
Regulation of Synaptic /Neuroligin Abundance by the /Nrf Stress Response Pathway Protects against Oxidative Stress
The Nrf family of transcription factors mediates adaptive responses to stress and longevity, but the identities of the crucial Nrf targets, and the tissues in which they function in multicellular organisms to promote survival, are not known. Here, we use whole transcriptome RNA sequencing to identify 810 genes whose expression is controlled by the SKN-1/Nrf2 negative regulator WDR-23 in the nervous system of Caenorhabditis elegans. Among the genes identified is the synaptic cell adhesion molecule nlg-1/neuroligin. We find that the synaptic abundance of NLG-1 protein increases following pharmacological treatments that generate oxidative stress or by the genetic activation of skn-1. Increasing nlg-1 dosage correlates with increased survival in response to oxidative stress, whereas genetic inactivation of nlg-1 reduces survival and impairs skn-1-mediated stress resistance. We identify a canonical SKN-1 binding site in the nlg-1 promoter that binds to SKN-1 in vitro and is necessary for SKN-1 and toxin-mediated increases in nlg-1 expression in vivo. Together, our results suggest that SKN-1 activation in the nervous system can confer protection to organisms in response to stress by directly regulating nlg-1/neuroligin expression.
Published in the journal:
. PLoS Genet 10(1): e32767. doi:10.1371/journal.pgen.1004100
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004100
Summary
The Nrf family of transcription factors mediates adaptive responses to stress and longevity, but the identities of the crucial Nrf targets, and the tissues in which they function in multicellular organisms to promote survival, are not known. Here, we use whole transcriptome RNA sequencing to identify 810 genes whose expression is controlled by the SKN-1/Nrf2 negative regulator WDR-23 in the nervous system of Caenorhabditis elegans. Among the genes identified is the synaptic cell adhesion molecule nlg-1/neuroligin. We find that the synaptic abundance of NLG-1 protein increases following pharmacological treatments that generate oxidative stress or by the genetic activation of skn-1. Increasing nlg-1 dosage correlates with increased survival in response to oxidative stress, whereas genetic inactivation of nlg-1 reduces survival and impairs skn-1-mediated stress resistance. We identify a canonical SKN-1 binding site in the nlg-1 promoter that binds to SKN-1 in vitro and is necessary for SKN-1 and toxin-mediated increases in nlg-1 expression in vivo. Together, our results suggest that SKN-1 activation in the nervous system can confer protection to organisms in response to stress by directly regulating nlg-1/neuroligin expression.
Introduction
Oxidative stress is generated in cells when an imbalance occurs between the production of electrophilic reactive species and the endogenous defenses against these harmful molecules [1]. Increased reactive oxygen species (ROS) can cause damage to cellular components such as lipids, DNA, and proteins, and can result in a myriad of detrimental effects, including protein aggregation, changes in cell signaling, or altered cell cycle progression. Given the nervous system's high metabolic demands, high lipid and iron content, and low regenerative ability, it is not surprising that oxidative stress is particularly detrimental to this tissue; indeed, elevated levels of reactive species have been implicated in a range of neurodegenerative diseases including Parkinson's disease, Alzheimer's disease and amyotrophic lateral sclerosis [2]–[4]. In multicellular organisms, investigating pathways that can mitigate the consequences of elevated oxidative stress on a cellular and organismal level is an ongoing area of investigation.
The Nrf (nuclear factor E2-related factor) family of transcription factors controls the primary response to oxidative and xenobiotic stress in mammals [5]–[7]. Studies of Nrf2 have established a critical role for the transcription factor in defending tissues from oxidative damage [7], [8]. During normal conditions, Nrf2 is sequestered in the cytoplasm by the kelch-domain containing protein Keap1; in response to stress, Keap1 releases Nrf2, allowing the transcription factor to translocate into the nucleus and initiate transcription of downstream targets by binding to the antioxidant response element (ARE) in the promoter region of stress response genes [6], [9].
Nrf2 is ubiquitously expressed, and in neurons, increased Nrf2 activity protects against toxicity by hydrogen peroxide, glutamate, and mitochondrial toxins [10], [11]. Microarray studies using neuronal cultures lacking Nrf2 have identified detoxifying, antioxidant and defense genes to be regulated by the transcription factor [10]–[12]. Interestingly, these studies have also found changes in the expression of genes involved in a variety of processes, including cell signaling and calcium homeostasis, as well as neuron-specific genes, but the functional significance of these changes has not been determined.
In C. elegans, the Nrf family homolog SKN-1 [13] confers cellular and organismal protection from a variety of environmental stressors. Although most studies have examined detoxification and stress response in the intestine, a role for SKN-1 in the nervous system is emerging. SKN-1 functions in a pair of neurons to promote longevity [14], and systemic knockdown of skn-1 increases dopaminergic neuron degeneration during methylmercury, aluminum and manganese toxicity [15]–[17]. Furthermore, loss of skn-1 enhances manganese-induced organismal death [18]. In C. elegans, the abundance of SKN-1 is negatively regulated in part by the DDB-1/CUL-4 ubiquitin ligase substrate targeting protein WDR-23, which is proposed to function in an analogous manner as Keap1 to promote degradation of SKN-1 during non-stressed conditions. [19], [20]. Mutants lacking wdr-23 have increased SKN-1 protein levels, express high levels of genes involved in antioxidant and xenobiotic responses, and are resistant to stress [19]–[21].
In this study, we find that SKN-1 is negatively regulated by WDR-23 in cholinergic motor neurons. Using whole transcriptome RNA sequencing (RNAseq) of wdr-23 mutants expressing functional wdr-23a in the nervous system, we identify 810 genes whose expression is likely to be regulated by SKN-1; one of these is the cell adhesion molecule nlg-1/neuroligin. Both nlg-1 expression in neurons and NLG-1 protein abundance at synapses increase in mutants with increased SKN-1 activity as well as in animals exposed to toxins that generate mitochondrial stress. Furthermore, increasing NLG-1 protein abundance enhances survival following toxin treatment, and loss of nlg-1 diminishes SKN-1-mediated toxin resistance. Together, these results support a role for SKN-1 in promoting organismal survival by regulating synaptic neuroligin abundance.
Results
WDR-23 regulates SKN-1 abundance in neurons
The skn-1 locus encodes three isoforms, skn-1a, skn-1b and skn-1c (wormbase.org), which differ in their N termini and utilize unique transcriptional start sites. skn-1c is primarily detected in the intestine; skn-1b, on the other hand, is expressed principally in a pair of sensory neurons and is involved in dietary restriction induced longevity [13], [14]. The expression of the longest isoform, skn-1a, however, has not been examined. To examine the expression of skn-1a, we generated a fluorescent reporter by using a 7.3 kb fragment upstream of the skn-1a start site to drive gfp fused to a nuclear localization signal (nls-gfp). skn-1a is a downstream gene within the bec-1/beclin operon [14], [22], [23], and the 7.3 kb fragment includes the bec-1 coding region and 3.3 kb upstream of bec-1 (Figure 1A). In animals expressing this construct, fluorescence was observed in many tissues, as previously reported [24], [25], as well as in cholinergic neurons of the ventral cord (labeled with the unc-17/VAChT promoter expressing mCherry) and GABAergic neurons (unlabeled ventral cord neurons; Figure 1B).

WDR-23 has two isoforms, WDR-23a and WDR-23b, which are expressed in neurons of the ventral cord [19]–[21]. In cholinergic neurons of the ventral cord, a WDR-23a::GFP fusion protein was found in cell bodies and in axons where it adopted a punctate pattern of fluorescence (Figure 1C), whereas WDR-23b::GFP localized exclusively to nuclei (Figure 1D). In axons, WDR-23a puncta co-localized with a synaptic vesicle marker (Figure 1C), and WDR-23a fluorescence became diffuse in animals lacking unc-104/kinesin (Figure 1E′), which is required for trafficking of organelles along microtubules in neurons [26]. This suggests that WDR-23a associates with presynaptic organelles. WDR-23a also co-localized with a mitochondrial marker at synapses (Figure 1E), and in unc-104 mutants, WDR-23a puncta remained co-localized with displaced mitochondria in axons (Figure 1E′). In mutants defective in drp-1/Drp1, a protein required for normal mitochondrial fission [27], both WDR-23 and mitochondrial markers became displaced in axons (Figure 1E″). Together, these results suggest that WDR-23a may associate with presynaptic organelles, including mitochondria, at synapses. Consistent with this, WDR-23a co-localizes with outer mitochondrial membrane markers when expressed in muscle cells [20].
To test whether WDR-23 regulates the abundance of SKN-1 in the nervous system, as it does in the intestine [19], [21], we examined the abundance of SKN-1a::GFP fusion proteins in motor neurons. We drove SKN-1a::GFP expression using the cholinergic unc-17/VAChT promoter since unc-17 is not transcriptionally regulated by wdr-23 [20], and therefore changes in SKN-1a::GFP should reflect changes in protein abundance and not skn-1 expression. In wild type animals, SKN-1a::GFP fluorescence was detected in 49% (n = 272) of ventral cord cholinergic neurons (Figure 2A), consistent with low levels of SKN-1 reported in the intestine in non-stressed animals [13]. In wdr-23 mutants, SKN-1a::GFP fluorescence was detected in a larger fraction of neurons compared to wild type animals (77%, n = 349). In addition, the average fluorescence intensity of SKN-1a::GFP in neuronal cell bodies of wdr-23 mutants increased compared to wild type controls (Figures 2A and 2B; mean fluorescence±sem wt: 44.7±2.8; wdr-23: 62.6±3.1; p<0.001, Student's t-test). Together, these results suggest that WDR-23 negatively regulates the abundance of SKN-1a in motor neurons.

A subset of neuronal genes is regulated by SKN-1
wdr-23 mutants have a variety of developmental and behavioral defects including reduced locomotion, resistance to stress, small size, and developmental delay. Each of these defects is suppressed by loss of skn-1 [19]–[21], suggesting that the primary function of WDR-23 is to negatively regulate SKN-1. We previously identified 2,285 transcripts that are significantly up-regulated in wdr-23 mutants compared to wild type controls using RNAseq [20]. To identify genes among these that are regulated by WDR-23 specifically in the nervous system, we performed RNAseq of wdr-23 mutants expressing an integrated array of full-length wdr-23a cDNA driven by the pan-neuronal snb-1 promoter (referred to as WDR-23 rescue). Of the 2,285 genes whose expression increased in wdr-23 mutants, transcripts of 810 were significantly reduced in the WDR-23 rescue animals (Figure 3A and Table S1). The average expression levels of the rescued genes in wdr-23 mutants was 20.74 fold above wild type which is similar to the average of 23.99 for all 2,285 genes, indicating that the rescue was not caused by a bias generated by rescuing specifically low expressing genes in wdr-23 mutants.

We used the Database for Annotation, Visualization and Integrated Discovery (DAVID) [28], to assign GO terms for the 810 rescued genes; of the 492 genes with predicted functions, many genes involved in stress, detoxification and metabolism were identified, consistent with the known stress response roles of skn-1 (Figure 3B). The expression of 177 genes has been experimentally examined (wormbase.org), and 93 of these are expressed in the nervous system (Figure 3C and Table S1). A subset of these is expressed exclusively in neurons (Table 1), including eight genes encoding insulins or FMRFamide related peptides, as well as three genes encoding proteins involved in neuropeptide processing—egl-3/proprotein convertase, egl-21/carboxypeptidase, and sbt-2/7B2. We also identified five genes encoding synaptic proteins, including unc-13/Munc13, nlg-1/neuroligin, pde-4/cyclic nucleotide phosphodiesterase, dlk-1/MAPKKK, and cab-1/NPDC-1 [29]–[32]. Together, these results suggest that neuronal WDR-23, possibly through SKN-1, regulates the expression of genes involved in neuroendocrine signaling and synaptic function.

To identify genes that may be direct SKN-1 targets, we examined the promoters of the neuronal genes for the presence of consensus SKN-1 binding sites. SKN-1 is predicted to bind the consensus sequence WWTDTCAT on either strand in the promoters of target genes. Using the web based program Regulatory Sequence Analysis Tools (RSAT), we scanned 1000 bp promoter fragments for each gene for the SKN-1 consensus [33]; the promoter fragments for 70 of the 93 neuronal genes contained at least one potential binding site (Table S1). We then cross-referenced the list of rescued neuronal genes with SKN-1 ChIP-seq datasets taken from L1, L3 and L4 stage animals (ModEncode and [34]) and found that 31 genes contained significant SKN-1 peaks within 2000 bp upstream or 500 bp downstream of the transcription start site (Table S1). These results indicate that a subset of the neuronal genes identified by RNAseq may be direct binding targets of SKN-1.
nlg-1 expression is positively regulated by SKN-1
Among the neuronal genes containing SKN-1 consensus sites in their promoter, the cell adhesion molecule nlg-1/neuroligin emerged as an interesting candidate since it has been implicated in C. elegans stress response and in synaptic function [30], [35], [36]. nlg-1 is the sole neuroligin family ortholog in C. elegans, and NLG-1 is expressed in cholinergic motor neurons where it localizes to presynaptic terminals [35], [37].
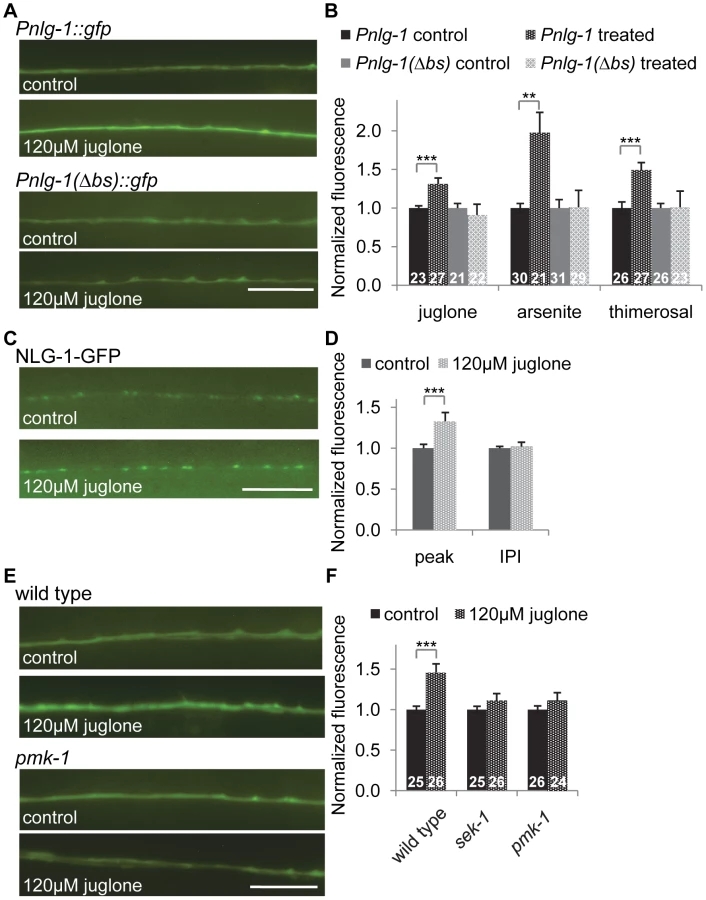
To examine the effects of skn-1 on nlg-1 expression, we constructed reporter strains consisting of a 3.6 kb nlg-1 rescuing promoter fragment [30] driving soluble gfp (Pnlg-1::gfp, vjIs47, and vjIs48; Figure 4A). In wild type animals, we detected Pnlg-1::gfp fluorescence in a few head neurons and in ventral nerve cord neurons, previously reported to be DA and VA class cholinergic motor neurons [30], [35]. nlg-1 reporter fluorescence has been reported at low levels in muscles [30], but we did not detect Pnlg-1::gfp in muscle cells in either transgenic line. In mutants lacking wdr-23, Pnlg-1::gfp fluorescence in motor neurons increased approximately 5 fold (Figure 4B and 4C), in agreement with the 5.6 fold increase in nlg-1 transcript levels detected by RNAseq [20]. Cell-specific expression of either wdr-23a or wdr-23b cDNA in nlg-1 expressing cells (using the nlg-1 promoter) fully rescued the increased Pnlg-1::gfp fluorescence to wild type levels (Figures 4B and 4C), indicating that the regulation of nlg-1 expression by wdr-23 is cell autonomous.

In skn-1 mutants lacking the skn-1a/c isoforms (zu67 mutants), Pnlg-1::gfp expression was similar to wild type. However, skn-1 is required for the increased Pnlg-1::gfp fluorescence caused by loss of wdr-23, since skn-1 mutations reduced the Pnlg-1::gfp reporter fluorescence of wdr-23 mutants to wild type levels (Figures 4D and 4E). Conversely, Pnlg-1::gfp fluorescence increased by ∼35% in mutants in which skn-1 is hyperactive (lax120gf or lax188gf; Figure 4F). lax120gf and lax188gf are thought to prevent SKN-1a/c interaction with mitochondrial docking proteins, resulting in an activated pool of SKN-1a/c [38]. Together, these results show that skn-1 is not necessary for baseline nlg-1 expression in motor neurons, but skn-1 activation positively regulates nlg-1 expression.
The nlg-1 promoter has four SKN-1 binding consensus sites within 500 bp upstream of the transcriptional start site (Figure S1). Promoter alignments between the nematode Caenorhabditis species elegans, briggsae, japonica and remanei revealed that one of these sites located 396 bp upstream of the C. elegans start is completely conserved in all species (Figure S1). This site has the sequence AATGTCAT, which matches the consensus perfectly. The underlined region is predicted to be a largely invariant sequence that directly interacts with SKN-1 [13], [39]. We mutated AATGTCAT at −396 to AACTGCAG in the Pnlg-1::gfp reporter to create a reporter with a deleted binding site (the Pnlg-1(Δbs)::gfp reporter; Figure 4A). Basal motor neuron fluorescence of transgenic animals expressing Pnlg-1(Δbs)::gfp was similar to transgenic animals expressing the Pnlg-1::gfp reporter (Figure 4B). However, Pnlg-1(Δbs)::gfp reporter fluorescence did not increase in either wdr-23 mutants or in skn-1(gf) mutants compared to wild type controls (Figure 4B, 4C and 4F), suggesting that this site is critical for skn-1-mediated increases in nlg-1 expression. To test whether SKN-1 binds to this site, we performed electrophoretic mobility shift assays. In vitro translated SKN-1a bound to labeled probes containing the putative SKN-1 binding site in the nlg-1 promoter, and binding was disrupted by the addition of excess unlabeled probe (Figure 4G). These results indicate that the SKN-1 binding site at −396 in the nlg-1 promoter can be bound by SKN-1 in vitro and is critical for skn-1-induced expression of nlg-1 in vivo.
Synaptic abundance of NLG-1/neuroligin levels is regulated by SKN-1 signaling
To determine whether the transcriptional regulation of nlg-1 by SKN-1 impacts NLG-1 protein levels at synapses, we examined synaptic levels of NLG-1 in animals expressing a fusion protein in which GFP was inserted near the C-terminus of NLG-1 (NLG-1-GFP, vjEx561 and vjIs105). This fusion protein is functional [30] and localizes to presynaptic terminals in motor neurons [35], [37]. NLG-1-GFP driven by the nlg-1 promoter adopted a punctate pattern of fluorescence in the dorsal and ventral cords, where presynaptic terminals of DA and VA class motor neurons are located, respectively (Figure 5A). We examined changes in the synaptic abundance of NLG-1-GFP by measuring the average punctal fluorescence intensity (peak fluorescence) and synapse number (interpunctal interval) [40], [41]. In skn-1(gf) mutants, the punctal fluorescence of NLG-1-GFP significantly increased in both the dorsal and ventral cords, while the interpunctal interval did not change (Figure 5A, 5B and Table S2). These results indicate that SKN-1 positively regulates synaptic NLG-1 protein abundance but does not affect synapse number.

Environmental stress induces NLG-1 expression
We next tested whether toxins that induce oxidative stress could increase neuroligin expression and abundance in motor neurons. We found that exposure to the mitochondrial stressors juglone or sodium arsenite, both of which have been shown to activate skn-1 [42], [43], robustly induced Pnlg-1::gfp fluorescence in motor neurons compared to untreated animals (Figure 6A and 6B). Treatment with an organic mercury (thimerosal) increased nuclear SKN-1::GFP in the intestine (Figure S2) and also increased Pnlg-1::gfp fluorescence (Figure 6B). However, these toxins had no effect on Pnlg-1(Δbs)::gfp fluorescence (Figure 6A and 6B). We found that juglone treatment significantly increased punctal fluorescence of NLG-1-GFP, without changing the interpunctal interval (Figure 6C, 6D and Table S2). These results suggest that activation of skn-1 by oxidative stress increases NLG-1 synaptic abundance by increasing nlg-1 expression in neurons.
Genetic regulation of nlg-1 expression
To further explore how SKN-1 activity is regulated in the nervous system, we tested the impact of altering insulin signaling, mitochondrial respiration or synaptic activity on Pnlg-1::gfp expression. The insulin-like signaling (IIS) pathway regulates SKN-1 in the intestine [44]; activation of DAF-2/insulin-like receptor leads to SKN-1 phosphorylation by SGK-1/SGK, resulting in decreased SKN-1 activity. We examined putative null sgk-1 mutants and found that Pnlg-1::gfp reporter fluorescence increased by ∼25% (Figure S3). Conversely, during conditions of stress, PMK-1/p38 MAPK phosphorylates SKN-1, resulting in nuclear SKN-1 translocation [45]. We found no change of baseline Pnlg-1::gfp fluorescence in mutants lacking either sek-1/MAPKK or pmk-1 (Figure 6E, 6F and S3). However, sek-1 and pmk-1 mutations suppressed the juglone-induced increase in Pnlg-1::gfp reporter fluorescence (Figure 6E and 6F).
C. elegans mutants with impaired mitochondrial respiration, for example the conserved clk-1/COQ7, necessary for the biosynthesis of coenzyme Q, and isp-1/ISP, a subunit of mitochorial complex III, are resistant to toxins that increase oxidative damage [46]. pink-1/PINK1, on the other hand, is predicted to act in conjunction with the E3 ligase Parkin to initiate mitophagy of damaged mitochondria [47], [48]. Pnlg-1::gfp expression increased mildly in mutants lacking clk-1, but not isp-1. Conversely, loss of pink-1 resulted in a significant decrease of Pnlg-1::gfp fluorescence (Figure S3).
Finally, in order to test whether neuronal activity regulates SKN-1 in the nervous system, we examined mutants with increased synaptic activity (dgk-1/diacylglycerol kinase or goa-1/Gαo) and mutants with decreased activity (unc-2/VGCC or unc-18/nSec1), as well as mutants lacking mef-2/MEF and mir-1/microRNA, which are involved in a retrograde synaptic signaling pathways that is dependent on nlg-1 [35]. We detected no change in reporter fluorescence in these mutants (Figure S3). Together, these results indicate that insulin signaling and mitochondrial metabolism contribute to the activation of SKN-1 in neurons, while changes in synaptic transmission do not seem to impact neuronal SKN-1 activity.
NLG-1 promotes organismal survival during SKN-1-dependent response to mitochondrial stress
We next sought to determine whether nlg-1 mediates the protective effects of skn-1 activation in response to environmental toxins. skn-1 mutants are hypersensitive to toxicity induced by arsenite and juglone treatment (Figure 7A and S4; [42], [43]). In addition, we found that skn-1 mutants were sensitive to thimerosal-induced toxicity (Figure S5), in agreement with studies showing SKN-1 protects from metal toxicity [15], [18]. In contrast, animals lacking wdr-23 were resistant to toxicity of both thimerosal and juglone, and resistance was completely blocked by loss of skn-1 (Figures 7A and S5; [43]). Similarly, hyperactive skn-1(gf) mutants were resistant to juglone toxicity (Figure 7B and S6). Interestingly, loss of wdr-23 did not confer protection against sodium arsenite (Figure S4), suggesting specificity in drug resistance in mutants lacking wdr-23.

nlg-1 mutants are hypersensitive to heavy metal toxicity by thimerosal and oxidative stress induced by paraquat [30]. We found that mutants lacking nlg-1 were also more sensitive to juglone toxicity (Figure 7A and S5). In contrast, transgenic animals over-expressing NLG-1-GFP were significantly more resistant to juglone-induced toxicity compared to non-transgenic controls (Figure 7C). To confirm that the juglone resistance caused by NLG-1-GFP transgenes was due to nlg-1 expression, we examined juglone responses of animals expressing Pnlg-1::gfp and found that they were not as resistant to juglone as NLG-1-GFP expressing animals (Figure S7). Finally, nlg-1 mutations dramatically reduced the ability of activated skn-1 to protect animals from the toxic effects of juglone: skn-1(gf);nlg-1 double mutants were significantly less resistant to juglone than skn-1(gf) mutants alone (Figure 7B). Together, these data suggest that the dosage of nlg-1 is a critical determinant of survival in response to stress, and that nlg-1 contributes to skn-1-dependent survival.
Discussion
In this study, we find that WDR-23 in neurons, possibly acting through SKN-1, regulates the expression of approximately 800 genes in multiple tissues, including the nervous system. One of these genes, the synaptic cell adhesion molecule nlg-1/neuroligin, contains a consensus SKN-1 binding site in its promoter necessary for SKN-1 binding in vitro and for nlg-1 expression following SKN-1 activation. Increasing SKN-1 activity elevates the abundance of NLG-1 protein at synaptic terminals, and changes in synaptic NLG-1 levels correlate with altered survival of animals in the presence of mitochondrial toxins. We propose a model in which, in response to mitochondrial stress, SKN-1 induces transcription of nlg-1, which in turn enhances organismal survival (Figure 7D).
SKN-1 transcriptional targets in neurons
Previous studies have demonstrated SKN-1/Nrf2 dependent transcriptional programs are initiated in response to oxidative and xenobiotic stress, and these programs are critical for organismal survival and longevity [42], [43], [49]. Among the genes regulated by SKN-1, few have been shown to mediate the protective effects of SKN-1, and fewer still have been shown to be direct binding targets. Furthermore, attempts to identify a comprehensive set of genes regulated by SKN-1/Nrf2 required to protect organisms from stress have been difficult due to the lack of tissue-level resolution.
Here, we have used comparative whole transcriptome RNA sequencing to identify an inclusive set of genes that are likely to be regulated by SKN-1 in the nervous system. For this analysis, we examined wdr-23 mutants expressing a rescuing wdr-23a transgene driven by the pan-neuronal snb-1 promoter. Genetic studies indicate that several distinct phenotypes displayed by wdr-23 mutants are completely suppressed by skn-1, indicating that SKN-1 is selectively activated in wdr-23 mutants, and a yeast-two hybrid screen identifies the only binding target of WDR-23 to be SKN-1 [19], indicating that SKN-1 is selectively activated in wdr-23 mutants. Interestingly, wdr-23 mutants appear to activate SKN-1 to a greater extent than skn-1(gf) mutations or toxin treatment. For example, loss of wdr-23 results in greater survival in response to juglone treatment than skn-1(gf) mutations. In addition, wdr-23 mutations increase nlg-1 expression in the ventral cord neurons fivefold, whereas skn-1(gf) or toxin treatment increased it by approximately 30%. Thus, wdr-23 mutants may provide increased sensitivity when used for transcriptional profiling, maximizing our ability to identify SKN-1 targets expressed in low abundance or in a small subset of cells. It is, however, possible that WDR-23 has functions beyond SKN-1 regulation, in which case some of the genes identified by this approach may not be targets of SKN-1. While the snb-1 promoter fragment drives expression of GFP strongly in the nervous system, the possibility that this promoter may be leaky raises the prospect that some of the genes identified here may be regulated by WDR-23 in other tissues in addition to the nervous system.
The 810 genes identified here most likely represent either direct SKN-1 targets or indirect targets of SKN-1 that are secondarily activated in neurons or other tissues. SKN-1 ChIP-seq of larval stage animals identified a list of approximately 3000 genomic peaks bound by SKN-1 in vivo [34]; we cross-referenced our gene list of rescued neuronal genes with SKN-1 ChIP-seq datasets taken from L1, L3 and L4 stage animals [34] and found that a subset of the genes we identified contain a significant SKN-1 peak near to their transcriptional start sites (Table S1), but many genes do not. Differences between these datasets may be a byproduct of using non-stressed animals for the ChIP-seq experiments, reflecting basal, but not stress-induced, promoter occupancy by SKN-1.
Among the genes we identified were several neuropeptides and insulins, as well as peptide processing enzymes reported to be expressed in neurons. We speculate that stress-induced peptide processing and release may be part of a humoral response to promote organismal survival. Identification of the precise peptidergic signaling pathways will help to elucidate the mechanisms by which SKN-1 confers survival. We also identified a handful of known synaptic genes, including two cell adhesion molecules, nlg-1 and ncam-1, indicating SKN-1 might play a role in maintaining the stability of neural networks. A theory has emerged suggesting axonal retraction might precede neuronal apoptosis in neurodegeneration, called dying back degeneration [50], and synaptic breakdown may occur prior to axonal retraction. The identification of cell adhesion molecules in this study suggests that prior to synaptic breakdown, neurons might initiate transcriptional programs to protect the integrity of the synapse. In support of this, recent evidence suggests that increased expression of NCAM in SH-SY5Y cultures prevents oxidative stress-induced apoptosis, and over-expression of a truncated NCAM molecule protects neuronal tissue in lesioned rats [51], [52].
SKN-1 as a mitochondrial stress sensor
Activation of SKN-1/Nrf2 can protect against cell toxicity induced by oxidative stress [53]–[55]. In primary neuronal cultures, for example, activation of Nrf2 prevents cell death in response to rotenone and MPP, potent inhibitors of mitochondrial respiration [11]. Loss of OPA1, a key regulator of the morphology of mitochondria, results in increased Nrf2 activation [56]. These studies and others collectively suggest a potential role for SKN-1/Nrf2 as a sensor for increased mitochondrial dysfunction in the nervous system. We found that genes involved in drug detoxification, including the glutathione precursors gst-4, gst-10, gcs-1, and gst-1 were reduced by neuronal expression of wdr-23a. Interestingly, gst-1 has also been shown to contribute to dopaminergic neuron survival after manganese treatment [16]. Given their confirmed expression in neurons, is possible that these genes have a role in directly enhancing neuronal protection.
SKN-1 associates with purified mitochondrial fractions, and the skn-1(gf) alleles are proposed to reduce mitochondrial association, suggesting that mitochondria may act as a sink for SKN-1 [38]. Our data is consistent with the idea that WDR-23a associates with presynaptic organelles, including mitochondria. First, WDR-23a localizes to presynaptic terminals, where mitochondria are abundant. Second, WDR-23a remains associated with mitochondria in mutants in which mitochondria are displaced. Third, WDR-23a localizes to the outer membrane of mitochondria in muscle cells [20]. We speculate that WDR-23 may be a mitochondrial stress sensor that regulates SKN-1 abundance.
Neuroligin as a neuronal target of SKN-1
Our data supports the idea that SKN-1 activation leads to increased NLG-1 abundance at synapses. We found that nlg-1 confers some, but not all, of the protective effects of activation of SKN-1, as nlg-1 mutations reduced, but did not eliminate, the resistance of skn-1(gf) mutants to juglone. Additional SKN-1 targets either in neurons or in other tissues are likely to contribute to organismal survival in response to juglone treatment. Interestingly, loss of nlg-1 did not suppress wdr-23 mutants resistance to thimerosal or juglone (Figure S5 and data not shown). This may be due to higher SKN-1 activity in wdr-23 mutants compared to skn-1(gf) mutants, which may compensate for the lack of nlg-1. Because nlg-1 mutants themselves are hypersensitive to stress, it is possible that nlg-1 and skn-1 function in parallel pathways to promote resistance; however, our data do not support this idea, but rather support the notion that nlg-1 is a direct transcriptional target of SKN-1. Basal nlg-1 transcription is not likely to be under skn-1 regulation, since nlg-1 reporter expression remains unchanged in skn-1 mutants. Consistent with this, SKN-1 does not occupy the SKN-1 binding site we identified at position −396 in the nlg-1 promoter in unstressed larval animals by ChIP-seq analysis.
How might increased expression of a synaptic cell adhesion molecule promote organismal survival in response to stress? Recent work has established the presence of “mitokines” in neurons—a signal produced in the neurons in response to dysfunctional mitochondrial electron transport [57]; release of these mitokines results in increased organismal survival. It is possible that nlg-1 may be required in neurons for proper release of mitokines. Furthermore, the neuropeptides identified in this study are candidates for being signals released in response to stress to promote resistance. nlg-1 expression has been detected in head neurons, motor neurons, and muscle cells [30]; thus, it is possible that nlg-1 functions in any of these tissues to convey protection.
In mammals, neuroligin is a post-synaptic cell adhesion molecule that binds to the presynaptic protein neurexin; this junction is necessary for maintaining mature synaptic connections and normal synaptic transmission [58], [59]. In humans, rare mutations in neuroligin are associated with autism and other cognitive disorders. Some of these mutations reduce neuroligin delivery to the cell surface, interfering with synapse development and synaptic transmission [60]–[62]. Interestingly, it has been suggested that oxidative stress and mitochondrial dysfunction may play a role in the pathogenesis of autism, as certain biomarkers for oxidative stress are elevated in autistic patients [63]–[65]. It is possible that individuals with these mutations are unable to increase synaptic neuroligin levels in response to stress, and it will be interesting to identify the cellular and molecular mechanisms underlying neuroligin-dependent survival in response to stress.
Materials and Methods
C. elegans strains
Strains were cultured at 20° using standard methods. All experiments were performed on young adult hermaphrodites unless otherwise indicated. The following strains were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR): sek-1(km4), pmk-1(km25), sgk-1(ok538), skn-1(zu67), nlg-1(ok259), clk-1(e2519), mef-2(gv1), mir-1(gk276), isp-1(gm150), and pink-1(ok3538). Strain wdr-23(tm1817) was provided by the National BioResource Project (Japan). The wild type reference strain was N2 Bristol. The following strains were also used: drp-1(tm1108), dgk-1(nu62), goa-1(sa734), unc-2(lj1), unc-18(md299), glo-1(zu391), unc-104(e1265), skn-1(lax120gf), skn-1(lax188gf), nuIs152[Pttx-3::RFP, Punc-129::GFP::SNB-1]II, nuIs321[Pmyo-2::GFP, Punc-17::mCherry], nuIs225[Pmyo-2::GFP, Psnb-1::WDR-23a], idIs7[rol-6(su1006), Pskn-1::skn-1b/c::GFP], yuIs25[Pmyo-2::GFP, Punc-129::mito-GFP]V, and vjIs26[Pmyo-2::GFP, Punc-129::WDR-23a::GFP]III. Mutant strains were outcrossed a minimum of 4 times; all integrants were outcrossed at least 8 times.
Molecular biology
C. elegans cDNA was used to clone all genes into pPD49.26 using standard molecular biology techniques, unless otherwise noted. Promoter elements were amplified from mixed stage genomic DNA. The following plasmids were generated: pTS147[Punc-129::invom::rfp], pTS31[Pbec-1::nls-gfp], pDS284[Pnlg-1::gfp], pDS286[Pnlg-1(ΔBS)::gfp], pKG8[Pnlg-1::nlg-1-gfp], pTS79[Punc-17::skn-1a::gfp], pDS139[Punc-129::snb-1::mCherry], pDS237[Psnb-1::wdr-23a::gfp], pTS85[Psnb-1::wdr-23b::gfp], pDS334[Pnlg-1::wdr-23a], pDS335[Pnlg-1::wdr-23b], and pTS199[T7::skn-1a].
Oligo sequences:
Pbec-1
oTS44: ccccccGGATCCcgacaattatacatgttcccc
oTS45: ccccccGCTAGCcgactgactggattatgatagatcc
Pnlg-1
oDS694: ccccccGCATGCtaagcccccgtacgctaacacc
oXL13: ccccccGGATCCgcctgttcacttccaaattcgc
Pnlg-1(Δbs)
oDS692: cctgttgccccccaaatgCTGCAGttacctcttttcctcccttctacc
oDS693 ggtagaagggaggaaaagaggtaaCTGCAGcatttggggggcaacagg
Transgenic lines
Transgenic strains were generated by injecting N2 with expression constructs (2.5–90 ng/µL) and the co-injection marker KP#708 (Pttx-3::rfp, 40 ng/µL) or KP#1106 (Pmyo-2::gfp, 10 ng/µL). Microinjection was performed using standard techniques as previously described [66]. At least three lines for each transgene were examined for expression, and representative lines were quantified. The following strains were made: vjEx7[Punc-129::wdr-23b::gfp] vjEx663[Punc-129::invom::rfp], vjEx254[Pbec-1::nls-gfp], vjEx391[Pnlg-1(Δbs)::gfp], vjEx756[Pnlg-1(Δbs)::gfp], vjEx561[Pnlg-1::nlg-1-gfp], vjEx339[Punc-129::snb-1::mCherry], vjEx423[Psnb-1::wdr-23a::gfp], vjEx426[Psnb-1::wdr-23b::gfp], vjEx447[Pnlg-1::wdr-23a], vjEx436[Pnlg-1::wdr-23b], vjIs45[Punc-17::skn-1a::gfp]II, vjIs47[Pnlg-1::gfp]IV, vjIs48[Pnlg-1::gfp]I, vjIs105[Pnlg-1::nlg-1-gfp]III.
Microscopy and analysis
To image animals, adult worms were paralyzed using 2,3-butanedione monoxime (BDM, 30 µg/µL; Sigma) and mounted on 2% agarose pads for imaging. Images were captured with a Nikon eclipse 90i microscope equipped with a Nikon PlanApo 60× or 100× objective (NA = 1.4) and a PhotometricsCoolsnap ES2 camera. For fluorescence imaging of synapses, images were captured either from the ventral or dorsal cord near the posterior gonadal bend of the worm, as indicated. Images of animals expressing nlg-1 reporters were captured at the ventral cord near the posterior gonadal bend of the worm. We found that skn-1 mutants expressing fluorescent integrants arrest at larval stages when the integrant is homozygous, making quantification of fluorescence markers challenging; however, skn-1 mutants expressing heterozygous integrants develop fully into adults.
Metamorph 7.0 software (Universal Imaging/Molecular Devices) was used to capture serial image stacks, and the maximum intensity projection was used for analysis of the dorsal and ventral cords. Line scans of the maximum intensity projection image were also recorded using Metamorph. The fluorescence intensity values were then quantified using Puncta 6.0 software written with Igor Pro (Wavemetrics), as previously described [67]. For all experiments, fluorescence values were normalized to the values of 0.5 µm FluoSphere beads (Invitrogen) captured during each imaging session. This was performed to provide a standard for comparing absolute fluorescence levels between animals from different sessions.
To quantify changes in neuronal SKN-1a::GFP, an anterior and posterior image was taken for each animal in both RFP and GFP channels. Cell bodies were identified by the presence of soluble mCherry. Average cell body fluorescence was calculated by outlining the entire cell body and taking the average intensity. Background values were determined by finding the average fluorescence of the area immediately adjacent to the cell body and were subtracted from each cells' average fluorescence. Cells were categorized as low, medium, or high expressing cells using arbitrary cut off levels after background subtraction—low expressing cells were those cells below the level of detection (less than 30 units different than background fluorescence), medium expressing cells were between 30–70 arbitrary units, and high expressing cells had a total fluorescence greater than 70 units.
Toxicity assays
Stock solutions of 50 mM juglone (Calbiochem) and 20 mM thimerosal (Enzo) were freshly dissolved in DMSO or water, respectively, prior to addition to molten NGM. Sodium arsenite (Ricca) was maintained in aqueous solution at 0.5% w/v and stored at room temperature. Plates were freshly made approximately 24 hours before use and seeded with concentrated OP50 the night before being used.
To assess longevity, age matched young adult animals were transferred to 100 mm NGM plates containing either drug or control and assayed over time. Animals which escaped the plates were excluded from the analysis. At least four replicates of n = 40 animals per genotype per stress were tested; final samples sizes reported in Table S3. Animals were stored at 20° except during time points. Animals were scored as dead if they did not respond to repeated light prodding. Percentages alive for each genotype were determined by averaging the fraction alive per plate at each time point and plotting graphically.
For fluorescence toxicity studies using the nlg-1 reporters, L4 stage animals were exposed to NGM plates supplemented with drug or control overnight for 14 hours; animals were allowed 2–4 hours recovery time before imaging. Concentrations were chosen that did not result in animal death after 14 hours. For juglone imaging, control plates were supplemented with an equal volume of DMSO, as the juglone was diluted to 50 mM in DMSO prior to addition to the test plates.
Statistical analysis
A Student's t test was used to determine significance when comparing fluorescence of nlg-1 reporters in different conditions, unless otherwise specified. Log rank tests with a Bonferroni correction were calculated by JMP Pro version 10.0 software and were used to determine significant differences between genotypes for toxicity studies; differences between genotypes are reported in Table S3.
RNA sequencing
Total RNA was isolated from approximately 10,000 mixed stage animals for wild type, wdr-23(tm1817) mutants and wdr-23;nuIs225 using Stat60 (Tel-test B, Texas). Transcriptome libraries were prepared using TruSeq RNA sample preparation kit (Illumina) according to manufacturer's TruSeq protocol as previously described [20]. Libraries were amplified by PCR and quality and quantity of libraries were evaluated on BioAnalyzer 2100 (Agilent). Sequencing was performed on HiSeq2000 (Illumina). Sequencing reads were aligned to the C. elegansgenome (release WS210) using TopHat [68]. Gene models were downloaded from ENSEMBL and quantified using Cufflinks. Differentially expressed genes at false discovery rate (FDR) of 0.05 were identified using the Cuffdiff module of the Cufflinks package.
Electrophoretic mobility shift assays
Full-length SKN-1a cDNA was cloned into a pBS backbone driven by the T7 promoter and expressed using TnT T7 Quick Coupled Transcription/Translation System for DNA (Promega). EMSA was performed using the LightShift Chemiluminescence EMSA Kit (Pierce) according to manufacturer's protocols. Complementary 5′ biotinylated oligonucleotides containing the pnlg-1 SKN-1 binding site were self-annealed and incubated with 1 µl of SKN-1 lysate, 1 µg Poly (dIdC) and 5 mM MgCl2 for 20 minutes at room temperature. Samples were separated on a 5% native polyacrylamide gel and blotted on Biodyne B nylon membranes (Pierce).
Pnlg-1 probes:
oTS300 : 5′ biotin-gttgccccccaaatgATGACATTacctcttttcctccc 3′
oTS310 : 5′ biotin-gggaggaaaagaggtAATGTCATcatttggIgggcaac 3′
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FinkelT, HolbrookNJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408 : 239–247.
2. HalliwellB (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97 : 1634–1658.
3. Abou-SleimanPM, MuqitMM, WoodNW (2006) Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci 7 : 207–219.
4. ZanaM, JankaZ, KalmanJ (2007) Oxidative stress: a bridge between Down's syndrome and Alzheimer's disease. Neurobiol Aging 28 : 648–676.
5. ItohK, ChibaT, TakahashiS, IshiiT, IgarashiK, et al. (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236 : 313–322.
6. KobayashiM, YamamotoM (2006) Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul 46 : 113–140.
7. SykiotisGP, BohmannD (2010) Stress-activated cap'n'collar transcription factors in aging and human disease. Sci Signal 3: re3.
8. LeeJM, LiJ, JohnsonDA, SteinTD, KraftAD, et al. (2005) Nrf2, a multi-organ protector? FASEB J 19 : 1061–1066.
9. ItohK, WakabayashiN, KatohY, IshiiT, IgarashiK, et al. (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13 : 76–86.
10. KraftAD, JohnsonDA, JohnsonJA (2004) Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci 24 : 1101–1112.
11. LeeJM, ShihAY, MurphyTH, JohnsonJA (2003) NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem 278 : 37948–37956.
12. LeeJM, CalkinsMJ, ChanK, KanYW, JohnsonJA (2003) Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem 278 : 12029–12038.
13. AnJH, BlackwellTK (2003) SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 17 : 1882–1893.
14. BishopNA, GuarenteL (2007) Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 447 : 545–549.
15. VanduynN, SettivariR, WongG, NassR (2010) SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol Sci 118 : 613–624.
16. SettivariR, VanduynN, LevoraJ, NassR (2013) The Nrf2/SKN-1-dependent glutathione S-transferase pi homologue GST-1 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of manganism. Neurotoxicology 38C: 51–60.
17. VanDuynN, SettivariR, LeVoraJ, ZhouS, UnrineJ, et al. (2013) The metal transporter SMF-3/DMT-1 mediates aluminum-induced dopamine neuron degeneration. J Neurochem 124 : 147–157.
18. BenedettoA, AuC, AvilaDS, MilatovicD, AschnerM (2010) Extracellular dopamine potentiates mn-induced oxidative stress, lifespan reduction, and dopaminergic neurodegeneration in a BLI-3-dependent manner in Caenorhabditis elegans. PLoS Genet 6: e1001084.
19. ChoeKP, PrzybyszAJ, StrangeK (2009) The WD40 repeat protein WDR-23 functions with the CUL4/DDB1 ubiquitin ligase to regulate nuclear abundance and activity of SKN-1 in Caenorhabditis elegans. Mol Cell Biol 29 : 2704–2715.
20. StaabTA, GriffenTC, CorcoranC, EvgrafovO, KnowlesJA, et al. (2013) The Conserved SKN-1/Nrf2 Stress Response Pathway Regulates Synaptic Function in Caenorhabditis elegans. PLoS Genet 9: e1003354.
21. HasegawaK, MiwaJ (2010) Genetic and cellular characterization of Caenorhabditis elegans mutants abnormal in the regulation of many phase II enzymes. PloS one 5: e11194.
22. wormbase.org Wormbase web site. Release ws236 ed.
23. BlumenthalT (2012) Trans-splicing and operons in C. elegans. WormBook 1–11.
24. MelendezA, TalloczyZ, SeamanM, EskelinenEL, HallDH, et al. (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301 : 1387–1391.
25. Takacs-VellaiK, VellaiT, PuotiA, PassannanteM, WickyC, et al. (2005) Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol 15 : 1513–1517.
26. HallDH, HedgecockEM (1991) Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell 65 : 837–847.
27. LabrousseAM, ZappaterraMD, RubeDA, van der BliekAM (1999) C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4 : 815–826.
28. DennisGJr, ShermanBT, HosackDA, YangJ, GaoW, et al. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: P3.
29. IwasakiK, ToyonagaR (2000) The Rab3 GDP/GTP exchange factor homolog AEX-3 has a dual function in synaptic transmission. EMBO J 19 : 4806–4816.
30. HunterJW, MullenGP, McManusJR, HeatherlyJM, DukeA, et al. (2010) Neuroligin-deficient mutants of C. elegans have sensory processing deficits and are hypersensitive to oxidative stress and mercury toxicity. Dis Model Mech 3 : 366–376.
31. KohnRE, DuerrJS, McManusJR, DukeA, RakowTL, et al. (2000) Expression of multiple UNC-13 proteins in the Caenorhabditis elegans nervous system. Mol Biol Cell 11 : 3441–3452.
32. CharlieNK, ThomureAM, SchadeMA, MillerKG (2006) The Dunce cAMP phosphodiesterase PDE-4 negatively regulates G alpha(s)-dependent and G alpha(s)-independent cAMP pools in the Caenorhabditis elegans synaptic signaling network. Genetics 173 : 111–130.
33. Thomas-ChollierM, SandO, TuratsinzeJV, JankyR, DefranceM, et al. (2008) RSAT: regulatory sequence analysis tools. Nucleic Acids Res 36: W119–127.
34. NiuW, LuZJ, ZhongM, SarovM, MurrayJI, et al. (2011) Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res 21 : 245–254.
35. HuZ, HomS, KudzeT, TongXJ, ChoiS, et al. (2012) Neurexin and neuroligin mediate retrograde synaptic inhibition in C. elegans. Science 337 : 980–984.
36. CalahorroF, AlejandreE, Ruiz-RubioM (2009) Osmotic avoidance in Caenorhabditis elegans: synaptic function of two genes, orthologues of human NRXN1 and NLGN1, as candidates for autism. J Vis Exp (34) 1616.
37. FeinbergEH, VanhovenMK, BendeskyA, WangG, FetterRD, et al. (2008) GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 57 : 353–363.
38. PaekJ, LoJY, NarasimhanSD, NguyenTN, Glover-CutterK, et al. (2012) Mitochondrial SKN-1/Nrf Mediates a Conserved Starvation Response. Cell Metab 16 : 526–537.
39. BlackwellTK, BowermanB, PriessJR, WeintraubH (1994) Formation of a monomeric DNA binding domain by Skn-1 bZIP and homeodomain elements. Science 266 : 621–628.
40. SieburthD, MadisonJM, KaplanJM (2007) PKC-1 regulates secretion of neuropeptides. Nat Neurosci 10 : 49–57.
41. DittmanJS, KaplanJM (2006) Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc Natl Acad Sci U S A 103 : 11399–11404.
42. OliveiraRP, Porter AbateJ, DilksK, LandisJ, AshrafJ, et al. (2009) Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 8 : 524–541.
43. PrzybyszAJ, ChoeKP, RobertsLJ, StrangeK (2009) Increased age reduces DAF-16 and SKN-1 signaling and the hormetic response of Caenorhabditis elegans to the xenobiotic juglone. Mech Ageing Dev 130 : 357–369.
44. TulletJM, HertweckM, AnJH, BakerJ, HwangJY, et al. (2008) Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132 : 1025–1038.
45. InoueH, HisamotoN, AnJH, OliveiraRP, NishidaE, et al. (2005) The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev 19 : 2278–2283.
46. ZubovychIO, StraudS, RothMG (2010) Mitochondrial dysfunction confers resistance to multiple drugs in Caenorhabditis elegans. Mol Biol Cell 21 : 956–969.
47. NarendraD, TanakaA, SuenDF, YouleRJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183 : 795–803.
48. YouleRJ, van der BliekAM (2012) Mitochondrial fission, fusion, and stress. Science 337 : 1062–1065.
49. ParkSK, TedescoPM, JohnsonTE (2009) Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging cell 8 : 258–269.
50. LuoL, O'LearyDD (2005) Axon retraction and degeneration in development and disease. Annual review of neuroscience 28 : 127–156.
51. FengZ, LiL, NgPY, PorterAG (2002) Neuronal differentiation and protection from nitric oxide-induced apoptosis require c-Jun-dependent expression of NCAM140. Mol Cell Biol 22 : 5357–5366.
52. KlementievB, NovikovaT, KorshunovaI, BerezinV, BockE (2008) The NCAM-derived P2 peptide facilitates recovery of cognitive and motor function and ameliorates neuropathology following traumatic brain injury. Eur J Neurosci 27 : 2885–2896.
53. ScapagniniG, VastoS, AbrahamNG, CarusoC, ZellaD, et al. (2011) Modulation of Nrf2/ARE pathway by food polyphenols: a nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Molecular neurobiology 44 : 192–201.
54. ChakrabortyS, AschnerM (2012) Altered manganese homeostasis: implications for BLI-3-dependent dopaminergic neurodegeneration and SKN-1 protection in C. elegans. Journal of trace elements in medicine and biology : organ of the Society for Minerals and Trace Elements 26 : 183–187.
55. JoshiG, JohnsonJA (2012) The Nrf2-ARE pathway: a valuable therapeutic target for the treatment of neurodegenerative diseases. Recent patents on CNS drug discovery 7 : 218–229.
56. BertholetAM, MilletAM, GuillerminO, DaloyauM, DavezacN, et al. (2013) OPA1 loss of function affects in vitro neuronal maturation. Brain : a journal of neurology 136 : 1518–1533.
57. DurieuxJ, WolffS, DillinA (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144 : 79–91.
58. SudhofTC (2008) Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455 : 903–911.
59. BottosA, RissoneA, BussolinoF, AreseM (2011) Neurexins and neuroligins: synapses look out of the nervous system. Cell Mol Life Sci 68 : 2655–2666.
60. ZhangC, MilunskyJM, NewtonS, KoJ, ZhaoG, et al. (2009) A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J Neurosci 29 : 10843–10854.
61. ComolettiD, De JacoA, JenningsLL, FlynnRE, GaiettaG, et al. (2004) The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J Neurosci 24 : 4889–4893.
62. ChihB, EngelmanH, ScheiffeleP (2005) Control of excitatory and inhibitory synapse formation by neuroligins. Science 307 : 1324–1328.
63. GhezzoA, ViscontiP, AbruzzoPM, BolottaA, FerreriC, et al. (2013) Oxidative Stress and Erythrocyte Membrane Alterations in Children with Autism: Correlation with Clinical Features. PLoS One 8: e66418.
64. GorrindoP, LaneCJ, LeeEB, McLaughlinB, LevittP (2013) Enrichment of elevated plasma F2t-isoprostane levels in individuals with autism who are stratified by presence of gastrointestinal dysfunction. PLoS One 8: e68444.
65. RossignolDA, FryeRE (2012) Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry 17 : 290–314.
66. MelloCC, KramerJM, StinchcombD, AmbrosV (1991) Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10 : 3959–3970.
67. Ch'ngQ, SieburthD, KaplanJM (2008) Profiling synaptic proteins identifies regulators of insulin secretion and lifespan. PLoS Genet 4: e1000283.
68. TrapnellC, WilliamsBA, PerteaG, MortazaviA, KwanG, et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28 : 511–515.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- GATA6 Is a Crucial Regulator of Shh in the Limb Bud
- Large Inverted Duplications in the Human Genome Form via a Fold-Back Mechanism
- Differential Effects of Collagen Prolyl 3-Hydroxylation on Skeletal Tissues
- Affects Plant Architecture by Regulating Local Auxin Biosynthesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy