis in miR-520c-3p overexpressing cells relative to the control cultures was demonstrated by polysome distribution profiles and incorporation of 35S-labeled amino acids into nascent protein in HeLa (Figure 1B and C, Figure S1A–C) and DLBCL (Figure 1D and E, Figure S1D–F) cell populations. Inversely, reduction of miR-520c-3p expression by miRZip-520c-3p transduction increased global protein synthesis levels. However, these observed differences were not as dramatic as the reduction after overexpression of mir-520c-3p (Figure S2A–D). This attenuated impact was likely due to the already low levels of miR-520c-3p present in the cells. Consistent with the repressed translation, the cell proliferation was reduced in all studied cell lines as fewer cells were observed in the miR-520c-3p upregulated populations compared to the control miRNA groups when examined by cell counting (Figure 1F and G) and measured in real-time by the xCELLigence System in HeLa cells (Figure 1H). Upon miR-520c-3p overexpression, all studied cell lines showed only slight or no increase in cell apoptosis, as measured by Annexin V staining (Figure 2A). Furthermore, flow cytometry analysis showed that seventy-two hours after transfection cells with increased miR-520c-3p expression delayed passage through G1 phase as compared to control-transfected populations (Figure 2B, Figure S3A and B). Interestingly, we discovered that HeLa cells overexpressing miR-520c-3p were enlarged, displayed a distinctive flat morphology, and had numerous vacuoles, which is characteristic of senescent cells. The induction of a senescent phenotype triggered by increased levels of miR-520c-3p was further supported by the enhanced activity of the senescence marker ß-galactosidase (Figure 2C) and was confirmed by other hallmarks of senescence such as elevated p16 and p53 protein levels and decreased HuR expression [36] in HeLa and DLBCL cells (Figure 2D). Altogether, these data indicate that overexpression of miR-520c-3p inhibited global translation and cell proliferation, and induced cellular senescence in cancer cells.


Elevated Levels of miR-520c-3p Decreased Translation of Its Target Genes
To better understand the molecular mechanisms whereby miR-520c-3p elicits global translational repression and to identify miR-520c-3p targets and pathways involved in this regulation, we performed microarray analysis of total mRNA and mRNA isolated from polysomal fractions (translational profiling). Significantly altered genes were further analyzed for representation of Gene Ontology (GO) terms to identify key biologically functional categories that were significantly changed in miR-520c-3p overexpressed compared to control transfected cells. Noticeably, GO analysis revealed mainly translation and translation related categories with greatest representations of genes which had decreased protein synthesis (lowest values in the most actively translating fractions [fractions 10–11], and highest values in the non-translating and low-translating fractions of the gradient [fractions 1–7]) in miR-520c-3p overexpressed versus control cells (Figure 3A, Figure S4). Additionally, genes affected by miR-520c-3p overexpression in the most translationally active polysomal fraction 10 were put in the context of the known molecular interactions by using Ingenuity Pathways Analysis (IPA). IPA revealed that the top five networks, with the highest number of involved genes, were molecular functions linked to RNA post-transcriptional modification, cellular development, cellular growth and proliferation, cell cycle, and cancer (Table S1). The effect of miR-520c-3p overexpression was furthermore verified for several mRNAs (Figure 3B) chosen from the most significantly altered in microarray data genes (Table S2), which were computationally predicted as direct targets of miR-520c-3p (by miRanda and Microcosm database) and, functionally represented above ranked pathways found by GO and IPA analysis. By comparing miR-520c-3p upregulated to control transfected cells by RT-qPCR analysis, we observed no influence of miR-520c-3p on total mRNA levels of most of the validated miR-520c-3p target transcripts (Figure 3C). Instead, we found that increased miR-520c-3p levels lowered protein expression of studied target genes (Figure 3D).
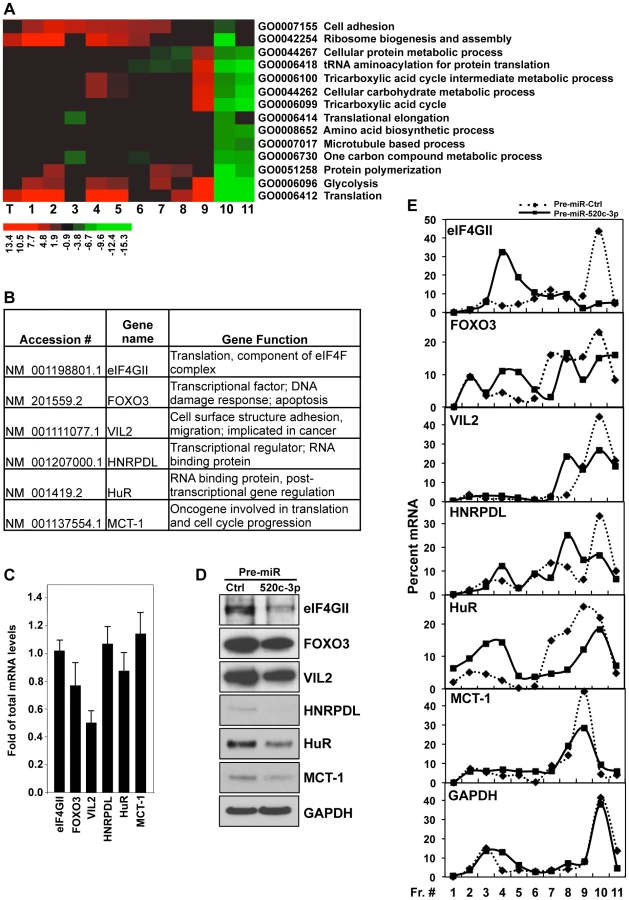
Given that microRNAs have been shown to influence translation as well as message stability [37], we investigated both potential post-transcriptional mechanisms (Figure 3E, Figure S5). We found only one message (Vil2 mRNA) to be affected on both the stability and translational level by miR-520c-3p. As shown in Figure 3E, corresponding with protein levels, we found decreased levels of all the validated transcripts in the actively translating fractions of the gradient (fractions 8–11), and elevated levels in the non-translating and low-translating fractions of the gradient (fractions 1–7) in groups transfected with miR-520c-3p. These differences were not observed when testing the distribution of the housekeeping GAPDH mRNA. The above data confirm that miR-520c-3p targets and decreases expression of many important genes involved in translational control, cell proliferation and cancer development.
eIF4GII Is a Specific Target of miR-520c-3p
Importantly, among the validated genes in which translation and protein levels were strongly decreased by miR-520c-3p was eukaryotic translation initiation factor 4 gamma, 3 (eIF4GII), one of the crucial components of the cap-dependent translation regulation complex eIF4F (Figure 3D and E). Therefore, we hypothesized that the influence of miR-520c-3p overexpression on global protein synthesis and cell phenotype might be mediated by eIF4GII. eIF4GII was predicted to have two miR-520c-3p binding sites in its 3′ untranslated region (3′UTR) (Figure 4A). The specific influence of miR-520c-3p on eIF4GII was initially confirmed by heterologous reporter constructs, which revealed diminished luciferase activity in the construct bearing the 3′ untranslated region (3′UTR) of eIF4GII mRNA suggesting that miR-520c-3p indeed repressed eIF4GII translation through its 3′UTR sequence (Figure 4B). Further, to corroborate that miR-520c-3p repressed eIF4GII translation specifically through the seed sites on its 3′UTR, heterologous GFP reporters were transfected in the conditions of either elevated miR-520c-3p or miR-Ctrl. These reporters were bearing segments with the predicted miR-520c-3p sites that were either wild type (wt) or with mutated (mut) seed region on eIF4GII 3′UTR (Figure 4C). As demonstrated in Figure 4D, increased miR-520c-3p levels efficiently reduced GFP wild type expression in both eIF4GII fragments, confirming miR-520c-3p's repressive ability on both studied sites. Mutation of the seed region 1 (mut1) only slightly increased GFP expression in miR-520c-3p overexpressing cells compared to the wild type. However, mutating the seed region 2 (mut2) in the eIF4GII 3′UTR strongly increased GFP levels in miR-520c-3p overexpressing populations. Significantly, GFP mRNA levels remained unchanged between transfection groups (Figure S6A). These data suggest that miR-520c-3p inhibits eIF4GII expression through two sites in eIF4GII 3′UTR but one site, seed region 1, may not be the true binding site for miR520c-3p. Instead seed region 2 is indeed functional; therefore mutation in site 2 prevents the downregulation of GFP protein.

The repression through the eIF4GII 3′UTR miR-520c-3p sites was additionally evaluated by rescue experiments. Transfection with miR-520c-3p efficiently repressed eIF4GII levels in vector transfected populations but did not affected the levels of overexpressed eIF4GII with deleted 3′UTR sequence, pCDH-eIF4GII(CR) (Figure 4E). Strikingly, overexpression of pCDH-eIF4GII(CR) also changed the levels of senescence markers and abolished miR-520c-3p triggered differences in their expression. On the contrary, knocking down miR-520c-3p with miRZip-520c-3p increased the expression of eIF4GII, was able to restore eIF4GII levels silenced by siRNA and modulated levels of studied senescence phenotype proteins (Figure 4F). Finally, we tested if miR-520c-3p overexpression affects the availability of eIF4GII for translation initiation and observed miR-520c-3p mediated reduction in the abundance of eIF4GII associated with the 5′ 7-methyl-GTP cap structure analog (Figure 4G). All together, these results strongly support the contention that the miR-520c-3p-elicited changes in protein translation and cell phenotype are indeed a consequence of modulated eIF4GII levels, via miR-520c-3p interaction with binding sites in the eIF4GII 3′UTR.
Contribution of eIF4GII in the Phenotype Triggered by miR-520c-3p
Given that mRNA translation is primarily regulated at the level of initiation and that eIF4G is a key scaffolding protein that forms a critical link between the mRNA cap structure and poly(A) tail during this process, we further postulated that the repression of the eIF4GII by miR-520c-3p might elicit a global mRNA translation inhibition upon miR-520c-3p overexpression. To test this possibility we silenced the eIF4GII protein levels by using eIF4GII-targeting siRNA. Indeed, knockdown of eIF4GII expression consistently repressed global translation as monitored by the global polysome distribution profiles (Figure 5A) and nascent protein synthesis assessed by incorporation of 35S-labeled amino acids (Figure 5B).

We also tested whether silencing of eIF4GII could affect the translation of specific mRNAs validated in Figure 3 as miR-520c-3p targets by monitoring the fraction of those mRNAs associated with the translational apparatus. Corresponding to the overexpression of miR-520c-3p, knockdown of eIF4GII decreased protein levels of the miR-520c-3p target genes (Figure 5C). As illustrated in Figure 5D, examined mRNA levels were very low in non-translating and low-translating fractions of the gradient (fractions 1–7). In the Ctrl siRNA group, most of the transcripts were abundant in the actively translating polysomal fractions of the gradient (fractions 8–10). Silencing of eIF4GII led to a leftward shift in mRNA distribution, indicating that the studied transcripts associated with smaller polysomes and further suggesting that eIF4GII siRNA suppressed the initiation of mRNA translation.
As we observed, overexpression of pCDH-eIF4GII(CR) construct affected the expression of senescence markers (Figure 4E), therefore we wished to determine if eIF4GII repression might also participate in the process of miR-520c-3p triggered formation of the senescent phenotype. Comparable to the cells with increased miR-520c-3p, silencing of eIF4GII by siRNA caused a significant decline in the cell proliferation (Figure 5E) but no significant increase in cellular apoptosis (Figure 5F). Moreover, eIF4GII knockdown initiated cell cycle arrest in G1 phase (Figure 5G, Figure S6B), induced ß-galactosidase activity (Figure 5H) and altered levels of the senescence markers p16, p53, and HuR (Figure 5I). Collectively, these findings indicate that the miR-520c-3p-elicited repression in global translation is facilitated through the influence of miR-520c-3p on eIF4GII expression, and further eIF4GII actively contributes to the miR-520c-3p triggered decrease in cell proliferation and the induction of senescence.
Both miR-520c-3p Overexpression and eIF4GII Knockdown Diminished the Clonogenic Capacity of Lymphoma Cells
Based on above data, we hypothesized that overexpression of miR-520c-3p can suppress tumor growth by decreasing protein translation, inhibiting proliferation, and promoting cell senescence. To address our hypothesis, we transfected Farage cells with either Pre-miRNA-Ctrl, Pre-miR-520c-3p (Figure 6A), siRNA targeting eIF4GII or control siRNA (Figure 6B) and carried out soft agar colony formation assays. As shown, either elevated miR-520c-3p levels or reduced eIF4GII levels significantly decreased the ability of cells to form colonies when compared to control transfections. These results provided further evidence that eIF4GII is implicated in miR-520c-3p mediated cells growth control.

Increase in miR-520c-3p Expression Resulted in Repressed Tumor Formation in a Human Xenograft Model
Finally, we examined whether overexpression of miR-520c-3p inhibits tumor formation in vivo. To this end, we established a preclinical xenograft mouse model with SUDHL4 cells transduced with either an empty vector (pCDH-Vector) or a vector overexpressing miR-520c-3p (pCDH-520c-3p). Upregulation of miR-520c-3p resulted in a significant repression of lymphoma tumors growth (Figure 6C and D, Figure S7A). Consistently, similar results were obtained in a xenograft model performed with Pre-miR-520c-3p transfected HeLa cells (Figure S7B). To test if the tumors growth repression was influenced by translational inhibition, polysome distribution profiles were analyzed in tumors harvested from xenografts (Figures 6E). Indeed, such profiles were consistent with our in vitro cell culture data (Figure 1B and D) showing a global translational decrease in miR-520c-3p overexpressing tumors. Likewise, Western blot analysis of examined senescence markers in xenografts confirmed that the decrease in tumors growth was also affected by induced cellular senescence (Figure 6F). In summary, our findings provide strong evidence that miR-520c-3p overexpression contributes to the observed inhibition of tumor formation in the human xenograft mouse model by maintaining a translationally repressed state, inhibiting proliferation and initiating cellular senescence in cancer cells. We propose that the observed decreased protein translation and premature senescence is mediated, at least in part, through a miR-520c-3p-mediated decrease in expression of translational factor, eIF4GII.
Reciprocal Expression of miR-520c-3p and eIF4GII in DLBCLs
Although eIF4GI has been found significantly elevated in a variety of cancers, to date, the tumorigenic potential of eIF4GII has not been investigated [16]–[19]. In the present study, we explored the expression of both miR-520c-3p levels and eIF4GII protein expression in a DLBCL model. As shown in Figure 1A, miR-520c-3p levels were consistently lower in both primary DLBCL cells and cultured DLBCL cell lines as compared to primary normal B-cells. Conversely, eIF4GII protein expression appeared to be low in normal B-cells and markedly elevated in all studied DLBCL cell lines (Figure 7A). To evaluate eIF4GII protein levels on primary DLBCL specimens and reactive lymph nodes, we performed immunohistochemistry analysis of tissue microarrays (TMA) on 49 primary DLBCL specimens (Table 1, Figure 7B). In agreement with Western blot analysis (Figure 7A), TMA data revealed that eIF4GII was markedly increased in 71% of all DLBCL (GCB and ABC subtype) compared to GCB of the reactive lymph nodes (14%) as summarized in Table 1 and representative sections shown in Figure 7B. Simultaneously, RNA from the same TMA samples was purified and expression of miR-520c-3p was measured by RT-qPCR (Table S3, Figure 7C). Despite the internal variability characteristic for primary samples derived from different patients we observed significant inverse relationship between eIF4GII protein expression and miR-520c-3p levels (Pearson correlation coefficient r = −0.38, p<0.02) in studied DLBCL cases. Altogether, this data support the role of miR-520c-3p and eIF4GII in the deregulation of protein synthesis in DLBCL and support the potential role of these molecules as specific therapeutic targets.


Discussion
Numerous studies have established that miRNAs are incorporated in major critical cellular pathways and can function in concert with other genes crucial for control of cellular growth [20], [21]. Nevertheless, to understand the role of these small regulatory molecules in tumorigenesis, it is essential to identify the gene/pathway that they regulate. Herein, we demonstrated that elevated levels of miR-520c-3p resulted in inhibition of global gene translation, cell proliferation and promoted senescence in HeLa and DLBCL cells. Microarray analysis revealed that miR-520c-3p regulates the expression of many translation, cell proliferation and cancer related genes. Among those identified by microarray analysis and validated miR-520c-3p target genes, we found, eIF4GII, a pivotal scaffold member of eIF4F, and a central organizing protein in the recruitment of mRNA during translational initiation. Furthermore we observed that, comparable to miR-520c-3p elevation, silencing of eIF4GII by siRNA repressed translation, cell proliferation, promoted senescence and diminished the clonogenic ability of cancer cells. In addition, the overexpression of miR-520c-3p reduced eIF4GII translation and consequently decreased its protein levels through the miR-520c-3p seed sequence on the eIF4GII 3′UTR. Hence, by decreasing eIF4GII protein expression, miR-520c-3p indirectly controls expression of many eIF4GII target genes and collaboratively promotes changes in the proteome of malignant cells.
Multiple lines of evidence have now been established that the disruption of the translational machinery is a major contributor to cancer development and progression [8], [10]. Translational deregulation can occur through modification in the expression of proteins involved in translational initiation and/or changes in miRNA expression [33]–[35]. In recent years, a great deal of research has been focused on investigating the mechanisms of translational regulation by miRNAs. However, the role of miRNAs in control of specific factors of the translation initiation complex eIF4F has not yet been thoroughly explored. To date, only one report has described a miRNA dependent mechanism for the targeting of eIF4E and subsequent inhibition of host protein synthesis [38]. Thus, our results provide the first evidence of a miRNA regulating the translational initiation complex through the targeting of eIF4GII, and thereby modulating protein synthesis in cancer cells. Although, one cannot exclude the role of multiple target genes contributing to the observed miR-520c-3p triggered phenotype, signaling pathways (kinases or phosphatases) or other miRNAs actions that may also be involved, our data strongly support a major role of eIF4GII in this control.
While translational deregulation appears to be an essential feature of tumorigenesis and cellular senescence is considered to be tumor-suppressor mechanism there are few studies to date that have directly addressed the link between translational control and senescence. The results from these studies are confounding with some suggesting little difference in global rates of translation following oncogene-induced senescence [39], while others have shown that rapamycin treatment can actually prolong the induction of oncogene-induced senescence [40]. Some data have even suggested that the initiation of autophagy through inhibition of mTOR signaling (and presumably inhibition of translation) is a necessary prerequisite for senescence [41], [42]. It is thus of broad interest to determine what role translation or translational factors can play in the induction and maintenance of the cellular senescence program.
Consistent with our in vitro results, we demonstrated in a human xenograft mouse model that overexpression of miR-520c-3p significantly inhibits tumor formation in vivo. Although miR-520c has previously been linked to cancer [43]–[45], to date its role in tumor progression remains controversial and the molecular mechanism of miR-520c's function still awaits further clarification. We have identified the miR-520c-3p as a negative regulator of tumor growth via specific downregulation of translational factor, eIF4GII. Based on the data gathered from our studies, we postulate that the main tumor-suppressive mechanisms of miR-520c-3p can be attributed through the repression of translation and induction of premature senescence, which is a direct function of reduced eIF4GII protein levels. Recent reports indicate that mechanisms inducing premature senescence of cancer cells are emerging as a promising anti-tumorigenic, back-up program to improve on an insufficient apoptotic response [46], [47]. In addition, the combination of senescence induced therapy and a conventional therapy is postulated as a rational approach to cancer treatment [48], [49]. Accordingly, corresponding inhibition of translation and induction of senescence by miRNAs and/or small molecule inhibitors that target eIF4GII hold promise as an innovative strategy for cancer therapy.
MicroRNAs play a fundamental role in cancer biology and the alteration of their expression can have substantial effects on cell phenotypes. Thus, the role of miRNAs in the cancer pathogenesis makes them novel biomarkers and important therapeutic tools [50]. Accordingly, the results of several studies offer the experimental basis for the use of these small molecules in a therapeutic approach. For example, miR-34a and miR-155 have been already established as promising targets for lymphoma treatment [29]–[31]. While existing expression profiling studies have identified numerous miRNAs differentially expressed in lymphoma malignancies, DLBCL was found to have the most heterogeneous miRNA signature [26], [32], [51], [52]. Aberrant expression of miRNA can be a consequence of various mechanisms such as structural alterations, chromatin remodeling, aberrant transcription factor activity, etc. Encoded on chromosome 19q13.42, miR-520c was identified as being associated with t(4;14) and t(11;14) translocations in multiple myeloma. In addition, several other miRNAs located on chromosome 19q13.42, such as miR-520g, miR-520h, and miR-520b, have been found to be downregulated in multiple myeloma [53]. Although recurrent translocations involving chromosome 19q13 such as t(14;19)(q32.3;q13.2) have been reported in B-cell malignancies, they are extremely rare [54], [55]. Given that epigenetics has a profound effect on the regulation of miRNA expression and DNA hypermethylation has been shown to be involved in the decrease of miRNA in lymphomas, we examined if the decreased expression of miR-520c in DLBCLs as compared to normal B cells could be due to hypermethylation of the genomic locus (Text S1, Figure S8, Table S4). However, no hypermethylation within the studied locus was detected in lymphomas. We observed focal areas of hypomethylation of miR-520 and 5′ from the coding sequence in lymphomas, which may represent a permissive state for the binding of putative transcriptional repressors. Since specific miRNA can be deregulated by multiple mechanisms, examining the regulation of miR-520c repression in DLBCL by alternative means is an opportunity for future investigation.
We found that reduced miR-520c-3p was correlated with elevated eIF4GII expression in DLBCL. While the other functionally complementary scaffolding factors eIF4E and eIF4GI have been described as significantly increased in a variety of cancers [8]–[10], [17]–[19], our studies are the first to implicate eIF4GII in a malignancy. In the light of increasing evidence that abnormal function of cellular eIF4F complex caused by elevated expression of initiation factors plays a major role in tumorigenesis [56], it is widely believed that targeting the molecules of this complex is a rational cancer therapy [11], [34], [57]. Recently, promising research has been done on targeting eIF4E by using eIF4E-specific antisense nucleotides [4]. Also, small-molecule inhibitors have been utilized to block interactions between the translation initiation factors eIF4E and eIF4G1, and to modulate eIF4A activity [12], [13]. Our findings that eIF4GII is overexpressed in DLBCL provides an additional component of the eIF4F complex consideration as a potential therapeutic target.
In summary, we revealed a tumor-suppressive function of miR-520c-3p and identified eIF4GII as a major effector of this action. Our findings demonstrate the ability of miR-520c-3p to repress the development of cancer malignancies through coordinated control of several significant mechanisms: (1) inhibition of eIF4GII and consequent repression of global translational machinery and induction of senescence in tumor cells, and (2) corresponding regulation of other target genes involved in cancer development. Whereas much extant research has demonstrated the role of miRNA-target gene interactions for a single pathway, only a limited number of studies have shown the interaction of a specific miRNA with a major regulatory node, which has the potential to enhance pleiotropic phenotypic effects. The data presented herein not only illustrates the complexity of the miR-520c-3p triggered post-transcriptional regulation network that regulates translation and cell growth, but also provides information about the cooperation between the functionally related mechanisms that results in tumor growth repression. These in vitro and in vivo results were confirmed in DLBCL patient samples, where miR-520c-3p levels were inversely correlated to eIF4GII protein expression. Our study established a novel link between regulation of translation and senescence in malignant cells and suggests that protein synthesis control is a crucial part of the complex mechanism leading malignant cells to senescence program.
Materials and Methods
Ethics Statement
Human sample studies have been approved by the University of Maryland Medical School Institutional Review Board and conform to the Declaration of Helsinki.
Animal procedures were performed in compliance with protocols approved by the Institutional Animal Care and Use Committee of the University of Maryland.
Cell Culture and Transfections
Human cervical carcinoma HeLa cells were cultured in DMEM (Gibco BRL, Gaithersburg, MD) supplemented with 10% FBS. Diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL) cell lines were cultured in RPMI medium 1640 (Gibco BRL, Gaithersburg, MD) containing 10% FBS. HeLa cells were transfected by using Oligofectamine (Invitrogen) with 50 nM small RNAs Pre-miR-520c-3p (AAAGUGCUUCCUUUUAGAGGGU), control Pre-miR-Ctrl (Ambion), or 20 nM siRNA targeting eIF4GII and control siRNA (Qiagen). Farage cells were transfected with Nucleofector Kit V (Amaxa, Inc., Germany). For transduction of SUDHL4 cells plasmids pCDH-Vector or pCDH-miR520c-3p (System Biosciences, CA) were transfected with PureFection (System Biosciences, CA) in HEK-293TN packaging cells. Afterward, SUDHL4 cells were co-cultivated with the viral particle producing HEK-293TN cells for 48 h, transferred to flasks and subjected to selection with 0.5 µg/ml puromycin. Plasmids pLenti-UTR-Luc-Blank or pLenti-UTR-Luc-eIF4GII(3′UTR) were purchased from Applied Biological Materials Inc. (BC, Canada), miRZip-Vector and miRZip-520c-3p constructs were from System Biosciences. Cloning of the eIF4GII 3′UTR fragments containing seed regions (wild-type or mutants) of miR-520c-3p in plasmid pcDNA3.1/NT-GFP-TOPO were performed using NT-GFP Fusion TOPO TA Expression Kit (Invitrogen). The EIF4G3 lentiviral expression vector was generated through amplification of EIF4G3's coding region (CD) from a human cDNA library and cloning the product into pCDH-EF1-MCS-IRES-Puro (Systems Bioscience).
Primary Cells
Human sample studies have been approved by the University of Maryland Medical School Institutional Review Board and conform to the Declaration of Helsinki. Primary normal B cells were purified from human peripheral blood mononuclear cells (PBMCs) obtained from Sanguine Biosciences, Inc. (Valencia, CA). B cells isolation was performed with using human B Cell Isolation Kit (B-CLL) from Miltenyi Biotec Inc (Auburn, CA) according to manufacturer's protocol. Collected B cells purity was determined by flow cytometry. DLBCL primary cells were obtained from lymph node biopsies, followed by cell density ficoll-gradient separation. Tonsils were provided by the UMGCC Pathology Biorepository and Research Core to serve as normal Germinal Center B-cell (GCB) controls. GCBs were isolated from tonsils as previously described [58] with purity greater than 95%, CD19+, CD38+ and IgD-.
Polyribosome Fractionation
Polysomal fractionations were carried out and microarray analysis was performed as previously described [59]. Briefly, cell lysates were fractionated by centrifugation through 10–50% linear sucrose gradients and divided into 11 fractions. RNA was purified from each fraction for microarray and RT-qPCR analysis.
Microarray Data Analysis
RNA purified from sucrose fractions was labeled using Illumina TotalPrep RNA Amplification Kit (Ambion; Austin, TX). Human HT-12 v1.0 gene expression BeadChips containing 48,000 RefSeq transcripts (Illumina, San Diego, CA), were used for microarray analysis. Raw microarray data were filtered by the detection p-value≤0.02, normalized by Z-score transformation and tested for significant differences in signal intensity. The sample quality was analyzed by scatter plot, principal component analysis (PCA), and gene sample z-scores based hierarchy clustering to exclude possible outliers. ANOVA test was used to eliminate the genes with larger variances within each comparison group. Genes were considered to be significantly changed after calculating Z-ratio, indicating fold difference (Z>1.5 or <−1.5), false discovery rate (fdr), which controls for the expected proportion of false rejected hypothesis (fdr≤0.3) and p<0.05. Genes differentially expressed in studied groups were analyzed for representation of Gene Ontology (GO) terms to identify key biologically functional categories that were significantly changed (p<0.05) in miR-520c-3p overexpressed versus control transfected cells as described before [60]. Ingenuity Pathways Analysis IPA (https://analysis.ingenuity.com/; Ingenuity Systems; Redwood City, CA) was performed to identify the top network functions amongst the genes translationally regulated by miR-520c-3p. Network analysis utilizes a curated knowledge base on known functional interactions and protein functions to algorithmically infer biochemical interactions. Significance of functions and pathways was calculated using the right-tailed Fisher's Test. See www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40489 for complete array results.
RNA Analysis
Total cellular RNA or RNA from polysomal fractions were purified by using Trizol (Invitrogen), reverse transcribed with the iScript cDNA synthesis kit (BioRad, Hercules, CA) and quantified by real-time qPCR analysis using gene-specific primer pairs. qPCR analysis was carried out by using iQ SYBR Green Supermix (Quanta) and BioRad iCycler instrument. RNA from tissue microarrays (TMAs) was isolated using RecoverAll Total Nucleic Acid Isolation Kit for FFPE (Life Technologies) according to manufacturer's protocol. Mature miR-520c-3p and RNU6B were quantified using TaqMan MicroRNA Assays (Applied Biosystems). Each reaction was carried out in triplicate and three independent experiments were run.
Oligonucleotides Used for Real Time RT-qPCR Analysis
For real-time qPCR quantification of selected miR-520c-3p targets following gene-specific primer pairs were used: CCCTTCAAGATGCACCTCAT and GTGGTCCACAGCATTTCCTT for eIF4GII mRNA, GATGCATTGCAGAGGCACTA and CCCTAATGCTGGAAGCACTC for FOXO3 mRNA, TGCAATCCAGCCAAATACAA and TTATCCAGCTTCAGCCAGGT for VIL2 mRNA, GGACCTTGGCCTCAGTGTTA and GAAAGTTGCAGGCACCAAAT for HNRPDL mRNA, TCCCCTTTCCCTGATCTTTT and TGGGATTCTGTGGGAGAAAG for HuR mRNA, TGCTATCATGGCAGAAGGAA and ATATGCCACAGCCCATCATT for MCT-1 mRNA and, CGGAGTCAACGGATTTGGTCGTAT and AGCCTTCTCCATGGTGGTGAAGAC for GAPDH mRNA.
Analysis of Newly Translated Protein
For analysis of de novo translation, 72 h after transfection 106 cells were incubated with 1 mCi L-[35S]methionine and L-[35S]cysteine (Easy Tag EXPRESS; PerkinElmer Life and Analytical Sciences, Waltham, MA) per 6-well plate for 20 minutes. Cell lysates were then resolved by SDS-PAGE and transferred onto PVDF membranes. Radiolabel incorporation was visualized by PhosphorImager (GE Healthcare).
Western Blot Analysis
10–50 µg of whole cell lysates were resolved by SDS-PAGE (Invitrogen, Carlsbad, CA) and transferred onto PVDF membranes. After incubation with monoclonal antibodies recognizing eIF4GII (Sigma), p53, HuR, VIL2, GFP (all from Santa Cruz), GAPDH and β-Actin (both from Abcam, Cambridge, UK), or polyclonal FOXO3 (Cell Signaling), p16, HNRPDL, eIF4E (all from Santa Cruz), blots were incubated with the secondary antibodies (Santa Cruz). Signals were detected by enhanced chemiluminescence (Pierce, Rockford, IL).
Assessment of Cell Cycle, Cell Proliferation, Cell Apoptosis and Senescence
72 h after transfections cells were fixed with 70% ethanol, washed with PBS and digested with RNAse (100 µm/ml) at 37 °C for 1 h. The cells were then stained with 50 µg/ml PI and analyzed for cell cycle with a flow cytometer. Cell proliferation in real-time was monitored by the xCELLigence System, RTCA DP Instrument (Roche Diagnostics GmbH, Germany). The cells were also stained with Trypan Blue (Sigma-Aldrich) and counted using a hemocytometer. The percentage of apoptotic cells was determined by flow cytometry with the Annexin V staining kit (BD Biosciences, CA). Senescence induction in the cells was tested by measurement of β-galactosidase activity with Senescence β-Galactosidase staining kit (Cell Signaling) and Western blot of p16, p53 and HuR senescence markers.
m7GTP Pull-Down Assay
For cap-binding assay, 500 µg total cell lysates were incubated with a 7-Methyl GTP-Sepharose beads (GE Healthcare, Little Chalfont, UK) overnight at 4°C. After washing of the beads, the bound proteins were analyzed by Western blot.
Colony Assay
Farage cells were transfected with Pre-miR-Ctrl, Pre-miR-520c-3p, eIF4GII siRNA or Ctrl siRNA and cultured in agarose/medium. 24 h later cells were plated at 7500 cells/ml in a 1∶3 agarose to DMEM mixture on the top of solidified 1∶1 agarose/DMEM feeder layer and covered with MDEM. Two weeks later colonies (>50 cells) were counted and pictures were taken with using Cell Colony Counter (Microbiology International).
Tissue Microarrays (TMA) Staining and Immunohistologic Analysis
Tissue microarrays, containing 10 reactive lymph nodes, 54 DLBCL and 6 other types of lymphoma were purchased from Biomax and stained with Vectastain Elite ABC kit (Vector Laboratories) as described before [61]. Anti-eIF4GII antibody (Abcam) was used at 1∶1000 dilution, 4°C, overnight. Hematoxylin-eosin (HE) staining was used for morphologic examination. The DLBCL was classified into GCB and ABC subtypes, according to well-accepted immunohistochemistry-based algorithms [61]. The immunohistochemical staining was evaluated according to the following criteria: negative, <25% neoplastic cells staining; low, 26–50% neoplastic cells staining; high, >50% neoplastic cells staining. The images were analyzed with a Nikon eclipse 50i microscope (Nikon Instruments).
In Vivo Tumor Growth in the Xenograft Models
Severe combined immunodeficiency (SCID) Beige mice were maintained in a pathogen–free environment under controlled conditions of light and humidity. Animal procedures were performed in compliance with protocols approved by the Institutional Animal Care and Use Committee of the University of Maryland. Six weeks old male mice were injected with 1.5×106 SUDHL4 cells, either expressing empty vector or overexpressing miR-520c-3p into the left and right dorsal flanks, resuspended in 100 µL PBS and mixed with an equal volume of Matrigel. Tumors were measured and volumes were calculated as previously described [61], [62]. Thirty-one days after injection, tumors were harvested for further analysis. The effect of miR-520c-3p overexpression on tumor growth was evaluated using analysis of variance (ANOVA) with time as a repeated measure factor.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Lazaris-KaratzasA, MontineKS, SonenbergN (1990) Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature 345 : 544–547.
2. ClemensMJ (2004) Targets and mechanisms for the regulation of translation in malignant transformation. Oncogene 23 : 3180–3188.
3. PolunovskyVA, BittermanPB (2006) The cap-dependent translation apparatus integrates and amplifies cancer pathways. RNA Biol 3 : 10–17.
4. GraffJR, KonicekBW, VincentTM, LynchRL, MonteithD, et al. (2007) Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest 117 : 2638–2648.
5. ShayJW, RoninsonIB (2004) Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 23 : 2919–2933.
6. GingrasAC, RaughtB, SonenbergN (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68 : 913–963.
7. CaronS, CharonM, CramerE, SonenbergN, Dusanter-FourtI (2004) Selective modification of eukaryotic initiation factor 4F (eIF4F) at the onset of cell differentiation: recruitment of eIF4GII and long-lasting phosphorylation of eIF4E. Mol Cell Biol 24 : 4920–4928.
8. WangS, RosenwaldIB, HutzlerMJ, PihanGA, SavasL, et al. (1999) Expression of the eukaryotic translation initiation factors 4E and 2alpha in non-Hodgkin's lymphomas. Am J Pathol 155 : 247–255.
9. MericF, HuntKK (2002) Translation initiation in cancer: a novel target for therapy. Mol Cancer Ther 1 : 971–979.
10. RuggeroD, MontanaroL, MaL, XuW, LondeiP, et al. (2004) The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med 10 : 484–486.
11. WendelHG, De StanchinaE, FridmanJS, MalinaA, RayS, et al. (2004) Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 428 : 332–337.
12. MoerkeNJ, AktasH, ChenH, CantelS, ReibarkhMY, et al. (2007) Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 128 : 257–267.
13. BordeleauME, RobertF, GerardB, LindqvistL, ChenSM, et al. (2008) Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest 118 : 2651–2660.
14. GradiA, ImatakaH, SvitkinYV, RomE, RaughtB, et al. (1998) A novel functional human eukaryotic translation initiation factor 4G. Mol Cell Biol 18 : 334–342.
15. ImatakaH, GradiA, SonenbergN (1998) A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. The EMBO Journal 17 : 7480–7489.
16. Fukuchi-ShimogoriT, IshiiI, KashiwagiK, MashibaH, EkimotoH, et al. (1997) Malignant transformation by overproduction of translation initiation factor eIF4G. Cancer Res 57 : 5041–5044.
17. BauerC, DiesingerI, BrassN, SteinhartH, IroH, et al. (2001) Translation initiation factor eIF-4G is immunogenic, overexpressed, and amplified in patients with squamous cell lung carcinoma. Cancer 92 : 822–829.
18. AvdulovS, LiS, MichalekV, BurrichterD, PetersonM, et al. (2004) Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 5 : 553–563.
19. TuL, LiuZ, HeX, HeY, YangH, et al. (2010) Over-expression of eukaryotic translation initiation factor 4 gamma 1 correlates with tumor progression and poor prognosis in nasopharyngeal carcinoma. Mol Cancer 9 : 78.
20. AmbrosV (2001) microRNAs: tiny regulators with great potential. Cell 107 : 823–826.
21. MeltzerPS (2005) Cancer genomics: small RNAs with big impacts. Nature 435 : 745–746.
22. VoliniaS, CalinGA, LiuCG, AmbsS, CimminoA, et al. (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103 : 2257–2261.
23. Nana-SinkamSP, CroceCM (2010) MicroRNA in chronic lymphocytic leukemia: transitioning from laboratory-based investigation to clinical application. Cancer Genet Cytogenet 203 : 127–133.
24. BenhamedM, HerbigU, YeT, DejeanA, BischofO (2012) Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nature Cell Biol 14 : 266–275.
25. BassoK, SumazinP, MorozovP, SchneiderC, MauteRL, et al. (2009) Identification of the human mature B cell miRNome. Immunity 30 : 744–752.
26. MalumbresR, SarosiekKA, CubedoE, RuizJW, JiangX, et al. (2009) Differentiation stage–specific expression of microRNAs in B lymphocytes and diffuse large B-cell lymphomas. Blood 113 : 3754–3764.
27. RaiD, KarantiS, JungI, DahiaPL, AguiarRC (2008) Coordinated expression of microRNA-155 and predicted target genes in diffuse large B-cell lymphoma. Cancer Genet Cytogenet 181 : 8–15.
28. MuP, HanYC, BetelD, YaoE, SquatritoM, et al. (2009) Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev 23 : 2806–2811.
29. CraigVJ, CogliattiSB, ImigJ, RennerC, NeuenschwanderS, et al. (2011) Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood 117 : 6227–6236.
30. CraigVJ, TzankovA, FloriM, SchmidCA, BaderAG (2012) Systemic microRNA-34a delivery induces apoptosis and abrogates growth of diffuse large B-cell lymphoma in vivo. Leukemia 26 : 2421–2424.
31. BabarIA, ChengCJ, BoothCJ, LiangX, WeidhaasJB, et al. (2012) Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci U S A 109: E1695–704.
32. Di LisioL, Sánchez-BeatoM, Gómez-LópezG, RodríguezME, Montes-MorenoS, et al. (2012) MicroRNA signatures in B-cell lymphomas. Blood Cancer J 2: e57.
33. StumpfCR, RuggeroD (2011) The cancerous translation apparatus. Curr Opin Genet Dev 21 : 474–483.
34. HagnerPR, SchneiderA, GartenhausRB (2010) Targeting the translational machinery as a novel treatment strategy for hematologic malignancies. Blood 115 : 2127–2135.
35. HorvilleurE, WilsonLA, WillisAE (2010) Translation deregulation in B-cell lymphomas. Biochem Soc Trans 38 : 1593–1597.
36. WangW, YangX, CristofaloVJ, HolbrookNJ, GorospeM (2001) Loss of HuR is linked to reduced expression of proliferative genes during replicative senescence. Mol Cell Biol 21 : 5889–5898.
37. FabianMR, SonenbergN, FilipowiczW (2010) Regulation of mRNA translation and stability by microRNAs. Annual Review of Biochemistry 79 : 351–379.
38. HoBC, YuSL, ChenJJ, ChangSY, YanBS, et al. (2011) Enterovirus-induced miR-141 contributes to shutoff of host protein translation by targeting the translation initiation factor eIF4E. Cell Host Microbe 9 : 58–69.
39. BellodiCN, KopmarN, RuggeroD (2010) Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J 29 : 1865–1876.
40. SerranoM (2012) Dissecting the role of mTOR complexes in cellular senescence. Cell Cycle 11 : 2231–2232.
41. Ramírez-ValleF, BraunsteinS, ZavadilJ, FormentiSC, SchneiderRJ (2008) eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J Cell Biol 181 : 293–307.
42. YoungAR, NaritaM (2010) Connecting autophagy to senescence in pathophysiology. Curr Opin Cell Biol 22 : 234–240.
43. HuangQ, GumireddyK, SchrierM, le SageC, NagelR, et al. (2008) The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol 10 : 202–210.
44. LiuP, WilsonMJ (2012) miR-520c and miR-373 upregulate MMP9 expression by targeting mTOR and SIRT1, and activate the Ras/Raf/MEK/Erk signaling pathway and NF-κB factor in human fibrosarcoma cells. J Cell Physiol 227 : 867–876.
45. KeklikoglouI, KoernerC, SchmidtC, ZhangJD, HeckmannD, et al. (2012) MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-κB and TGF-β signaling pathways. Oncogene 31 : 4150–4163.
46. ShayJW, RoninsonIB (2004) Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 23 : 2919–2933.
47. EwaldJA, DesotelleJA, WildingG, JarrardDF (2010) Therapy-induced senescence in cancer. J Natl Cancer Inst 102 : 1536–1546.
48. LleonartME, CarneroA, PaciucciR, WangZQ, ShomronN (2011) Cancer, senescence, and aging: translation from basic research to clinics. J Aging Res 692301 doi: 10.4061/2011/692301
49. NardellaC, ClohessyJG, AlimontiA, PandolfiPP (2011) Pro-senescence therapy for cancer treatment. Nat Rev Cancer 11 : 503–551.
50. CalinGA, CroceCM (2006) MicroRNA signatures in human cancers. Nature Reviews Cancer 6 : 857–866.
51. LawrieCH, ChiJ, TaylorS, TramontiD, BallabioE, et al. (2009) Expression of microRNAs in diffuse large B cell lymphoma is associated with immunophenotype, survival and transformation from follicular lymphoma. J Cell Mol Med 13 : 1248–1260.
52. Mazan-MamczarzK, GartenhausRB (2013) Role of microRNA deregulation in the pathogenesis of diffuse large B-cell lymphoma (DLBCL). Leuk Res 37 : 1420–1428.
53. ChiJ, BallabioE, ChenXH, KušecR, TaylorS, et al. (2011) MicroRNA expression in multiple myeloma is associated with genetic subtype, isotype and survival. Biol Direct 6 : 23.
54. IbrahimHA, AmenF, ReidAG, NareshKN (2011) BCL3 rearrangement, amplification and expression in diffuse large B-cell lymphoma. Eur J Haematol 87 : 480–485.
55. MichauxL, DierlammJ, WlodarskaI, BoursV, Van den BergheH (1997) t(14;19)/BCL3 rearrangements in lymphoproliferative disorders: a review of 23 cases. Cancer Genet Cytogenet 94 : 36–43.
56. De BenedettiA, GraffJR (2004) eIF-4E expression and its role in malignancies and metastases. Oncogene 23 : 3189–3199.
57. WendelHG, SilvaRL, MalinaA, MillsJR, ZhuH, et al. (2007) Dissecting eIF4E action in tumorigenesis. Genes Dev 21 : 3232–3237.
58. GreeveJ, PhilipsenA, KrauseK, KlapperW, HeidornK, et al. (2003) Expression of activation-induced cytidine deaminase in human B-cell non-Hodgkin lymphomas. Blood 101 : 3574–8350.
59. Mazan-MamczarzK, HagnerPR, ZhangY, DaiB, LehrmannE, et al. (2011) ATM regulates a DNA damage response posttranscriptional RNA operon in lymphocytes. Blood 117 : 2441–2450.
60. Mazan-MamczarzK, HagnerPR, DaiB, WoodWH, ZhangY, et al. (2008) Identification of transformation-related pathways in a breast epithelial cell model using a ribonomics approach. Cancer Res 68 : 7730–7735.
61. DaiB, ZhaoXF, Mazan-MamczarzK, HagnerP, CorlS, et al. (2011) Functional and molecular interactions between ERK and CHK2 in diffuse large B-cell lymphoma. Nat Commun 2 : 402.
62. DaiB, ZhaoXF, HagnerP, ShapiroP, Mazan-MamczarzK, et al. (2009) Extracellular signal-regulated kinase positively regulates the oncogenic activity of MCT-1 in diffuse large B-cell lymphoma. Cancer Res 69 : 7835–7843.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- GATA6 Is a Crucial Regulator of Shh in the Limb Bud
- Large Inverted Duplications in the Human Genome Form via a Fold-Back Mechanism
- Differential Effects of Collagen Prolyl 3-Hydroxylation on Skeletal Tissues
- Affects Plant Architecture by Regulating Local Auxin Biosynthesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy